DOI:
10.1039/D6RA01721E
(Paper)
RSC Adv., 2026,
16, 21953-21966
A sustainable one-pot multi-component approach toward the synthesis of symmetric and unsymmetrical spiro heterocycles catalyzed by the Brønsted acidic ionic liquid [CMMIM][BF4]
Received
27th February 2026
, Accepted 7th April 2026
First published on 28th April 2026
Abstract
An efficient and sustainable one-pot protocol is developed for the synthesis of a broad range of isatin- and acenaphthoquinone-based spiroxanthene and spirochromenecarboxylate derivatives in aqueous ethanol. This approach involves the condensation of isatin- and acenaphthoquinone-based 1, 2 diketones with cyclic and acyclic active-methylene compounds, catalyzed by 5–6 mol% of the Brønsted acidic ionic liquid [CMMIM][BF4]. The versatility of the protocol is demonstrated by the successful synthesis of 20 isatin- and acenaphthoquinone-derived spiroxanthene and spirochromenecarboxylate derivatives. This sustainable methodology offers a short reaction time and good to excellent yield (up to 97%), with easy product isolation, avoiding column chromatographic purification and providing valuable green chemistry metrics for symmetric 8a (E-factor: 0.15 and RME: 86.92%) and unsymmetrical 9a (E-factor: 0.08 and RME: 92.5%) derivatives. This study highlights the potential activity of Brønsted acidic ionic liquids [CMMIM][BF4] as green and reusable catalysts for the atom-economical, environmentally benign construction of bioactive spiro-heterocycles.
Introduction
Heterocyclic compounds play a vital role in the design of medicines and natural products.1 Their chemical diversity and structural flexibility support the development of molecules with biological activity.2,3 Over the past two decades, nitrogen-containing heterocycles have become increasingly important in various organic synthesis approaches.4 Isatin is acknowledged as a crucial privileged skeleton for bioactive molecule development.5 It was first synthesized in 1840 by Erdmann and Laurent through the oxidation of indigo dye.6 Isatin consists of a fused benzene ring with a five-membered nitrogen-containing ring, making it a versatile building block in organic chemistry. It can be easily modified at the N-1, C-3, C-5, and C-7 positions to create a new skeleton with tailored activities.7 It has unique reactivity due to the presence of a carbonyl group at the C-3 position.8 The development of spiro-oxindole derivatives through an isatin-mediated multicomponent approach represents a significant breakthrough in rapidly generating drug-like compound libraries with extensive scaffold diversity.9 Isatin derivatives are promising molecules in medicinal chemistry owing to their diverse biological activities, including anticancer, antiviral, antioxidant, antimicrobial, and central nervous system activity.10–12 Xanthenes exhibit similar biological activities. Xanthene can also be used as a fluorescent dye in photodynamic therapy, chemoanalytics, and bioanalytics.13–16 The potentially bioactive spirooxiindoles and xanthenes are shown in Fig. 1 and 2.
 |
| | Fig. 1 Potentially bioactive spirooxindole compounds. | |
 |
| | Fig. 2 Potentially bioactive xanthene derivatives. | |
Recent advances highlight the importance of designing synthetic routes that combine efficiency, resource stability, and environmental sustainability.17–20 Minimizing waste is a core goal of organic synthetic chemistry; in this respect, MCRs are a remarkable example of sustainable and environmentally benign processes. Multicomponent reactions (MCRs) are a cornerstone of synthetic organic chemistry, with a legacy extending over a century.21 Although the precise origin of MCR chemistry is difficult to trace, one of the earliest documented examples is the Hantzsch synthesis of dihydropyridines, first reported in 1882. This was soon followed by the Biginelli three-component condensation in 1893, further illustrating the foundational role of MCRs in constructing complex molecular architectures from simple precursors.22 In recent years, MCRs have become a major focus of current chemistry research. In MCRs, three or more reactants are added simultaneously in a one-pot manner. MCRs are also known as sequential or tandem reactions, in which the intermediate formed in one step reacts with additional reagents in the medium, yielding a final product that is a combination of all the atoms of the starting components.23–27 MCRs have several advantages compared to linear synthetic protocols, as they can be carried out under solvent-free, aqueous, aqueous ethanol, or ionic liquid conditions and sometimes at room temperature or under microwave irradiation, reducing energy and solvent consumption.28 Since all the reactants are combined in one pot, MCRs reduce the number of synthetic steps and purification steps, so there is no need to isolate the reaction intermediate, which minimizes reliance on toxic solvents and reagents, saving time and resources. MCRs are highly atom economical, as almost all the carbon atoms from the starting materials are incorporated into the final product.26 In MCRs, multiple bonds are formed simultaneously, so they reduce the number of synthetic steps but require careful control of selectivity.
In the context of the green chemistry principle, ionic liquids are types of salts that exist in the liquid state at or below 100 °C. It is composed of a large organic cation and a small organic/inorganic anion, which results in a low melting point.29 Their remarkable properties make them of potential interest to all chemists, as they consist of at least two tunable components (the anion and cation).30 Hence, the terminology “designer solvents” is now widely used.31 In 1914, Walden reported the synthesis of [EtNH3][NO3] ionic liquid with a melting point of 12 °C, which is recognized as the origin of ionic liquid chemistry.29 Ionic liquid has a range of unique and tunable properties, including high thermal and chemical stability, excellent solvation ability, non-volatility, non-flammability, low vapor pressure, high ionic conductivity and electrical conductivity.32–34
Task-specific ionic liquids are one of the most prominent subclasses of ionic liquids in which functional groups are introduced in either the anion or cation of the ionic liquid. The introduction of such functional anions or cations leads to the formation of wide acidic or basic ionic liquids suited for various organic reactions.35,36 By altering the cation and the anion as well as the length of the alkyl groups on the cation, the physical and chemical properties of ionic liquid may be finely tuned; such structural modification endows these materials with the dual capability to act as both solvents and catalytic species, thus enhancing catalytic performance, selectivity, and recyclability in alignment with green chemistry principles.36,37
Because of the biologically active nature of both isatins and xanthenes, the concept of merging them through the synthesis of spirooxindoles bearing a xanthene moiety holds great promise for generating valuable heterocyclic structures.38 However, current research on these compounds remains scarce. In recent years significant efforts have been explored to the synthesise of symmetric and unsymmetrical spiroindoline using various catalyst including p-TSA,39 SBA-15-Pr-SO3H,40 ZnO,41 LTTM,42 Mg(ClO4)2,43 solid acids as catalyst,44 CAN,45 g-Fe2O3@SiO2-EC-Zn,46 DIL@GO,47 MNPs@SiO2@PUF@Zn,48 ionic liquid 1-methylimidazolium chloride,49 β-cyclodextrin-SO3H,50 PS-PTSA,51 ZnCl2 + urea,52 FeCl3,53 DES,54 GO/Cys–Cu(II),55 MoO3/BF3 in ball milling,56 and in Zr-MOF/rGO-nanocomposites.57 Although certain methods demonstrate acceptable performance, many are hindered by severe reaction conditions, long reaction durations, poor product yields, costly catalysts, use of metal-supported catalysts and the reliance on hazardous solvents, which complicate post-reaction processing. Therefore, there is an urgent demand for the development of more efficient approaches that employ affordable and reusable catalysts to advance environmentally sustainable synthesis practices. With a strong emphasis on green chemistry principles, our research group has focused on developing efficient and environmentally responsible synthetic methodologies for constructing pharmacologically important heterocyclic systems using a one-pot tandem reaction in green solvents.60–62 Herein, we report a one-pot three-component synthesis of isatin- and acenaphthoquinone-based spiroindoline derivatives using [CMMIM][BF4], a reusable green catalyst.
