DOI:
10.1039/D5RA09260D
(Paper)
RSC Adv., 2026,
16, 12702-12712
Benzo[d]imidazole anchored oxadiazole derivatives: synthesis, characterization, biological evaluation, in silico docking and ADME-T analysis
Received
30th November 2025
, Accepted 20th February 2026
First published on 6th March 2026
Abstract
A novel series of 1,2,4-oxadiazoles based on benzo[d]imidazole-1,3,4-oxadiazole fused 1,2,4-oxadiazoles, were synthesised. The proposed chemical structures of the novel hybrids were confirmed by a variety of spectroscopic methods, including HRMS, NMR (1H/13C), and infrared. The antibacterial properties of these compounds showed that compound 8b (37 ± 1.20 mm) and compound 8i (6.9 ± 1.01 µM) had the highest zone of inhibition (ZI) and lowest minimum inhibitory (MIC) values against S. aureus ATCC 29213, which was verified by in silico evaluation. Compound 8f exhibited considerably better antibacterial activity (31 ± 0.19 mm) against S. epidermidis ATCC 12228, and scaffold 8f showed a significantly higher antibacterial effect (31 ± 0.19 mm) against S. pyogenes ATCC 19615 compared to moxifloxacin (29 ± 0.16 mm). Finally, in silico research that includes molecular modelling also validates an in vitro investigation and explains the strong binding pattern of 8b, 8f, 8i, and 8k against E. coli Topoisomerase IV (PDB: 3FV5) and S. aureus GyrB (PDB: 4URN). Extending our exploration, an analysis of the ADME-Tox profiling confirmed the safe use of these newly synthesized scaffolds, paving the way for promising therapeutic applications in the field of antimicrobial therapy.
1 Introduction
As a rising concern to world health, antibiotic resistance increases morbidity, mortality, and medical expenses.1 Since many bacterial isolates are becoming more resistant to numerous drugs, bacterial illnesses are seen as a global issue. Every year, almost 700![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people die from diseases that cannot be cured.2 Experts estimate that more than 10 million people would die from various antibiotic resistances globally by 2050 if the problem is not resolved. Severe infections, which greatly worsen suffering in people with compromised immune systems and result in major morbidity and mortality, are commonly caused by a variety of pathogenic microorganisms, including bacteria, fungi, yeasts, and viruses.3 Antimicrobial resistance (AMR), which has been exacerbated by the COVID-19 pandemic, has posed a serious danger to public health and global economic development.4 While there are several reasons for this, the increase in HIV-positive patients and the number of cancer patients undergoing chemotherapy are two important ones.5 Thankfully, other antibiotics have been approved for commercial use through structural modification of tetracyclines, quinolones and aminoglycosides, and other compounds, such as plazomicin, delafloxacin, moxifloxacin, and gemifloxacin. At the same time, certain natural antibacterial compounds have been the subject of clinical studies.6 As a result, researchers are now looking for novel molecular targets for antimicrobial drugs. Therefore, there is a continuing need for new antimicrobial drugs that can specifically target various bacteria without interfering with the metabolic functions of the host.
000 people die from diseases that cannot be cured.2 Experts estimate that more than 10 million people would die from various antibiotic resistances globally by 2050 if the problem is not resolved. Severe infections, which greatly worsen suffering in people with compromised immune systems and result in major morbidity and mortality, are commonly caused by a variety of pathogenic microorganisms, including bacteria, fungi, yeasts, and viruses.3 Antimicrobial resistance (AMR), which has been exacerbated by the COVID-19 pandemic, has posed a serious danger to public health and global economic development.4 While there are several reasons for this, the increase in HIV-positive patients and the number of cancer patients undergoing chemotherapy are two important ones.5 Thankfully, other antibiotics have been approved for commercial use through structural modification of tetracyclines, quinolones and aminoglycosides, and other compounds, such as plazomicin, delafloxacin, moxifloxacin, and gemifloxacin. At the same time, certain natural antibacterial compounds have been the subject of clinical studies.6 As a result, researchers are now looking for novel molecular targets for antimicrobial drugs. Therefore, there is a continuing need for new antimicrobial drugs that can specifically target various bacteria without interfering with the metabolic functions of the host.
Numerous researchers in medicinal chemistry have been drawn to the promising findings in the chemistry, structure–activity relationship, and biological activities of various heterocycles, particularly interested in the synthesis of benzimidazole and its derivatives. Heterocyclic compounds, especially nitrogen heterocycles, make up a substantial class of chemicals utilized in the pharmaceutical business and account for almost 60% of all pharmacological medications.7 Benzimidazole scaffold exhibit high aromaticity and metabolic stability, which is advantageous for drug availability and efficacy. Its heteroatoms facilitate key interactions, such as hydrogen bonding and hydrophobic interactions, with various biological targets, including enzymes (e.g., β-tubulin, ChE inhibitors) and α-amylase and α-glucosidase inhibitors. The thiadiazole moiety can also inhibit DNA/RNA synthesis, contributing to its broad-spectrum of activity observed in several FDA-approved drugs like albendazole, tiabendazole, benomyl, enviradene, and bendamustine have been used as fungicidal agents in the market (Fig. 1). An extensive pharmacological framework of benzimidazole has been reported in a number of publications. These include antibacterial,8 antifungal,9 antituberculosis,10 antiviral,11 antimalarial,12 antiulcer,13 antileishmanial,14 anti-inflammatory,15 antidiabetic,16 antiprotozoal,17 anti-convulsant,18 antioxidant,19 anti-hypertensive,20 anti-Alzheimer, and analgesic.21
 |
| | Fig. 1 Commercial medications chemical structures based on 1,3,4-oxadiazole, 1,2,4-oxadiazoles, and benzo[d]imidazole. | |
The analogues of 1,3,4-oxadiazole and imidazole are the basic building blocks of many medications.22 This class of heterocycles contains compounds that have demonstrated a wide range of therapeutic potential, including antibacterial,23 antiepileptic,24 anticancer,25 antiviral,26 and anti-inflammatory properties.27 Commercial medications such as Raltegravir (anti-HIV), Furamizole (antimicrobial), Zibotentan (anticancer), Tiodazosin (antihypertensive), Nesapidil (vasodilator), and Fenadiazole (sedative) all contain the 1,3,4-oxadiazole molecule, which has proven pharmacophore status (Fig. 1).28 The hydrophilic and electron-donating characteristics oxadiazole ring allows for non-covalent binding with enzyme and other biomolecules, nitrogenated-based heterocycles, like 1,2,4-oxadiazole-derived molecules, exhibit encouraging biological activity.29 Numerous bioactive qualities, such as antibacterial,30 anticancer,31 antitubercular,32 anti-inflammatory, and antiparasitic activities,33 have since been shown by studies on 1,2,4-oxadiazole derivatives. Additionally, a 1,2,4-oxadiazole moiety is present in the molecular structure of clinically used medications like Oxolamine, Prenoxdiazine, Irrigor, Fasiplon (GABA-receptor), and Butalamine (vasodilatation agent), respectively. This emphasizes how crucial this moiety is to drug discovery and design (Fig. 1).34
Recent studies highlight that 1,3,4-oxadiazole derivatives are increasingly important as privileged scaffolds in medicinal chemistry, with advancements focused on overcoming drug resistance and improving the efficiency of synthesis. The novelty in recent literature lies in the shift toward conventional/microwave synthetic methodologies that produce high yields, as well as the design of hybrid molecules that target complex, multidrug-resistant infections and cancer cell lines. Molecular hybridization is a powerful strategy in medicinal chemistry and drug discovery where two or more pharmacophoric units from different bioactive substances are combined into a single molecule. This approach aims to create a new hybrid compound with enhanced affinity, improved efficacy, altered selectivity profiles, and/or dual modes of action, often resulting in superior properties compared to the parent compounds or simple combination therapies. In this work, a series of benzo[d]imidazole derivatives with 1,2,4-oxadiazole and 1,3,4-oxadiazole groups are synthesized and evaluated, in silico modeling and biological evaluation.35 We anticipate that this study will increase our understanding of how the pharmacological action of oxadiazole derivatives of benzimidazole is influenced by their chemical structure. The aromatic rings benzimidazole and bis-oxadiazole both add to the molecule's overall rigidity and planarity, which is advantageous for attaching to biological targets. The structural flexibility and varied substituent patterns on both rings increase the potential for fine-tuning the compounds for specific therapeutic outcomes. The findings of this study may contribute for novel antimicrobial drugs with enhanced selectivity and efficacy, expanding the range of chemotherapeutic alternatives for the treatment of bacteria. The present heterocyclic work offers significant practical and conceptual advantages by addressing the need for more efficient, sustainable, and functionalized chemical synthesis in the pharmaceutical and materials science industries.
