DOI:
10.1039/D5RA09131D
(Paper)
RSC Adv., 2026,
16, 6377-6393
Ascorbic acid-mediated solid-state synthesis of cavity-engineered CuO–Cu2O–Cu heterostructures: structural transformation mechanism and photocatalytic detoxification of Cr(VI)
Received
26th November 2025
, Accepted 22nd January 2026
First published on 30th January 2026
Abstract
The discharge of toxic Cr(VI) into water presents environmental health hazards, requiring efficient, sustainable photocatalysts for its reduction to Cr(III). In this work, a simple, environmentally friendly solvent-free solid-state route was developed to synthesize CuO–Cu2O–Cu heterostructures, where the novel use of solid ascorbic acid (AA) as a green reductant enables both the solid-state formation of Cu species and controlled reduction of CuO to Cu2O and Cu by adjusting the AA amount. Structural analyses confirmed the coexistence of Cu, Cu2O, and CuO phases, while FESEM and HRTEM revealed cavity-like core–shell architectures with dense heterointerfaces facilitating charge transfer. The optimized CuO–A1.1 showed a narrower band gap (1.42 eV vs. 1.67 eV for CuO) and a higher Cu+/Cu2+ ratio with oxygen vacancies, boosting its photocatalytic performance. The photocatalytic detoxification of Cr(VI) under visible light showed that all heterostructures were active. The CuO–Cu2O–Cu sample prepared by reducing 1 g of CuO with 1.1 g of ascorbic acid (CuO–A1.1) was the most efficient. It achieved complete Cr(VI) reduction within 30 min. Its pseudo-first-order rate constant was 0.1036 min−1, which is nearly 50 times higher than that of pristine CuO (0.0021 min−1). OFAT screening identified pH, Cr(VI) concentration, and catalyst dosage as key factors. One-way ANOVA with Tukey's test showed that pH (F = 762.06, p < 0.0001, R2 = 0.9965) and concentration (F = 92.46, p < 0.001, R2 = 0.97) significantly affected reduction efficiency, while catalyst dosage showed no significant effect (p > 0.05). Levene's test confirmed variance homogeneity, validating the results. Radical scavenging confirmed electrons and ˙O2− as key reactive species. Post-reaction XPS analyses and EPR measurements verified the proposed mechanism in which interfacial charge transfer among Cu, Cu2O, and CuO promotes electron migration and suppresses recombination. These results establish CuO–Cu2O–Cu heterostructure photocatalysts for Cr(VI) reduction and provide mechanistic insights into phase–function relationships in copper oxide.
1 Introduction
In recent decades, rapid industrialization and economic growth have led to increasingly severe environmental contamination. Industrial activities release large volumes of wastewater containing heavy metals into aquatic systems, causing water scarcity and contamination.1,2 The treatment of hazardous wastewater containing heavy metals presents a major challenge for environmental protection.3,4 Heavy metal pollution poses global threats to both aquatic ecosystems and human health,5 originating primarily from industrial processes,6 manufacturing sectors,7 and metal processing.8 Unlike organic pollutants, heavy metals are non-biodegradable and persistently accumulate in ecosystems,1 infiltrating food chains and concentrating at higher trophic levels.9 Among these, chromium (Cr), especially hexavalent chromium (Cr(VI)), is a notable industrial contaminant due to its high solubility and mobility, which facilitates membrane penetration,10,11 rendering it ∼100 times more toxic than trivalent chromium (Cr(III)).12 Consequently, converting Cr(VI) to Cr(III) is essential for mitigating chromium pollution.13
Various strategies, including chemical reduction, ion exchange, bacterial degradation, and adsorption, have been employed for Cr(VI) remediation.14–16 However, these approaches can be costly and may produce secondary pollution,17 highlighting the need for eco-friendly, efficient, and practical water treatment methods. Photocatalysis has emerged as a promising solution due to its strong redox capability and lack of secondary pollution.18 In particular, nanomaterial-based photocatalysts offer simplicity, cost-effectiveness, and high efficiency for water purification.19 Semiconductor photocatalysts that utilize light to generate photogenerated electrons and reactive oxygen species at room temperature can effectively reduce highly toxic Cr(VI) to less hazardous Cr(III).20,21 Transition metal oxides (TMOs) such as TiO2, ZnO, WO3, CuO, and Cu2O have been extensively investigated for photocatalytic environmental remediation owing to their variable oxidation states, chemical stability, and ability to generate reactive species under light irradiation.22,23 Copper-based oxides, specifically CuO and Cu2O, are gaining attention due