DOI:
10.1039/D5RA09296E
(Paper)
RSC Adv., 2026,
16, 19334-19351
Selective sensing characteristics of Fe doped and (Fe, N, O) co-doped molybdenum disulfide toward CO, CO2, and NH3 gases: a first-principles investigation
Received
2nd December 2025
, Accepted 7th April 2026
First published on 13th April 2026
Abstract
This work investigates the effect of supercell size and effective dopant concentration on Fe-doped and Fe–N/Fe–O co-doped MoS2 systems toward CO, CO2, NH3 adsorption using first-principles calculations. The results show that supercell size is a key physical factor governing the predicted properties of these systems. As the supercell expands from a 2 × 2 × 1 supercell to a 4 × 4 × 1 supercell, the effective dopant concentration decreases markedly, leading to substantial changes in the electronic structure, while the magnetic character remains largely preserved. In particular, Fe–MoS2, Fe–N–MoS2, and Fe–O–MoS2 systems evolve from metallic or half-metallic behavior in smaller supercells toward semiconducting states in the 4 × 4 × 1 model. Adsorption energies also vary with supercell size, although less systematically than the electronic properties. Among the studied systems, Fe–N–MoS2 shows the most noticeable response toward NH3 in the high-concentration 2 × 2 × 1 regime, with an adsorption energy of −0.264 eV, an adsorption distance of 1.5267 Å, evident Fe-3d/N-2p hybridization, and stronger charge transfer. In addition, the calculated sensing descriptors indicate a measurable NH3 response, including a maximum selectivity of 2.24%, an electronic sensitivity of 98.9% at the Fermi level, and a recovery time on the order of 10−8 s at 300 K. Overall, these results demonstrate that the predicted sensing behavior of doped MoS2 is strongly dependent on supercell size, with the effective dopant concentration governing both the electronic structure and adsorption characteristics of these two-dimensional systems.
1. Introduction
Air pollution driven by anthropogenic activities has intensified worldwide, creating an urgent demand for reliable technologies that can continuously monitor hazardous gases in the environment.1 Toxic and reactive species such as carbon monoxide (CO), carbon dioxide (CO2), and ammonia (NH3) are particularly important targets because they are ubiquitous in industrial, transportation, and domestic settings and can critically affect air quality and public safety. These practical concerns motivate the development of sensing platforms capable of detecting CO, CO2, NH3 with high accuracy, rapid response, and strong selectivity.2,3 Continued innovation in material design and system integration remains essential to achieve robust and sustainable sensing technologies. Against this backdrop, two-dimensional (2D) materials have gained considerable attention owing to their large specific surface area, tunable bandgap, and versatile electronic structures.4–14 In addition, their electronic structures are highly tunable by external perturbations such as adsorption, strain, defect engineering, and chemical doping, allowing large modulation of conductivity and surface potential upon gas interaction. Within this family, monolayer MoS2 is especially attractive owing to its intrinsic semiconducting behavior and chemical stability, while its electronic properties can be engineered over a broad range via defect and dopant control.15 Despite these advantages, pristine MoS2 often exhibits weak interactions with small and relatively stable molecules such as CO and CO2, and it may yield limited charge-transfer signatures toward NH3 when adsorption occurs on ideal basal-plane sites. Consequently, pristine MoS2 alone is frequently insufficient to deliver the combination of sensitivity, selectivity, and operational robustness required for practical CO, CO2, NH3 monitoring.16–19
To overcome this bottleneck, defect engineering and substitutional doping, co-doping has been widely adopted to reconfigure the local electronic environment of MoS2, create chemically active adsorption centers, and enhance charge-transfer capability between gas molecules and the sensing channel.15,16,20–53 Such modifications can introduce defect-derived states near the Fermi level (Ef), tune band-edge alignment, and modify the surface dipole and work function factors that directly influence adsorption energetics and the magnitude or sign of sensing response. Among commonly used dopants, N and O have been reported to induce favorable electronic modulation in monolayer MoS2 for sensing. N can markedly increase conductivity even pushing the system toward metallic-like behavior, while O typically alters bonding and band alignment and modulates work function, thereby changing the surface electrostatic response and sensitivity.16,17,27,31 These observations imply that co-doping can provide a more controllable route than single-dopant strategies to simultaneously tune adsorption energetics and electronic readout.
From a mechanistic standpoint, incorporating a transition-metal dopant such as Fe is particularly appealing because localized d-states close to the Ef can act as efficient adsorption-activation centers and amplify adsorption-induced redistribution in the sensing channel. Coupling Fe with a second dopant offers an additional handle to tailor Fe's local coordination and electronic character. In an Fe–N environment, N lone-pair electrons can reshape the local electric field and modulate the direction/magnitude of charge transfer, while in an Fe–O environment, O's high electronegativity can stabilize the defect complex and regulate surface dipole/work function—key descriptors for surface-sensitive detection.16,17,27,31 Although many studies have examined pristine and heteroatom-doped MoS2 systems20–53 systematic and side-by-side comparisons of Fe-based co-doping configurations (Fe–N vs. Fe–O) toward multiple gases (CO, CO2, NH3) under a unified computational framework remain limited, making it difficult to extract transferable design principles linking dopant chemistry to sensing-relevant descriptors.
In this work, we employ spin-polarized density functional theory (DFT) calculations to comparatively investigate CO, CO2, and NH3 adsorption on Fe-doped MoS2 (Fe–MoS2), Fe–N co-doped MoS2 (Fe–N–MoS2), and Fe–O co-doped MoS2 (Fe–O–MoS2). Using a consistent modeling and evaluation protocol, we correlate adsorption energetics and adsorption-induced geometrical signatures with charge-transfer behavior, surface electrostatic response, and transport-relevant electrical sensitivity, together with stability and recovery considerations. By establishing direct links between dopant configuration and sensing descriptors for three technologically important gases, this study aims to clarify atomistic sensing mechanisms and provide practical design guidelines for MoS2-based chemiresistive sensors with improved selectivity, stability, and operational reliability.
2. Computational methods
This study investigates the structural, electronic, and gas–adsorption properties of monolayer MoS2 using DFT calculations, employing a 2 × 2 × 1 supercell containing 12 atoms (4 Mo and 8 S). Fe substitutes for a Mo site and N or O substitutes for an S site. In a 2 × 2 × 1 MoS2 supercell containing 4 Mo sites and 8 S sites, one Fe substitution corresponds to Fe–Mo = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4, 25%, and one N or O substitution corresponds to N or O–S = 1:8, 12.5%. Because these concentrations are relatively high, the results obtained from the 2 × 2 × 1 model should be understood as representing a concentrated doping regime rather than a dilute-defect limit. To clarify the effect of supercell size and effective dopant concentration, additional calculations were performed using larger 3 × 3 × 1 supercell and 4 × 4 × 1 supercell. As the supercell size increases, the effective dopant concentration decreases substantially, which can modify the electronic structure and adsorption behavior of the doped systems. Therefore, the larger supercells were included to explicitly assess concentration-dependent trends and to provide a more reliable reference for comparison, particularly in the more dilute doping limit. The corresponding results are discussed separately in the section on the effect of supercell size and effective dopant concentration. A vacuum thickness of 40 Å is introduced along the z direction to eliminate spurious interactions between periodic images of the monolayer. For all adsorption configurations, the gas molecules are initially placed 3 Å above the surface before structural relaxation.
:4, 25%, and one N or O substitution corresponds to N or O–S = 1:8, 12.5%. Because these concentrations are relatively high, the results obtained from the 2 × 2 × 1 model should be understood as representing a concentrated doping regime rather than a dilute-defect limit. To clarify the effect of supercell size and effective dopant concentration, additional calculations were performed using larger 3 × 3 × 1 supercell and 4 × 4 × 1 supercell. As the supercell size increases, the effective dopant concentration decreases substantially, which can modify the electronic structure and adsorption behavior of the doped systems. Therefore, the larger supercells were included to explicitly assess concentration-dependent trends and to provide a more reliable reference for comparison, particularly in the more dilute doping limit. The corresponding results are discussed separately in the section on the effect of supercell size and effective dopant concentration. A vacuum thickness of 40 Å is introduced along the z direction to eliminate spurious interactions between periodic images of the monolayer. For all adsorption configurations, the gas molecules are initially placed 3 Å above the surface before structural relaxation.
Density functional theory (DFT) calculations are performed to explore the structural, electronic, and gas-sensing characteristics of CO, CO2, and NH3 adsorption on Fe-doped MoS2 (Fe–MoS2), Fe–N co-doped MoS2 (Fe–N–MoS2), and Fe–O co-doped MoS2 (Fe–O–MoS2), using the Vienna Ab initio Simulation Package (VASP).54 Exchange–correlation effects are described within the generalized gradient approximation (GGA) using the Perdew–Burke–Ernzerhof (PBE) functional,55 while electron–ion interactions are treated with the projector augmented-wave (PAW) method.56 To properly capture the localized nature of Fe-3d electrons, the DFT + U method is employed, where the effective on-site Coulomb parameter Ueff = U − J is applied to Fe-3d orbitals, with U = 3 eV and J = 0.5 eV.57,58 Long-range dispersion interactions, which are critical in gas adsorption, are included using Grimme's semi-empirical DFT-D3 correction.59 A plane-wave kinetic energy cutoff of 500 eV is adopted to ensure reliable convergence of the total energy and electronic structure. The Brillouin zone is sampled using Γ-centered k-point meshes of 5 × 5 × 1 for structural optimizations and AIMD calculations, and 30 × 30 × 1 for self-consistent electronic calculations. Structural relaxations are carried out until the total energy change between two consecutive ionic steps is less than 10−5 eV, and the maximum Hellmann–Feynman force on each atom is below 0.01 eV Å−1.