2 Results and discussion
This study used the Vilsmeier reaction sequence shown in Scheme 1 to synthesize a new series of 1,2,4-oxadiazole based on benzo[d]imidazole-1,3,4-oxadiazole. We have prepared compounds number 1–3 based on previous literature.36 Methyl 3-(1,5-dimethyl-1H-benzo[d]imidazole-2-yl)propanoate (1) and hydrazine hydrate were refluxed for approximately 10 hours at 75 °C with methanol present. 5-[2-(1,5-Dimethyl-1H-1,3-benzodiazol-2-yl)ethyl]-1,3,4-oxadiazole-2-thiol (3) was obtained by refluxing the reaction mixture for eight hours after the hydrazide (2) further reacted with carbon disulphide and potassium hydroxide in ethanol. The yield was excellent, at 90%. Additionally, in the presence of potassium carbonate at room temperature, the oxadiazole (3) was transformed into propargylation in the presence of 2-bromoacetonitrile as a light-brown solid (4). The key intermediate (5) was synthesized by the reaction of (4) in DCM in the presence of mCPBA at 0 °C for 2 h. (Z)-2-{[5-[2-(1,5-Dimethyl-1H-benzimidazol-2-yl)ethyl]-1,3,4-oxadiazol-2-yl]sulfonyl}-N′-hydroxyethanimidamide (6) was create when a combination of TEA and hydroxylamine hydrochloride was added dropwise to DCM at rt. O-Acylation and cyclization steps were conducted in a one-pot reaction with the carboxylic acids in the presence of coupling reagent 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC.HCl). Next, 1,2,4-oxadiazoles were obtained through reflux of the acylated intermediate in ethanol in the presence of sodium acetate (Scheme 1).37 The 1,2,4-oxadiazole derivatives were then formed as a result of component (6) and the corresponding acids (7a–7k) in refluxing DCM for eight hours in the presence of EDC.HCl. Ultimately, the obtained intermediate was refluxed in the presence of sodium acetate in ethanol for 3–4 hours. Reaction progress was monitored by TLC (2:8; ethyl acetate and hexane). After completion, the reaction solvent was distilled out, then 2 N NaOH was added, followed by extraction with ethyl acetate. The separated organic layer was distilled, and the crude was purified by flash column chromatography to produce compounds (8a–8k) (67–92%) (Fig. 2).
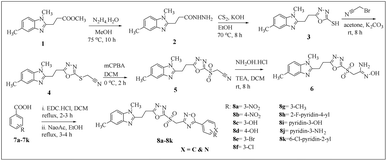 |
| | Scheme 1 Synthesis of benzo[d]imidazole-substituted oxadiazole derivatives (8a–8k). | |
 |
| | Fig. 2 Synthesized oxadiazole scaffolds. | |
2.1 Antibacterial activity
Three Gram-negative strains, Escherichia coli ATCC 25922, Klebsiella pneumonia ATCC 13883, and Pseudomonas aeruginosa ATCC 27853, as well as three Gram-positive strains, Streptococcus pyogenes ATCC 19615, Staphylococcus epidermidis ATCC 12228, and Staphylococcus aureus ATCC 29213, were used to test the titled derivatives (8a–8k) in vitro antibacterial activity. Title substances antibacterial inhibitory activity was tested using a previously established agar wall diffusion technique. The diameter zone of inhibition (ZI) and minimal inhibitory concentration (MIC) values for the investigated derivatives are displayed in Tables 1 and 2.38 The results indicated that the derivatives antibacterial activity was greatly impacted by the sort and makeup of the substituents on the oxadiazole moiety and phenyl ring. The tested compounds (8a–8k) showed moderate to good antibacterial effectiveness against Gram-negative bacteria as compared to Gram-positive bacteria. When compared to the conventional moxifloxacin and gemifloxacin, all compounds showed moderate antibacterial activity. Oxadiazole compound 8b, which had a zone of inhibition of 37 ± 1.20 mm and significant antibacterial activity against S. aureus ATCC 29213, was used as a positive control based on the findings of antibacterial screening. The results revealed that the highest S. pyogenes ATCC 19615 inhibition potential is exhibited by compound 8f with ZI value of 31 ± 0.19 mm. Additionally, compounds 8b (IC50 32 ± 0.30 mm) and 8i (ZI = 30 ± 0.38 mm) also showed potent activity against the S. epidermidis ATCC 12228 strain. The other derivatives have significant inhibitory antibacterial activity on S. epidermidis ATCC 12228 within the ZI values (8 ± 0.87–24 ± 1.08 mm), whereas compounds 8a, 8f and 8k did not show antibacterial inhibition activity against S. epidermidis.