to their advantageous properties, such as narrow band gaps (1.6–2.3 eV),24 low cost, and strong visible-light absorption.25 These characteristics enable their effective use in solar energy applications and catalysis.26 The dynamic redox interconversion between Cu2+, Cu+, and Cu0 states enhances their catalytic versatility, making them suitable for various reduction and oxidation reactions.26,27 However, copper oxides also face significant challenges that hinder their long-term efficiency. Key issues include photo-corrosion, rapid electron–hole recombination, and structural instability under prolonged illumination.28 These factors limit their practical applications in photocatalysis, necessitating innovative strategies to enhance their performance.29,30 Combining two or more phases with differing band gaps can enhance photocatalytic efficiency by improving charge separation and broadening the photoexcitation range.31,32 CuO/Cu2O heterostructures, in particular, demonstrate enhanced visible-light photocatalytic activity33 and are typically prepared via thermal or chemical oxidation of metallic Cu.34–37 However, oxidation-based methods, such as calcination-induced Cu2O/CuO composites, provide limited control over phase composition and interfacial architecture because temperature fluctuations can affect phase ratios and photocatalytic behavior.38
Recently, controlled reduction strategies have emerged as more versatile approaches for heterojunction construction. Kumar et al. reported CuO–Cu2O–Cu nanocomposites prepared via controlled reduction, yielding multi-phase systems with internal heterojunctions that enhanced photocatalytic H2 evolution and dye degradation.39 Similarly, CuO–Cu2O–Cu heterostructures produced via chemical precipitation and glucose-mediated reduction exhibited improved hydrogen generation.40 Despite numerous studies on CuO and Cu2O photocatalysts, most reported synthesis methods rely on wet-chemical or hydrothermal techniques that require controlled atmospheres, surfactants, or complex multistep procedures. These methods often provide limited control over the Cu0/Cu+/Cu2+ balance and interfacial architecture, which are critical for optimizing charge separation. Starting with CuO and selectively reducing it to Cu2O or metallic Cu offers a more predictable and controllable pathway for phase evolution but remains underexplored. Solid-state approaches for such transformations are especially appealing, offering straightforward, solvent-free, and potentially scalable alternatives to wet-chemical methods.
To the best of our knowledge, this study addresses the environmental challenge of toxic Cr(VI) contamination by developing efficient photocatalysts for its reduction to Cr(III). Copper-based semiconductors (CuO, Cu2O) were selected for their low cost and visible-light activity. CuO–Cu2O–Cu heterostructures were engineered using a green, ascorbic acid-mediated solid-state reduction method that enables precise control of phase composition and particle size. Compared with conventional hydrothermal or wet-chemical synthesis routes, the ascorbic acid-mediated solid-state method developed in this work is environmentally benign, solvent-free, and energy-efficient. It enables precise phase control and high-yield synthesis of CuO–Cu2O–Cu heterostructures using a green reductant, providing a scalable pathway for sustainable photocatalyst fabrication. Comprehensive characterization (XRD, FTIR, XPS, SEM, TEM, and UV-Vis DRS) was employed to elucidate phase evolution and band gap tuning associated with stepwise reduction. Photocatalytic activity toward Cr(VI) reduction under visible light was systematically evaluated, including recyclability studies to assess stability. Post-reaction XPS and XRD analyses provided direct evidence of structural and surface changes during catalysis, while theoretical band position calculations and EPR measurements supported the proposed charge transfer mechanism.
2 Experimental and analytical methods
2.1 Materials
All chemicals used in this study were of analytical grade and used without further purification. Copper nitrate (Cu(NO3)2·3H2O ≥99%), citric acid (C6H8O7 ≥99%), ethylene glycol (C2H6O2, 99.8%), ascorbic acid (AA) (C6H8O6 ≥99%), 1,5-diphenylcarbazide ACS reagent (DPC), and potassium dichromate (K2Cr2O7 ≥99%) were purchased from Sigma-Aldrich. Hydrochloric acid (HCl, 32%) and sodium hydroxide (NaOH ≥98%) were obtained from Merck. Methanol (CH3OH ≥ 99%) was obtained from Piochem. Deionized (DI) water was used in all preparation and processing steps. All chemicals were stored in airtight containers at room temperature in a dry environment. Glassware was cleaned with 1 M HNO3 and rinsed with DI water before use.