3. Results and discussion
3.1. Geometric structure
A 2 × 2 × 1 MoS2 supercell is constructed, in which Fe is introduced by substitution at Mo sites and N/O is introduced by substitution at S sites because of their relatively similar atomic radius, which is expected to promote structural stability. A total of 25 possible adsorption configurations are considered for the target gases, including CO (9 sites), CO2 (8 sites), and NH3 (8 sites). These sites comprise A (on top of S/N/O atoms), B (Mo/Fe–S bridge), C (center of the MoS2 hexagon), and D (on top of the Fe atom), as illustrated in Fig. S1–S4. Specifically, after examining 25 plausible adsorption configurations for each substrate-Fe–MoS2, Fe–N–MoS2, and Fe–O–MoS2-upon adsorption of CO, CO2, and NH3, the most stable adsorption sites were determined based on the formation energy. The results indicate that the most stable configurations for Fe–MoS2–CO, Fe–MoS2–CO2, and Fe–MoS2–NH3 are C1, D1, and D1, respectively; for Fe–N–MoS2–CO, Fe–N–MoS2–CO2, and Fe–N–MoS2–NH3 are B1, A1, and A1, respectively; and for Fe–O–MoS2–CO, Fe–O–MoS2–CO2, and Fe–O–MoS2–NH3 are A1, A1, and A1, respectively. These stable configurations are summarized in Table 1. Formation energy or binding energy are essential quantities for identifying the most stable adsorption sites on a material surface, where the most stable configuration corresponds to the most negative energy value. Specifically, the formation energy (Eform) is calculated as follows eqn (1):60| |
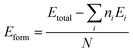 | (1) |
While Ei is the reference energy per atom, ni denotes the number of atoms of species i, and N is the total number of atoms in the system.
Table 1 Formation (Binding) energy of 25 possible (initial) adsorption configurations of Fe–MoS2, Fe–N–MoS2 and Fe–O–MoS2
| Configuration |
A1 |
B1 |
C1 |
D1 |
A2 |
B2 |
C2 |
D2 |
C3 |
| Fe–MoS2–CO |
−4.2108 |
−4.2109 |
−4.2110 |
−4.2106 |
−4.2105 |
−4.2108 |
−4.2106 |
−4.2106 |
−4.2106 |
| Fe–MoS2–CO2 |
−4.1482 |
−4.1484 |
−4.1484 |
−4.1485 |
−4.1473 |
−4.1476 |
−4.1475 |
−4.1474 |
— |
| Fe–MoS2–NH3 |
−4.0359 |
−4.0385 |
−4.0358 |
−4.0386 |
−4.0383 |
−4.0383 |
−4.0383 |
−4.0387 |
— |
| Fe–N–MoS2–CO |
−4.2728 |
−4.2729 |
−4.2668 |
−4.2642 |
−4.2644 |
−4.2726 |
−4.2726 |
−4.2625 |
−4.2625 |
| Fe–N–MoS2–CO2 |
−4.2089 |
−4.2088 |
−4.2087 |
−4.2032 |
−4.1987 |
−4.1990 |
−4.2038 |
−4.2037 |
— |
| Fe–N–MoS2–NH3 |
−4.0954 |
−4.0953 |
−4.0949 |
−4.0847 |
−4.0950 |
−4.0951 |
−4.0953 |
−4.0876 |
— |
| Fe–O–MoS2–CO |
−4.0730 |
−4.0730 |
−4.0728 |
−4.0666 |
−4.0668 |
−4.0729 |
−4.0723 |
−4.0652 |
−4.0723 |
| Fe–O–MoS2–CO2 |
−4.0238 |
−4.0237 |
−4.0233 |
−4.0192 |
−4.0143 |
−4.0148 |
−4.0191 |
−4.0131 |
— |
| Fe–O–MoS2–NH3 |
−4.3143 |
−4.3143 |
−4.3143 |
−4.3050 |
−4.3138 |
−4.3139 |
−4.3132 |
−4.3072 |
— |
After identifying the most stable adsorption configurations, calculations of the electronic properties and sensing characteristics were subsequently carried out. Overall, the buckling height (h) shows only negligible changes from pristine-MoS2 (1.564 Å) to Fe-doped MoS2 (1.566 Å), and to the co-doped systems Fe–N (1.439 Å) and Fe–O (1.5465 Å), indicating good structural stability at 0 K. In addition, the stability of h is consistently maintained during gas adsorption across all structures, as shown in Fig. 1(a–l). Furthermore, the internal bond lengths within the lattice vary only slightly, with average values around 2.4 Å (see l1 and l2 in Table 2). Taken together, these structural parameters confirm that Fe doping as well as Fe–N and Fe–O co-doping preserve the overall structural stability. In contrast, the role of Fe doping and Fe–N/O co-doping becomes clear from the adsorption energies and adsorption distances. Notably, all gas molecules tend to be drawn closer to the substrate under Fe–N/O co-doping, accompanied by more negative adsorption energies (Table 2). For CO and CO2, the adsorption distances to the surface are very similar: approximately 3.1 Å for Fe–MoS2, whereas they decrease to about 2.4–2.6 Å for Fe–N/O systems. Moreover, this similarity in adsorption distance can be attributed to the similarity in adsorption energy, the adsorption energy is defined in eqn (2).61
| | |
Eads = Etotal − Egas − Esub
| (2) |
where
Etotal is the total energy of the combined slab-gas system,
Egas is the energy of the isolated gas molecule, and
Esub is the energy of the isolated substrate (either doped or co-doped) prior to adsorption.
 |
| | Fig. 1 Configurations of the systems (a) Fe–MoS2; (b) Fe–MoS2–CO–C1; (c) Fe–MoS2–CO2–D1; (d) Fe–MoS2–NH3–D1; (e) Fe–N–MoS2; (f) Fe–N–MoS2–CO–B1; (g) Fe–N–MoS2–CO2–A1; (h) Fe–N–MoS2–NH3–A1; (i) Fe–O–MoS2; (j) Fe–O–MoS2–CO–A1; (k) Fe–O–MoS2–CO2–A1; (l) Fe–O–MoS2–NH3–A1. Here, h is the substrate buckling (Å), and d is the adsorption distance measured from the gas molecule to the topmost layer of the substrate (Å). Element colors: S (yellow), Mo (violet), N (light gray), H (pink), C (brown), O (red), Fe (dark yellow). | |
Table 2 Structural parameters of Fe–MoS2, Fe–N–MoS2 and Fe–O–MoS2 before and after CO, CO2, and NH3 adsorption, including adsorption energy (Eads), adsorption distance (d), buckling height (h), and Mo–S longest (l1) and shortest (l2) bond lengths, magnetic moment (Mag), charge transfer (ΔQ) and the electronic state (bandgap, metal or half-metal)
| Configurations |
Eads (eV) |
d (Å) |
h (Å) |
l1 (Å) |
l2 (Å) |
Mag (µB) |
ΔQ (e) |
Electronic state |
| Fe–MoS2 |
— |
— |
1.5660 |
2.4122 |
2.4052 |
2.0000 |
— |
Half-metal |
| Spin-up: 0 eV |
| Spin-dw: 1.07 eV |
| Fe–MoS2–CO–C1 |
−0.247 |
3.088 |
1.5641 |
2.4099 |
2.4006 |
2.0000 |
−0.009 |
Half-metal |
| Spin-up: 0 eV |
| Spin-dw: 1.04 eV |
| Fe–MoS2–CO2–D1 |
−0.248 |
3.1708 |
1.5632 |
2.4093 |
2.3998 |
2.0000 |
−0.018 |
Half-metal |
| Spin-up: 0 eV |
| Spin-dw: 1.05 eV |
| Fe–MoS2–NH3–D1 |
−0.263 |
2.6248 |
1.5698 |
2.4102 |
2.4004 |
2.0000 |
−0.004 |
Half-metal |
| Spin-up: 0 eV |
| Spin-dw: 1.01 eV |
| Fe–N–MoS2 |
— |
— |
1.5439 |
2.4481 |
2.4121 |
1.735 |
— |
Metal |
| Fe–N–MoS2–CO–B1 |
−0.150 |
2.5449 |
1.539 |
2.4444 |
2.4086 |
1.7788 |
0.00003 |
Metal |
| Fe–N–MoS2–CO2–A1 |
−0.272 |
2.5864 |
1.5408 |
2.4425 |
2.4094 |
1.7341 |
−0.0126 |
Metal |
| Fe–N–MoS2–NH3–A1 |
−0.264 |
1.5267 |
1.5438 |
2.4438 |
2.4076 |
1.7273 |
0.0126 |
Metal |
| Fe–O–MoS2 |
— |
— |
1.5465 |
2.4308 |
2.3952 |
2.0001 |
— |
0.03 eV |
| Fe–O–MoS2–CO–A1 |
−0.190 |
2.418 |
1.5421 |
2.4284 |
2.3926 |
2.0011 |
−0.0055 |
0.03 eV |
| Fe–O–MoS2–CO2–A1 |
−0.272 |
2.6197 |
1.5423 |
2.4282 |
2.3934 |
2.0001 |
−0.0131 |
0.03 eV |
| Fe–O–MoS2–NH3–A1 |
−0.260 |
1.5994 |
1.5408 |
2.428 |
2.3913 |
2.0000 |
0.0086 |
0.03 eV |
Specifically, CO exhibits only a modest variation in adsorption energy, with the most negative value on Fe–MoS2 (−0.247 eV) and the least negative value on Fe–N–MoS2 (−0.150 eV). In contrast, CO2 shows stable adsorption energetics, maintaining an adsorption energy of approximately −0.272 eV on both Fe–N–MoS2 and Fe–O–MoS2. Likewise, NH3 displays the most consistent adsorption behavior, with an adsorption energy of around −0.26 eV across all three adsorption systems. These results indicate that CO2 and NH3 interact favorably with the studied substrates.