Table 1 Antibacterial activity of benzo[d]imidazolyl-oxadiazole derivatives (8a–8k)a
| Mean diameter of zones of inhibition (mean ± SEM) (mm) |
| Entry |
Gram-positive strains |
Gram-negative strains |
| S. pyogenes ATCC 19615 |
S. epidermidis ATCC 12228 |
S. aureus ATCC 29213 |
K. pneumonia ATCC 13883 |
E. coli ATCC 25922 |
P. aeruginosa ATCC 27853 |
| MXF: moxifloxacin; GMF: gemifloxacin; NA: not active; each value is an average of three replicates, ± denotes standard deviation among triplicates. |
| 8a |
14 ± 0.14 |
NA |
20 ± 0.33 |
13 ± 0.20 |
NA |
31 ± 0.78 |
| 8b |
28 ± 0.10 |
32 ± 0.30 |
37 ± 1.20 |
NA |
34 ± 1.12 |
13 ± 1.17 |
| 8c |
10 ± 0.55 |
19 ± 1.03 |
NA |
22 ± 0.11 |
25 ± 0.05 |
NA |
| 8d |
NA |
8 ± 0.87 |
17 ± 0.04 |
19 ± 0.24 |
11 ± 0.09 |
9 ± 0.62 |
| 8e |
27 ± 0.99 |
22 ± 0.55 |
16 ± 0.80 |
11 ± 0.08 |
NA |
NA |
| 8f |
31 ± 0.19 |
NA |
30 ± 0.37 |
29 ± 0.83 |
28 ± 0.10 |
12 ± 0.89 |
| 8g |
NA |
12 ± 0.73 |
15 ± 0.57 |
17 ± 0.28 |
NA |
17 ± 1.10 |
| 8h |
11 ± 0.43 |
9 ± 0.79 |
NA |
20 ± 0.05 |
10 ± 0.83 |
23 ± 0.90 |
| 8i |
28 ± 0.22 |
30 ± 0.38 |
26 ± 0.92 |
28 ± 0.73 |
36 ± 0.13 |
NA |
| 8j |
NA |
24 ± 1.08 |
21 ± 1.04 |
NA |
14 ± 1.35 |
30 ± 0.75 |
| 8k |
26 ± 0.32 |
NA |
32 ± 0.80 |
NA |
25 ± 0.51 |
28 ± 1.10 |
| MXF |
29 ± 0.16 |
31 ± 0.08 |
34 ± 0.62 |
30 ± 0.28 |
33 ± 0.23 |
26 ± 0.79 |
| GMF |
30 ± 0.94 |
28 ± 0.37 |
31 ± 0.16 |
29 ± 0.28 |
35 ± 0.77 |
29 ± 0.40 |
Table 2 MIC values of the most active analogues against various microbial species (µM)
| Mean diameter of zones of inhibition (mean ± SEM) (mm) |
| Entry |
Gram-positive strains |
Gram-negative strains |
| S. pyogenes ATCC 19615 |
S. epidermidis ATCC 12228 |
S. aureus ATCC 29213 |
K. pneumonia ATCC 13883 |
E. coli ATCC 25922 |
P. aeruginosa ATCC 27853 |
| 8b |
14.7 ± 1.20 |
9.2 ± 0.09 |
>50 |
27.4 ± 0.70 |
30.3 ± 1.20 |
>50 |
| 8f |
42.8 ± 0.91 |
>50 |
40.2 ± 1.23 |
8.0 ± 1.03 |
>50 |
10.5 ± 1.22 |
| 8i |
>50 |
18.9 ± 1.15 |
6.9 ± 1.01 |
19.3 ± 1.65 |
7.0 ± 1.35 |
>50 |
| 8k |
7.3 ± 0.85 |
>50 |
37.1 ± 0.67 |
>50 |
20.6 ± 1.60 |
22.9 ± 0.55 |
| MXF |
8.2 ± 1.10 |
10.4 ± 0.80 |
7.3 ± 0.92 |
9.1 ± 1.18 |
8.9 ± 1.07 |
12.8 ± 1.02 |
| GMF |
10.4 ± 0.14 |
10.0 ± 1.06 |
8.5 ± 1.12 |
10.5 ± 1.01 |
8.5 ± 0.87 |
11.3 ± 0.54 |
Notably, 3-chlorophenyl (8f), pyridin-3-ol (8i), and 6-chloropyridin-2-yl (8k) linked to benzo[d]imidazole showed the highest inhibitory effect against S. aureus ATCC 29213, with ZI values of 30 ± 0.37, 26 ± 0.92, and 32 ± 0.80 mm (GMF at 31 ± 0.16 mm). Against K. pneumoniae ATCC 13883, however, compound 4-hydroxyphenyl-1,2,4-oxadiazole (8d), 3-tolyl-1,2,4-oxadiazole (8g), and 3-fluoropyridin-4-yl-1,2,4-oxadiazole (8h) showed moderate activity (ZI = 19 ± 0.24, 17 ± 0.28, and 20 ± 0.05 mm). In comparison to MXF (ZI = 33 ± 0.23 mm), compound 8b, which has a 4-nitro group at the 4th position with phenyl, and compound 8i, which has a hydroxy group at the 3rd position with pyridine, are the lead compounds of the series. They have the highest inhibitory action against E. coli ATCC 25922 (ZI values of 34 ± 1.12 and 36 ± 0.13 mm). Then, in 1,2,4-oxadiazole analogues 8a, 8h, and 8j, we found 3-nitrophenyl, 3-fluoropyridin-4-yl, and pyridin-2-amine with ZI values of 31 ± 0.78, 23 ± 0.90, and 30 ± 0.75 mm against P. aeruginosa ATCC 27853. These compounds demonstrated superior potency in comparison to MXF (ZI = 26 ± 0.79 mm). The ATCC 27853 strain of P. aeruginosa was not susceptible to the antibacterial activity of compounds 8c, 8e, and 8i. Compared to counterparts with electron-withdrawing substituents like NO2 and F, those with electron-donating substituents (such as Br, NH2, and CH3) show less antibacterial activity. Compared to a nitro group at the third position in 8a, the presence of a nitro group at the fourth position on the phenyl ring in 8b demonstrated the strongest antibacterial activity. The antibacterial activity of substituted halogen compounds increased when the chlorine atom (8f) replaced the bromine atom (8e). The results showed that the amine substituent pyridin-2-yl moiety (8j) was less active than the chloro substituent pyridin-2-yl on the 1,2,4-oxadiazole (8k).