Results and discussion
Initially, our study commenced with the synthesis of [CMMIM][BF4] 4 following a previously reported protocol by our research group (Scheme 1).58 The synthesized ionic liquid [CMMIM][BF4] 4 was extensively characterized using 1H and 13C NMR and FT-IR spectroscopy and compared with those described in the literature. According to literature reports, ionic liquid [CMMIM][BF4] 4 exhibits a pKa value of 2.59
 |
| | Scheme 1 Synthesis of Brønsted acid ionic liquid [CMMIM][BF4] 4. | |
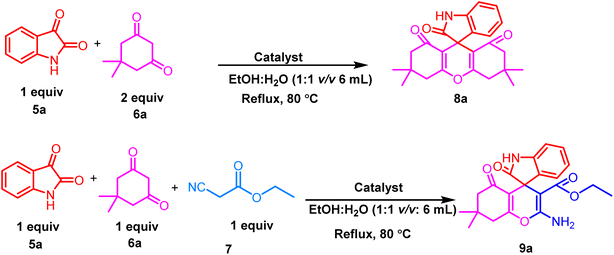
The catalytic potential of ionic liquid [CMMIM][BF4] 4 was investigated to construct nitrogen-containing heterocyclic compounds, including spiro xanthene 8 and spiro chromene 9 derivatives. We initiated our investigation to explore a range of reaction variables to acquire the most efficient reaction conditions for the construction of 8 and 9 derivatives using a one-pot reaction, as shown in Table 1. The starting materials for our model reaction are isatin 5a and dimedone 6a in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio for the construction of spiro xanthene derivative 8a and isatin 5a, dimedone 6a and ethyl cyanoacetate 7, all of which are in equimolar ratios for the construction of spiro chromeno derivative 9a. We started optimization using a range of polar protic and polar aprotic solvents under reflux conditions. The desired products 8a and 9a were obtained up to 45–55% in 24 h under reflux conditions upon the addition of polar aprotic solvents, including acetonitrile (CH3CN), dimethylformamide (DMF), and tetrahydrofuran (THF) (Table 1, entry 1–3). Using chlorinated solvents, such as dichloromethane (DCM) and chloroform (CHCl3), the yield of the product increases to 52% under reflux conditions in 24 h (Table 1, entry 4–5). Later, the use of polar protic solvents like methanol (MeOH), ethanol (EtOH), and isopropyl alcohol (IPA) gratifyingly increases the desired product yield to 78% at the same time and under the same conditions (Table 1, entry 6–8). Interestingly, we obtain a major change in the yield when an aqueous ethanol solution is used to optimize the reaction conditions in a short time. Later, we conduct the reaction by changing the ratio of the aqueous ethanol (1:1) to obtain the most effective condition for the desired products 8a and 9a (Table 1, entries 9–11).
:2 molar ratio for the construction of spiro xanthene derivative 8a and isatin 5a, dimedone 6a and ethyl cyanoacetate 7, all of which are in equimolar ratios for the construction of spiro chromeno derivative 9a. We started optimization using a range of polar protic and polar aprotic solvents under reflux conditions. The desired products 8a and 9a were obtained up to 45–55% in 24 h under reflux conditions upon the addition of polar aprotic solvents, including acetonitrile (CH3CN), dimethylformamide (DMF), and tetrahydrofuran (THF) (Table 1, entry 1–3). Using chlorinated solvents, such as dichloromethane (DCM) and chloroform (CHCl3), the yield of the product increases to 52% under reflux conditions in 24 h (Table 1, entry 4–5). Later, the use of polar protic solvents like methanol (MeOH), ethanol (EtOH), and isopropyl alcohol (IPA) gratifyingly increases the desired product yield to 78% at the same time and under the same conditions (Table 1, entry 6–8). Interestingly, we obtain a major change in the yield when an aqueous ethanol solution is used to optimize the reaction conditions in a short time. Later, we conduct the reaction by changing the ratio of the aqueous ethanol (1:1) to obtain the most effective condition for the desired products 8a and 9a (Table 1, entries 9–11).
Table 1 Solvent screening for the syntheses of 8aa and 9aa
Upon initiating the reaction in EtOH:H2O (1:1 v/v: 6 mL), the desired products 8a and 9a were obtained in a yield of up to 97% in 3 h at 80 °C. When we conduct the same reaction at room temperature in EtOH:H2O (1:1), the yield of the product decreased to 53% in 12 h (Table 1, entry 12). When water is used as a solvent, it decreases the reaction yield because of the non-solubility of isatin in water (Table 1, entry 13). Without any solvent, we obtain a yield of up to 45% in 12 h (Table 1, entry 14).
To prove the efficiency of the catalyst, initially, the reaction was conducted without a catalyst; the results showed that the product is formed with a very low yield (Table 2, entry 1). To confirm the effective involvement of catalyst 4 in the reaction, the model reaction was investigated with various amounts of catalyst 4 (Table 2, entries 2–6). The results showed that the best amounts of catalyst 4 are 6 mol% and 5 mol% for 8a and 9a, respectively (Table 2, entries 3–4). When the loading of catalyst 4 is increased, there is no significant change in the reaction duration, and the yield of the product (Table 2, entries 5–6). Furthermore, the reaction was carried out in a series of acid catalysts, including p-TSA, CH3COOH, and H2SO4, to compare the yield and reaction time with our ionic liquid catalyst 4. We have observed that there is no significant increment in the yield (Table 2, entries 7–9). Based on our investigation, we concluded that the use of 5–6 mol% of catalyst 4 at 80 °C in ethanol:water (1:1 v/v 6 mL) was assumed to be optimal. We intended to explore the potential of this strategy for the construction of a series of spiro xanthene 8 derivatives and spiro chromeno 9 derivatives using halide-substituted isatin 5, acenaphthenequinone 5e, and active-methylene compounds, such as 1,3 cyclohexanedione, dimedone and 3-methyl-1-phenyl-5-pyrazolone 6, along with ethyl cyanoacetate 7 (Fig. 3). All these reactions successfully afforded the desired products 8a–8g and 9a–9m in excellent yields, irrespective of the substituent nature of isatin 5 and active-methylene compound 6 (Fig. 4 and 5). All these substituents afforded the maximum yields. All the resulting spiro derivatives 8a–8g and 9a–9m were isolated by the simple filtration of the reaction mixture. The crude product was washed with 20–30% ethyl acetate/hexane and diethyl ether and then dried under vacuum. The synthesized spiro derivatives were characterized by 1H NMR, 13C NMR, and FTIR spectroscopy and were confirmed to be similar to those described in the literature.
Table 2 Catalyst screening and catalyst loading mol% for the syntheses of 8aa and 9aa
|
| Entry |
Catalyst |
Time (h) |
Yield (%) |
| 8a |
9a |
| Reaction was conducted using 5a (1 equiv.), 6a (2 equiv.) for the synthesis of compound 8a and 5a (1 equiv.), 6a (1 equiv.), 7 (1 equiv.) for the synthesis of compound 9a in various catalyst mol%. |
| 1 |
No catalyst |
24 |
Trace |
Trace |
| 2 |
[CMMIM][BF4] (3 mol%) |
18 |
75 |
78 |
| 3 |
[CMMIM][BF4] (5 mol%) |
3 |
93 |
97 |
| 4 |
[CMMIM][BF4] (6 mol%) |
3 |
95 |
97 |
| 5 |
[CMMIM][BF4] (7 mol%) |
3 |
95 |
97 |
| 6 |
[CMMIM][BF4] (10 mol%) |
3 |
95 |
97 |
| 7 |
p-TSA (20 mol%) |
24 |
55 |
57 |
| 8 |
CH3COOH (20 mol%) |
24 |
50 |
53 |
| 9 |
H2SO4 (20 mol%) |
24 |
60 |
64 |
 |
| | Fig. 3 Substrate scope. | |
 |
| | Fig. 4 Library of the synthesized spiroxanthene derivatives 8a–8g. | |
 |
| | Fig. 5 Library of the synthesized spiro chromeno derivatives 9a–9m. | |
Additionally, the proposed structures of products 8c, 9a and 9i were confirmed using single crystal XRD analysis. These crystals were obtained from a DCM:MeOH (1:1) solvent mixture. The obtained crystal structures 8c (CCDC no. 2498521), 9a (CCDC no. 2500027) and 9i (CCDC no. 2515684) are shown in Fig. 6–8.63 The summary of this crystallographic information is provided in Tables S2–S4 of the SI.
 |
| | Fig. 6 ORTEP diagram of compound 8c (CCDC no. 2498521). | |
 |
| | Fig. 7 ORTEP diagram of compound 9a (CCDC no. 2500027). | |
 |
| | Fig. 8 ORTEP diagram of compound 9i (CCDC no. 2515684). | |
Plausible reaction mechanism
The plausible reaction mechanism for the synthesis of spiro xanthene 8 derivatives and spiro chromeno 9 derivatives is proposed with respect to the structural characteristics of the catalyst and the previously reported studies.39–42 To produce spiro xanthene initially, the carbonyl group of isatin was activated using Brønsted acidic catalyst 4 through hydrogen bonding, thereby facilitating its reactivity towards nucleophilic attack of enolizable dimedone to generate the aldol condensation product. Meanwhile, the process continues with the Michael addition of another enolizable C–H activated dimedone or 1,3-cyclohexadione, followed by intramolecular cyclization and dehydration, to obtain the desired product 8 (Fig. 9).