3.2 Thermal stability
Ab initio molecular dynamics (AIMD) simulations were employed to evaluate the thermal stability of the Fe–MoS2, Fe–N–MoS2, and Fe–O–MoS2 systems at finite temperatures. Each structure was equilibrated at 300 K and 700 K for a total simulation time of 10 ps, as illustrated in Fig. 2. At 300 K, all systems exhibit stable energy fluctuations without noticeable bond breaking or structural distortion, as shown in Fig. 2(b), (d), and (f). The total energy oscillates around an equilibrium value with only minor variations, and comparison of the atomic configurations before and after the simulation reveals negligible structural changes. When the temperature is increased to 700 K, larger energy fluctuations are observed, which is expected due to enhanced thermal motion. However, the overall lattice framework remains intact throughout the simulation time. Although slight variations in bond lengths appear at 700 K, no structural collapse, dopant segregation, or desorption events are detected within the 10 ps time window. These results indicate short-time thermal stability in AIMD simulations within the 300–700 K range and suggest structural robustness under the simulated conditions. It should be noted that the present AIMD simulations represent short-time dynamic stability under idealized environments and do not account for long-term degradation mechanisms, oxidative conditions, humidity effects, or dopant migration at extended timescales. Therefore, the thermal robustness discussed here is limited to the simulated conditions and warrants further experimental verification.
 |
| | Fig. 2 The AIMD spectra of the Fe–MoS2 (a and b), Fe–N–MoS2 (c and d) and Fe–O–MoS2 (e and f) systems are simulated at temperatures of 300 K and 700 K over a time span of 10 ps. | |
3.3 Electronic properties
The most significant effect of doping and co-doping is the modification of intrinsic material properties. Both Fe doping and Fe–N/O co-doping exhibit a pronounced impact by transforming MoS2 from a typical wide-bandgap semiconductor (1.8 eV) into a metallic or half-metallic system in all considered configurations.17 Here, a material is classified as metallic when both spin-up and spin-down bands intersect the Ef, whereas a half-metal is defined when only one of the two spin channels (spin-up or spin-down) crosses Ef (the system is metallic for one spin channel but remains semiconducting for the other).
Accordingly, Fe–MoS2 with CO, CO2, and NH3 adsorption shows a clear half-metallic character: in the spin-up channel (black curves), the bands cross Ef, indicating metallic behavior, while in the spin-down (spin-dw) channel (red curves), no bands intersect Ef, preserving a semiconducting character. In addition, Fig. 3(a–d) highlights the distinctive role of Fe, which provides the dominant contribution to the electronic structure. This confirms that Fe is the key factor responsible for converting MoS2 from a conventional semiconductor into a half-metal. In contrast, CO, CO2, and NH3 are not the primary driving factors, as their contributions are either weak (C and O) or located deep within the energy spectrum (N and H). For Fe–O–MoS2, Fe–O co-doping narrows the band gap, transforming pristine semiconducting MoS2 with a band gap of about 1.8 eV into a material with a very small gap of 0.03 eV, in which Fe remains the main contributor. Meanwhile, O enhances the activation center by introducing an additional dispersive band very close to Ef in the spin-down channel, as shown in Fig. 3(i–l). By contrast, Fe–N co-doping exhibits a markedly different synergistic effect compared to Fe–O, converting MoS2 into a fully metallic system, with both spin-up and spin-down channels crossing Ef, accompanied by noticeable contributions from N. This difference is rationalized by the much higher electronegativity of O compared with N, which enables O to attract electrons more strongly, thereby increasing the local electron density and promoting Fe as a new activation site with a stronger tendency to accept electrons. Overall, doping and co-doping induce substantial changes in the electronic properties across all three systems. Among the adsorbates, NH3 contributes more noticeably to the band structure, whereas CO and CO2 do not introduce significant electronic states near the Ef. In contrast, the s (DOS) analysis provides deeper insight into the nature of orbital hybridization and the interactions among atoms within the material.
 |
| | Fig. 3 Electronic band structures of (a) Fe–MoS2; (b) Fe–MoS2–CO–C1; (c) Fe–MoS2–CO2–D1; (d) Fe–MoS2–NH3–D1; (e) Fe–N–MoS2; (f) Fe–N–MoS2–CO–B1; (g) Fe–N–MoS2–CO2–A1; (h) Fe–N–MoS2–NH3–A1; (i) Fe–O–MoS2; (j) Fe–O–MoS2–CO–A1; (k) Fe–O–MoS2–CO2–A1; (l) Fe–O–MoS2–NH3–A1. The black and red lines illustrate spin-up and spin-dw, respectively. | |
Overall, all configurations exhibit strong hybridization among Fe-3d, S-2p, and Mo-4d states near the Ef, as evidenced by their substantial orbital overlap in this energy region. In addition, Fe-3d plays a dominant role in hybridized states located deeper in the electronic structure (approximately 1–2 eV below the Ef), indicating that Fe is the key contributor responsible for altering the electronic properties of the host material. Specifically, for the Fe–MoS2 system, CO and CO2 show almost no participation in hybridization with the substrate, as no notable orbital overlap is observed. By contrast, NH3 exhibits very strong hybridization: the N-2p states of NH3 overlap strongly with S-2p and Mo-4d states at around −1 eV, consistent with Fig. 3. A similar behavior is also observed for the Fe–N–MoS2 and Fe–O–MoS2 substrates, which helps explain the relatively stable adsorption energies and the pronounced reduction in adsorption distances reported in Table 2. Furthermore, Fig. 4(e) and (i) reveal the contributions of N and O to the electronic structure, showing relatively weak hybridization due to the limited overlap of their states. This further clarifies their role in shaping the metallic or half-metallic character of Fe–N–MoS2 and Fe–O–MoS2, respectively. Therefore, the features observed in Fig. 4(f), (j), (g), and (h) that involve N and O contributions are mainly associated with the dopant atoms within the lattice, rather than N and O originating from the adsorbed gas molecules.
 |
| | Fig. 4 The density of states of (a) Fe–MoS2; (b) Fe–MoS2–CO–C1; (c) Fe–MoS2–CO2–D1; (d) Fe–MoS2–NH3–D1; (e) Fe–N–MoS2; (f) Fe–N–MoS2–CO–B1; (g) Fe–N–MoS2–CO2–A1; (h) Fe–N–MoS2–NH3–A1; (i) Fe–O–MoS2; (j) Fe–O–MoS2–CO–A1; (k) Fe–O–MoS2–CO2–A1; (l) Fe–O–MoS2–NH3–A1. | |
To further clarify the interaction mechanism between the gas molecules and the substrate, COHP calculations were performed to investigate the hybridization characteristics and bonding nature of the adsorbate–substrate interaction.62 This method allows the bonding and antibonding contributions between selected atomic pairs to be decomposed, thereby providing direct evidence for the presence or absence of significant bond formation. In the COHP plots, positive values generally represent bonding states, indicating that orbital overlap contributes to stabilizing the interaction between two atoms, whereas negative values correspond to antibonding states, reflecting a tendency to weaken or destabilize the bond. In particular, the states located near the Ef are more important for evaluating the interaction nature, as these electronic states directly participate in electrical conduction and determine the effective bond strength in the system. If pronounced bonding peaks appear and remain significant near the Ef, this usually suggests strong hybridization and the possible formation of a clear chemical bond between the gas molecule and the substrate. By contrast, if the contributions near the Ef are small or predominantly antibonding, the interaction is generally weaker and more characteristic of physisorption. In this way, the COHP analysis serves as a complement to the adsorption-energy and charge-transfer results, providing a more comprehensive understanding of the interactions of CO, CO2, and NH3 with Fe–MoS2, Fe–N–MoS2, and Fe–O–MoS2. Overall, the results show that the hybridization between the gas molecules and the substrate is relatively weak in the vicinity of the Ef, whereas more distinct bonding features mainly appear deeper within the occupied energy region, approximately from −3 eV to −7 eV (Fig. 5). The bonding and antibonding intensities show a very pronounced imbalance across all investigated systems, with antibonding states being dominant in most cases, indicating the intrinsically weak nature of the interaction. This is fully consistent with the adsorption energies, which are not strongly negative, and with the adsorption distances, which decrease only slightly. This indicates that the gas-surface interaction is not strong enough to form stable chemical bonds near the Ef, but instead mainly reflects orbital overlap in lower-energy states. This trend is fully consistent with the adsorption energies, adsorption distances, and density of states (DOS) results obtained in the present study, thereby further supporting the conclusion that the adsorption process in these systems is predominantly weak to moderate in nature rather than strong chemisorption.
 |
| | Fig. 5 The COHP calculations for CO (a, d and i), CO2 (b, e and g) and NH3 (c, f and h) adsorption on Fe–MoS2, Fe–N–MoS2, and Fe–O–MoS2. | |
3.4. Charge density difference
To gain deeper insight into the adsorption mechanism, the charge density difference (CDD) distributions along the z direction for CO, CO2, and NH3 molecules interacting with all considered substrates are analyzed. The CDD is defined in eqn (3) as follows:63| | |
Δρ = ρtotal − ρsub − ρgas
| (3) |
where, ρtotal, ρsub, ρgas are the charge densities of the gas-adsorbed configuration, the clean substrate, and the isolated gas molecule, respectively.