The results of in vitro assessment of the antimicrobial efficacy of most active benzo[d]imidazoles 8b, 8f, 8i and 8k using MICs are tabulated in Table 2 and Fig. 1. MICs against S. pyogenes ATCC 19615, which indicate that the tested oxadiazoles are most effective at 7.3 ± 0.85 µM and that the lowest concentration considerably inhibits their development, followed by MXF at 8.2 ± 1.10 µM. On the other hand, the MICs of the investigated oxadiazole composites that had bactericidal effects on isolates of S. aureus ATCC 29213 and S. epidermidis ATCC 12228 were 9.2 ± 0.09 and 6.9 ± 1.01 µM for 8b and 8i, respectively. Similarly, MICs showed that the most effective analogues for K. pneumoniae ATCC 13883 were 8f and 8i, with 8.0 ± 1.03 and 19.3 ± 1.65 µM, respectively, followed by 8b, with 27.4 ± 0.70 µM. The 8i and 8k oxadiazole composites were found to be the most effective at 7.0 ± 1.35 and 20.6 ± 1.60 µM, respectively, according to MICs against E. coli ATCC 25922. The 8b compound came in second at 30.3 ± 1.20 µM. S. aureus ATCC 29213 was less susceptible to the antibacterial activity of compounds 8f and 8k (MIC = 40.2 ± 1.23, 37.1 ± 0.67 µM) with m-chloro and o-chloro on the phenyl ring of the 1,2,4-oxadiazole portion. When tested against P. aeruginosa ATCC 27853, compounds 8b and 8i (MIC > 50 µM) that had a nitro or hydroxy group on the phenyl ring of the 1,2,4-oxadiazole portion did not show antibacterial efficacy. The nitration and chlorination of the phenyl moiety at position 4th and 3rd with oxadiazole ring resulted in compounds that displayed good antimicrobial on both Gram-positive and Gram-negative strains. The lower activity of compounds bearing bulky groups meta-bromo and meta-nitro (e.g., 8a and 8e) suggests steric hindrance or reduced interaction potential. This consistent correlation across in vitro, SAR, and docking data underscores the impact of electronic and steric features on biological efficacy. Additionally, heteroaryl substitutions, particularly pyridine-based analogues (8i and 8k), showed improved antibacterial potency. This may be attributed to the presence of heteroatoms that facilitate additional hydrogen bonding and favorable interactions with bacterial enzymes, as supported by docking studies. The introduction of EWG (NO2, Cl) substituents does contribute good activity than EDG (CH3 and NH2) groups. The ortho and meta positions of the phenyl group substituents were well tolerated, when compared to other substitution via para groups more beneficial and showed excellent antibacterial activity (Fig. 3).
 |
| | Fig. 3 Antibacterial efficacy of the tested benzo[d]imidazoles on the S. aureus and E. coli. | |
It should be noted that the present biological evaluation is limited to planktonic antibacterial assays, which represents a limitation of the current study. Biofilm-associated and fungal infections are clinically more challenging and are not addressed here. However, previous reports have demonstrated that benzimidazole and oxadiazole scaffolds possess promising antifungal and antibiofilm activities, attributed to their ability to disrupt microbial membranes and interfere with biofilm formation pathways. Therefore, the benzo[d]imidazole-oxadiazole hybrids reported in this study may also hold potential for antifungal and antibiofilm applications, which will be explored in future investigations.
2.2 Docking analysis
Molecular docking was done to make sure that strong ligands had an inhibitory mechanism. This research makes a substantial contribution to our knowledge of molecular biological profiles. We conducted a molecular docking study on the most active ligands that were found to be the most effective inhibitors in biological evaluation. We used the particular identifier codes 4URN for S. aureus GyrB and 3FV5 for E. coli Topoisomerase IV to retrieve the protein molecules from the RCSB Protein Data Bank (PDB).39–42 Polar hydrogens were added after co-crystallized ligand and water molecules were removed from both proteins to prepare them for docking. Using the π-system of the enzyme, ligand molecules attach to the enzymes in the active site by π-cation, π-cation, π-donor H-bond, π-alkyl, π-loan pair, and π–π interactions, according to the results of the docking study.
In addition to these bonds, the potent compounds also displayed various forms of H-bonding, van der Waals, carbon H-bond, and alkyl–alkyl interactions (Fig. 4, 5, and 6). According to the dissociation constant of 3.65 µM and negative score energy values of −7.42 kcal mol−1, respectively, compound 8b formed the most stable complex with the S. aureus (PDB ID: 4URN) target. This molecule showed one electrostatic alkyl–alkyl interaction with LeuA:157 and one strong π-donor H-bond with LysA:5 (bond distance = 2.75 Å). Additionally, it demonstrated four hydrophobic van der Waals contacts with the S. aureus target's active site residue: ProA:91, IleA:85, GlyA:84, and GlnA:6 (Table 3 and Fig. 4). There are four strong conventional H-bonds that the compound's sulfonyl group established with TyrA:143 [O28⋯OD1], LysA:159 [O28⋯NZ], AsnA:4 [O21⋯OD1], and AsnA:141 [O21⋯NH] at bond distances of 3.13 Å, 3.28 Å, 2.86 Å, and 2.41 Å, respectively. Additionally, the π-sulfur bonding interaction seen with TyrA:143 can be related to the sulfonyl group. Since the sulfonyl gatekeeper residue frequently plays a critical role in inhibitor selectivity, these interactions are very intriguing. Extending from the core structure, the 1,3,4-oxadiazole group seems to establish two hydrogen connections with LysA:5 [N14⋯O, 3.09 Å] and AsnA:7 [N13⋯N, 3.16 Å], further solidifying the ligand's location [Fig. 6]. At the same time, 1,2,4-oxadiazole of this ligand formed one H-bond interaction via AsnA:4 residue [N22⋯NZ, 2.95 Å] and π-anion interaction via AspA:139 [3.18 Å] in the active site S. aureus target. A possibly high affinity and specificity for S. aureus is suggested by the compound's capacity to interact with numerous residues through its varied functional groups.