 |
| | Fig. 9 Plausible reaction mechanism for the synthesis of 8a. | |
Similarly, for the synthesis of spiro-chromeno 9 derivatives, the carbonyl group of isatin was activated by catalyst 4 through hydrogen bonding, thereby facilitating its reactivity towards the nucleophilic attack of active methylene compound (ethyl cyanoacetate) 7 to generate intermediate B. Knoevenagel condensation of intermediate B, leading to the formation of intermediate C. Subsequently, the Michael addition of the intermediate C with enolizable C–H activated dimedone or 1,3-cyclohexadione 6a, followed by intramolecular cyclization, dehydration and subsequent tautomerization, to afford the final product 9 (Fig. 10). The structure of intermediate C was confirmed by 1H and 13C NMR, HRMS and FT-IR spectroscopy; the corresponding spectra are included in the SI.
 |
| | Fig. 10 Plausible reaction mechanism for the synthesis of 9a. | |
Catalyst recovery and recyclability
Due to its hydrophilic nature, the catalyst [CMMIM][BF4] 4 dissolves in aqueous ethanol and can be easily recovered. After completing the reaction, the solid product was isolated by filtration, and the filtrate containing ionic liquid catalyst 4 was evaporated under reduced pressure to recover [CMMIM][BF4] ionic liquid catalyst 4. The recovered catalyst was washed with diethyl ether and then dried under a high vacuum to reuse it in successive cycles. After each cycle, the efficiency of the recovered catalyst 4 was checked and reused for a minimum of five successive cycles for both model reactions without any noticeable loss in catalytic activity, as shown in Fig. 11 and 12.
 |
| | Fig. 11 Recyclability of catalyst 4 for 8a. | |
 |
| | Fig. 12 Recyclability of catalyst 4 for 9a. | |
Gram-scale synthesis
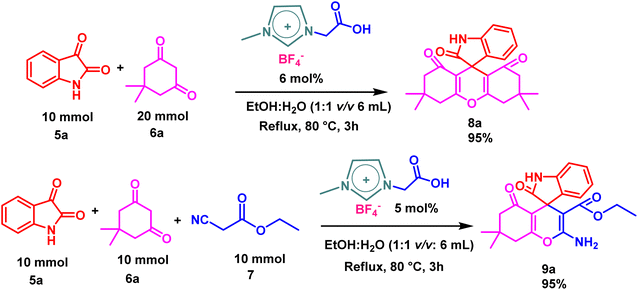 Gram-scale synthesis was performed to demonstrate the practical applicability of the optimized reaction conditions for the synthesis of spiro derivatives. Compound 8a was obtained with 95% yields using 10 mmol of isatin 5a and 20 mmol of dimedone 6a in the presence of 6 mol% catalyst 4 in aqueous ethanol at 80 °C for 3 h. Similarly, compound 9a was synthesized with 95% yields by employing 10 mmol of isatin 5a, 10 mmol of dimedone 6a, and 10 mmol of ethyl cyanoacetate 7 with 5 mol% catalyst 4 in aqueous ethanol at 80 °C for 3h.
Gram-scale synthesis was performed to demonstrate the practical applicability of the optimized reaction conditions for the synthesis of spiro derivatives. Compound 8a was obtained with 95% yields using 10 mmol of isatin 5a and 20 mmol of dimedone 6a in the presence of 6 mol% catalyst 4 in aqueous ethanol at 80 °C for 3 h. Similarly, compound 9a was synthesized with 95% yields by employing 10 mmol of isatin 5a, 10 mmol of dimedone 6a, and 10 mmol of ethyl cyanoacetate 7 with 5 mol% catalyst 4 in aqueous ethanol at 80 °C for 3h.
Conclusions
In summary, we developed a sustainable, efficient and versatile protocol for the construction of diverse spiro-heterocyclic frameworks, including spiroxanthene and spirochromeno derivatives, using task-specific ionic liquid [CMMIM][BF4] 4 as a catalyst in aqueous ethanol media. A series of 20 derivatives was successfully obtained with high yields ranging from 88% to 97% and characterized by 1H NMR, 13C NMR and FT-IR spectroscopy. The present synthetic approach shows several advantages, including mild reaction conditions, short reaction time, transition-metal-free synthesis, low catalyst loading mol percentage, simple product isolation without column chromatography and excellent yields. In both model reactions, catalyst 4 could be reused for at least five successive cycles without any noticeable loss in catalytic activity, and the method was successfully scaled up to the gram level, confirming its practicality. Furthermore, spiroxanthene and spirochromeno derivatives 8c, 9a and 9i were confirmed using single-crystal XRD analysis. This methodology demonstrates greater environmental performance, evidenced by a low E-factor 8a (0.15), a high RME (86.9%), a low E-factor 9a (0.08) and a high RME (92.5%), expressively outperforming conventional routes. Green chemistry matrices were also calculated, as included in the SI.
Experimental section
Materials and methods
All chemicals were purchased from different commercial companies and their purity has been included here. Isatin (98%) from Avra, 5-bromoisatin (88%) from Avra, 5-fluoroisatine (98%) from Sigma-Aldrich, 5-chloroisatine (98%) from Merck, acenaphthenequinone (95%) from Hyma, ethyl cyanoacetate (95%) from Hyma, 3-methyl-1-phenyl-5-pyrazolone (98%) from TCI, dimedone (95%) from Sigma-Aldrich, cyclohexane-1,3-dione (99.85%) from BLD pharm, 1-methylimidazole (98%) from Hyma, and 2-chloroacetic acid (99%) from TCI, and all organic solvents were acquired from commercial suppliers and utilized without any additional purification. Analytical thin-layer chromatography (TLC) was carried out using 0.25 mm silica gel-coated Kieselgel 60 F254 plates. 1H NMR (400 MHz), 13C NMR (100 MHz), and DEPT-135 NMR (100 MHz) spectra were obtained using a Bruker AVANCE III 400 MHz spectrometer. Chemical shifts and coupling constants are expressed in parts per million (ppm) and Hertz (Hz), respectively, using tetra-methyl silane (TMS) as an internal standard. The residual solvent peak appears at 2.5 ppm for DMSO-d6, while D2O resonates at 4.79 ppm. The multiplicities was written as singlet (s), doublet (d), doublet of doublet (dd), multiplet (m), and triplet (t). The FT-IR spectra were collected by employing the PerkinElmer RX-I FTIR spectrometer in ATR mode.
General procedure for the synthesis of 1-carboxymethyl 3-methyl imidazolium tetrafluoroborate 4
In a 250 mL round-bottom flask, 2.0 g (1.0 equiv., 2.43 mmol) of 1-methylimidazole 1 was added to 2.5 g (1.1 equiv., 2.68 mmol) of chloroacetic acid 2. The mixture was stirred for 2 hours at 80 °C. Then, the reaction mixture was left to cool, and the pale-yellow viscous liquid was washed several times with ether and then dried under a vacuum. The resulting [BMMIM][Cl] ionic-liquid 3 was mixed with NaBF4 (2.94 g, 1.1 equiv., 2.68 mmol), diluted in 20 mL of dry acetonitrile, and stirred for 36 hours at room temperature. The resultant white precipitate (NaCl) was filtered and washed frequently with acetonitrile (3 × 30 mL). The concentration of cumulative filtrates provides 1-carboxymethyl 3-methyl imidazolium tetrafluoroborate [CMMIM][BF4] 4 as a pale-yellow viscous liquid, with 97% yield. The product was characterized by 1H NMR, 13C NMR, DEPT-135, and FT-IR spectra and was discovered to be the same as those mentioned in the literature.
Characterization data for product 4
1-Carboxymethyl 3-methyl imidazolium tetrafluoroborate [CMMIM][BF4−] 4 (ref. 58). Pale yellow liquid, 97% yield; FT-IR (ATR mode, cm−1): 3330, 1760, 1030, 759, 625, 523; 1H NMR (400 MHz, D2O, ppm) δ 8.66 (s, 1H), 7.42 (s, 1H), 7.41 (s, 1H), 5.03 (s, 2H), 3.86 (s, 3H); 13C NMR (100 MHz, D2O, ppm) δ 172.41, 139.83, 125.96, 52.28, 38.34; 19F NMR (400 MHz, D2O, ppm) δ −150.36, −150.41; 11B NMR (128 MHz, D2O, ppm) δ −1.43.
General procedure for the synthesis of 3′,3′,6′,6′-tetramethyl-3′,4′,6′,7′-tetrahydrospiro[indoline-3,9′-xanthene]-1′,2,8′(2′H,5′H)-trione 8a
To a dry 25 mL round-bottom flask, isatin 5a (1 equiv.), dimedone 6a (2 equiv.) and 6 mol% of [CMMIM][BF4−] ionic liquid 4 were successively added to 6 mL of ethanol: water (1:1 v/v 6 mL) and refluxed at 80 °C for 3 h. The progress of the reaction was monitored using TLC. After the completion of the reaction, the resulting solid precipitate was filtered out and then washed with 30% ethyl acetate (10 mL × 3) and ether without any column chromatography to achieve the desired product 8a, with 95% yield. The obtained products were analyzed using 1H NMR, 13C NMR, and FT-IR spectra and were found to resemble those previously published in the literature.