The CDD data reveal charge transfer between atoms and are crucial for elucidating interatomic interactions at the atomic scale, particularly in doped or adsorbed configurations. Regions of charge accumulation appear as orange areas surrounding the atoms, while blue areas indicate charge depletion; the corresponding planar-averaged electrostatic potential along the z axis is also plotted. Co-doping is expected to enhance the charge-exchange capacity of the system through heteroatom-induced electronic responses and the resulting electronic imbalance within the substrate. Overall, the CDD analysis indicates a pronounced enhancement for the Fe–N–MoS2 system, which exhibits differences of up to 0.015 e, 0.022 e, and 0.518 e for CO (Fig. 6(d)), CO2 (Fig. 6(e)), and NH3 (Fig. 6(f)), respectively. These results demonstrate that Fe–N–MoS2 enables highly efficient charge transfer with the gas molecules, consistent with its metallic nature and higher electron concentration, whereas Fe–MoS2 and Fe–O–MoS2 behave as half-metals with a slightly lower electron density. Notably, the Fe–N–MoS2–NH3 configuration shows the strongest charge-transfer capability, presenting the largest charge-density difference among all investigated systems, which highlights the promising potential of Fe–N co-doped MoS2 for NH3 sensing applications.
 |
| | Fig. 6 The plots show the charge density difference and the plane-averaged electrostatic potential along the z-direction for (a) Fe–MoS2–CO–C1; (b) Fe–MoS2–CO2–D1; (c) Fe–MoS2–NH3–D1; (d) Fe–N–MoS2–CO–B1; (e) Fe–N–MoS2–CO2–A1; (f) Fe–N–MoS2–NH3–A1; (i) Fe–O–MoS2–CO–A1; (g) Fe–O–MoS2–CO2–A1; (h) Fe–O–MoS2–NH3–A1. The orange and green regions indicate charge accumulation and charge depletion, respectively. | |
In addition, to quantify charge donation/acceptance, Bader-charge analysis64 is performed for all doped and co-doped adsorption configurations; the results are summarized in Table 2. The net charge on atom j is defined by eqn (4):65
| | |
ΔqBaderj = Zvalj − NBaderj
| (4) |
where
Zvalj is the number of valence electrons and
NBaderj is the Bader-assigned electron count and the total charge on the adsorbed molecule. The Bader charges are computed as follows
eqn (5):
65| |
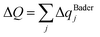 | (5) |
Bader-charge analysis is performed to quantify charge transfer between the adsorbed gas molecules and the substrates. In this convention, ΔQ > 0 indicates that the gas molecule donates electrons to the substrate (donor behavior), while the substrate accepts electrons (acceptor behavior), and vice versa for ΔQ < 0. Overall, Fe–MoS2 exhibits an intrinsic half-metallic character with a relatively high electron density in the substrate; consequently, electrons tend to be transferred to the gas molecules, leading to negative ΔQ values for all adsorption cases. In addition, for Fe–N–MoS2 and Fe–O–MoS2, CO and CO2 adsorption shows almost exclusively electron acceptance from the substrate, further supporting the chemically inert nature of CO and CO2, which is associated with their robust sp and sp2 hybridized bonding. In contrast, NH3 displays a distinct interaction with Fe–N–MoS2, where NH3 is willing to donate 0.0126 electrons to the substrate. This behavior is consistent with the trends observed in adsorption energy, adsorption distance, band structures, DOS, and CDD analyses.
3.5. Selectivity analysis
The change in Φ between the gas-adsorbed surface and the pristine surface is a crucial criterion for evaluating gas-sensor sensitivity. In particular, Φ represents the minimum energy needed to remove an electron from the material surface into the vacuum, as defined in eqn (6):66where Evac is the vacuum level (the electrostatic potential in the far-vacuum region above the surface, where the potential flattens).
Specifically, the work-function change is a surface-sensitive quantity that responds strongly to adsorption and doping, and it serves as a key descriptor of gas selectivity among different molecules. As defined in eqn (6), the variation in Φ is commonly employed as a descriptor of selectivity and can be written as eqn (7):66
| |
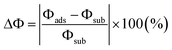 | (7) |
where
Φads is the
Φ of the gas-covered surface and
Φsub is the
Φ of the pristine substrate. The work function is a highly surface-sensitive descriptor reflecting changes in the surface electrostatic environment, and the relative work-function difference among different gases, referenced to the pristine (gas-free) substrate, serves as an indicator of selectivity. In this context, a larger separation in the work-function response corresponds to higher gas selectivity.
Fig. 7 presents the variation of the electrostatic potential and the corresponding work function (Φ) along the z-direction for all Fe–MoS2, Fe–N–MoS2, and Fe–O–MoS2 cases. Overall, no substantial differences in work-function values are observed among the systems. The work function remains around 5.3 eV for gas-adsorbed Fe–MoS2, approximately 5.6 eV for gas-adsorbed Fe–N–MoS2, and about 5.4 eV for gas-adsorbed Fe–O–MoS2. This suggests that Fe/N/O-doped and co-doped systems adsorbing CO, CO2, and NH3 may not exhibit sufficiently distinct selectivity.
 |
| | Fig. 7 The Φ for electron emission from the substrate (a) Fe–MoS2; (b) Fe–MoS2–CO–C1; (c) Fe–MoS2–CO2–D1; (d) Fe–MoS2–NH3–D1; (e) Fe–N–MoS2; (f) Fe–N–MoS2–CO–B1; (g) Fe–N–MoS2–CO2–A1; (h) Fe–N–MoS2–NH3–A1; (i) Fe–O–MoS2; (j) Fe–O–MoS2–CO–A1; (k) Fe–O–MoS2–CO2–A1; (l) Fe–O–MoS2–NH3–A1. | |
Accordingly, Fe–MoS2 exhibits poor gas selectivity, as it is difficult to distinguish these gases via electrical signals because the work-function variations are negligible. In contrast, Fe–N–MoS2 and Fe–O–MoS2 show improved, yet not outstanding, selectivity. The largest relative change is only 2.24% for Fe–N–MoS2–NH3 and 1.93% for Fe–O–MoS2–CO as present in Fig. 8 Specifically, the Fe–MoS2 configuration upon adsorption of CO, CO2, and NH3 is almost indistinguishable among these gases because the selectivity values are highly similar, namely 0.15%, 0.19%, and 0.32%, respectively. In contrast, Fe–O–MoS2 exhibits better selectivity than Fe–MoS2, with selectivity values of 1.93%, 1.24%, and 0.77% for CO, CO2, and NH3, respectively; therefore, Fe–O–MoS2 can discriminate between CO and NH3. Moreover, Fe–N–MoS2 shows relatively good selectivity by distinguishing CO and CO2 from NH3, with corresponding selectivity values of 0.32%, 0.59%, and 2.24%. Although these values are relatively small, they are still sufficient for discrimination in the context of modern technologies, and sensor performance should be evaluated not only in terms of selectivity but also sensitivity, recovery time, and related metrics.
 |
| | Fig. 8 Comparison of selectivity based on work function differences of substrate configuration and adsorption configuration. | |
3.6. Electrical sensitivity
To obtain a more quantitative and impartial assessment of the gas-sensing behavior in the doped configurations, the energy-dependent modulation of the electronic conductivity within the semiclassical Boltzmann transport formalism is explored (Fig. 9). In this study, the electrical conductivity is evaluated using the semiclassical Boltzmann transport framework in VASP 6.5.0 under the electron-phonon transport mode. Specifically, the transport coefficients are derived from the band structure through the group velocities and the Fermi-Dirac distribution; scattering effects are incorporated via the constant relaxation time approximation (CRTA), in which the conductivity scales linearly with the carrier relaxation time.
 |
| | Fig. 9 Electronic conductivity change based on Boltzmann transport for (a) Fe–MoS2, (b) Fe–N–MoS2, and (c) Fe–O–MoS2 upon adsorption gases. | |
The conductivity variation (Δσ) is defined as the difference between the gas-adsorbed and substrate-MoS2, as given by the following expression (eqn (8)):67
| |
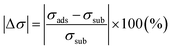 | (8) |
Here,
σads is the electronic conductivity of the gas-adsorbed system, and
σsub is the electronic conductivity of the clean (non-adsorbed) substrate.