 |
| | Fig. 4 The active sites of S. aureus GyrB [PDB code: 4URN] are bound by ligands 8b and 8k, respectively, demonstrating ionic interactions, hydrophobic contacts, and hydrogen bonds. | |
 |
| | Fig. 5 Compound 8i binds to the E. Coli topoisomerase IV active site (PDB code: 3FV5), as shown in (A) and (B), which display 8i's 3D and 2D interactions, respectively. | |
 |
| | Fig. 6 The Ligplot interactions of 8b, 8i, and 8k with the antibacterial protein active site are depicted in the figure, highlighting comparable important interactions inside the active site pockets. | |
Table 3 Different docking pose interactions in DNA GyrB and topoisomerase
| Entry |
Gram-positive strains |
| ΔG (kcal mol−1)/(kl) |
Interacting residues |
Bond distance [Å] |
| Antibacterial drug candidate S. aureus [4URN] |
| 8b |
−7.42/3.65 µM |
Hydrogen bonding: AsnA:7, AsnA:4 × 2, AsnA:141, TyrA:143, LysA:159 |
|
| π-donor hydrogen bonding: LysA:5 alkyl–alkyl: LeuA:157 |
|
| π-anion: AspA:12, AspA:139 |
>50 |
| π-sulfur: ThrA:143 |
|
| π-loan pair: ThrA:83 |
|
| van der Waals: ProA:91, IleA:85, GlyA:84, GlnA:6 |
|
| 8k |
−8.10/32.22 µM |
Hydrogen bonding: ThrA:166 × 2, GlyA:140, AspA:139 |
2.97, 3.04, 2.86, 3.06 |
| AsnA:146 × 2, ThrA:147, ArgA:117 × 2 |
|
| Carbon H-bonding: ThrA:166 |
|
| π-alkyl & alkyl–alkyl: ArgA:138, AlaA:2, ArgA:79 |
|
| π-donor: H-bond: AspA:139 |
|
| π-π stacking: AspA:139 × 2 |
|
| van der Waals: AlaA:2, MetA:3, LysA:165, LysA:164, LysA:162, ThrA:163, AsnA:4 |
3.22, 2.80 |
|
| Antibacterial drug candidate E. coli [3FV5] |
| 8i |
−7.15/31.07 µM |
Hydrogen bonding: HisA:37, ThrA:147 × 2, AsnA:146, ArgA:117 × 2 |
2.70, 3.18, 2.77, 2.76, 3.29,2.82 |
| Carbon H-bonding: AspA:56, AsnA:146 |
|
| π-cation: ArgA:40 × 2 |
|
| π-anion: ArgA:117, AspA:56 |
|
| van der Waals: AlaA:38, GlyA:57, LysA:39, AspA:42, AspA:55, IleA:54, ArgA:145 |
2.60, 2.89 |
Notably, the compound 8k complex with the S. aureus target (PDB code: 4URN) had a good dissociation constant of 32.22 µM and the best negative energy value of −8.10 kcal mol−1. With the antibacterial target's active site, this molecule established three electrostatic π-alkyl and alkyl–alkyl ArgA:79, ArgA:138, and AlaA:2 contacts. Interestingly, these studies have shown that the amino acid AspA:139 × 2 [distance = 3.22 Å and 2.80 Å] plays a significant role in amide π-stacking in the inhibition of S. aureus receptors. GlyA:140 formed a hydrogen bond with the sulfonyl group's oxygen (O29, 2.86 Å) during the co-crystallized ligand's interaction. Two more π-donor hydrogen bonding contacts between AspA:139 and the 1,3,4-oxadiazole core and ThrA:167 and the sulfonyl moiety were discovered. The contact between the co-crystallized ligand entailed two bonds of hydrogen involving the region of the hinge (ThrA:166) with the 1,3,4-oxadiazole and benzo[d]imidazole rings at a distance 3.04 and 2.97 Å accordingly (Fig. 4).
In addition, one extra hydrogen bonding interaction have been found between AspA:139 [3.06 Å] and the nitrogen of the 1,3,4-oxadiazole moiety (Fig. 6). In the binding pocket of E. coli Topoisomerase IV (PDB code: 3FV5), the N-methyl group of the benzo[d]imidazole ring of ligand8i connected with AspA:56 via a carbon H-bond. Furthermore, at a distance of 2.60 and 2.89 Å, the benzo[d]imidazole ring demonstrated π-cation interactions with the ArgA:40 × 2 residue. The antibacterial known inhibitor's 1,3,4-oxadiazole ring interacted with ArgA:117 via π-anion and H-bond interactions. Furthermore, hydroxy pyridine core of compound 8i exhibited two strong hydrogen bonds (conventional types) with the residue HisA:37, and ThrA:147 respectively (bond distance = 2.96 Å and 2.75 Å) (Fig. 5). Seven electrostatic interactions (van der Waals) were detected with ArgA:145, GlyA:57, AlaA:38, LysA:39, AspA:42, AspA:55, and IleA:54, respectively. As a result, two typical H-bond contacts between the residues, ThrA:147 and AsnA:146, were formed between 1,2,4-oxadiazole and sulfonyl.
2.3 ADME-T analysis outcomes
The https://biosig.lab.uq.edu.au/pkcsm/prediction; and https://tox.charite.de/protox3/index.php?site=compound_input online servers were used to calculate ADME-T parameters.43–45 In silico ADME-T (Absorption, Distribution, Metabolism, Excretion, and Toxicity) predictions are a crucial, cost-effective, and rapid method for screening compounds during the early stages of drug discovery. These computational tools allow for the assessment of drug-likeness and potential safety issues before synthesis, facilitating the selection of better lead candidates and reducing late-stage attrition. No more than one infraction of Lipinski's rule of five (ROF) is present in any of the synthesized benzo[d]imidazole-oxadiazole derivatives, which is acceptable. However, as evidenced by their low values for qualitative human oral absorption, these compounds are beyond the optimal range for expected aqueous solubility (ESOL). In spite of this, the synthesized oxadiazole shows significant MlogP values [0.81 to 2.87] and low Caco-2 permeability values [−0.03 to −0.50], suggesting the possibility of intestinal absorption. The SwissADME website calculates other metrics that fit within the recommended limitations, including the blood–brain partition coefficient, affinity to HIA, and topological polar surface area (TPSA), Ghose, Veber, Egan, Muegge, and Lead like ness (Table 4). Fig. 7 shows the bioavailability radar chart for the tested derivatives. Out of the six examined parameters—lipophilicity (LIPO), size, polarity, insolubility (INSOL), insaturation (INSATU), and flexibility (FLEX)—compounds 8a and 8d only exhibit one violation, and that was in the polarity parameter (Fig. 7). A BOILED-Egg, a two-dimensional figure that used estimated TPSA and A WLOGP features, was used to show the results. The most promising compound absorption across the blood–brain barrier (BBB) and human intestinal absorption (HIA) was shown in graphs. A BOILED-Egg chart was made, where the white region represented gastrointestinal absorption and the non-mutually exclusive yellow area represented BBB penetration. A blue hue indicates a high probability that the tested drug is a PGP substrate, whereas a red color indicates a lower probability. PGP is an efflux protein that is in charge of the uptake and efflux of several pharmaceuticals. All of the recently produced chemicals 8a–8k are seen to lie inside the gray region of Fig. 8, suggesting that they are likely to be poorly absorbed via the GI tract by passive diffusion. Although they are outside the plot's range and may not be able to reach the brain, compounds 8a, 8b, 8i, and 8j have the ability to cross the blood–brain barrier. Furthermore, the Boiled-egg plot's red spots (8e and 8f) indicate that the candidates are not P-glycoprotein (PGP) substrates, indicating good bioavailability as these compounds may be able to stop the reflux impact.
