DOI:
10.1039/D6RA00786D
(Paper)
RSC Adv., 2026,
16, 19626-19635
Process optimization and characterization of fatty acids and lipid concomitants in the aqueous extraction of camellia oil
Received
29th January 2026
, Accepted 31st March 2026
First published on 14th April 2026
Abstract
This study established an aqueous-based method for extracting camellia oil (CO). The optimized process, determined on the basis of bulk oil yield, consisted of sequential procedures: steaming (20 min), enzymatic hydrolysis (1% cellulase, 1% alkaline protease) and aqueous extraction. This process was named steaming aqueous enzymatic extraction (SAEE). Compared with the unheated sample, steaming pretreatment contributed to a 17.56% enhancement in the extraction rate of bulk oil, which was attributed to its degrading effect on cell wall structure and its reduction effect on cellulose crystallinity. In addition, CO was further evaluated in terms of fatty acid profile, deterioration indicators and lipid concomitants. Oleic, linoleic, palmitic and stearic acids were the dominant fatty acids, and oleic acid had the highest content (77.34% ± 0.38% to 77.58% ± 0.24%). The acid values ranged from 0.30 ± 0.02 to 0.35 ± 0.03 mg KOH per g, and the peroxide values were between 0.03 ± 0.00 and 0.04 ± 0.00 g per 100 g. Moreover, phenolics, α-tocopherol, squalene and phytosterols were present in the CO, and α-tocopherol was the distinguishing component, ranging from 277.62 ± 0.77 to 281.33 ± 4.86 mg kg−1. These results suggested that SAEE could be a promising method for producing high-grade CO.
1 Introduction
The camellia oil (CO) extracted from oil tea camellia seeds (CS) is characterized by a high unsaturated fatty acid content (>80%) consisting of oleic, linoleic and linolenic acids.1 It has been demonstrated that these unsaturated fatty acids are capable of providing health benefits to humans.2 In addition, CO contains multitudinous phytochemicals, including polyphenols, vitamin E, squalene and phytosterols, which also exert multiple health effects on humans.3
The extraction process has been identified as a key factor determining the chemical composition of CO.4 Notably, the aqueous extraction process (AEP) is promoted due to its low operating temperature and water-based environment.5 In AEP, water is used as the extraction medium, which is capable of disrupting the binding between triglycerides and proteins. Afterwards, the bulk oil is collected on account of the density difference between oil and water.6 Moreover, an enzymatic hydrolysis procedure is incorporated into the AEP, resulting in the aqueous enzymatic extraction (AEE) method, in which cell wall components and proteins associated with lipid bodies are degraded by enzymes.7,8 Currently, AEE is widely applied in multifold oilseed extraction, offering various advantages. Rice bran oil obtained via AEE exhibits lower acid and peroxide values and higher contents of vitamin E, phytosterols and squalene than the oil obtained through solvent extraction.9 Furthermore, hazardous components such as glycidyl esters and 3-chloropropanol esters are absent in peanut oil prepared by AEE, while they are present in the corresponding oil extracted by pressing or solvent extraction.10 Studies have demonstrated that AEE can not only effectively extract CO but also preserve its bioactive components, resulting in higher contents of vitamin E and squalene compared to solvent extraction.11 However, the application of AEE still faces some challenges in oilseed processing. The intact cell wall structure severely hinders enzymatic efficiency,12 and severe emulsification during extraction leads to the reduction of oil yield.13 Notably, steaming pretreatment facilitates oil release by various effects, such as disrupting cellular structures, increasing material porosity, promoting oil body aggregation and inactivating endogenous enzymes,14–16 which exhibited a higher oil yield when extracting corn germ and date seed.14,17
In this study, an aqueous-based method was established, which sequentially consisted of steaming, cellulase-alkaline protease hydrolysis and aqueous extraction, and was named SAEE. The principle of excellent efficiency of SAEE was investigated by monitoring the chemical structure, crystallinity and surface structure of CS. In addition, the features of CO were evaluated from aspects of fatty acid profile, lipid concomitants and oil degradation indicators. The research route is shown in Fig. 1.
 |
| | Fig. 1 Research route. | |
2 Experimental section
2.1 Chemicals and materials
CS was purchased from the local market in Guizhou Province, China. Standards of β-sitosterol, 5α-cholestane and lanosterol were obtained from Aladdin Co., Ltd (Shanghai, China), and standards of methyl undecanoate, squalene, squalane and α-tocopherol were purchased from Sigma-Aldrich (Saint Louis, MO, USA). In addition, standards of cycloartenol and β-amyrin were purchased from Bvant Co., Ltd (Shanghai, China). Gallic acid and alkaline protease (200 U mg−1) were purchased from Solarbio Science Technology Co., Ltd (Beijing, China), and cellulase (50 U mg−1) was purchased from Rhawn Science Co., Ltd (Shanghai, China). Chromatographic grade n-hexane was obtained from Aladdin Co., Ltd (Shanghai, China). n-Heptane and methanol were of chromatography grade, which were purchased from Kermel Chemical Reagent Co., Ltd (Tianjin, China) and Tedia Company, Inc (Fairfield, OH, USA), respectively. Other chemicals and reagents were of analytical grade.
2.2 Pretreatment of CS
Aliquots of CS were manually dehulled and steamed for 0, 10, 20, 40, 60 and 80 minutes, respectively. Subsequently, these steamed CS samples were dehydrated at 50 °C prior to undergoing a sequential process consisting of crushing and sieving (40-mesh), and the resulting sieved CS were named as S0, S10, S20, S40, S60 and S80, respectively. The preprocessed CS was stored at 4 °C for further analysis.
2.3 Extraction of CO using SAEE
The SAEE procedure was based on a previous study with minor modifications.18 The steamed CS powder was mixed with distilled water at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 (w/v), and the mixture was mixed with HCl (2 mol L−1) to adjust the pH to 4.8 ± 0.2 prior to adding 1% (w/w) cellulase, which subsequently underwent a sequential process consisting of enzymatic hydrolysis (50 °C, 1 h), thermal inactivation (90 °C, 10 min) and cooling. Afterwards, the pH of the mixture was elevated to 9.0 ± 0.2 using NaOH (2 mol L−1) before adding 1% (w/w) alkaline protease; the following process was carried out in the same manner as that for cellulase hydrolysis. The digesta was then separated by centrifugation (6500 g, 20 min) into bulk oil, emulsion and residue. To recover the oil from the emulsion, it was demulsified through the sequential process of freezing (−18 °C, 12 h), thawing (40 °C, 30 min) and centrifuging (6500 g, 10 min). The recovered oil was designated as demulsified oil. The residue was recovered by freeze-drying. The individual sample was subjected to steaming aqueous enzymatic extraction in triplicate. The flow diagram of SAEE is shown in Fig. 2.