Overall, most of the gas-adsorbed systems exhibit a reasonably good electronic-conductivity response because they show relatively large deviations in electronic conductivity compared with the corresponding pristine, non-adsorbed substrates. Fe–MoS2 displays an attractive electronic sensitivity, with an average sensitivity of approximately 50% over most of the investigated energy range. In addition, at the Ef, Fe–MoS2 shows sensitivities of 92.3% for CO, 52.5% for CO2, and only 11.9% for NH3, indicating a clear influence of the adsorbed gas molecules on the conductivity of the substrate. Meanwhile, Fe–N–MoS2 exhibits a slightly higher average electronic sensitivity than Fe–MoS2, with an average response of around 60%. Moreover, the sensitivities directly at the Ef are 40.1% for CO, 62.4% for CO2, and 98.9% for NH3. These results indicate that N co-doping significantly enhances the response toward NH3, while still maintaining a moderate response toward CO2, thereby making Fe–N–MoS2 the most prominent system for NH3 detection among the investigated substrates. In contrast, for Fe–O–MoS2, the sensitivities at the Fermi level are 42.3% for CO, 4.6% for CO2, and 8.5% for NH3, suggesting that O co-doping mainly preserves a moderate response to CO, whereas the responses to CO2 and NH3 remain weak. In general, among all the systems considered, the highest sensitivity at the Ef is observed for Fe–N–MoS2–NH3 (98.9%), followed by Fe–MoS2–CO (92.3%). Fe–MoS2 also exhibits a noticeable response toward CO2, while Fe–N–MoS2 shows a relatively balanced response toward both CO2 and NH3. By contrast, Fe–O–MoS2 demonstrates a more selective behavior, but its response intensity is not uniformly strong, since only the CO-adsorbed system shows a moderate sensitivity. Therefore, based on the equilibrium response at the Ef, Fe–N–MoS2 can be regarded as the most promising substrate for NH3 sensing, whereas Fe–MoS2 appears to be more favorable for CO detection and still retains a moderate sensitivity toward CO2.
3.7. Recovery time
Another essential metric for assessing gas sensors is the recovery time, which denotes the period needed for the substrate to release the adsorbed gas and thus revert to its original state and inherent characteristics. If this value is too large, desorption proceeds too slowly for practical sensing,68 whereas an excessively short recovery time yields unstable signals, since the adsorbed species detach almost immediately. The recovery time τ can be written as eqn (9):69,70| |
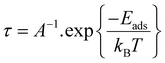 | (9) |
where A is the attempt frequency (typically 1012 s−1), kB is the Boltzmann constant (8.62 × 10−5 eV K−1) and T (K) is the system temperature.
The recovery time was evaluated over the temperature range of 300–700 K, consistent with the thermally stable behavior confirmed by the AIMD results in Fig. 2. As listed in Table 3, all investigated systems exhibit very short recovery times, and a clear temperature-dependent trend is observed: the desorption time decreases systematically as the temperature increases from 300 to 700 K, typically from the 10−8–10−9 s range down to the 10−11 s range. This behavior is physically reasonable because higher temperature provides additional thermal energy to overcome the adsorption interaction, thereby facilitating faster desorption. A closer comparison among the systems reveals non-negligible differences in gas-substrate interaction strength. For pristine Fe–MoS2, the recovery times for CO, CO2, and NH3 at 300 K are 1.4 × 10−8, 1.5 × 10−8, and 2.6 × 10−8 s, respectively, indicating that NH3 is desorbed slightly more slowly than CO and CO2. For Fe–N–MoS2, CO shows an exceptionally short recovery time of 3.2 × 10−11 s at 300 K, which is almost two orders of magnitude shorter than those of CO2 (3.7 × 10−8 s) and NH3 (2.7 × 10−8 s). This suggests that CO interacts much weaklier and reversibly with Fe–N–MoS2, whereas CO2 and NH3 remain on the surface for a relatively longer time. A similar tendency is also found for Fe–O–MoS2, where CO exhibits a shorter recovery time (1.6 × 10−9 s at 300 K) than CO2 (3.7 × 10−8 s) and NH3 (2.3 × 10−8 s). Therefore, although all systems recover rapidly, the co-doped substrates still retain distinct desorption characteristics depending on the adsorbed gas. From the sensing perspective, these results indicate excellent reversibility, since the adsorbed molecules can leave the surface very quickly once the external gas atmosphere is removed. However, the recovery times are also extremely short, especially at elevated temperatures, implying that the adsorption-induced signal may decay rapidly. In practice, this means that while the studied systems are favorable for fast-response and reusable sensing platforms, they may also require sufficiently rapid signal acquisition and stable readout conditions to avoid loss of detectable response. Thus, the recovery-time results should be interpreted as evidence of highly reversible adsorption rather than as a standalone indicator of superior sensing performance.
Table 3 Recovery time τ(s) versus temperature 300 K–700 K for Fe–MoS2, Fe–N–MoS2, and Fe–O–MoS2 upon adsorption of CO, CO2 and NH3
| Configuration |
300 K |
400 K |
500 K |
600 K |
700 K |
| Fe–MoS2–CO |
1.4 × 10−8 |
1.3 × 10−9 |
3.1 × 10−10 |
1.2 × 10−10 |
6.0 × 10−11 |
| Fe–MoS2–CO2 |
1.5 × 10−8 |
1.3 × 10−9 |
3.2 × 10−10 |
1.2 × 10−10 |
6.1 × 10−11 |
| Fe–MoS2–NH3 |
2.6 × 10−8 |
2.0 × 10−9 |
4.5 × 10−10 |
1.6 × 10−10 |
7.8 × 10−11 |
| Fe–N–MoS2–CO |
3.2 × 10−10 |
7.7 × 10−11 |
3.2 × 10−11 |
1.8 × 10−11 |
1.2 × 10−11 |
| Fe–N–MoS2–CO2 |
3.7 × 10−8 |
2.6 × 10−9 |
5.5 × 10−10 |
1.9 × 10−10 |
9.0 × 10−11 |
| Fe–N–MoS2–NH3 |
2.7 × 10−8 |
2.1 × 10−9 |
4.6 × 10−10 |
1.7 × 10−10 |
8.0 × 10−11 |
| Fe–O–MoS2–CO |
1.6 × 10−9 |
2.5 × 10−10 |
8.3 × 10−11 |
4.0 × 10−11 |
2.3 × 10−11 |
| Fe–O–MoS2–CO2 |
3.7 × 10−8 |
2.6 × 10−9 |
5.5 × 10−10 |
1.9 × 10−10 |
9.0 × 10−11 |
| Fe–O–MoS2–NH3 |
2.3 × 10−8 |
1.9 × 10−9 |
4.1 × 10−10 |
1.5 × 10−10 |
7.4 × 10−11 |
3.8. Effect of supercell size and effective dopant concentration on electronic and adsorption properties
Moreover, to clarify the influence of supercell size and effective dopant concentration on the fundamental properties of the material, including adsorption energy, magnetism, and electronic structure, we performed additional calculations using 3 × 3 × 1 and 4 × 4 × 1 supercells for Fe–MoS2, Fe–N–MoS2, and Fe–O–MoS2, together with one representative adsorbed gas molecule (CO), as summarized in Table 4. In the 2 × 2 × 1 MoS2 supercell containing 4 Mo sites and 8 S sites, one substituted Fe atom corresponds to Fe–Mo = 1:4 (25%), while one substituted N or O atom corresponds to N/O–S = 1:8 (12.5%). When the supercell size increases to 3 × 3 × 1, these concentrations decrease to 1:9 (11.11%) and 1:18 (5.56%), respectively, and further to 1:16 (6.25%) and 1:32 (3.125%) in the 4 × 4 × 1 supercell. Therefore, increasing the supercell size directly reduces the effective dopant concentration and provides a physical basis for the differences observed among the calculated models.
Table 4 Effect of supercell size on the binding energy, magnetic moment, adsorption energy, and electronic states of Fe–MoS2, Fe–N–MoS2, and Fe–O–MoS2 before and after CO adsorption in the 2 × 2 × 1, 3 × 3 × 1, and 4 × 4 × 1 supercells
| Supercell |
Configurations |
Binding energy (eV) |
Magnetic moment (µB) |
Adsorption energy (eV) |
Electronic states |
| 2 × 2 × 1 |
Fe–MoS2 |
−4.6653 |
2.0000 |
✗ |
Half-metal |
| Fe–MoS2–CO |
−4.2110 |
2.0000 |
−0.247 |
Half-metal |
| Fe–N–MoS2 |
−4.4535 |
2.4121 |
✗ |
Metal |
| Fe–N–MoS2–CO |
−4.2729 |
2.4086 |
−0.150 |
Metal |
| Fe–O–MoS2 |
−4.2368 |
2.3952 |
✗ |
0.03 |
| Fe–O–MoS2–CO |
−4.0730 |
2.3926 |
−0.190 |
0.03 |
| 3 × 3 × 1 |
Fe–MoS2 |
−4.8808 |
2.0000 |
✗ |
Half-metal |
| Fe–MoS2–CO |
−4.8598 |
2.0000 |
−0.279 |
Half-metal |
| Fe–N–MoS2 |
−4.7903 |
1.0000 |
✗ |
0.26 |
| Fe–N–MoS2–CO |
−4.7609 |
1.0000 |
−0.222 |
0.27 |
| Fe–O–MoS2 |
−4.7052 |
2.0000 |
✗ |
0.12 |
| Fe–O–MoS2–CO |
−4.6788 |
2.0000 |
−0.203 |
0.11 |
| 4 × 4 × 1 |
Fe–MoS2 |
−4.9667 |
2.0000 |
✗ |
0.22 |
| Fe–MoS2–CO |
−5.0856 |
2.0000 |
−0.540 |
0.21 |
| Fe–N–MoS2 |
−4.9201 |
1.0000 |
✗ |
0.25 |
| Fe–N–MoS2–CO |
−5.0262 |
1.0000 |
−0.105 |
0.23 |
| Fe–O–MoS2 |
−4.8683 |
2.0000 |
✗ |
0.22 |
| Fe–O–MoS2–CO |
−4.9754 |
2.0000 |
−0.107 |
0.20 |
The additional calculations reveal a clear supercell-size dependence. The binding energy remains within approximately −5 to −4 eV (Table 4), indicating that all systems remain thermodynamically stable. The magnetic moment also varies with supercell size, although it remains relatively large in all cases, suggesting that the magnetic character is preserved. The adsorption energy changes as well, but less systematically than the electronic structure. By contrast, the electronic character shows the strongest dependence on supercell size. As shown in Fig. 10, Fe–MoS2 retains half-metallic behavior in the 2 × 2 × 1 and 3 × 3 × 1 supercells but becomes semiconducting in the 4 × 4 × 1 model with a band gap of about 0.22 eV. A similar transition is observed for Fe–MoS2–CO, which changes from half-metallic behavior in the smaller supercells to a semiconducting state with a band gap of about 0.21 eV in the 4 × 4 × 1 model. Likewise, Fe–N–MoS2 and Fe–N–MoS2–CO evolve from metallic behavior in the 2 × 2 × 1 supercell to semiconducting states in the larger supercells, with 4 × 4 × 1 band gaps of about 0.25 and 0.23 eV, respectively. Fe–O–MoS2 and Fe–O–MoS2–CO show the same concentration-dependent trend, with the band gap increasing from about 0.03 eV in the 2 × 2 × 1 model to about 0.12/0.11 eV in the 3 × 3 × 1 supercell and further to about 0.22/0.20 eV in the 4 × 4 × 1 model. These results indicate that the 4 × 4 × 1 supercell should not be regarded merely as an auxiliary calculation, but rather as an essential lower-concentration reference for clarifying how effective dopant concentration governs the predicted properties of the doped MoS2 systems.