Table 4 Physicochemical and lead likeness properties of oxadiazole compounds (8a–8k)
| Entry |
TPSA |
IlogP |
MlogP |
logS (ESOL) |
Lipinski |
Ghose |
Veber |
Egan |
Muegge |
Leadlike ness |
Bio. score |
Brenk |
| 8a |
184.0 |
2.38 |
1.50 |
−4.70 |
No |
No |
No |
No |
No |
No |
0.17 |
2 |
| 8b |
184.0 |
2.38 |
1.50 |
−4.70 |
No |
No |
No |
No |
No |
No |
0.17 |
2 |
| 8c |
158.4 |
2.56 |
1.79 |
−4.51 |
Yes |
No |
No |
No |
No |
No |
0.55 |
0 |
| 8d |
158.4 |
3.17 |
1.79 |
−4.51 |
Yes |
No |
No |
No |
No |
No |
0.55 |
0 |
| 8e |
138.1 |
3.33 |
2.87 |
−5.55 |
Yes |
No |
Yes |
No |
Yes |
No |
0.55 |
0 |
| 8f |
138.1 |
3.10 |
2.76 |
−5.24 |
Yes |
No |
Yes |
No |
Yes |
No |
0.55 |
0 |
| 8g |
138.1 |
3.77 |
2.50 |
−4.95 |
Yes |
Yes |
Yes |
No |
Yes |
No |
0.55 |
0 |
| 8h |
151.0 |
2.65 |
1.68 |
−4.14 |
Yes |
No |
No |
No |
No |
No |
0.55 |
0 |
| 8i |
171.3 |
2.76 |
0.81 |
−3.86 |
Yes |
No |
No |
No |
No |
No |
0.55 |
0 |
| 8j |
171.0 |
2.14 |
0.81 |
−3.65 |
Yes |
No |
No |
No |
No |
No |
0.55 |
0 |
| 8k |
151.0 |
2.79 |
1.79 |
−4.80 |
Yes |
No |
No |
No |
No |
No |
0.55 |
1 |
 |
| | Fig. 7 Bioavailability radar of benzo[d]imidazole compounds 8a, and 8d. | |
 |
| | Fig. 8 The intestinal absorption and blood–brain barrier penetration of 1,3,4-oxadiazoles are represented by the boiled egg [8a–8k]. | |
It also evaluates the bioavailability score and looks at cytochromes P450 molecular interactions (CYP). Since substances like 1A2, 2C19, 2C9, 2D6, 3A4, and 2 × 101 were expected to have minimal inhibitory activity on different CYP450 isozymes, drug–drug interactions are unlikely to happen. Crucially, no drug exhibited CYP2E1 inhibition, indicating a more specific metabolic profile. With the exception of compounds 8c, 8d, 8i, 8j, and 8k, excretion predictions showed that none of the compounds were substrates for renal OCT2. The Ames test prediction evaluates the potential for genetic damage. A positive AMES alert in silico indicates a potential mutagenic risk. During optimization, such compounds are usually deprioritized, or the specific functional group responsible for the alert is identified and replaced to ensure the final candidate is non-mutagenic. All of the compounds' toxicity predictions were encouraging; none of them (except 8a, 8b, 8i, and 8j) displayed any indications of AMES toxicity, indicating a low probability of mutagenic consequences used to predict the toxicological endpoints and organ toxicities of ligands by the Protox-II service. The web server chose toxicity targets and computed the acute toxicity using selected models. The six different targets linked to pharmacological side effects served as the foundation for the anticipated amount of toxicity. The compounds' cytotoxicity, carcinogenicity, neurotoxicity, cardiotoxicity, hepatotoxicity, and mutagenicity were evaluated (Table 5). In silico, hepatotoxicity is often predicted based on structural alerts or machine learning models (e.g., HepG2 cytotoxicity). If a lead compound shows high hepatotoxicity potential, it suggests that structural modifications (e.g., reducing lipophilicity or modifying metabolically active sites) may be required to decrease liver toxicity. All the oxadiazole compounds showed hepatotoxicity, while compounds 8d and 8h were showed to be non-carcinogenic. Furthermore, compounds 8a and 8b shown mutagenicity, while compounds 8a, 8b, 8e, and 8f, respectively, demonstrated ecotoxicity (Table 4). The active cluster of physicochemical regions for optimal oral bioavailability (hepatotoxicity, neurotoxicity, cardiotoxicity, etc.) is shown by the blue color radar, whereas the inactive cluster is represented by the orange color radar (Fig. 9 and 10). All things considered, these ADME-T predictions demonstrate the advantageous pharmacokinetic advantageous and safety characteristics of compounds 8a–8k, which make them attractive options for additional therapeutic agent development.
Table 5 Absorption, metabolism, excretion and toxicity results of benzo[d]imidazole compounds (8a–8k)
| Entry |
8a |
8b |
8c |
8d |
8e |
8f |
8g |
8h |
8i |
8j |
8k |
| Absorption |
| Caco-2 |
−0.49 |
−0.50 |
−0.27 |
−0.30 |
−0.32 |
−0.32 |
0.33 |
−0.03 |
−0.08 |
−0.05 |
−0.16 |
| Skin per. |
−2.73 |
−2.73 |
−2.73 |
−2.73 |
−2.73 |
−2.73 |
−2.73 |
−2.73 |
−2.73 |
−2.73 |
−2.73 |
| Pgp |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
| VDss |
−0.5 |
−0.49 |
−0.62 |
−0.61 |
−0.55 |
−0.56 |
−0.55 |
−0.68 |
−0.62 |
−0.64 |
−0.69 |
| BBB |
−1.96 |
−1.96 |
−1.57 |
−1.60 |
−1.64 |
−1.63 |
−1.45 |
−1.90 |
−1.81 |
−1.72 |
−1.86 |
| CNS |
−3.63 |
−3.63 |
−3.74 |
−3.72 |
−3.44 |
−3.45 |
−3.47 |
−4.06 |
−4.22 |
−4.13 |
−3.84 |
|
| Metabolism |
| CYP1A2 |
No |
No |
No |
No |
No |
No |
No |
No |
No |
No |
No |
| CYP2C19 |
No |
No |
No |
No |
No |
No |
No |
No |
No |
No |
No |
| CYP2C9 |
No |
No |
No |
No |
Yes |
Yes |
No |
No |
No |
No |
Yes |
| CYP2D6 |
No |
No |
No |
No |
No |
No |
No |
No |
No |
No |
No |
| CYP3A4 |
No |
No |
No |
No |
No |
No |
No |
No |
No |
No |
No |
| CYP2E1 |
No |
No |
No |
No |
No |
No |
No |
No |
No |
No |
No |
|
| Excretion |
| OCT 2 sub |
Yes |
Yes |
No |
No |
Yes |
Yes |
Yes |
Yes |
No |
No |
No |
| T1/2 |
−0.003 |
0.032 |
0.25 |
0.28 |
0.15 |
0.20 |
0.25 |
0.27 |
0.21 |
0.27 |
0.08 |
|
| Toxicity |
| AMES |
Yes |
Yes |
No |
No |
No |
No |
No |
No |
Yes |
Yes |
No |
| hERG I |
No |
No |
No |
No |
No |
No |
No |
No |
No |
No |
No |
| hERG II |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
| Skin. Sen |
No |
No |
No |
No |
No |
No |
No |
No |
No |
No |
No |
| Hep. Tox. |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