Characterization data for products 8a–8g
3′,3′,6′,6′-Tetramethyl-3′,4′,6′,7′-tetrahydrospiro[indoline-3,9′-xanthene]-1′,2,8′(2′H,5′H)-trione, 8a. White solid, 95% yield; mp (°C): >300;64 FT-IR (ATR mode, cm−1): 3363, 2966, 1712, 1666, 1616, 1469, 1307, 1170, 1126, 1018, 748, 524; 1H NMR (400 MHz, DMSO-d6, ppm) δ 10.28 (s, 1H), 7.06 (t, J = 7.5 Hz, 1H), 6.84 (d, J = 7.0 Hz, 1H), 6.77 (t, J = 7.4 Hz, 1H), 6.71 (d, J = 7.6 Hz, 1H), 2.64 (d, J = 17.6 Hz, 2H), 2.57–2.48 (m, 2H), 2.19 (d, J = 15.9 Hz, 2H), 2.03 (d, J = 15.9 Hz, 2H), 1.03 (s, 6H), 0.95 (s, 6H); 13C NMR (100 MHz, DMSO-d6, ppm) δ 195.45, 178.69, 163.92, 144.30, 134.51, 128.19, 122.67, 121.13, 113.46, 108.93, 51.00, 45.67, 32.12, 28.40, 27.06; DEPT-135 (100 MHz, DMSO-d6, ppm) δ 128.19, 122.67, 121.13, 108.93, 51.00, 40.59, 28.40, 27.06.
5-Fluoro-3′,3′,6′,6′-tetramethyl-3′,4′,6′,7′-tetrahydrospiro[indoline-3,9′-xanthene]-1′,2,8′(2′H,5′H)-trione, 8b. White solid, 92% yield; mp (°C):289–291;64 FT-IR (ATR mode, cm−1): 3370, 2972, 1728, 1666, 1620, 1485, 1340, 1172, 1114, 1033, 817, 582; 1H NMR (400 MHz, DMSO-d6, ppm) δ 10.34 (s, 1H), 6.88 (t, J = 8.9 Hz, 1H), 6.78 (d, J = 6.4 Hz, 1H), 6.68 (dd, J = 8.1, 4.2 Hz, 1H), 2.62 (d, J = 17.6 Hz, 2H), 2.54 (d, J = 18.1 Hz, 2H), 2.19 (d, J = 15.9 Hz, 2H), 2.07 (d, J = 15.9 Hz, 2H), 1.02 (s, 6H), 0.97 (s, 6H); 13C NMR (100 MHz, DMSO-d6, ppm) δ 195.11, 178.16, 163.74, 156.51, 140.22, 135.61, 135.54, 113.80, 113.57, 112.48, 110.27, 110.03, 108.77, 50.46, 45.69, 31.62, 27.70, 26.86.
3′,4′,6′,7′-Tetrahydrospiro[indoline-3,9′-xanthene]-1′,2,8′(2′H,5′H)-trione 8c. Pale yellow solid, 90% yield; mp (°C): >300;64 FT-IR (ATR mode, cm−1): 3360, 2951, 1732, 1674, 1620, 1477, 1303, 1207, 1130, 983, 748, 621, 532; 1H NMR (400 MHz, DMSO-d6, ppm) δ 10.28 (s, 1H), 7.05 (t, J = 7.5 Hz, 1H), 6.86 (d, J = 7.0 Hz, 1H), 6.76 (t, J = 7.3 Hz, 1H), 6.69 (d, J = 7.4 Hz, 1H), 2.66 (d, J = 5.4 Hz, 4H), 2.27–2.09 (m, 4H), 1.88 (d, J = 5.5 Hz, 4H); 13C NMR (100 MHz, DMSO-d6, ppm) δ 195.26, 178.50, 165.19, 143.82, 134.34, 127.70, 122.71, 120.73, 114.15, 108.32, 45.41, 37.05, 26.95, 19.77.
5-Chloro-3′,4′,6′,7′-tetrahydrospiro[indoline-3,9′-xanthene]-1′,2,8′(2′H,5′H)-trione 8d. Pale yellow solid, 88% yield; mp (°C): >300;64 FT-IR (ATR mode, cm−1): 3359, 2958, 1724, 1670, 1620, 1481, 1303, 1219, 1126, 979, 837, 736, 628, 540; 1H NMR (400 MHz, DMSO-d6, ppm) δ 10.44 (s, 1H), 7.10 (dd, J = 8.2, 2.2 Hz, 1H), 6.97 (d, J = 2.1 Hz, 1H), 6.70 (d, J = 8.2 Hz, 1H), 2.67 (t, J = 6.1 Hz, 4H), 2.22 (dd, J = 11.0, 6.2 Hz, 4H), 1.92–1.87 (m, 4H); 13C NMR (100 MHz, DMSO-d6, ppm) δ 19.78, 27.08, 29.13, 37.07, 45.79, 109.73, 113.64, 123.01, 124.78, 127.66, 136.40, 142.99, 165.87, 178.48, 195.68.
5-Bromo-3′,4′,6′,7′-tetrahydrospiro[indoline-3,9′-xanthene]-1′,2,8′(2′H,5′H)-trione 8e. Pale yellow solid, 89% yield; mp (°C): >300;64 FT-IR (ATR mode, cm−1): 3356, 2958, 1735, 1658, 1608, 1469, 1350, 1303, 1207, 1134, 987, 833, 617, 540; 1H NMR (400 MHz, DMSO-d6, ppm) δ 10.45 (s, 1H), 7.23 (dd, J = 8.2, 2.0 Hz, 1H), 7.08 (d, J = 1.8 Hz, 1H), 6.66 (d, J = 8.2 Hz, 1H), 2.67 (t, J = 6.1 Hz, 4H), 2.26–2.18 (m, 4H), 1.91 (dd, J = 12.5, 6.2 Hz, 4H); 13C NMR (100 MHz, DMSO-d6, ppm) δ 19.79, 27.09, 29.14, 37.08, 45.76, 110.36, 112.53, 113.65, 125.64, 130.55, 136.81, 143.41, 165.92, 178.40, 195.73.
5-Fluoro-3′,4′,6′,7′-tetrahydrospiro[indoline-3,9′-xanthene]-1′,2,8′(2′H,5′H)-trione 8f. White solid, 93% yield; mp (°C): >300; FT-IR (ATR mode, cm−1): 3359, 2958, 1724, 1689, 1666, 1485, 1300, 1219, 1130, 979, 794, 601, 532; 1H NMR (400 MHz, DMSO-d6, ppm) δ 10.39 (s, 1H), 6.93 (t, J = 9.1 Hz, 1H), 6.87 (d, J = 8.1 Hz, 1H), 6.73 (dd, J = 8.3, 4.3 Hz, 1H), 2.72 (t, J = 5.9 Hz, 4H), 2.34–2.20 (m, 4H), 2.01–1.88 (m, 4H); 13C NMR (100 MHz, DMSO-d6, ppm) δ 195.48, 178.59, 165.61, 158.99, 156.65, 140.24, 135.94, 113.84, 113.69, 113.61, 110.92, 110.68, 108.74, 108.66, 45.98, 37.05, 27.04, 19.75.
3′,5′-Dimethyl-1′,7′-diphenyl-1′H,2H,7′H-spiro[acenaphthylene-1,4′-pyrano[2,3-c:6,5-c′]dipyrazol]-2-one 8g. Yellow solid, 95% yield; mp (°C): 202–204;65 FT-IR (ATR mode, cm−1): 3050, 2796, 1728, 1624, 1566,1492, 1396, 1361, 1303, 1215, 1118, 995, 783, 732, 690, 582, 543; 1H NMR (400 MHz, DMSO-d6, ppm) δ 8.29 (d, J = 8.0 Hz, 1H), 8.12–7.93 (m, 2H), 7.88 (dt, J = 14.9, 7.5 Hz, 1H), 7.78–7.46 (m, 6H), 7.51–7.25 (m, 4H), 7.19 (dd, J = 24.0, 17.1 Hz, 2H), 2.00 (d, J = 25.9 Hz, 6H); 13C NMR (100 MHz, DMSO-d6, ppm) δ 13.34, 53.31, 118.30, 119.21, 121.48, 122.14, 123.83, 124.84, 128.14, 128.68, 128.90, 129.12, 130.39, 131.39, 133.19, 137.44, 140.26, 141.94, 147.85, 203.07.