 |
| | Fig. 10 Effect of supercell size on the electronic band structure of Fe–MoS2, Fe–N–MoS2, and Fe–O–MoS2 before and after CO adsorption in the 2 × 2 × 1, 3 × 3 × 1, and 4 × 4 × 1 supercells. In which, from left to right: (a) Fe–MoS2, (b) Fe–N–MoS2, (c) Fe–O–MoS2, (d) Fe–MoS2–CO, (e) Fe–N–MoS2–CO, and (f) Fe–O–MoS2–CO for the 2 × 2 × 1, 3 × 3 × 1, and 4 × 4 × 1 supercells. | |
4. Conclusion
In this study, first-principles calculations were systematically employed to elucidate how supercell size and the corresponding effective dopant concentration govern the predicted properties of Fe-doped and Fe–N/Fe–O co-doped MoS2 with the CO, CO2, NH3 adsorption, thereby establishing a clearer physical basis for interpreting concentration-dependent behavior in doped two-dimensional systems. The results show that enlarging the supercell from 2 × 2 × 1 to 3 × 3 × 1 and 4 × 4 × 1 markedly reduces the effective dopant concentration and induces pronounced changes in the electronic structure, while the magnetic moment remains comparatively stable and the adsorption energy varies less systematically. In particular, Fe–MoS2, Fe–N–MoS2, and Fe–O–MoS2, together with their corresponding CO-adsorbed configurations, evolve from metallic or half-metallic states in the smaller supercells toward semiconducting behavior in the 4 × 4 × 1 model, with band gaps reaching about 0.22, 0.25, and 0.22 eV, respectively, confirming that the 4 × 4 × 1 supercell should be regarded as an essential lower-concentration reference rather than a secondary verification model. At the same time, the 2 × 2 × 1 supercell remains physically meaningful as a concentrated-doping regime, within which stronger dopant–dopant and dopant–host interactions generate more pronounced electronic responses. Under this regime, Fe–N–MoS2 exhibits the most noticeable tendency toward NH3 adsorption, as reflected by an adsorption energy of −0.264 eV, a shortened adsorption distance of 1.5267 Å, evident Fe-3d/N-2p hybridization, stronger charge redistribution, a maximum selectivity of 2.24%, and a calculated electronic sensitivity of 98.9% at the Fermi level. Overall, the present work highlights the novelty that supercell size in doped MoS2 is not merely a computational setting but a physically decisive parameter that controls the balance among electronic structure, magnetic character, and adsorption behavior, and thus provides a more rigorous framework for evaluating and designing doped and co-doped MoS2-based gas-sensing materials.
Author contributions
Thi H. Ho: investigation, data curation, formal analysis, writing – original draft. Khiet An Vuong: data curation, visualization. Le Phuong Truong: writing – reviewing and editing. Nguyen Hai Dang: writing – reviewing and editing. Minh-Thuan Pham: writing – reviewing and editing. D. M. Hoat: formal analysis, writing – reviewing and editing. Huynh Anh Huy: software, formal analysis, writing – reviewing and editing. Duy Khanh Nguyen: supervision, software, conceptualization, validation, resources, funding acquisition, formal analysis, writing – reviewing and editing.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability
The data that supports the findings of this study are available within the article.
Supplementary information (SI): investigated configurations of Fe-doped and Fe–N/Fe–O co-doped MoS2 systems with the CO, CO2, NH3 adsorptions. See DOI: https://doi.org/10.1039/d5ra09296e.
Acknowledgements
Thi H. Ho and Duy Khanh Nguyen gratefully acknowledge the support of Van Lang University.
References
- P. Saxena and P. Shukla, A Review on Recent Developments and Advances in Environmental Gas Sensors to Monitor Toxic Gas Pollutants, Environ. Prog. Sustainable Energy, 2023, 42, e14126 CrossRef CAS.
- R. A. Potyrailo, Multivariable Sensors for Ubiquitous Monitoring of Gases in the Era of Internet of Things and Industrial Internet, Chem. Rev., 2016, 116(19), 11877–11923, DOI:10.1021/acs.chemrev.6b00187.
- C. Hu, W. Zhang, J. Yang, Y. Pei, X. Tan, B. Dong, H. Song and L. Xu, Chemiresistive Gas Sensors for Intelligent Sensing: Design Strategies{,} Emerging Applications and Future Challenges, Chem. Soc. Rev., 2025, 54, 11302–11367, 10.1039/D5CS00979K.
- D. K. Nguyen, D. M. Hoat, A. Bafekry, V. Van On, J. F. Rivas-Silva, M. Naseri and G. H. Cocoletzi, Theoretical Prediction of the PtOX (X = S and Se) Monolayers as Promising Optoelectronic and Thermoelectric 2D Materials, Phys. E Low-dimens. Syst. Nanostruct., 2021, 131, 114732, DOI:10.1016/J.PHYSE.2021.114732.
- D. K. Nguyen, J. Guerrero-Sanchez, T. V. Vu, R. Ponce-Pérez and D. M. Hoat, Electronic and Magnetic Properties of the WSO Janus Monolayer Engineered by Intrinsic Defects, Surf. Interfaces, 2022, 32, 102114, DOI:10.1016/J.SURFIN.2022.102114.
- D. K. Nguyen, C. V. Ha, J. Guerrero-Sanchez and D. M. Hoat, Doping Janus MoSSe Monolayer with Al/Ga and P/As Atoms, and Their Clusters: Effective Methods for the Band Structure and Magnetism Engineering, RSC Adv., 2025, 15(7), 5096–5104, 10.1039/d5ra00561b.
- D. K. Nguyen, D. Q. Hoang and D. M. Hoat, Exploring the Sensing Ability of B- and Si-Doped WS2 Monolayer toward CO and NO Gas, Int. J. Quantum Chem., 2022, 122(15), 1–10, DOI:10.1002/qua.26916.
- N. T. T. Tran, G. Gumbs, D. K. Nguyen and M. F. Lin, Fundamental Properties of Metal-Adsorbed Silicene: A DFT Study, ACS Omega, 2020, 5(23), 13760–13769, DOI:10.1021/acsomega.0c00905.
- D. K. Nguyen, N. T. T. Tran, Y. H. Chiu and M. F. Lin, Concentration-Diversified Magnetic and Electronic Properties of Halogen-Adsorbed Silicene, Sci. Rep., 2019, 9(1), 1–15, DOI:10.1038/s41598-019-50233-w.
- D. K. Nguyen, D. Q. Hoang and D. M. Hoat, Exploring a Silicene Monolayer as a Promising Sensor Platform to Detect and Capture NO and CO Gas, RSC Adv., 2022, 12(16), 9828–9835, 10.1039/d2ra00442a.
- Q. Hu, Y. Li, L. Xu and Y. Gui, Adsorption Performance of Cu, Zn-Decorated Ti3C2O2 for Harmful Gas Molecules in Greenhouse Environments: A DFT Study, Diam. Relat. Mater., 2025, 158, 112674, DOI:10.1016/J.DIAMOND.2025.112674.
- S. Chen, L. Lin and Y. Gui, Ptn (N=1-3) Cluster-Doped Ti3C2O2 as an Adsorbent Material for Eliminating Dissolved Gases (CO, C2H2, CH4, C2H4) within Transformer Oil: A Density Functional Theory (DFT) Investigation, Surf. Interfaces, 2025, 72, 107270, DOI:10.1016/J.SURFIN.2025.107270.
- T. Zhang, G. He, Y. Gui, S. Liu, Y. Li and L. Cao, Investigating the Adsorption Performance of Agricultural Greenhouses Hazardous Gases on Rh Doped HfX2 (X=S, Se) Monolayers through DFT for Potential Gas Sensor Applications, Surf. Interfaces, 2024, 52, 104914, DOI:10.1016/J.SURFIN.2024.104914.