| Neuro Tox. |
No |
No |
No |
No |
No |
No |
No |
No |
No |
No |
No |
| Cardi. Tox. |
No |
No |
No |
No |
No |
No |
No |
No |
No |
No |
No |
| Carcino. city |
Yes |
Yes |
Yes |
No |
Yes |
Yes |
Yes |
No |
Yes |
Yes |
Yes |
| Muta. city |
Yes |
Yes |
No |
No |
No |
No |
No |
No |
No |
No |
No |
| Cytotox. |
No |
No |
No |
No |
No |
No |
No |
No |
No |
No |
No |
| BBB-bar. |
Yes |
Yes |
No |
No |
Yes |
Yes |
Yes |
Yes |
No |
Yes |
Yes |
| Eco. Tox. |
Yes |
Yes |
No |
No |
Yes |
Yes |
No |
No |
No |
No |
No |
| Clinical Tox. |
No |
No |
No |
No |
No |
No |
No |
No |
No |
No |
No |
| ER-alpha |
No |
No |
No |
No |
No |
No |
No |
No |
No |
No |
No |
| GABA |
No |
No |
No |
No |
No |
No |
No |
No |
No |
No |
No |
| Pregnane-X |
No |
No |
No |
No |
No |
No |
No |
Yes |
No |
No |
No |
 |
| | Fig. 9 Comparison of input compound (8a) with dataset compounds. | |
 |
| | Fig. 10 Toxicity model report of triazole 8a and 8i. | |
3 Conclusion
A set of innovative benzo[d]imidazole-oxadiazole derivatives (8a–8k) was developed and produced using six step component procedures. Compound structures were verified via HRMS characterizations, 1H-NMR, 13C-NMR, and FT-IR. The antibacterial efficiency of the studied molecules was assessed, and the majority of the compounds under investigation showed encouraging antimicrobial qualities. According to an in vitro assessment of antibacterial activity, molecule 8f showed encouraging potency as an inhibitor of K. pneumonia and P. aeruginosa, with an MIC value of 8.0 ± 1.03, 10.5 ± 1.22 µM compared to GMF (MIC = 10.5 ± 1.01 & 11.3 ± 0.54 µM). Furthermore, it was observed that molecule 8b, 8f and 8i exhibited an excellent inhibitory antibacterial effect on E. coli ATCC 25922, as indicated by its ZI values of 34 ± 1.12, 28 ± 0.10, and 36 ± 0.13 mm accordingly. The compound 8k produced more potent antibacterial activity on S. pyogenes, and S. aureus (ZI = 26 ± 0.32 & 32 ± 0.80 mm), respectively, in comparison to MXF and GMF. Compounds having electron-withdrawing groups, on the other hand, exhibit more antibacterial action. By illustrating interactions such π-cation, π-sigma, H-bonds, CH-bonding, halogen bonds, van der Waals, π-alkyl, and the π-anion cloud, the results corroborate the experimental findings. Moreover, molecules 8b, 8k, and 8i demonstrated the highest docking scores of −7.42, −8.10, and −7.15 kcal mol−1 in in silico tests conducted on the most potent compounds with S. aureus (4URN) and E. coli (3FV5) enzymes. Likewise, in silico ADME/Tox discovered the drug-like properties of the proposed molecule, demonstrating its novelty as a potential lead. None of the compounds violated the Lipinski and Veber standards, and all of them, with the exception of 8a and 8b, fell within the expected range. Collectively, these findings underscore the potential of bis-oxadiazole hybrids, particularly compounds 8b and 8f, as promising leads for the development of new antimicrobial agents.
Author contributions
Yellamanda Rao Salluri contributed the synthetic work and data analysis. Kalyani Chepuri has contributed the biological evaluation. SA has contributed ideology and concept.
Conflicts of interest
There are no conflicts to declare.
Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5ra09260d.
Acknowledgements
The authors YRS and SA thankful to VFSTR for providing the infrastructure to conduct the research work. SA greatly acknowledges ANRF (i.e., formerly DST-SERB) for providing financial support under TARE (TAR/2022/000207). The author YRS is thankful to Curia for providing NOC as well as extensive support.
References
- N. R. Kumar, T. A. Balraj, S. N. Kempegowda and A. Prashant, Antibiotics., 2024, 13, 46 CrossRef CAS PubMed.
- W. Xue, X. Zuo, X. Zhao, X. Wang, X. Zhang, J. Xia, M. Cheng and H. Yang, Bioorg. Chem., 2024, 147, 107314 CrossRef CAS PubMed.
- M. Chalkha, M. Akhazzane, F. Z. Moussaid, O. Daoui, A. Nakkabi, M. Bakhouch, S. Chtita, S. Elkhattabi, A. I. Housseini and M. El. Yazidi, New J. Chem., 2022, 46, 2747 RSC.
- R. Lucas, Y. Hadizamani, J. Gonzales, B. Gorshkov, T. Bodmer, Y. Berthiaume, U. Moehrlen, H. Lode, H. Huwer, M. Hudel, M. A. Mraheil, H. A. F. Toque, T. Chakraborty and J. Hamacher, Toxins, 2020, 12, 223 CrossRef CAS PubMed.
- S. Kumar, S. Gupta, V. Rani and P. Sharma, Med. Chem., 2022, 18, 831 CrossRef CAS PubMed.
- J. S. Lim, Y. Y. Chai, W. X. Ser, A. V. Haeren, Y. H. Lim, T. Raja, J. B. Foo, S. Hamzah, R. Sellappans and H. Y. Yow, Iran. J. Basic Med. Sci., 2024, 27, 134 Search PubMed.
-
(a) C. E. Theodore, S. B. B. Prasad, K. Y. Kumar, M. S. Raghu, F. Alharethy, M. K. Prashanth and B.-H. Jeon, J. Mol. Struct., 2024, 1302, 137521 CrossRef CAS;
(b) T. Jain, P. R. Muktapuram, K. Sharma, O. Ravi, G. Pant, K. Mitra, S. R. Bathula and D. Banerjee, Bioorg. Med. Chem. Lett., 2018, 28, 1776 CrossRef CAS PubMed;
(c) T. Jain, P. R. Muktapuram, S. Pochampalli, K. Sharma, G. Pant, K. Mitra, S. R. Bathula and D. Banerjee, J. Med. Microbiol., 2017, 66, 1706 CrossRef CAS PubMed.
- A. Baron, C. L. Sann and J. Mann, Bioorg. Med. Chem., 2022, 58, 116656 CrossRef CAS PubMed.
- R. S. Kankate, P. S. Gide and D. P. Belsare, Arabian J. Chem., 2019, 12, 2224 CrossRef CAS.
- M. Ashfaq, S. S. Shah, T. Najam, M. M. Ahmad, R. Tabassum and G. Rivera, Curr. Med. Chem., 2014, 21, 911 CrossRef CAS PubMed.
- N. Su, Q. Wang, H. Y. Liu, L. M. Li, T. Tian, J. Y. Yin, W. Zheng, Q. X. Ma, T. T. Wang, T. Li, T. L. Yang, J. M. Li, N. C. Diao, K. Shi and R. Du, Front. Vet. Sci., 2023, 9, 1086180 CrossRef PubMed.
- Y. Bansal and O. Silakari, Bioorg. Med. Chem., 2012, 20, 6208 CrossRef CAS PubMed.