:7 (w/v), and the mixture was mixed with HCl (2 mol L−1) to adjust the pH to 4.8 ± 0.2 prior to adding 1% (w/w) cellulase, which subsequently underwent a sequential process consisting of enzymatic hydrolysis (50 °C, 1 h), thermal inactivation (90 °C, 10 min) and cooling. Afterwards, the pH of the mixture was elevated to 9.0 ± 0.2 using NaOH (2 mol L−1) before adding 1% (w/w) alkaline protease; the following process was carried out in the same manner as that for cellulase hydrolysis. The digesta was then separated by centrifugation (6500 g, 20 min) into bulk oil, emulsion and residue. To recover the oil from the emulsion, it was demulsified through the sequential process of freezing (−18 °C, 12 h), thawing (40 °C, 30 min) and centrifuging (6500 g, 10 min). The recovered oil was designated as demulsified oil. The residue was recovered by freeze-drying. The individual sample was subjected to steaming aqueous enzymatic extraction in triplicate. The flow diagram of SAEE is shown in Fig. 2.
 |
| | Fig. 2 Flow diagram of steaming aqueous enzymatic extraction. | |
The extraction rate of bulk oil (Yb, %) and demulsified oil (Yd, %) were calculated referring to eqn (1) and (2), respectively. The oil extraction rate (Y, %) was calculated referring to eqn (3).
| |
 | (1) |
| |
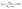 | (2) |
and
where
mb and
md indicate the quantity of bulk oil and demulsified oil obtained from SAEE (g), respectively,
mc indicates the dry weight of CS powder (g), and
a represents the oil content in CS kernel (%).
2.4 Determination of chemical structure using fourier transform infrared (FTIR) spectroscopy
The chemical structure of the preprocessed CS was determined using a FTIR spectrophotometer (Nicolet 6700, Thermo, USA).12,19 The preprocessed CS, prepared as described in Section 2.2, was degreased using petroleum ether extraction prior to crushing and sieving (100-mesh). Afterwards, the CS powder was ground with KBr at a ratio of 1:100 (w/w) before pressing into a thin tablet. Eventually, the tablet was analysed in the spectral range between 500 and 4000 cm−1 at a resolution of 4 cm−1; the scanning count was set at 32. These raw data underwent ordered processing composed of baseline correction and result normalization (referring to the peak of 1504 cm−1) prior to plotting the spectra. The individual sample was measured in triplicate.
2.5 Determination of crystallinity using X-ray diffraction (XRD)
The crystallinity of the preprocessed CS was determined using XRD (Empyrean, PANalytical B. V).12,20,21 The preprocessed CS, prepared as described in Section 2.2, was subjected to sequential procedures of degreasing, crushing and sieving (100-mesh), and then, the CS powder was evenly tiled on the sample rack for XRD scanning over a 2θ range between 5 and 40° at a scan rate of 4° min−1, with the step size set at 0.02°. The crystallinity of the sample was calculated referring the eqn (4).22 The individual sample was analysed in triplicate.| |
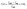 | (4) |
where CrI indicates crystallinity (%), I200 indicates the intensity of the highest peak near 22°, and Iam indicates the intensity of the lowest peak near 18° (amorphous region).
2.6 Determination of surface structure using scanning electron microscopy (SEM)
The surface structure of the CS powder (Section 2.2) and the residue (Section 2.3) was observed using SEM. The powder was glued to a black conductive tape attached to the specimen stub, and then a conductive coating was sprayed on the surface of the sample. The microstructure of sample was monitored using a scanning electron microscope (EM-30, Coxem, South Korea) at a magnification of 1000×.
2.7 Extraction of CO using solvent extraction (SE)
The CS was ground into powder using a crusher prior to mixing with n-hexane at the ratio of 1:6 (w/v), and the oil extraction was carried out with a magnetic stirrer (50 °C, 6 h). The crude oil was collected by sequential procedures of filtrating and solvent removal by a rotary evaporator (45 °C), which was subsequently centrifugated (8000 g, 10 min) for purification, and the purified CO (SEO) was stored at −18 °C for further analysis.23 The individual sample was prepared and analysed in triplicate.
2.8 Extraction of CO using mechanical pressing (MP)
The CS was mechanically pressed using an expeller (Guangzhou Dehaiwei Industrial Equipment Co., Ltd, China) coupled with auxiliary heating at 100 °C, and the crude oil was centrifuged (8000 g, 10 min) to remove physical impurities. The purified CO (MPO) was preserved in a refrigerator at −18 °C for further analysis. The individual sample was prepared and analysed in triplicate.
2.9 Analysis of fatty acid profile
The fatty acid profile of CO was determined according to the protocol established by our group.1 The CO was subjected to sequential procedures of saponification and methylation. The fatty acid methyl esters were analysed using a gas chromatograph (7820 A, Agilent Technologies, USA), and the results were expressed as a percentage of total fatty acids.
2.10 Estimation of acid value (AV)
The AV of CO was determined according to the National Food Safety Standard of China, GB 5009.229. The CO was thoroughly dissolved in an ether–isopropanol solution (1:1, v/v), and then the solution was titrated with KOH. The AV was expressed as milligrams (mg) of KOH per gram of oil.
2.11 Estimation of peroxide value (POV)
The POV of CO was determined in accordance with the National Food Safety Standard of China, GB 5009.227. The CO was thoroughly dissolved in a chloroform–acetic acid solution (2:3, v/v), and then the peroxide content in CO was quantified via potassium iodide oxidation and iodine reduction reactions. The POV was expressed as grams of iodine per 100 grams of CO.
2.12 Determination of total phenolics
Total phenolics in CO were determined using the Folin–Ciocalteu method, referring to a previous study.24 Briefly, the CO (2.0000 g) was dissolved in n-hexane (6 mL), and phenolic compounds were separated using a solid-phase extraction (SPE) column at a flow rate of 1.0 mL min−1. The column was first rinsed with n-hexane to remove impurities and then eluted with methanol to recover the phenolic fraction. The methanolic eluate was dried under a nitrogen stream at 45 °C, and the residue was reconstituted in 2 mL of a methanol–water mixture (1:1, v/v). After being stored at −18 °C for 16 h, the solution was centrifuged at 6500 g for 5 min at 4 °C, and the supernatant was collected. One milliliter of the supernatant was mixed with 0.5 mL of Folin–Ciocalteu reagent. After 5 min of reaction, 2.0 mL of 7.5% sodium carbonate solution was added, and the volume was adjusted to 10 mL with distilled water. The mixture was allowed to react in the dark for 1 h, and absorbance was measured at 765 nm using a spectrophotometer (UV-5200 PC, Yuanxi Instrument Co., Ltd, Shanghai). A gallic acid standard was subjected to the above procedures, and total phenolics were calculated referring to the standard curve and expressed as mg of gallic acid equivalent (GAE) per kilogram (kg) of CO (mg GAE per kg).