General procedure for the synthesis of ethyl 2-amino-7,7-dimethyl-2′,5-dioxo-5,6,7,8-tetrahydrospiro[chromene-4,3′-indoline]-3-carboxylate 9a
To a dry 25 mL round-bottom flask, isatin 5a (1 equiv.), dimedone 6a (1 equiv.), ethyl cyanoacetate 7 (1 equiv.) and 5 mol% of [CMMIM][BF4] ionic liquid 4 were successively added to 6 mL of ethanol:water (1:1 v/v) and refluxed at 80 °C for 3 h. The progress of the reaction was monitored using TLC. After the completion of the reaction, the resulting solid precipitate was filtered out and then washed with 30% ethyl acetate (10 mL × 3) and ether without column chromatography to achieve the desired product 9a, with 97% yield. The obtained products were analyzed using 1H NMR, 13C NMR, and FT-IR spectra and were found to resemble those published previously in the literature.
Characterization data for products 9a–9m
Ethyl 2-amino-7,7-dimethyl-2′,5-dioxo-5,6,7,8-tetrahydrospiro[chromene-4,3′-indoline]-3-carboxylate 9a. Ivory solid, 95% yield; mp (°C): 255–257;67 FT-IR (ATR mode, cm−1): 3375, 3186, 1696, 1681, 1639, 1527, 1469, 1342, 1292, 1222, 1138, 1029, 748, 675, 601; 1H NMR (400 MHz, DMSO-d6, ppm) δ 10.15 (s, 1H), 7.86 (s, 2H), 7.04 (t, J = 7.5 Hz, 1H), 6.84 (d, J = 7.1 Hz, 1H), 6.76 (t, J = 7.4 Hz, 1H), 6.68 (d, J = 7.6 Hz, 1H), 3.70 (td, J = 6.9, 4.2 Hz, 2H), 2.59 (d, J = 17.5 Hz, 1H), 2.49 (d, J = 17.1 Hz, 1H), 2.16 (d, J = 15.8 Hz, 1H), 2.02 (d, J = 15.8 Hz, 1H), 1.02 (s, 3H), 0.95 (s, 3H), 0.80 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, DMSO-d6, ppm) δ 194.64, 179.77, 167.63, 162.39, 159.09, 144.03, 135.97, 127.15, 122.21, 120.51, 113.08, 108.10, 76.30, 58.82, 50.63, 46.59, 31.53, 27.77, 26.66, 13.08; DEPT-135 (100 MHz, DMSO-d6, ppm) δ 127.15, 122.21, 120.51, 108.09, 58.81, 50.62, 40.10, 27.77, 26.65, 13.08.
Ethyl 2-amino-5′-chloro-7,7-dimethyl-2′,5-dioxo-5,6,7,8-tetrahydrospiro[chromene-4,3′-indoline]-3-carboxylate 9b. Ivory solid, 97% yield; mp (°C): 250–251;67 FT-IR (ATR mode, cm−1): 3387, 3285, 1724, 1678, 1651, 1516, 1481, 1300, 1222, 1168, 1041, 636, 551; 1H NMR (400 MHz, DMSO-d6, ppm) δ 10.31 (s, 1H), 7.94 (s, 2H), 7.10 (dd, J = 8.2, 2.0 Hz, 1H), 6.91 (d, J = 1.8 Hz, 1H), 6.68 (d, J = 8.2 Hz, 1H), 3.76–3.71 (m, 2H), 2.53 (d, J = 14.2 Hz, 2H), 2.11 (q, J = 15.8 Hz, 2H), 1.01 (s, 3H), 0.97 (s, 3H), 0.83 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, DMSO-d6, ppm) δ 194.84, 179.52, 167.43, 162.88, 159.18, 143.13, 138.12, 126.98, 124.38, 122.37, 112.47, 109.36, 75.66, 58.95, 50.56, 46.89, 27.42, 27.08, 13.12.
Ethyl 2-amino-5′-bromo-7,7-dimethyl-2′,5-dioxo-5,6,7,8-tetrahydrospiro[chromene-4,3′-indoline]-3-carboxylate 9c. White solid, 95% yield; mp (°C): 295–298;68 FT-IR (ATR mode, cm−1): 3390, 3282, 1720, 1685, 1616, 1516, 1477, 1350, 1296, 1211, 1165, 1053, 1018, 821, 632, 536; 1H NMR (400 MHz, DMSO-d6, ppm) δ 10.32 (s, 1H), 7.94 (s, 2H), 7.23 (dd, J = 8.2, 1.9 Hz, 1H), 7.01 (d, J = 1.6 Hz, 1H), 6.66 (t, J = 11.2 Hz, 1H), 3.82–3.66 (m, 2H), 2.51 (s, 2H), 2.11 (q, J = 15.8 Hz, 2H), 0.99 (d, J = 15.5 Hz, 6H), 0.83 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, DMSO-d6, ppm) δ 194.86, 179.39, 167.42, 162.90, 159.18, 143.53, 138.50, 129.84, 125.05, 112.48, 112.04, 109.99, 75.66, 58.97, 50.55, 46.84, 31.56, 27.40, 27.09, 13.12.
Ethyl 2-amino-5′-fluoro-7,7-dimethyl-2′,5-dioxo-5,6,7,8-tetrahydrospiro[chromene-4,3′-indoline]-3-carboxylate 9d. White solid, 97% yield; mp (°C): 291–292;69 FT-IR (ATR mode, cm−1): 3383, 3275, 1724, 1689, 1647, 1481, 1354, 1296, 1165, 1053, 802, 536; 1H NMR (400 MHz, DMSO-d6, ppm) δ 10.18 (s, 1H), 7.91 (s, 2H), 6.90–6.83 (m, 1H), 6.77 (dd, J = 8.1, 2.5 Hz, 1H), 6.64 (dd, J = 8.3, 4.3 Hz, 1H), 3.82–3.65 (m, 2H), 2.54 (dd, J = 21.1, 11.6 Hz, 2H), 2.19–2.01 (m, 2H), 1.02 (s, 3H), 0.97 (s, 3H), 0.81 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, DMSO-d6, ppm) δ 194.74, 179.75, 167.51, 162.73, 159.15, 140.41, 113.18, 112.95, 112.57, 110.26, 110.02, 108.33, 75.79, 58.89, 50.60, 47.15, 31.53, 27.58, 26.93, 13.12.
Ethyl 2-amino-2′,5-dioxo-5,6,7,8-tetrahydrospiro[chromene-4,3′-indoline]-3-carboxylate 9e. White solid, 93% yield; mp (°C): 252–254;66 FT-IR (ATR mode, cm−1): 3363, 3248, 1685, 1651, 1612, 1516, 1469, 1292, 1226, 1076, 1029, 929, 740, 621, 605, 500; 1H NMR (400 MHz, DMSO-d6, ppm) δ 10.21 (s, 1H), 7.92 (s, 2H), 7.10 (t, J = 7.5 Hz, 1H), 6.91 (d, J = 7.1 Hz, 1H), 6.82 (t, J = 7.4 Hz, 1H), 6.73 (d, J = 7.6 Hz, 1H), 3.87–3.68 (m, 2H), 2.70 (t, J = 5.8 Hz, 2H), 2.36–2.15 (m, 2H), 2.08–1.78 (m, 2H), 0.94–0.76 (m, 3H); 13C NMR (100 MHz, DMSO-d6, ppm) δ 194.81, 179.88, 167.66, 164.20, 158.98, 144.02, 136.10, 127.12, 122.43, 120.51, 114.22, 108.02, 76.36, 58.82, 46.71, 37.09, 26.93, 19.65, 13.09.
Ethyl 2-amino-5′-chloro-2′,5-dioxo-5,6,7,8-tetrahydrospiro[chromene-4,3′-indoline]-3-carboxylate 9f. White solid, 95% yield; mp (°C): 280–282;71 FT-IR (ATR mode, cm−1): 3348, 3178, 1700, 1651, 1527, 1477, 1300, 1219, 1141, 1033, 806, 586, 532; 1H NMR (400 MHz, DMSO-d6, ppm) δ 10.31 (s, 1H), 7.93 (s, 2H), 7.09 (dd, J = 8.1, 1.9 Hz, 1H), 6.94 (d, J = 1.5 Hz, 1H), 6.68 (d, J = 8.2 Hz, 1H), 3.82–3.65 (m, 2H), 2.64 (t, J = 6.0 Hz, 2H), 2.29–2.12 (m, 2H), 1.95–1.84 (m, 2H), 0.82 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, DMSO-d6, ppm) δ 195.01, 179.63, 167.46, 164.78, 159.06, 143.10, 138.22, 126.94, 124.37, 122.62, 113.57, 109.28, 75.73, 58.96, 46.98, 36.99, 26.96, 19.57, 13.12.