- Y. Wang, Y. Gui, J. Yang, G. Jin, P. Yang, M. Gao and H. Huang, DFT Study of Metal (Ag, Au, Pt)-Modified SnS2 for Adsorption of SF6 Decomposition Gases in Gas-Insulated Switchgear, Langmuir, 2024, 40(13), 7049–7059, DOI:10.1021/acs.langmuir.4c00110.
- K. F. Mak, C. Lee, J. Hone, J. Shan and T. F. Heinz, Atomically Thin MoS2: A New Direct-Gap Semiconductor, Phys. Rev. Lett., 2010, 105(13), 2–5, DOI:10.1103/PhysRevLett.105.136805.
- H. A. Huy, V. Khiet An, P. T. Le, H. D. Nguyen, M.-T. Pham, O. Khyzhun, D. D. Vo and D. K. Nguyen, First-Principles Investigation of B, O and B, N Co-Doped MoS2 Monolayers toward CO, CO2, and NH3 Gases, ACS Appl. Nano Mater., 2026, 9(4), 2105–2116, DOI:10.1021/acsanm.5c05571.
- K. A. Vuong, N. H. Dang, L. P. Truong, M. T. Pham, D. M. Hoat, M. T. Dang, G. P. Chang-Chien and D. K. Nguyen, Enhanced Sensing Characteristics of N Doped and (N, O) Co-Doped Molybdenum Disulfide to Detect Toxic Gases: A Comprehensive First-Principles Study, RSC Adv., 2026, 16(4), 3662–3680, 10.1039/d5ra08975a.
- D. Zhang, Y. Sun, P. Li and Y. Zhang, Facile Fabrication of MoS2-Modified SnO2 Hybrid Nanocomposite for Ultrasensitive Humidity Sensing, ACS Appl. Mater. Interfaces, 2016, 8(22), 14142–14149, DOI:10.1021/acsami.6b02206.
- C. Guo, L. Liu, Z. Cai, Y. Wang, K. Guan, F. Sun, W. Zhou, X. Gou, X. He, X. Wang and T. Zhang, Two-Dimensional SnS2 Single Crystal for Sensitive NO2 Detection at Room Temperature, Microsyst. Nanoeng., 2025, 11, 246, DOI:10.1038/s41378-025-01054-7.
- G. Yang, X. Wang, Y. Li, Z. Zhang, J. Huang, F. Zheng, Q. Pan, H. Wang, Q. Li and Y. Cai, Self-Supporting Network-Structured MoS2/Heteroatom-Doped Graphene as Superior Anode Materials for Sodium Storage, RSC Adv., 2023, 13(18), 12344–12354, 10.1039/d2ra08207a.
- B. Cheng, H. Wang, S. Xiang, S. Lu and B. Ren, MoS2-Based Composites for Electrochemical Detection of Heavy Metal Ions: A Review, Nanomaterials, 2025, 15(21), 1–27, DOI:10.3390/nano15211639.
- P. Tripathi, D. Singh, A. Pathak, A. K. Verma, A. S. K. Sinha and S. Singh, Engineering MoS2-Based Electrocatalysts for Water Splitting: A Comprehensive Review of Doping{,} Heterostructures{,} and Support Integration Strategies, J. Mater. Chem. A, 2025, 13, 39603–39659, 10.1039/D5TA05734E.
- M. Qu, S. Xu, A. Du, C. Zhao and Q. Sun, CO2 Capture, Separation and Reduction on Boron-Doped MoS2, MoSe2 and Heterostructures with Different Doping Densities: A Theoretical Study, ChemPhysChem, 2021, 22(23), 2392–2400, DOI:10.1002/CPHC.202100377.
- M. J. Szary and P. Radomski, Unveiling the Chemical Underpinnings behind the Enhanced Adsorption Interaction of NO2 on MoS2, MoSe2, and MoTe2 Transition Metal Dichalcogenides, J. Phys. Chem. C, 2023, 127(43), 21374–21386, DOI:10.1021/acs.jpcc.3c05101.
- C. Shen, Y. Q. Chen and W. Zhao, Enhanced VOCs Adsorption on Group VIII Transition Metal-Doped MoS2: A DFT Study, Chem. Phys., 2025, 589, 112497, DOI:10.1016/J.CHEMPHYS.2024.112497.
- S. R. Arnab, J. Halder and M. S. Islam, Detecting Decomposition Gases with Ni-Doped MoS2: A First-Principles DFT Calculation, Chem. Phys., 2025, 593, 112621, DOI:10.1016/J.CHEMPHYS.2025.112621.
- A. Vaidyanathan, S. Lakshmy, G. Sanyal, S. Joseph, N. Kalarikkal and B. Chakraborty, Nitrobenzene Sensing in Pristine and Metal Doped 2D Dichalcogenide MoS2: Insights from Density Functional Theory Investigations, Appl. Surf. Sci., 2021, 550, 149395, DOI:10.1016/J.APSUSC.2021.149395.
- A. Wu, Q. Song and H. Liu, Oxygen Atom Adsorbed on the Sulphur Vacancy of Monolayer MoS2: A Promising Method for the Passivation of the Vacancy Defect, Comput. Theor. Chem., 2020, 1187, 112906, DOI:10.1016/J.COMPTC.2020.112906.
- S. Zhao, J. Xue and W. Kang, Gas Adsorption on MoS2 Monolayer from First-Principles Calculations, Chem. Phys. Lett., 2014, 595–596, 35–42, DOI:10.1016/j.cplett.2014.01.043.
- Y. Linghu and C. Wu, Gas Molecules on Defective and Nonmetal-Doped MoS2 Monolayers, J. Phys. Chem. C, 2020, 124(2), 1511–1522, DOI:10.1021/acs.jpcc.9b10450.
- L. J. Kong, G. H. Liu and L. Qiang, Electronic and Optical Properties of O -Doped Monolayer MoS2, Comput. Mater.
Sci., 2016, 111, 416–423, DOI:10.1016/j.commatsci.2015.10.001.
- H. Yang, Y. Liu, C. Gao, L. Meng, Y. Liu, X. Tang and H. Ye, Adsorption Behavior of Nucleobases on Doped MoS2 Monolayer: A DFT Study, J. Phys. Chem. C, 2019, 123(51), 30949–30957, DOI:10.1021/acs.jpcc.9b08018.
- F. Tan, J. Li, X. Fang and L. Guan, The Optical Properties of Few-Layer MoS2 by DFT Calculations, Phys. E Low-dimens. Syst. Nanostruct., 2024, 155, 115813, DOI:10.1016/J.PHYSE.2023.115813.
- J. Maksymiuk, I. A. Wrona, R. Szczesniak and A. P. Durajski, Tunable Optical and Electronic Properties of Monolayer MoS2 via Substitutional Doping, Surf. Sci., 2025, 761, 122788, DOI:10.1016/J.SUSC.2025.122788.
- J.-Y. Ji, W. Zhang, C. Li, Y. Cao, J. Xue, H. Gu and J.-P. Lang, In Situ Fe/Co/B Codoped MoS2 Ultrathin Nanosheets Enable Efficient Electrocatalytic Nitrogen Reduction, ACS Appl. Mater. Interfaces, 2024, 16(32), 41734–41742, DOI:10.1021/acsami.4c09370.
- Y. Jing, X. Tan, Z. Zhou and P. Shen, Tuning Electronic and Optical Properties of MoS2 Monolayer via Molecular Charge Transfer, J. Mater. Chem. A, 2014, 2(40), 16892–16897, 10.1039/C4TA03660C.
- P. Zhao, H. Wang, Z. Huang, B. Fan, Y. Liu, Z. Yin, Y. Wang, X. Zheng, Y. Deng and X. Fan, High-Throughput Screening of Nitrogen Reduction Reaction on Single Atom@1T’-MoS2, Appl. Surf. Sci., 2023, 631, 157480, DOI:10.1016/J.APSUSC.2023.157480.
- T. E. Gber, H. Louis, A. E. Owen, B. E. Etinwa, I. Benjamin, F. C. Asogwa, M. M. Orosun and E. A. Eno, Heteroatoms (Si, B, N, and P) Doped 2D Monolayer MoS2 for NH3 Gas Detection, RSC Adv., 2022, 12(40), 25992–26010, 10.1039/d2ra04028j.
- M. Shakil, A. Naz, I. Zeba, S. S. A. Gillani, M. Rafique, S. Ahmed and M. Zafar, Structural and Magnetic Behavior of MoS2 on Doping of Transition Metals: A DFT Study, J. Supercond. Novel Magn., 2021, 34(12), 3441–3453, DOI:10.1007/s10948-021-06074-8.
- H. Yan, H. Chen, X. Cui, Q. Guan, B. Wang and Y. Cai, Unraveling Energetics and States of Adsorbing Oxygen Species with MoS2 for Modulated Work Function, Nanoscale Horiz., 2025, 10(2), 359–368, 10.1039/D4NH00441H.
- R. Liu, H. L. Fei and G. L. Ye, Recent Advances in Single Metal Atom-Doped MoS2 as Catalysts for Hydrogen Evolution Reaction, Tungsten, 2020, 2(2), 147–161, DOI:10.1007/s42864-020-00045-7.