2.13 Analysis of α-tocopherol content
The α-tocopherol content was determined using HPLC (1260 infinity, Agilent Technologies, USA), as described by a previous study.24 The CO (0.5000 g) was thoroughly dissolved in n-hexane, of which the volume was adjusted to 10 mL with n-hexane. Afterwards, the sample was analysed by HPLC coupled with the ZorBax RX-SIL column (250 mm × 4.6 mm, 5 µm) at 30 °C, with isocratic elution at 1 mL min−1 (n-hexane/isopropanol, v/v = 98.5:1.5). α-Tocopherol was identified using a fluorescence detector with excitation and emission wavelengths of 294 nm and 328 nm, respectively, and the content was quantified by referring to the standard curve and expressed as mg of α-tocopherol per kg of CO (mg kg−1).
2.14 Analysis of squalene content
The squalene content was determined using GC, as described by a previous study.24 The CO (1.0000 g) was vigorously mixed with an internal standard consisting of squalane (300 µL, 1 mg mL−1) and 5α-cholestane (300 µL, 1 mg mL−1), and then, the mixture was saponified by ethanolic potassium hydroxide solution (40 mL, 1 mol L−1) at 80 °C for 50 min. The saponified mixture was extracted for two times by a biphasic system composed of n-hexane (50 mL) and distilled water (50 mL). The n-hexane phase was collected using a separatory funnel, and the aqueous phase was extracted for two times using n-hexane. These n-hexane phases were combined and then washed to neutrality using ethanol solution (ethanol/water, v/v = 1:9, 50 mL). The total n-hexane phase was dehydrated over anhydrous sodium sulfate prior to removing the solvents by a rotary evaporator at 40 °C, and the extract was reconstituted to 10 mL using n-hexane.
One microliter of the extract was injected into a GC outfitted with an HP-5 quartz capillary column (30 m × 0.32 mm × 0.25 µm) and a flame ionization detector (FID) (300 °C), with a split ratio of 1:10. The oven temperature initiated from 160 °C and increased to 220 °C at a rate of 15 °C min−1, where it was held for 2 min, and then it was sequentially raised to 280 °C at a rate of 5 °C min−1 and held for 5 min. Eventually, it was ascended to 300 °C at a rate of 5 °C min−1 and maintained for 5 min. Squalene was identified in accordance with the retention time of the standard, and the content was quantified referring to the internal standard. Results were expressed as mg of squalene per kg of CO (mg kg−1).
2.15 Analysis of the phytosterol compound profile
The phytosterol contents were determined according to previous studies,24,25 using the same n-hexane extract prepared in Section 2.14. One microliter of the extract was injected into the injector (250 °C) of the GC with the split ratio of 1:10. Separation of the extract was performed on an HP-5 quartz capillary column coupled with a heating program. The initial temperature of the oven was set at 160 °C for 1 min, which was then ascended to 280 °C at 15 °C min−1 and held for 5 min. It was eventually raised to 300 °C at 5 °C min−1 and maintained for 8 min. The FID was employed to detect phytosterol compounds at 330 °C. Phytosterol compounds were identified in accordance with the retention time of the standard, and 5α-cholestane served as the internal standard for calculating the contents, which were expressed as mg of phytosterols per kg of CO (mg kg−1).
2.16 Statistical analysis
Data are presented as means ± standard deviation (SD) (n = 3), and the ANOVA and Tukey's multiple comparisons (IBM SPSS Statistics 26) were adopted to test significant differences at the level of p < 0.05.
3 Results and discussion
3.1 Extraction of CO using SAEE
3.1.1 Extraction efficiency of CO. In terms of AEP, a significant challenge for oil release is interfering substances, including the cell wall structure and amphiphilic components.12,18 Fortunately, thermal treatment has the potential to disrupt the cell wall and cleave covalent bonds between lipids and the cell matrix, which promotes the aggregation of oil.26,27 Additionally, cellulases and proteases also show significant efficiency for hydrolysing the cell wall structure of oilseeds.28 Accordingly, an aqueous-based extraction method was established to extract CO from CS, which was sequentially composed of steaming, cellulases-protease hydrolysis and aqueous extraction. Oil directly released from CS was referred to as bulk oil, and oil associated with amphiphilic substances was present as an emulsion. Hence, a demulsification procedure was incorporated into the SAEE process. The extraction efficiencies were between 74.45% ± 1.70% and 87.06% ± 1.51% depending on steaming time, which was comparable to the 82.95% efficiency reported by Zhu et al.,13 and the steaming procedure ascended the extraction efficiency, as shown in Fig. 3. Notably, the aliquot processed by steaming for 20 min presented the highest rate of bulk oil relative to total oil. Specifically, the rates of bulk oil relative to total oil ranged from 10.09% ± 2.16% to 27.65% ± 3.74%, and the rates of demulsified oil relative to total oil ranged from 55.52% ± 1.76% to 66.56% ± 5.52%.
 |
| | Fig. 3 Extraction rate of camellia oil using SAEE (steaming aqueous enzymatic extraction) with different steaming times. Within the same color, error bars with no letters in common indicate significant differences (p < 0.05). | |
Steaming pretreatment and enzymatic hydrolysis synergistically promoted CO release. Protein and cellulose are the dominant components of CS, which were efficiently degraded by protease and cellulase.13 Moreover, heat treatment modified the lipoprotein membrane of oilseeds, promoting oil release due to the reduction in oil viscosity,26 and the porosity of oilseeds was increased under heating conditions by rupturing the cell wall and cleaving covalent bonds between lipid and the cellular matrix,26,27 which further enhanced oil release and enzyme infiltration.