Ethyl 2-amino-5′-bromo-2′,5-dioxo-5,6,7,8-tetrahydrospiro[chromene-4,3′-indoline]-3-carboxylate 9g. White solid, 96% yield; mp (°C): 261–263;66 FT-IR (ATR mode, cm−1): 3359, 3190, 1685, 1651, 1527, 1480, 1300, 1222, 1134, 1029, 813, 578; 1H NMR (400 MHz, DMSO-d6, ppm) δ 10.32 (s, 1H), 7.93 (s, 2H), 7.22 (d, J = 8.0 Hz, 1H), 7.04 (s, 1H), 6.64 (d, J = 8.1 Hz, 1H), 3.85–3.58 (m, 2H), 2.64 (t, J = 5.7 Hz, 2H), 2.35–2.08 (m, 2H), 2.02–1.77 (m, 2H), 0.83 (t, J = 7.0 Hz, 3H); 13C NMR (100 MHz, DMSO-d6, ppm) δ 195.03, 179.51, 167.45, 164.82, 159.06, 143.50, 138.62, 129.81, 125.27, 113.58, 112.08, 109.91, 75.75, 58.98, 46.94, 37.00, 26.96, 19.57, 13.13.
Ethyl 2-amino-5′-fluoro-2′,5-dioxo-5,6,7,8-tetrahydrospiro[chromene-4,3′-indoline]-3-carboxylate 9h. White solid, 97% yield; mp (°C): 223–224;69 FT-IR (ATR mode, cm−1): 3375, 3192, 1689, 1662, 1527, 1477, 1350, 1296, 1188, 1083, 1029, 806, 682, 597, 505; 1H NMR (400 MHz, DMSO-d6, ppm) δ 10.18 (s, 1H), 7.91 (s, 2H), 6.91–6.82 (m, 1H), 6.78 (dd, J = 8.2, 2.4 Hz, 1H), 6.63 (dd, J = 8.3, 4.3 Hz, 1H), 3.80–3.63 (m, 2H), 2.64 (t, J = 6.0 Hz, 2H), 2.36–2.11 (m, 2H), 1.89 (dd, J = 12.3, 6.1 Hz, 2H), 0.90–0.74 (m, 3H); 13C NMR (100 MHz, DMSO-d6, ppm) δ 194.95, 179.90, 167.55, 164.63, 159.05, 156.55, 140.40, 137.91, 113.68, 113.16, 110.52, 108.36, 75.89, 58.92, 47.28, 37.06, 27.00, 19.61, 13.14.
Ethyl 6′-amino-3′-methyl-2-oxo-1′-phenyl-1′H-spiro[indoline-3,4′-pyrano[2,3-c]pyrazole]-5′-carboxylate 9i. White solid, 96% yield; mp (°C): 208–210;70 FT-IR (ATR mode, cm−1): 3356, 3217, 1689, 1639, 1508, 1377, 1284, 1222, 1134, 1045, 929, 744, 675, 601; 1H NMR (400 MHz, DMSO-d6, ppm) δ 10.52 (s, 1H), 8.22 (s, 2H), 7.82 (d, J = 8.1 Hz, 2H), 7.52 (t, J = 7.9 Hz, 2H), 7.34 (t, J = 7.4 Hz, 1H), 7.18 (t, J = 7.5 Hz, 1H), 6.97 (d, J = 7.2 Hz, 1H), 6.88 (dd, J = 16.7, 7.7 Hz, 2H), 3.93–3.64 (m, 2H), 1.59 (s, 3H), 0.91–0.57 (m, 3H); 13C NMR (100 MHz, DMSO-d6, ppm) δ 179.25, 167.87, 161.32, 144.20, 143.94, 142.18, 137.35, 135.78, 129.41, 127.75, 126.32, 123.17, 121.74, 119.93, 108.82, 98.19, 74.57, 58.99, 47.47, 13.09, 11.71.
Ethyl 6′-amino-5-bromo-3′-methyl-2-oxo-1′-phenyl-1′H-spiro[indoline-3,4′-pyrano[2,3-c]pyrazole]-5′-carboxylate 9j. White solid, 96% yield; mp (°C): 254–256;68 FT-IR (ATR mode, cm−1): 3344, 3190, 1698, 1643, 1516, 1400, 1284, 1134, 1041, 810, 744, 690, 543; 1H NMR (400 MHz, DMSO-d6, ppm) δ 10.67 (s, 1H), 8.27 (s, 2H), 7.82 (d, J = 8.0 Hz, 2H), 7.52 (t, J = 7.8 Hz, 2H), 7.35 (t, J = 7.8 Hz, 2H), 7.23 (s, 1H), 6.83 (d, J = 8.2 Hz, 1H), 3.90–3.65 (m, 2H), 1.63 (s, 3H), 0.92–0.69 (m, 3H); 13C NMR (100 MHz, DMSO-d6, ppm) δ 178.91, 167.67, 161.43, 144.08, 143.97, 141.50, 138.24, 137.31, 130.51, 126.40, 126.20, 120.10, 113.42, 110.80, 97.51, 74.04, 59.11, 47.68, 13.16, 11.78.
Ethyl 6′-amino-5-fluoro-3′-methyl-2-oxo-1′-phenyl-1′H-spiro[indoline-3,4′-pyrano[2,3-c]pyrazole]-5′-carboxylate 9k. White solid, 90% yield; mp (°C): 199–200;70 FT-IR (ATR mode, cm−1): 3387, 3190, 1720, 1685, 1635, 1485, 1384, 1276, 1122, 1033, 802, 694, 586; 1H NMR (400 MHz, DMSO-d6, ppm) δ 10.55 (s, 1H), 8.26 (s, 2H), 7.82 (d, J = 8.3 Hz, 2H), 7.52 (t, J = 7.8 Hz, 2H), 7.35 (t, J = 7.3 Hz, 1H), 7.08–6.91 (m, 2H), 6.85 (dd, J = 8.3, 4.2 Hz, 1H), 3.91–3.64 (m, 2H), 1.62 (s, 3H), 0.81 (m, J = 14.1, 7.1 Hz, 3H); 13C NMR (100 MHz, DMSO-d6, ppm) δ 179.29, 167.73, 161.40, 157.14, 144.07, 138.37, 137.33, 129.40, 126.38, 120.00, 114.03, 111.25, 111.01, 109.52, 97.64, 74.15, 59.05, 48.01, 13.14, 11.73.
Ethyl 2′-amino-7′,7′-dimethyl-2,5′-dioxo-5′,6′,7′,8′-tetrahydro-2H-spiro[acenaphthylene-1,4′-chromene]-3′-carboxylate 9l. White solid, 95% yield; mp (°C): 244–246;69 FT-IR (ATR mode, cm−1): 3387, 3278, 2966, 1720, 1654, 1523, 1338, 1303, 1219, 1080, 771, 597, 509; 1H NMR (400 MHz, DMSO-d6, ppm) δ 8.18 (d, J = 7.8 Hz, 1H), 8.03 (s, 2H), 7.85 (m, J = 21.2, 13.6, 7.7 Hz, 3H), 7.68–7.57 (m, 1H), 7.33 (d, J = 6.8 Hz, 1H), 3.42–3.35 (m, 2H), 2.69 (dd, J = 41.4, 17.6 Hz, 2H), 2.09 (dd, J = 60.2, 16.0 Hz, 2H), 1.10 (s, 3H), 1.03 (s, 3H), 0.04 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, DMSO-d6, ppm) δ 205.17, 195.24, 167.43, 162.93, 159.37, 145.28, 140.61, 135.97, 129.41, 129.23, 128.20, 127.69, 123.75, 119.11, 118.99, 114.84, 77.19, 58.38, 50.68, 50.00, 31.68, 27.76, 26.69, 12.18.
Ethyl 2′-amino-7′,7′-dimethyl-2,5′-dioxo-5′,6′,7′,8′-tetrahydro-2H-spiro[acenaphthylene-1,4′-chromene]-3′-carboxylate 9m. White solid, 94% yield; mp (°C): 261–263;69 FT-IR (ATR mode, cm−1): 3390, 3302, 1716, 1678, 1604, 1519, 1342, 1292, 1249, 1195, 1080, 1018, 887, 786, 536, 500; 1H NMR (400 MHz, DMSO-d6, ppm) δ 8.19 (d, J = 7.7 Hz, 1H), 8.02 (s, 2H), 7.95–7.75 (m, 3H), 7.63 (t, J = 7.6 Hz, 1H), 7.35 (d, J = 6.8 Hz, 1H), 3.46–3.24 (m, 3H), 2.79 (t, J = 5.7 Hz, 2H), 2.33–2.07 (m, 2H), 1.98 (dd, J = 11.6, 5.9 Hz, 2H), 0.04 (t, J = 7.0 Hz, 3H); 13C NMR (100 MHz, DMSO-d6, ppm) δ 205.45, 195.63, 167.61, 164.97, 159.39, 145.55, 140.70, 136.11, 129.54, 129.30, 128.38, 127.83, 123.85, 119.35, 119.21, 116.08, 77.40, 58.55, 50.91, 36.56, 26.90, 19.81, 12.30.