2.2 Synthesis of pristine CuO
Pristine CuO was synthesized by a Pechini route with minor modifications.41 Copper nitrate, ethylene glycol, and citric acid were combined in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 molar ratio and stirred at room temperature until homogeneous. The viscous solution was heated to 100 °C for 3 h to form a polymeric gel; the gel was dried at 110 °C for 12 h to remove volatile components and then calcined in air at 400 °C for 6 h (heating rate 5 °C min−1). The dried calcined powder was ground and stored in a desiccator.
:4 molar ratio and stirred at room temperature until homogeneous. The viscous solution was heated to 100 °C for 3 h to form a polymeric gel; the gel was dried at 110 °C for 12 h to remove volatile components and then calcined in air at 400 °C for 6 h (heating rate 5 °C min−1). The dried calcined powder was ground and stored in a desiccator.
2.3 Synthesis of CuO–Cu2O–Cu heterostructures
CuO–Cu2O–Cu heterostructures were prepared by a solid-state ascorbic acid–mediated reduction process (Scheme 1). In a typical procedure, 1.0 g of CuO powder was mixed thoroughly with different amounts of ascorbic acid (0.2, 0.5, 0.8, and 1.1 g) using a mortar and pestle for 30 min to ensure homogeneity, then transferred to a ceramic crucible under an air-limited environment (ceramic crucible covered with a lid) and calcined in air at 350 °C (5 °C min−1 ramp) for 1 h. After calcination, the powders were washed sequentially with water and ethanol several times using vacuum filtration to remove soluble by-products, then dried at 70 °C for 12 h and stored in a desiccator. Reaction atmosphere was nominally air, but the local microenvironment during AA decomposition is reducing due to CO/CO2/H2O/CH4 evolution, which enables partial reduction without an inert gas (as described in detail in Section 3.1.2). The final samples were designated as CuO–A0.2, CuO–A0.5, CuO–A0.8, and CuO–A1.1, corresponding to the CuO/ascorbic acid ratios. The solid-state reduction process exhibited high recovery, with yields ranging from 91% to 98% depending on the ascorbic acid content. The detailed yield values are presented in Table S2 (SI). The chemical rationale for the solid-state reduction (CuO → Cu2O → Cu) is reported in detail in Section 3.1.2 (reaction pathway and mechanism of CuO–Cu2O–Cu Formation). All experiments were repeated three times, and the same yield and phase evolution patterns were consistently observed, confirming the reliability and robustness of the procedure. Full details regarding the characterization, electrochemical impedance spectroscopy measurements, and statistical analysis of the CuO–Cu2O–Cu heterostructures can be found in the SI.
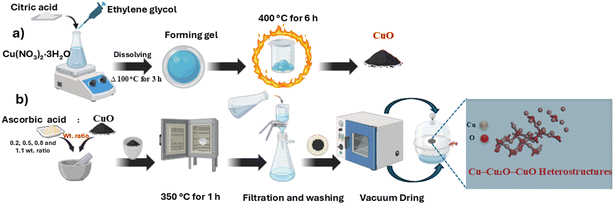 |
| | Scheme 1 Schematic illustration of the synthetic route. (a) Preparation of phase-pure CuO nanoparticles by the Pechini route. (b) Subsequent formation of CuO–Cu2O–Cu heterostructures via the ascorbic acid-mediated solid-state reduction of CuO by varying the ascorbic acid weights. | |
2.4 Photocatalytic reduction of Cr(VI)
The photocatalytic activity of the CuO–Cu2O–Cu heterostructures was evaluated by the reduction of Cr(VI) under visible-light irradiation. Photocatalytic reduction experiments were carried out at room temperature (25 ± 1 °C) using a glass reactor with an internal volume of 50 cm3. In a typical experiment, 20 mg of the synthesized photocatalyst was dispersed in 40 mL of an aqueous Cr(VI) solution with a concentration of 25 mg L−1. Methanol (1 mM) was added as a hole scavenger, and the pH = 2.5. To ensure uniform dispersion, the suspension was first sonicated for 1 minute and then stirred at 500 rpm in the dark for 30 minutes to establish adsorption–desorption equilibrium. Illumination was provided by a visible LED lamp (2 × 15 W), positioned approximately 20 cm above the reactor, corresponding to an estimated light intensity of ca. 84 mW cm−2. During irradiation, the suspension was continuously stirred to maintain homogeneity. During irradiation, 3 mL aliquots were withdrawn at regular time intervals, centrifuged at 5000 rpm for 10 min, and the resulting supernatant was analyzed to determine the residual Cr(VI) concentration. To evaluate the influence of different parameters on photocatalytic performance, a series of experiments was conducted by varying the initial Cr(VI) concentration (15–45 mg L−1), solution pH (2.5–8), and photocatalyst dosage (20–50 mg). All experimental runs were conducted in triplicate (n = 3) under each tested condition. In this study, pH values of the reaction system were adjusted by using HCl (1 M) or NaOH (1 M) solution. In addition, radical scavenger tests were performed using isopropanol (IPA), benzoquinone (BQ), and silver nitrate (AgNO3) to selectively quench hydroxyl radicals (˙OH), superoxide radicals (˙O2−), and photogenerated electrons (e−), respectively,42 thereby providing deeper insight into the active species governing the photocatalytic reduction of Cr(VI). The scavengers were added at a 20-fold molar concentration relative to the initial Cr(VI) concentration. Furthermore, recyclability experiments were carried out over multiple consecutive runs, in which the catalyst was recovered by centrifugation, thoroughly washed several times with distilled water, dried, and reused under identical conditions to evaluate its long-term stability and reusability.