3.1.2 Changes in the chemical structure of CS induced by steaming. During the steaming process, the chemical structures of oilseed components were prone to collapse, which can be characterized by identifying functional groups using FTIR spectroscopy. The absorption peaks of these samples were observed at wavenumbers of 3390, 2930, 1743, 1650, 1413, 1250, 1109, 1053, 925 and 839 cm−1, as shown in Fig. 4. The absorption peaks at 2930 cm−1 and 1743 cm−1 corresponded to the C–H stretching vibrations of methyl or methylene groups in cellulose molecules and the free C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations in hemicellulose, respectively.21,29,30 The absorbances of these peaks were higher in steamed samples than in their unsteamed counterparts, which was attributed to the rearrangement of C–H groups and exposure of CO groups induced by steaming.12,31 Additionally, the absorption peaks at 3390 cm−1 and 1053 cm−1 related to the O–H and C–O–C stretching vibrations in t cellulose and hemicellulose molecule, respectively.21,29 The absorbances of both peaks decreased after steaming, indicating partial breakage of intermolecular hydrogen bonds and glycosidic linkages within cellulose and hemicellulose molecules.21 These structural alterations may contribute to the formation of a loosened and porous matrix, enhancing enzyme accessibility.12,21 Furthermore, the absorption peaks at 1650 cm−1 and 839 cm−1 corresponded to the CO stretching vibration of carboxyl groups in pectin and the ring vibration of α-bonding between galacturonic acid units in the backbone of most pectin, respectively.21,32 With respect to the absorbances of both peaks, a gradual descending trend was observed with prolonged steaming, which may suggest that steaming treatment led to the degradation of pectin. In addition, the absorption peaks at 1109 cm−1 and 1413 cm−1 corresponded to the C–O–C tensile vibrations and the C–H bending vibrations of methyl or methylene groups in cellulose, respectively,29,32 and absorbances of both showed a small amount of decrease after 80 min. The absorption peaks at 1250 cm−1 and 925 cm−1 were associated with the C–O tensile vibrations in lignin and the ring vibrations in hemicellulose, respectively.21,33 The absorbance of both remained constant throughout the whole process, as shown in Table 1. To sum up, these results suggest that the steaming process may promote the degradation of cellulose, hemicellulose and pectin in oilseeds by means of breaking intermolecular chemical bonds within the cell wall matrix. The breakdown of these cell wall components converted the intact cellular structure into a loosened and porous matrix, improving enzyme accessibility, which may account for the elevated extraction efficiency of CO. These results are consistent with the study by Guo et al., who reported that microwave pretreatment of peanut powder could degrade cellulose, hemicellulose as well as pectin and disrupt the crystalline structure of cellulose.12
O stretching vibrations in hemicellulose, respectively.21,29,30 The absorbances of these peaks were higher in steamed samples than in their unsteamed counterparts, which was attributed to the rearrangement of C–H groups and exposure of CO groups induced by steaming.12,31 Additionally, the absorption peaks at 3390 cm−1 and 1053 cm−1 related to the O–H and C–O–C stretching vibrations in t cellulose and hemicellulose molecule, respectively.21,29 The absorbances of both peaks decreased after steaming, indicating partial breakage of intermolecular hydrogen bonds and glycosidic linkages within cellulose and hemicellulose molecules.21 These structural alterations may contribute to the formation of a loosened and porous matrix, enhancing enzyme accessibility.12,21 Furthermore, the absorption peaks at 1650 cm−1 and 839 cm−1 corresponded to the CO stretching vibration of carboxyl groups in pectin and the ring vibration of α-bonding between galacturonic acid units in the backbone of most pectin, respectively.21,32 With respect to the absorbances of both peaks, a gradual descending trend was observed with prolonged steaming, which may suggest that steaming treatment led to the degradation of pectin. In addition, the absorption peaks at 1109 cm−1 and 1413 cm−1 corresponded to the C–O–C tensile vibrations and the C–H bending vibrations of methyl or methylene groups in cellulose, respectively,29,32 and absorbances of both showed a small amount of decrease after 80 min. The absorption peaks at 1250 cm−1 and 925 cm−1 were associated with the C–O tensile vibrations in lignin and the ring vibrations in hemicellulose, respectively.21,33 The absorbance of both remained constant throughout the whole process, as shown in Table 1. To sum up, these results suggest that the steaming process may promote the degradation of cellulose, hemicellulose and pectin in oilseeds by means of breaking intermolecular chemical bonds within the cell wall matrix. The breakdown of these cell wall components converted the intact cellular structure into a loosened and porous matrix, improving enzyme accessibility, which may account for the elevated extraction efficiency of CO. These results are consistent with the study by Guo et al., who reported that microwave pretreatment of peanut powder could degrade cellulose, hemicellulose as well as pectin and disrupt the crystalline structure of cellulose.12
 |
| | Fig. 4 Fourier transform infrared (FTIR) spectra of oil tea camellia seeds prepared by steaming for different times. | |
Table 1 Peak absorbances in the FTIR spectra of CS prepared by steaming for different timesa
| Wave number (cm−1) |
Absorbance |
| S0 |
S10 |
S20 |
S40 |
S60 |
S80 |
| Values (means ± SD, n = 3) with different lowercase letters within the same row indicate significant differences (p < 0.05). FTIR indicates fourier transform infrared spectroscopy; CS indicates oil tea camellia seeds. |
| 3390 |
5.69 ± 0.31 a |
5.33 ± 0.13 ab |
5.48 ± 0.26 ab |
5.36 ± 0.18 ab |
5.04 ± 0.23 ab |
4.45 ± 0.99 b |
| 2930 |
2.09 ± 0.03 b |
2.65 ± 0.35 a |
2.47 ± 0.04 ab |
2.37 ± 0.02 ab |
2.54 ± 0.14 a |
2.33 ± 0.30 ab |
| 1743 |
0.61 ± 0.02 c |
0.88 ± 0.06 b |
0.87 ± 0.01 b |
0.81 ± 0.03 b |
0.97 ± 0.05 a |
0.97 ± 0.05 a |
| 1650 |
3.42 ± 0.16 a |
3.29 ± 0.05 ab |
3.26 ± 0.13 ab |
3.26 ± 0.06 ab |
3.16 ± 0.08 ab |
2.76 ± 0.52 b |
| 1413 |
2.00 ± 0.04 a |
2.09 ± 0.01 a |
2.07 ± 0.04 a |
2.05 ± 0.00 a |
2.00 ± 0.02 a |
1.84 ± 0.26 a |
| 1250 |
1.43 ± 0.02 a |
1.45 ± 0.08 a |
1.46 ± 0.01 a |
1.45 ± 0.00 a |
1.44 ± 0.04 a |
1.35 ± 0.15 a |
| 1109 |
2.95 ± 0.04 a |
2.96 ± 0.08 a |
2.99 ± 0.06 a |
2.99 ± 0.01 a |
2.93 ± 0.06 a |
2.61 ± 0.55 a |
| 1053 |
4.13 ± 0.06 a |
4.03 ± 0.12 a |
4.12 ± 0.10 a |
4.11 ± 0.04 a |
3.99 ± 0.07 a |
3.50 ± 0.85 a |
| 925 |
0.92 ± 0.00 a |
0.94 ± 0.25 a |
0.94 ± 0.04 a |
0.90 ± 0.00 a |
0.89 ± 0.14 a |
0.87 ± 0.09 a |
| 839 |
0.62 ± 0.06 a |
0.53 ± 0.45 a |
0.53 ± 0.05 a |
0.51 ± 0.03 a |
0.47 ± 0.15 a |
0.47 ± 0.02 a |
3.1.3 Changes in the crystallinity of CS induced by steaming. The crystalline structure mainly originates from cellulose components arranged in a fiber crystal array, which not only determines the arrangement of atoms in the crystalline regions but also is responsible for the thermal stability and mechanical properties of the matrix.12,30 Meanwhile, crystallinity reflects the proportion of these ordered regions within the matrix and can be detected using XRD. The changes in crystallinity of CS with increasing steaming time are shown in Fig. 5. In this study, the crystallinity of CS was calculated by using the highest peak intensity of the point near 22° and the lowest peak intensity of the point near 18°, where the peak near 22° corresponds to type I cellulose,21 and the peak near 18° is attributed to amorphous scattering.12 The crystallinity of raw CS was significantly higher (p < 0.05) than that of steamed CS, as shown in Fig. 5B. This result is in accordance with a previous study suggesting that steaming treatment reduces cellulose content and crystallinity in Cocos nucifera,34 which may be due to the fragmentation of crystalline grains during steaming. It induced the deformation of the crystal structure, increasing the amorphous regions.35 Similarly, Guo et al. reported that the crystallinity of peanut powder also exhibited a decreasing trend with prolonged microwave heating, which was in favour of enzymatic accessibility.12 This study suggests that the steaming process significantly reduces the crystallinity of cellulose in CS by disrupting its crystalline structure, which resulted in a decrease in thermal stability.