Author contributions
Rujuma Begum: formal analysis, investigation, writing-original manuscript. Barnali Maiti: conceptualization, funding acquisition, supervision, and writing-reviewing and editing. All authors have approved the final version of the manuscript.
Conflicts of interest
The authors declare no competing interests.
Data availability
CCDC 2498521, 2500027 and 2515684 contain the supplementary crystallographic data for this paper.72a–c
The data that supports the findings of this study are available in the supplementary information (SI). Supplementary information: the analytical data, including 1H NMR, 13C NMR, FT-IR spectroscopy and SXRD. See DOI: https://doi.org/10.1039/d6ra01721e.
Acknowledgements
The authors thank the DST-Govt of India for the funding provided through DST-SERB-YSS/2015/000450, the Chancellor of VIT for providing an opportunity to carry out this study, and VIT for providing the VIT SEED GRANT (SG20220031 and SG20240023) for carrying out this research work. We sincerely acknowledge the VIT-SIF for providing the instrumental facilities. We acknowledge the scientific and technical assistance of the Single Crystal XRD facility supported jointly by DST and VIT under the DST-FIST Scheme at VIT, Vellore.
References
- A. R. Katritzky, Introduction: Heterocycles, Chem. Rev., 2004, 104, 2125–2126, DOI:10.1021/cr0406413.
- P. N. Kalaria, S. C. Karad and D. K. Raval, Eur. J. Med. Chem., 2018, 158, 917–936, DOI:10.1016/j.ejmech.2018.08.040.
- D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394, DOI:10.1038/s41557-018-0021-z.
- L. Sun, T. Huang, A. Dick, M. E. Meuser, W. A. Zalloum, C.-H. Chen, X. Ding, P. Gao, S. Cocklin, K.-H. Lee, P. Zhan and X. Liu, Eur. J. Med. Chem., 2020, 190, 112085, DOI:10.1016/j.ejmech.2020.112085.
- G. S. Singh and Z. Y. Desta, Chem. Rev., 2012, 112, 6104–6155, DOI:10.1021/cr300135y.
- V. Varun, S. Sonam and R. Kakkar, MedChemComm, 2019, 10, 351–368, 10.1039/C8MD00585K.
- M. A. Alshams, M. S. Nafie, H. F. Ashour and A. S. A. Yassen, RSC Adv., 2025, 15, 32188–32231, 10.1039/D5RA05002B.
- P. Brandão, C. Marques, A. J. Burke and M. Pineiro, Eur. J. Med. Chem., 2021, 211, 113102, DOI:10.1016/j.ejmech.2020.113102.
- H. A. Younus, M. Al-Rashida, A. Hameed, M. Uroos, U. Salar, S. Rana and K. M. Khan, Expert Opin. Ther. Pat., 2021, 31, 267–289, DOI:10.1080/13543776.2021.1858797.
- P. Pakravan, S. Kashanian, M. M. Khodaei and F. J. Harding, Pharmacol. Rep., 2013, 65, 313–335, DOI:10.1016/S1734-1140(13)71007-7.
- S. Chowdhary, Shalini, A. Arora and V. Kumar, Pharmaceuticals, 2022, 15, 536, DOI:10.3390/ph15050536.
- A. J. Boddy and J. A. Bull, Org. Chem. Front., 2021, 8, 1026–1084, 10.1039/D0QO01085E.
- M. Maia, D. I. S. P. Resende, F. Durães, M. M. M. Pinto and E. Sousa, Eur. J. Med. Chem., 2021, 210, 113085, DOI:10.1016/j.ejmech.2020.113085.
- F. Laneri, N. Licciardello, Y. Suzuki, A. C. E. Graziano, F. Sodano, A. Fraix and S. Sortino, Pharmaceutics, 2022, 15, 96, DOI:10.3390/pharmaceutics15010096.
- M. Aslam, S. Mohandoss, P. Subramanian, S. You, W.-G. Yang, S. H. Kim and Y. R. Lee, Org. Lett., 2021, 23, 1383–1387, DOI:10.1021/acs.orglett.1c00042.
- S. G. Keller, M. Kamiya and Y. Urano, Molecules, 2020, 25, 5964, DOI:10.3390/molecules25245964.
- H. Cao, X. Xiao, X. Zhang, Y. Zhang and L. Yu, Chin. Chem. Lett., 2025, 36, 110924, DOI:10.1016/j.cclet.2025.110924.
- H. Liu, L. Liang, F. Tian, X. Xi, Y. Zhang, P. Zhang, X. Cao, Y. Bai, C. Zhang and L. Dong, Angew. Chem., Int. Ed., 2024, 63, e202402509, DOI:10.1002/anie.202402509.
- M. Zhang, J. Wang, L. Peng, W. Yang, T. Zhao, B. Chen, Z. Zhao and N. Dang, Prog. Org. Coat., 2026, 213, 109956, DOI:10.1016/j.porgcoat.2026.109956.
- G. Tian, Z. Li, C. Zhang, X. Liu, X. Fan, K. Shen, H. Meng, N. Wang, H. Xiong, M. Zhao, X. Liang, L. Luo, L. Zhang, B. Yan, X. Chen, H. J. Peng and F. Wei, Nat. Commun., 2024, 15, 3037, DOI:10.1038/s41467-024-47270-z.
- B. Ganem, Acc. Chem. Res., 2009, 42, 463–472, DOI:10.1021/ar800214s.
- J. E. Biggs-Houck, A. Younai and J. T. Shaw, Curr. Opin. Chem. Biol., 2010, 14, 371–382, DOI:10.1016/j.cbpa.2010.03.003.
- B. H. Rotstein, S. Zaretsky, V. Rai and A. K. Yudin, Chem. Rev., 2014, 114, 8323–8359, DOI:10.1021/cr400615v.
- S. E. John, S. Gulati and N. Shankaraiah, Org. Chem. Front., 2021, 8, 4237–4287, 10.1039/D0QO01480J.
- T. Zarganes-Tzitzikas, A. L. Chandgude and A. Dömling, Chem. Rec., 2015, 15, 981–996, DOI:10.1002/tcr.201500201.
- C. S. Graebin, F. v. Ribeiro, K. R. Rogério and A. E. Kümmerle, Curr. Org. Synth., 2019, 16, 855–899, DOI:10.2174/1570179416666190718153703.
- H. Yazdani, S. E. Hooshmand and R. S. Varma, ACS Sustainable Chem. Eng., 2021, 9, 16556–16569, DOI:10.1021/acssuschemeng.1c04361.
- A. Dömling, W. Wang and K. Wang, Chem. Rev., 2012, 112, 3083–3135, DOI:10.1021/cr100233r.
- Z. Xue, L. Qin, J. Jiang, T. Mu and G. Gao, Phys. Chem. Chem. Phys., 2018, 20, 8382–8402, 10.1039/C7CP07483B.
- Z. Lei, B. Chen, Y.-M. Koo and D. R. MacFarlane, Chem. Rev., 2017, 117, 6633–6635, DOI:10.1021/acs.chemrev.7b00246.
- A. S. Amarasekara, Chem. Rev., 2016, 116, 6133–6183, DOI:10.1021/acs.chemrev.5b00763.
- P. Nockemann, B. Thijs, T. N. Parac-Vogt, K. van Hecke, L. van Meervelt, B. Tinant, I. Hartenbach, T. Schleid, V. T. Ngan, M. T. Nguyen and K. Binnemans, Inorg. Chem., 2008, 47, 9987–9999, DOI:10.1021/ic801213z.
- S. Saikia, H. Borah, P. Deka and R. R. Dutta, in Handbook of Ionic Liquids, Wiley, Hoboken, NJ, 2024, DOI:10.1002/9783527839520.ch2.
- R. R. Hawker, R. S. Haines and J. B. Harper, Org. Biomol. Chem., 2018, 16, 3453–3463, 10.1039/C8OB00651B.