- A. Kushwaha, N. R. Bharti, A. Sharma, S. K. Kedia, G. Gupta and N. Goel, Enhanced NO2 Gas Sensing in Nanocrystalline MoS2 via Swift Heavy Ion Irradiation: An Experimental and DFT Study, ACS Sens., 2024, 9(11), 5966–5975, DOI:10.1021/acssensors.4c01812.
- Y. Jiang, S. Li, F. Zhang, W. Zheng, L. Zhao and Q. Feng, Metal-Semiconductor 1T/2H-MoS2 by a Heteroatom-Doping Strategy for Enhanced Electrocatalytic Hydrogen Evolution, Catal. Commun., 2021, 156, 106325, DOI:10.1016/j.catcom.2021.106325.
- R. Du and W. Wu, Adsorption of Gas Molecule on Rh, Ru Doped Monolayer MoS2 for Gas Sensing Applications: A DFT Study, Chem. Phys. Lett., 2022, 789, 139300, DOI:10.1016/J.CPLETT.2021.139300.
- Y. Li, C. Yu, Y. Gan, P. Jiang, J. Yu, Y. Ou, D. F. Zou, C. Huang, J. Wang, T. Jia, Q. Luo, X. F. Yu, H. Zhao, C. F. Gao and J. Li, Mapping the Elastic Properties of Two-Dimensional MoS2 via Bimodal Atomic Force Microscopy and Finite Element Simulation, npj Comput. Mater., 2018, 4, 49, DOI:10.1038/s41524-018-0105-8.
- F. M. Enujekwu, Y. Zhang, C. I. Ezeh, H. Zhao, M. Xu, E. Besley, M. W. George, N. A. Besley, H. Do and T. Wu, N-Doping Enabled Defect-Engineering of MoS2 for Enhanced and Selective Adsorption of CO2: A DFT Approach, Appl. Surf. Sci., 2021, 542, 148556, DOI:10.1016/j.apsusc.2020.148556.
- Y. Gui, Y. Lin, C. Ji, P. Luo and X. Chen, Density Functional Theory Calculations for the Adsorption Property of Hazardous Industrial Gasses on Transition-Metal-Modified MoS2 Nanosheets, ACS Appl. Nano Mater., 2022, 5(8), 11111–11118, DOI:10.1021/acsanm.2c02283.
- R. Costa-Amaral, A. Forhat, N. A. M. S. Caturello and J. L. F. D. Silva, Unveiling the Adsorption Properties of 3d, 4d, and 5d Metal Adatoms on the MoS2 Monolayer: A DFT-D3 Investigation, Surf. Sci., 2020, 701, 121700, DOI:10.1016/J.SUSC.2020.121700.
- Z. Li, D. Zhou and J. Xiangke, Highly Selective Adsorption on Monolayer MoS2 Doped with Pt, Ag, Au and Pd and Effect of Strain Engineering: A DFT Study, Sens. Actuators, A Phys., 2021, 322, 112637, DOI:10.1016/j.sna.2021.112637.
- A. M. Homayounfard, M. Maleki, H. Ghanbari, M. H. Kahnamouei and B. Safaei, Growth of Few-Layer Flower-like MoS2 on Heteroatom-Doped Activated Carbon as a Hydrogen Evolution Reaction Electrode, Int. J. Hydrogen Energy, 2024, 55, 1360–1370, DOI:10.1016/J.IJHYDENE.2023.11.123.
- M. Yin, K. Wang, C. Gao, R. Yang, Y. Huang and L. Yu, Synthesis and Insights into the Gas Sensing Mechanisms of N-Doped MoS2 Hierarchical Structures with Superior Gas Sensing Properties at Room Temperature, Mater. Res. Bull., 2024, 179, 112943, DOI:10.1016/J.MATERRESBULL.2024.112943.
- D. Zhang, J. Wu, P. Li and Y. Cao, Room-Temperature SO2 Gas-Sensing Properties Based on a Metal-Doped MoS2 Nanoflower: An Experimental and Density Functional Theory Investigation, J. Mater. Chem. A, 2017, 5(39), 20666–20677, 10.1039/C7TA07001B.
- J. Bai, C. Tang, S. Zhao, S. Liu, W. Wang, C. Han, Q. Zhao and Y. Shen, Few-Layer Molybdenite-Derived MoS2 Nanosheets and SnO2 Nanostructures Heterojunctions for High-Performance NH3 Detection: Insights from Experimental and DFT Analysis, Chem. Eng. J., 2025, 509, 161374, DOI:10.1016/J.CEJ.2025.161374.
- J. Hafner, Materials Simulations Using VASP—a Quantum Perspective to Materials Science, Comput. Phys. Commun., 2007, 177(1–2), 6–13, DOI:10.1016/J.CPC.2007.02.045.
- L. O. Wagner, T. E. Baker, E. M. Stoudenmire, K. Burke and S. R. White, Kohn-Sham Calculations with the Exact Functional, Phys. Rev. B Condens. Matter, 2014, 90(4), 1–15, DOI:10.1103/PhysRevB.90.045109.
- G. Kresse and D. Joubert, From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method, Phys. Rev. B, 1999, 59, 1758 CrossRef CAS.
- S. Dudarev and G. Botton, Electron-Energy-Loss Spectra and the Structural Stability of Nickel Oxide: An LSDA+U Study, Phys. Rev. B Condens. Matter, 1998, 57(3), 1505–1509, DOI:10.1103/PhysRevB.57.1505.
- Y. R. Wang, S. Li and J. B. Yi, Transition Metal-Doped Tin Monoxide Monolayer: A First-Principles Study, J. Phys. Chem. C, 2018, 122(8), 4651–4661, DOI:10.1021/acs.jpcc.7b12282.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu, J. Chem. Phys., 2010, 132(15), 154104, DOI:10.1063/1.3382344.
- J. Ma, J. He, D. Mazumdar, K. Munira, S. Keshavarz, T. Lovorn, C. Wolverton, A. W. Ghosh and W. H. Butler, Computational Investigation of Inverse Heusler Compounds for Spintronics Applications, Phys. Rev. B, 2018, 98(9), 94410, DOI:10.1103/PhysRevB.98.094410.
- J. Wu, X. Li, L. Huang, T. Liang, X. Xing and A. Luo, SnSe Monolayer-Based Heavy Metal Sensors with High Sensitivity, Selectivity, and Reusability: Insights from First Principle Calculation, Results Phys., 2023, 53, 106973, DOI:10.1016/j.rinp.2023.106973.
- V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, Crystal Orbital Hamilton Population (COHP) Analysis as Projected from Plane-Wave Basis Sets, J. Phys. Chem. A, 2011, 115(21), 5461–5466, DOI:10.1021/jp202489s.
- L. Ju, X. Tang, X. Li, B. Liu, X. Qiao, Z. Wang and H. Yin, NO2 Physical-to-Chemical Adsorption Transition on Janus WSSe Monolayers Realized by Defect Introduction, Molecules, 2023, 28, 1644 CrossRef CAS PubMed.
- R. F. W. Bader, A Quantum Theory of Molecular Structure and Its Applications, Chem. Rev., 1991, 91(5), 893–928, DOI:10.1021/cr00005a013.
- K. Wang, M. Kapitzke, L. Green and B. Paulus, Modulating Electronic and Optical Properties of Monolayered MoS2 by Covalent Mono- and Bisfunctionalization, J. Mater. Chem. C, 2022, 10(15), 6009–6016, 10.1039/d2tc00391k.
- A. Kahn, Fermi Level, Work Function and Vacuum Level, Mater. Horiz., 2016, 3(1), 7–10, 10.1039/c5mh00160a.
- J. Prasongkit, V. Shukla, A. Grigoriev, R. Ahuja and V. Amornkitbamrung, Ultrahigh-Sensitive Gas Sensors Based on Doped Phosphorene: A First-Principles Investigation, Appl. Surf. Sci., 2019, 497, 143660, DOI:10.1016/j.apsusc.2019.143660.
- R. Gao, Y. Yong, X. Yuan, S. Hu, Q. Hou and Y. Kuang, First-Principles Investigation of Adsorption Behaviors and Electronic, Optical, and Gas-Sensing Properties of Pure and Pd-Decorated GeS2Monolayers, ACS Omega, 2022, 7(50), 46440–46451, DOI:10.1021/acsomega.2c05142.
- Y. H. Zhang, Y. B. Chen, K. G. Zhou, C. H. Liu, J. Zeng, H. L. Zhang and Y. Peng, Improving Gas Sensing Properties of Graphene by Introducing Dopants and Defects: A First-Principles Study, Nanotechnology, 2009, 20, 185504, DOI:10.1088/0957-4484/20/18/185504.
- B. A. Kalwar, W. Fangzong, A. M. Soomro, M. R. Naich, M. H. Saeed and I. Ahmed, Highly Sensitive Work Function Type Room Temperature Gas Sensor Based on Ti Doped HBN Monolayer for Sensing CO2, CO, H2S, HF and NO. A DFT Study, RSC Adv., 2022, 12(53), 34185–34199, 10.1039/d2ra06307g.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Khiet An Vuongc,
Le Phuong Truongd,
Nguyen Hai Dange,
Minh-Thuan Phamf,
D. M. Hoatgh,
Huynh Anh Huy
ab,
Khiet An Vuongc,
Le Phuong Truongd,
Nguyen Hai Dange,
Minh-Thuan Phamf,
D. M. Hoatgh,
Huynh Anh Huy