 |
| | Fig. 5 X-ray diffraction (XRD) spectra (A) and crystallinity (B) of oil tea camellia seeds prepared by steaming for different times. Error bars with no letters in common indicate significant differences (p < 0.05). | |
3.1.4 Surface structure of CS and its residue. The surface structure of raw and steamed CS (20 min) and their residues were monitored using SEM. The raw CS exhibited a smooth surface composed of numerous spherical and lumpy particles, as shown in Fig. 6A. In contrast, the steamed CS showed a rough surface consisted of abundant irregular pores (Fig. 6B), which was similar to a previous study reporting that obvious porosity and irregular cavities were present in CS processed by microwave heating.36
 |
| | Fig. 6 Scanning electron microscopy (SEM) images of CS and residues. (A) and (C) show the raw camellia seeds and the corresponding residue, respectively, while (B) and (D) represent the steamed camellia seeds (20 min) and the corresponding residue, respectively. IS indicates intracellular substances. | |
The surface structures of raw and steamed CS residues after the SAEE process were also monitored. Compared with raw CS, the surface structure of the corresponding residue exhibited a significant deterioration, characterized by larger cavities and folds, and a small amount of intracellular substances was present in the CS residue (Fig. 6C). These results suggest that the CS cell wall was disrupted during the SAEE process, and the CO as well as intracellular substances (proteins and starch granules) were simultaneously extracted in this process. This finding is in accordance with the study reported by Xu et al.9 Analogously, compared with steamed CS, the surface of its residue exhibited a further increase in the number and apparent size of surface cavities (Fig. 6D). In addition, the steamed CS residue exhibited more pronounced structural disruption than the raw CS residue, which may facilitate oil release during extraction. Zhang et al. suggested that the elevated oil yield originates from the impact of the steaming process, which promotes the formation of a looser surface structure and the aggregation of oil droplets in oilseeds.16
3.2 Assessment of CO quality
The CO observed from SAEE was assessed from aspects of fatty acid profile and deterioration indicators (AV and POV). Notably, it was separately determined for bulk and demulsified fractions, and the MPO and SEO (oils prepared from MP and SE process, respectively) served as controls.
3.2.1 Fatty acid profile. Fatty acids are the dominant components in edible oil, and their profiles are associated with their nutritional value. The CO prepared by SAEE contained palmitic, stearic, oleic, linoleic and linolenic acids, and oleic acid made a dominant contribution (between 77.34 ± 0.38 and 77.58% ± 0.24%) to the total content (Table 2). Oleic acid is capable of regulating cholesterol levels and preventing arteriosclerosis.37 These results are consistent with those reported by Wang et al. (77.12% ± 0.09% and 79.49% ± 0.01%). In addition, a few fatty acids were identified in CO including palmitoleic acid (C16:1), eicosadienoic acid (C20:2) and docosadienoic acid (C22:2).38 The individual fatty acid contents in the bulk fraction were comparable to those in the demulsified fraction, which is consistent with the result reported by Zhu et al.13
Table 2 Acid value, peroxide value and fatty acid content of COa
| Indicators |
CO prepared from SAEE |
SEO |
MPO |
| Bulk fraction |
Demulsified fraction |
| Values (means ± SD, n = 3) with different lowercase letters within the same row indicate significant differences (p < 0.05). CO indicates camellia oil, SAAE indicates steaming aqueous enzymatic extraction, SEO indicates CO prepared from solvent extraction, and MPO indicates CO prepared from mechanical pressing. |
| Acid value (mg KOH per g) |
0.30 ± 0.02 c |
0.35 ± 0.03 c |
2.24 ± 0.03 a |
0.73 ± 0.06 b |
| Peroxide value (g per 100 g) |
0.04 ± 0.00 a |
0.03 ± 0.00 b |
0.01 ± 0.00 c |
0.01 ± 0.00 c |
| Fatty acid content (%) |
|
|
|
|
| Palmitic acid |
11.14 ± 0.06 a |
11.22 ± 0.25 a |
11.17 ± 0.07 a |
11.03 ± 0.07 a |
| Stearic acid |
2.76 ± 0.06 a |
2.83 ± 0.11 a |
2.82 ± 0.01 a |
2.74 ± 0.19 a |
| Oleic acid |
77.58 ± 0.24 ab |
77.34 ± 0.38 b |
77.12 ± 0.07 b |
77.93 ± 0.08 a |
| Linoleic acid |
7.85 ± 0.07 b |
7.95 ± 0.02 b |
8.18 ± 0.06 a |
7.69 ± 0.06 c |
| Linolenic acid |
0.66 ± 0.06 ab |
0.66 ± 0.03 ab |
0.71 ± 0.00 a |
0.62 ± 0.06 b |
| Saturated fatty acids |
13.91 ± 0.12 a |
14.05 ± 0.34 a |
13.99 ± 0.07 a |
13.77 ± 0.16 a |
| Unsaturated fatty acids |
86.09 ± 0.12 a |
85.95 ± 0.34 a |
86.01 ± 0.07 a |
86.23 ± 0.16 a |
| Polyunsaturated fatty acids |
8.52 ± 0.13 a |
8.61 ± 0.05 a |
8.89 ± 0.06 a |
8.30 ± 0.12 a |
3.2.2 AV and POV of CO. The AV and POV are major indicators for evaluating the deterioration level of edible oils. The AV indicates the content of free fatty acids in edible oil,39 and the AV values of the bulk and demulsified fractions were 0.30 ± 0.02 and 0.35 ± 0.03 mg KOH per g, respectively, both of which were significantly lower than those of SEO and MPO, as shown in Table 2. This is consistent with the results reported by Zhang et al., who found that the AV of CO prepared by aqueous enzymatic extraction was lower than that obtained from hexane extraction and cold pressing.40 Similarly, the acid value of gardenia fruit oil extracted by aqueous enzymatic extraction was significantly lower than that obtained from pressing and solvent extraction.23 The mild and alkaline circumstances in the aqueous enzymatic extraction process accounted for the low AV of oil.41 The POV indicates the content of primary oxidation products present in edible oil,42 and the POV of CO was similar to those of MPO and SEO, as shown in Table 2.
3.3 Lipid concomitant profile of CO
CO contains multitudinous concomitants, consisting of phenolic compounds, α-tocopherol, squalene and phytosterols,43 which also serve as indicators for evaluating oil quality due to their multiple functional properties.3 These compounds not only exhibit bioactive properties including antioxidant, anti-inflammatory and hypolipidemic activities, but also play crucial roles in delaying oil oxidation.3,44–46 Therefore, the concomitant profile is of great importance for improving CO quality.