- K. P. Barot, S. Nikolova, I. Ivanov and M. D. Ghate, Mini-Rev. Med. Chem., 2013, 13, 1421 CrossRef CAS PubMed.
- R. S. Keri, A. Hiremathad, S. Budagumpi and B. M. Nagaraja, Chem. Biol. Drug Des., 2015, 86, 19 CrossRef PubMed.
- W. Akhtar, M. F. Khan, G. Verma, M. Shaquiquzzaman, M. A. Rizvi and S. H. Mehdi, Eur. J. Med. Chem., 2017, 126, 705 CrossRef CAS PubMed.
- B. Dik, D. Coskun, E. Bahcivan and K. Uney, Turk. J. Med. Sci., 2021, 51, 1579 Search PubMed.
- A. B. Popov, L. Krstulovic, S. Kostrun, D. Jelic, A. Bokulic, M. R. Stojkovic, I. Zonjic, M. C. Taylor, J. M. Kelly and M. Bajic, Eur. J. Med. Chem., 2020, 207, 112802 CrossRef CAS PubMed.
- B. Bhrigu, N. Siddiqui, D. Pathak, M. S. Alam, R. Ali and B. Azad, Acta Pol. Pharm., 2012, 69, 53 CAS.
- S. V. Bhandari, O. G. Nagras, P. V. Kuthe, A. P. Sarkate, K. S. Waghamare, D. N. Pansare, S. Y. Chaudhari, S. N. Mawale and M. C. Belwate, J. Mol. Struct., 2023, 1276, 134747 CrossRef CAS.
- S. Z. B. Bohurudeen, A. Ambala, T. R. Allaka, M. Z. Ahmed, B. Thirunavukkarasu, R. Tirumalareddy and S. Venkatesan, J. Mol. Struct., 2025, 1322, 140251 CrossRef CAS.
- Z. Wu, M.-B. Xia, D. Bertsetseg, Y.-H. Wang, X.-L. Bao, W.-B. Zhu, X. Tao, P.-R. Chen, H.-S. Tang, Y.-J. Yan and Z.-L. Chen, Bioorg. Chem., 2020, 101, 104042 CrossRef CAS PubMed.
- R. K. Salahuddin and J. K. Sahu, Curr. Drug Res. Rev., 2021, 13, 90 CrossRef PubMed.
- G. R. Venkateswara, T. R. Allaka, M. K. Gandla, V. N. P. K. Veera, S. R. Pindi, P. R. R. Vaddi and H. B. Bollikolla, J. Heterocycl. Chem., 2023, 60, 1666 CrossRef.
- S. Wang, H. Liu, X. Wang, K. Lei, G. Li, J. Li, R. Liu and Z. Quan, Eur. J. Med. Chem., 2020, 206, 112672 CrossRef CAS PubMed.
- T. Glomb, K. Szymankiewicz and P. Swiatek, Molecules, 2018, 23, 3361 CrossRef PubMed.
- F. Peng, T. Liu, Q. Wang, F. Liu, X. Cao, J. Yang, L. Liu, C. Xie and W. Xue, J. Agric. Food Chem., 2021, 69, 11085 CrossRef CAS PubMed.
- G. Chawla, B. Naaz and A. A. Siddiqui, Mini-Rev. Med. Chem., 2018, 18, 216 CAS.
- B. A. Khan, S. S. Hamdani, S. Jalil, S. A. Ejaz, J. Iqbal, A. M. Shawky, A. M. Alqahtani, G. A. Gabr, M. A. A. Ibrahim and P. A. Sidhom, Pharmaceuticals, 2023, 16, 11 CrossRef CAS PubMed.
- T. M. Dhameliya, S. J. Chudasma, T. M. Patel and B. P. Dave, Mol. Diversity, 2022, 26, 2967 CrossRef CAS PubMed.
- P. H. Parikh, J. B. Timaniya, M. J. Patel and K. P. Patel, Med. Chem. Res., 2020, 29, 538 CrossRef CAS.
- M. Zhou, J. C. Boulos, E. A. Omer, S. M. Klauck and T. Efferth, Molecules, 2023, 28, 5658 CrossRef CAS PubMed.
- U. A. Atmaram, P. K. Gadekar, H. Sivaramakrishnan, N. Naik, V. M. Khedkar, D. Sarkar, A. Choudhari and S. R. Mohana, Bioorg. Chem., 2019, 86, 507 CrossRef PubMed.
- D. K. Meher, A. K. Nayak, A. K. Singh and M. Pathak, J. Pharmacogn. Phytochem., 2020, 9, 1307 CrossRef CAS.
- F. S. Fernandes, H. Santos, S. R. Lima, C. Conti, M. T. Rodrigues, L. A. Zeoly, L. L. G. Ferreira, R. Krogh, A. D. Andricopulo and F. Coelho, Eur. J. Med. Chem., 2020, 201, 112418 CrossRef CAS PubMed.
- S. Gandamalla, S. Mavurapu, K. Sambaru and T. R. Allaka, J. Chin. Chem. Soc., 2025, 72, 721 CrossRef CAS.
- A. Tejeswara Rao, K. Bhaskar, P. Naveen, K. Naveen, C. Kalyani and A. Jaya Shree, Mol. Diversity, 2022, 26, 1581 CrossRef PubMed.
- S. C. Karad, V. B. Purohit, R. P. Thummar, B. K. Vaghasiya, R. D. Kamani, P. Thakor, V. R. Thakkar, S. S. Thakkar, A. Ray and D. K. Raval, Eur. J. Med. Chem., 2017, 126, 894 CrossRef CAS PubMed.
- S. Mirzayi, M. Kakanj, S. Sepehri, B. Alavinejad, Z. Bakherad and M. Ghazi-Khansari, Phosphorus, Sulfur Silicon Relat. Elem., 2021, 196, 1109 CrossRef CAS.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 16, 2785 CrossRef PubMed.
- J. Lu, S. Patel, N. Sharma, S. M. Soisson, R. Kishii, M. Takei, Y. Fukuda, K. J. Lumb and S. B. Singh, ACS Chem. Biol., 2014, 9, 2023 CrossRef CAS PubMed.
- R. A. Laskowski, J. Jabłońska, L. Pravda, R. S. Vařeková, J. M. J. B. Baell and G. A. Holloway, J. Med. Chem., 2010, 53, 2719 CrossRef PubMed.
- N. M. O'Boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch and G. R. Hutchison, J. Cheminf., 2011, 3, 33 Search PubMed.
- A. B. Reddy, T. R. Allaka, V. S. R. Avuthu, P. V. V. N. Kishore and H. Nagarajaiah, ChemistrySelect, 2024, 9, e202401989 CrossRef CAS.
- A. Daina, O. Michielin and V. J. S. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- P. Banerjee, A. O. Eckert, A. K. Schrey and R. Preissner, Nucleic Acids Res., 2018, 46, W257 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Kalyani Chepuri
a,
Kalyani Chepuri