- M. Llaver, E. F. Fiorentini, P. Y. Quintas, M. N. Oviedo, M. B. Botella Arenas and R. G. Wuilloud, Adv. Sample Prep., 2022, 1, 100004, DOI:10.1016/j.sampre.2022.100004.
- A. D. Sawant, D. G. Raut, N. B. Darvatkar and M. M. Salunkhe, Green Chem. Lett. Rev., 2011, 4, 41–54, DOI:10.1080/17518253.2010.500622.
- C. Yue, D. Fang, L. Liu and T.-F. Yi, J. Mol. Liq., 2011, 163, 99–121, DOI:10.1016/j.molliq.2011.09.001.
- D. Steinsits, B. Rávai, Z. Kelemen, L. Hackler, V. Vernyik, L. G. Puskás and E. Bálint, J. Org. Chem., 2025, 90, 6454–6467, DOI:10.1021/acs.joc.5c00270.
- S. Ahadi, H. R. Khavasi and A. Bazgir, Chem. Pharm. Bull., 2008, 56, 1328–1330, DOI:10.1248/cpb.56.1328.
- G. M. Ziarani, N. Lashgari and A. R. Badiei, Sci. Iran., 2013, 20, 580–586, DOI:10.1016/j.scient.2012.11.018.
- J. Kothandapani, A. Ganesan, G. K. Mani, A. J. Kulandaisamy, J. B. B. Rayappan and S. Selva Ganesan, Tetrahedron Lett., 2016, 57, 3472–3475, DOI:10.1016/j.tetlet.2016.06.094.
- D. R. Chandam, A. A. Patravale, S. D. Jadhav and M. B. Deshmukh, J. Mol. Liq., 2017, 240, 98–105, DOI:10.1016/j.molliq.2017.05.070.
- C. Chen, C. Lv, J. Liang, J. Jin, L. Wang, C. Wu and R. Shen, Molecules, 2017, 22, 1295, DOI:10.3390/molecules22081295.
- K. Niknam, A. Ebrahimpour, A. Barmak and G. Mohebbi, Monatsh. Chem., 2018, 149, 73–85, DOI:10.1007/s00706-017-2076-8.
- R. Joshi, A. Kumawat, S. Singh, T. K. Roy and R. T. Pardasani, J. Heterocycl. Chem., 2018, 55, 1783–1790, DOI:10.1002/jhet.3217.
- M. S. Ghasemzadeh and B. Akhlaghinia, ChemistrySelect, 2018, 3, 3161–3170, DOI:10.1002/slct.201703189.
- N. Patel, U. Patel and A. Dadhania, Res. Chem. Intermed., 2021, 47, 2189–2206, DOI:10.1007/s11164-021-04405-x.
- M. A. Bodaghifard and Z. Mousavi, Appl. Organomet. Chem., 2020, 34, e5859, DOI:10.1002/aoc.5859.
- R. Westphal, E. Venturini Filho, L. B. Loureiro, C. F. Tormena, C. Pessoa, C. d. J. Guimarães, M. P. Manso, R. G. Fiorot, V. R. Campos, J. A. L. C. Resende, F. Medici and S. J. Greco, Molecules, 2022, 27, 8051, DOI:10.3390/molecules27228051.
- F. Xu, Y. Wang, F. He, Z. Li, S. Guo, Y. Xie, D. Luo and J. Wu, Green Chem. Lett. Rev., 2022, 15, 139–152, DOI:10.1080/17518253.2021.2023660.
- A. S. Kumari, C. Sudhakar and K. Swarupa, Russ. J. Org. Chem., 2023, 59, 1747–1754, DOI:10.1134/S107042802310010X.
- V. B. Nishtala and S. Basavoju, Synth. Commun., 2019, 49, 2342–2349, DOI:10.1080/00397911.2019.1620784.
- S. Balaji, K. Thirugnanasambandham, M. S. Pandian and R. Ganesamoorthy, J. Indian Chem. Soc., 2025, 102, 101673, DOI:10.1016/j.jics.2025.101673.
- N. Azizi, S. Dezfooli and M. Mahmoudi Hashemi, J. Mol. Liq., 2014, 194, 62–67, DOI:10.1016/j.molliq.2014.01.009.
- M. Toorbaf, L. Moradi and A. Dehghani, J. Mol. Struct., 2023, 1294, 136335, DOI:10.1016/j.molstruc.2023.136335.
- D. Mallah, B. B. F. Mirjalili, H. Basharnavaz and A. Bamoniri, RSC Adv., 2025, 15, 25949–25964, 10.1039/D5RA01991E.
- G. Kumar, A. Dutta, M. Goswami, B. Meena, S. Parasuboyina, R. Nongkhlaw and D. T. Masram, J. Mol. Struct., 2023, 1287, 135653, DOI:10.1016/j.molstruc.2023.135653.
- S. Das, B. Choudhury, B. Maiti and K. Chanda, Org. Biomol. Chem., 2025, 23, 2000–2009, 10.1039/D4OB01625D.
- Z. Fei, D. Zhao, T. J. Geldbach, R. Scopelliti and P. J. Dyson, Chem.–Eur. J., 2004, 10, 4886–4893, DOI:10.1002/chem.200400145.
- S. D. Pasuparthy, P. Somkuwar, V. Kali, A. K. Somanahalli Kalleshappa and B. Maiti, New J. Chem., 2024, 48, 14904–14923, 10.1039/D4NJ02651A.
- V. Kali and B. Maiti, J. Mol. Struct., 2024, 1299, 137053, DOI:10.1016/j.molstruc.2023.137053.
- U. Dasmahapatra, B. Maiti and K. Chanda, Org. Biomol. Chem., 2024, 22, 8459–8471, 10.1039/D4OB01261E.
- CCDC no. 2498521 (8c), CCDC no. 2500027 (9a) and CCDC no. 2515684 (9i) contains the supplementary crystallographic data for this paper.
- N. Patel, U. Patel and A. Dadhania, Res. Chem. Intermed., 2021, 47, 2189–2206, DOI:10.1007/s11164-021-04405-x.
- A. Dandia, S. L. Gupta, A. Indora, P. Saini, V. Parewa and K. S. Rathore, Tetrahedron Lett., 2017, 58, 1170–1175, DOI:10.1016/j.tetlet.2017.02.014.
- N. Chouha, T. Boumoud, B. Boumoud and A. Debache, J. Chem. Pharm. Res., 2018, 10, 113–117 CAS.
- F. Xu, Y. Wang, F. He, Z. Li, S. Guo, Y. Xie, D. Luo and J. Wu, Green Chem. Lett. Rev., 2022, 15, 139–152, DOI:10.1080/17518253.2021.2023660.
- S. F. Hojati, A. S. Kaheh, M. Moosavifar and F. Daghestani, J. Cluster Sci., 2022, 33, 1387–1397, DOI:10.1007/s10876-021-02063-y.
- A. Gharib, N. N. Pesyan, B. R. H. Khorasani, M. Roshani and J. H. W. Scheeren, Bulg. Chem. Commun., 2013, 45, 371–378 CAS.
- Z. Karimi-Jaberi and A. Fereydoonnezhad, Iran. Chem. Commun., 2017, 5(364–493), 407–416 Search PubMed.
- R. Azimi and Z. Lasemi, Res. Chem. Intermed., 2022, 48, 1615–1630, DOI:10.1007/s11164-022-04667-z.
-
(a) CCDC 2498521: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2pvxgy;
(b) CCDC 2500027: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2pxh15;
(c) CCDC 2515684: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2qfs32.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*



![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :
:











 Gram-scale synthesis was performed to demonstrate the practical applicability of the optimized reaction conditions for the synthesis of spiro derivatives. Compound 8a was obtained with 95% yields using 10 mmol of isatin 5a and 20 mmol of dimedone 6a in the presence of 6 mol% catalyst 4 in aqueous ethanol at 80 °C for 3 h. Similarly, compound 9a was synthesized with 95% yields by employing 10 mmol of isatin 5a, 10 mmol of dimedone 6a, and 10 mmol of ethyl cyanoacetate 7 with 5 mol% catalyst 4 in aqueous ethanol at 80 °C for 3h.
Gram-scale synthesis was performed to demonstrate the practical applicability of the optimized reaction conditions for the synthesis of spiro derivatives. Compound 8a was obtained with 95% yields using 10 mmol of isatin 5a and 20 mmol of dimedone 6a in the presence of 6 mol% catalyst 4 in aqueous ethanol at 80 °C for 3 h. Similarly, compound 9a was synthesized with 95% yields by employing 10 mmol of isatin 5a, 10 mmol of dimedone 6a, and 10 mmol of ethyl cyanoacetate 7 with 5 mol% catalyst 4 in aqueous ethanol at 80 °C for 3h.