3.3.1 Total phenolics. Phenolic compounds are secondary metabolites of CS. Subgroups, such as phenolic acids, catechins and other flavonoids, are present in CO,47 and these phenolics are capable of delaying CO deterioration by quenching lipid radicals while providing health benefits to humans.48,49 Total phenolics in the bulk and demulsified oils were 11.93 ± 0.97 and 12.07 ± 1.13 mg GAE per kg, respectively, as shown in Table 3, which are higher than the 8.98 mg kg−1 reported for CO extracted by AEE in a previous study.44 The enzymatic hydrolysis in SAEE facilitates the release of these phenolics by disrupting the interactions between antioxidants and the seed matrix, such as polysaccharides, proteins and pectin.8,50 In addition, phenolic compounds in CO obtained from AEE contain rutin, quercetin, apigenin, catechins and benzoic acid.44,51
Table 3 Lipid concomitant profile of COa
| Lipid concomitants |
CO prepared from SAEE |
SEO |
MPO |
| Bulk fraction |
Demulsified fraction |
| Values (means ± SD, n = 3) with different lowercase letters within the same row indicate significant differences (p < 0.05). CO indicates camellia oil, SAAE indicates steaming aqueous enzymatic extraction, SEO indicates CO prepared from solvent extraction, and MPO indicates CO prepared from mechanical pressing. |
| Total phenolics (mg GAE per kg) |
11.93 ± 0.97 b |
12.07 ± 1.13 b |
14.07 ± 2.70 b |
19.31 ± 3.21 a |
| α-Tocopherol (mg kg−1) |
281.33 ± 4.86 a |
277.62 ± 0.77 a |
278.68 ± 3.86 a |
266.95 ± 0.56 b |
| Squalene (mg kg−1) |
198.35 ± 3.72 a |
190.15 ± 7.06 ab |
185.12 ± 5.02 b |
198.61 ± 6.88 a |
| Phytosterols (mg kg−1) |
|
|
|
|
| β-Sitosterol (mg kg−1) |
274.02 ± 2.80 ab |
265.84 ± 7.31 b |
287.89 ± 11.42 a |
261.83 ± 7.63 b |
| Lanosterol (mg kg−1) |
350.91 ± 9.68 a |
344.42 ± 6.31 a |
335.75 ± 12.71 a |
345.38 ± 8.63 a |
| β-Amyrin (mg kg−1) |
825.75 ± 20.27 a |
809.99 ± 8.51 a |
794.25 ± 31.88 a |
816.42 ± 10.00 a |
| Cycloartenol (mg kg−1) |
28.35 ± 3.48 ab |
27.30 ± 1.23 ab |
30.75 ± 2.80 a |
23.68 ± 1.42 b |
| Total phytosterols (mg kg−1) |
1479.03 ± 23.37 a |
1447.54 ± 9.78 a |
1448.65 ± 53.24 a |
1447.32 ± 22.17 a |
3.3.2 α-Tocopherol content. Tocopherol is a lipid-soluble phytochemical composed of four isomers (α-, β-, γ- and δ-).45 α-Tocopherol is the predominant form of tocopherol in CO, accounting for 98% of total tocopherol.52 Previous studies have reported that α-tocopherol is the sole tocopherol present in CO, with contents ranging from 187.54 to 330.51 mg kg−1.53 It is considered an indicator for evaluating the tocopherol level in CO in the present study. α-Tocopherol not only delays oil oxidation but also provides health benefits to humans, including reducing the risk of cardiovascular diseases and certain cancers.45 There was no significant difference (p > 0.05) in α-tocopherol content among the bulk fraction, demulsified fraction and SEO, which ranged from 277.62 ± 0.77 to 281.33 ± 4.86 mg kg−1; notably, these values were higher than those of MPO, as shown in Table 3. Fang et al. also suggested that the α-tocopherol content in CO obtained by aqueous enzymatic extraction was higher than that obtained by physical pressing.51 Tocopherols are susceptible to thermal and may be degraded during physical pressing.54 In addition, α-tocopherol contents in CO from roasting and microwave-assisted aqueous enzymatic extraction were 134.9 ± 0.4 mg kg−1 and 135.7 ± 0.9 mg kg−1, respectively,40 both of which are lower than the present results.
3.3.3 Squalene content. Squalene is a lipid-soluble component composed of six isoprene units,55 and it is employed as a precursor in the synthesis of phytosterols, steroids and vitamin D.56 Squalene also possesses anti-inflammatory and antioxidant effects.57 The squalene contents in the bulk and demulsified fractions were 198.35 ± 3.72 and 190.15 ± 7.06 mg kg−1, respectively, and were comparable to those of MPO and SEO, as shown in Table 3. Our previous study showed that the squalene content in different CO ranged from 45.8 ± 0.8 to 184.1 ± 5.5 mg kg−1,24 which were lower than those reported in the present study. This difference may be primarily attributed to variations in extraction methods. The present results were lower than that (215.5 ± 3.2 mg kg−1) reported by Yuan et al.,58 which may be attributed to differences in CS varieties and extraction processes.
3.3.4 Phytosterol content. Phytosterols are composed of a sterol nucleus and an alkyl chain,45 and they not only provide health benefits, including cholesterol-lowering, anti-inflammatory, antibacterial and antitumor,59 but also possess antioxidant activity in edible oil.60 The present results showed that total phytosterols in the bulk and demulsified fractions were 1479.03 ± 23.37 and 1447.54 ± 9.78 mg kg−1, respectively, with no significant difference (p > 0.05) compared to MPO and SEO. Phytosterols consisted of β-sitosterol, lanosterol, β-amyrin and cycloartenol. Notably β-amyrin made a dominant contribution to total phytosterols, accounting for 55.0% and 55.9% of total phytosterols in the bulk and demulsified fractions, respectively, followed by lanosterol, β-sitosterol and cycloartenol, as shown in Table 3. In this study, the β-amyrin contents of CO from SAEE were 809.99 ± 8.51 and 825.75 ± 20.27 mg kg−1 in the bulk and demulsified fractions, respectively, which are consistent with the results reported by Wang et al. (607.24 ± 4.49–913.15 ± 10.04 mg kg−1), while the β-sitosterol content (265.84 ± 7.31–274.02 ± 2.80 mg kg−1) was higher than that reported in a previous study (106.96 ± 2.98–240.12 ± 7.94 mg kg−1).38 This variation may be attributed to differences in CS cultivars and regional climatic conditions. The contents of individual phytosterols in CO obtained from SAEE and the control (MPO and SEO) showed no significant differences.It should be noted that a refining process is essential for edible oil obtained by pressing and solvent extraction before consumption, which results in a reduction (50–90%) of bioactive compounds, such as α-tocopherol, squalene and polyphenols.61 In contrast, CO prepared from SAEE meets edible oil standards after moisture removal,61 avoiding the loss of bioactive components. The present study showed that CO prepared from SAEE presented a lower acid value and comparable levels of α-tocopherol, squalene and phytosterols compared to MPO and SEO. This suggests that the SAEE process may be a promising approach for producing high-grade CO.
4 Conclusions
It was established that the CO extraction method consist of steaming (20 min), enzymatic hydrolysis (1% cellulase, 1% alkaline protease) and aqueous extraction, which exhibited excellent efficiency in terms of bulk oil yield. CO was obtained in bulk and emulsion forms, and the steaming pretreatment facilitated CO extraction, ascribing to its destroying effect on cellulose, hemicellulose and pectin in the oilseeds, as well as the reduction effect on cellulose crystallinity. The levels of these deterioration indicators for CO were within acceptable ranges. Moreover, the CO contained multitudinous concomitants including phenolics, α-tocopherol, squalene and phytosterols, which suggests that SAEE can be employed to produce high-grade CO. Nevertheless, several limitations remain in the present study. The pore structure of samples subjected to different steaming times is not quantitatively characterized, and the formation mechanism of emulsions during the extraction process remains to be further clarified. Furthermore, the profiles of minor concomitants and the flavor quality of CO require systematic investigation. Future studies should focus on elucidating the influence of steaming pretreatment on the pore structure of oilseeds by means of BET analysis. In addition, the mechanism of emulsion formation during extraction should be further clarified based on interfacial chemistry. A more comprehensive characterization of lipid concomitants and flavor properties of CO is also warranted.
Author contributions
Haiqing Fan: data curation, formal analysis, writing – original draft, writing – review and editing, investigation, methodology, visualization, conceptualization. Yong Zhu: funding acquisition, investigation, writing – review and editing, project administration, methodology, resources, conceptualization, supervision, validation. Likang Qin: conceptualization, resources. Haiyan Zhong: conceptualization, resources. Yue Chen: formal analysis. Qianrui Wang: methodology. Wenxin Zha: methodology. Shimao Chen: methodology.
Conflicts of interest
The authors have declared no conflicts of interest.
Data availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (Grant No. 32460410) and the Guizhou Science and Technology Program, China [Qian Ke He zhong da zhuan xiang zi (2024) 025; ZSYS (2025) 032].
References
- C. He, L. Qin, J. Yang and Y. Zhu, LWT--Food Sci. Technol., 2023, 173, 114293 CrossRef CAS.
- J. Chen and H. Liu, Int. J. Mol. Sci., 2020, 21, 5695 CrossRef CAS PubMed.
- P. Qin, J. Shen, J. Wei and Y. Chen, Curr. Res. Food Sci., 2024, 8, 100753 CrossRef CAS PubMed.
- G. Li, L. Ma, Z. Yan, Q. Zhu, J. Cai, S. Wang, Y. Yuan, Y. Chen and S. Deng, Processes, 2022, 10, 1489 CrossRef CAS.
- S. Wang, Y. Guo, D. Xie, L. Zheng, X. Liu and Z. Wang, Food Chem., 2024, 447, 138934 CrossRef CAS PubMed.
- A. Rosenthal, D. L. Pyle and K. Niranjan, Food Bioprod. Process., 1998, 76, 224–230 CrossRef CAS.
- C. Benkirane, A. Ben Moumen, A. Allay, Y. Rbah, M. Barkaoui, H. Serghini Caid, A. Elamrani and F. Mansouri, Biocatal. Agric. Biotechnol., 2024, 61, 103354 CrossRef CAS.
- J. Jiao, Z. Li, Q. Gai, X. Li, F. Wei, Y. Fu and W. Ma, Food Chem., 2014, 147, 17–24 CrossRef CAS PubMed.
- D. Xu, J. Hao, Z. Wang, D. Liang, J. Wang, Y. Ma and M. Zhang, LWT--Food Sci. Technol., 2021, 149, 111817 CrossRef CAS.
- P. Li, PhD thesis, Jiangnan University, 2017, in Chinese.
- X. Fang, X. Fei, H. Sun and Y. Jin, Eur. J. Lipid Sci. Technol., 2016, 118, 244–251 CrossRef CAS.
- X. Guo, B. Wu, Y. Jiang, Y. Zhang, B. Jiao and Q. Wang, Food Hydrocoll., 2024, 147, 109407 CrossRef CAS.
- F. Zhu, R. Wu, B. Chen, F. Zhang, Y. Chen, F. Cao, P. Yu and E. Su, Ind. Crop. Prod., 2024, 212, 118392 CrossRef CAS.
- S. Amigh and S. Taghian Dinani, Heat Mass Tran., 2020, 56, 2345–2354 CrossRef CAS.
- Y. Gao, Z. Ding, Y. Liu and Y. Xu, Trends Food Sci. Technol., 2024, 144, 104315 CrossRef CAS.
- Z. Zhang, H. Jia, H. Qin, Y. Wei, B. Li, Y. Liu, A. Wei, W. Zhu and Y. Wang, LWT--Food Sci. Technol., 2023, 184, 114985 CrossRef CAS.
- L. C. Dickey, M. J. Kurantz and N. Parris, Ind. Crop. Prod., 2008, 27, 303–307 CrossRef CAS.
- Y. Lu, R. Hou, S. Shao, J. Li, N. Yu, X. Nie and X. Meng, Food Chem., 2023, 414, 135681 CrossRef CAS PubMed.
- T. Raj, R. Gaur, B. Y. Lamba, N. Singh, R. P. Gupta, R. Kumar, S. K. Puri and S. S. V. Ramakumar, Bioresour. Technol., 2018, 249, 139–145 CrossRef CAS PubMed.
- S. Chen, R. Liu, Y. Lei, J. J. Morrell and L. Yan, J. Mater. Res. Technol., 2021, 11, 1637–1644 CrossRef CAS.
- H. Ouyang, L. Wu, Y. Hu, L. Li, Z. Li, H. He, Z. Jiang, Q. Li, H. Ni and M. Zheng, LWT--Food Sci. Technol., 2023, 184, 114963 CrossRef CAS.
- L. Segal, J. J. Creely, A. E. Martin and C. M. Conrad, Text. Res. J., 1959, 29, 786–794 CrossRef CAS.
- D. Wang, Y. Yuan, T. Xie, G. Tang, G. Song, L. Li, T. Yuan, F. Zheng and J. Gong, Ind. Crop. Prod., 2023, 191, 116021 CrossRef CAS.
- Q. Li, Y. Zhu and L. Qin, J. Oleo Sci., 2024, 73, 943–952 CrossRef CAS PubMed.
- S. Zhong, B. Huang, T. Wei, Z. Deng, J. Li and Q. Wen, Foods, 2023, 12, 374 CrossRef CAS PubMed.
- S. Cao, F. Xiang, S. Li, X. Ma, H. Hu, Q. Guo, B. Jiao, D. Agyei, Q. Wang and A. Shi, LWT--Food Sci. Technol., 2024, 206, 116596 CrossRef CAS.
- K. Suri, B. Singh and A. Kaur, Food Chem., 2022, 368, 130777 CrossRef CAS PubMed.
- X. Meng, H. Ge, Q. Ye, L. Peng, Z. Wang and L. Jiang, J. Am. Oil Chem. Soc., 2018, 95, 29–37 CrossRef CAS.
- X. Tian, J. Liu, C. Qiao, Z. Cheng, N. Wu and B. Tan, J. Cereal. Sci., 2024, 118, 103938 CrossRef CAS.
- Z. Wang, E. Xu, F. Fu, L. Lin and S. Yi, Ind. Crop. Prod., 2022, 187, 115341 CrossRef CAS.
- J. Qian, F. Zhao, J. Gao, L. Qu, Z. He and S. Yi, Ultrason. Sonochem., 2021, 77, 105672 CrossRef CAS PubMed.
- S. Beluns, S. Gaidukovs, O. Platnieks, G. Gaidukova, I. Mierina, L. Grase, O. Starkova, P. Brazdausks and V. K. Thakur, Ind. Crop. Prod., 2021, 170, 113780 CrossRef CAS.
- M. Szymanska-Chargot, M. Chylinska, B. Kruk and A. Zdunek, Carbohydr. Polym., 2015, 115, 93–103 CrossRef CAS PubMed.
- H. Kolya and C. Kang, Original Research, 2021 Search PubMed.
- J. Jiang, J. Wang, X. Zhang and M. Wolcott, Ind. Crop. Prod., 2017, 109, 498–508 CrossRef CAS.
- M. Ye, H. Zhou, J. Hao, T. Chen, Z. He, F. Wu and X. Liu, Ind. Crop. Prod., 2021, 161, 113193 CrossRef CAS.
- M. Pieszka, W. Migdał, R. Gąsior, M. Rudzińska, D. Bederska-Łojewska, M. Pieszka and P. Szczurek, J. Chem., 2015, 2015, 659541 CrossRef.
- X. Wang, Q. Zeng, V. Verardo and M. D. M. Contreras, Food Chem., 2017, 233, 302–310 CrossRef CAS PubMed.
- L. Zhou, Y. Peng, Z. Xu, J. Chen, N. Zhang, T. Liang, T. Chen, Y. Xiao, S. Feng and C. Ding, Molecules, 2024, 29, 1864 CrossRef CAS PubMed.
- H. Zhang, P. Gao, Y. Mao, J. Dong, W. Zhong, C. Hu, D. He and X. Wang, LWT--Food Sci. Technol., 2023, 173, 114396 CrossRef CAS.
- S. B. Zhang, Q. Y. Lu, H. Yang, Y. Li and S. Wang, J. Am. Oil Chem. Soc., 2011, 88, 727–732 CrossRef CAS.
- N. K. Mohammed, A. S. Meor Hussin, C. P. Tan, M. Y. Abdul Manap and A. M. Alhelli, Int. J. Food Prop., 2017, 20, S2395–S2408 CrossRef CAS.
- T. Shi, G. Wu, Q. Jin and X. Wang, Trends Food Sci. Technol., 2020, 97, 88–99 CrossRef CAS.
- Y. Lu, R. Hou, M. Li, N. Yu, W. Huan, X. Nie and X. Meng, Eur. Food Res. Technol., 2023, 249, 1875–1885 CrossRef CAS.
- H. Schwartz, V. Ollilainen, V. Piironen and A. Lampi, J. Food Compos. Anal., 2008, 21, 152–161 CrossRef CAS.
- S. Zhang and X. Li, Int. J. Biol. Macromol., 2018, 115, 811–819 CrossRef CAS PubMed.
- X. Wang, Q. Zeng, M. Del Mar Contreras and L. Wang, Food Res. Int., 2017, 102, 184–194 CrossRef CAS PubMed.
- F. Zhang, F. Zhu, B. Chen, E. Su, Y. Chen and F. Cao, Food Res. Int., 2022, 156, 111159 CrossRef CAS PubMed.
- Y. Zhu, J. Yang, L. Qin, C. He and S. Zhou, LWT--Food Sci. Technol., 2024, 201, 116222 CrossRef CAS.
- A. Ranalli, A. Malfatti, L. Lucera, S. Contento and E. Sotiriou, Food Res. Int., 2005, 38, 873–878 CrossRef CAS.
- X. Fang, M. Du, F. Luo and Y. Jin, Food Sci. Technol. Res., 2015, 21, 779–785 CrossRef CAS.
- X. Chen, T. Li, W. Sun, S. Mao, B. Wafae, L. Zhang, Y. Xiang, J. Xu, Q. Zhou, C. Wu, S. Yan, D. Zhou, G. Fan, X. Li and X. Li, Innov. Food Sci. Emerg. Technol., 2024, 92, 103579 CrossRef CAS.
- W. Zeng, X. Liu, Y. Chao, Y. Wu, S. Qiu, B. Lin, R. Liu, R. Tang, S. Wu, Z. Xiao and C. Li, Food Chem., 2024, 447, 139046 CrossRef CAS PubMed.
- P. Gao, Z. Zhou, S. Wang, Y. Zheng, C. Liu, W. Zhong, J. Yin and M. J. T. Reaney, LWT--Food Sci. Technol., 2024, 208, 116715 CrossRef CAS.
- Z. Huang, Y. Lin and J. Fang, Molecules, 2009, 14, 540–554 CrossRef CAS PubMed.
- S. Y. Park, S. J. Choi, H. J. Park, S. Y. Ma, Y. I. Moon, S. Park and M. Y. Jung, Food Sci. Biotechnol., 2020, 29, 769–775 CrossRef CAS PubMed.
- P. Zhang, N. Liu, M. Xue, M. Zhang, Z. Xiao, C. Xu, Y. Fan, W. Liu, J. Qiu, Q. Zhang and Y. Zhou, Int. J. Mol. Sci., 2023, 24, 8518 CrossRef CAS PubMed.
- S. Yuan, F. Wu, X. Yang, W. Min, Z. He, C. Wu, X. Liu and P. Wang, Food Chem., 2024, 461, 140888 CrossRef CAS PubMed.
- T. H. J. Beveridge, T. S. C. Li and J. C. G. Drover, J. Agric. Food Chem., 2002, 50, 744–750 CrossRef CAS PubMed.
- M. Yang, F. Huang, C. Liu, C. Zheng, Q. Zhou and H. Wang, Food Bioprocess Technol., 2013, 6, 3206–3216 CrossRef CAS.
- J. Yang, J. Li, M. Wang, X. Zou, B. Peng, Y. Yin and Z. Deng, Eur. J. Lipid Sci. Technol., 2019, 121, 1800431 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Haiyan Zhongb,
Yue Chenb,
Qianrui Wanga,
Wenxin Zhaa,
Shimao Chena and
Yong Zhu
a,
Haiyan Zhongb,
Yue Chenb,
Qianrui Wanga,
Wenxin Zhaa,
Shimao Chena and
Yong Zhu