2.5 Analysis of Cr(VI)
The photoreduction of Cr(VI) during the experiments was monitored spectrophotometrically by the diphenylcarbazide (DPC) colorimetric procedure,43 where 0.5 mL of a filtered sample was transferred to a 10 mL colorimetric tube, mixed with 1.5 mL of 0.25% DPC in acetone and 1 mL of 2 M H2SO4, and diluted to 5 mL with distilled water. After a reaction time of 5 minutes, absorbance was measured at 540 nm using a UV-vis spectrophotometer. The photocatalytic efficiency was calculated as follows:| |
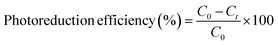 | (1) |
where C0 is the initial concentration, and Ct is the concentration after the photoreduction.
3 Results and discussion
3.1 Characterization of CuO–Cu2O–Cu heterostructures
3.1.1 XRD analysis. CuO–Cu2O–Cu heterostructures were prepared via a solid-state ascorbic acid–mediated reduction of pristine CuO. By varying the ascorbic acid content from 0.2 to 1.1 g per gram of CuO, the relative proportions of CuO, Cu2O, and metallic Cu could be systematically tuned, providing a controllable pathway for heterostructure formation. The crystalline structures of the resulting samples were analyzed by X-ray diffraction (XRD) (Fig. 1(a)). The pristine CuO sample exhibited sharp, well-defined diffraction peaks at 32.6, 35.7, 38.8, 48.9, 53.5, 58.2, 61.6, 66.4, and 68.1°, corresponding to the monoclinic phase planes (110), (002), (111), (202), (020), (202), (113), (022), and (200), in agreement with standard JCPDS data,44 confirming the crystallinity of the synthesized material. Upon gradual introduction of ascorbic acid, additional diffraction peaks emerged, indicating progressive reduction of CuO. At low ascorbic acid concentration (CuO–A0.2), weak reflections at 43.3°, 50.6°, and 74.3° appeared, corresponding to the (111), (200), and (220) planes of metallic Cu (JCPDS No. 04-0836), confirming the onset of CuO reductionIncreasing the ascorbic acid content (CuO–A0.5 and higher) led to the appearance of Cu2O peaks at 36.6° and 41.7°, corresponding to the (111) and (200) planes of cubic Cu2O (JCPDS No. 05-0667). The coexistence of CuO, Cu2O, and Cu phases confirms the successful formation of CuO–Cu2O–Cu heterostructures, with their relative intensities evolving in direct correlation with the amount of reducing agent. In the highly reduced CuO–A1.1 sample, Cu and Cu2O peaks dominate, indicating extensive conversion of CuO into reduced phases. Crystallite sizes were calculated from XRD peak broadening using the Scherrer equation:| |
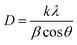 | (2) |
where D is the average crystallite size, K = 0.9, λ = 1.5406 Å (Cu Kα), β = FWHM, and θ = Bragg angle. Characteristic reflections were selected for each phase: CuO (002, 111), Cu2O (111, 200), and Cu (111, 200). The calculated results are summarized in Table 1.
 |
| | Fig. 1 XRD patterns (a) and FTIR spectra (b) of CuO, CuO–A0.2, CuO–A0.5, CuO–A0.8, and CuO–A1.1 samples. | |
Table 1 Crystallite sizes of CuO, Cu2O, and Cu phases in CuO–Cu2O–Cu heterostructures determined by Scherrer analysis
| Sample |
Phase |
(hkl) |
2θ (°) |
Crystallite size average (nm) |
| Pristine CuO |
CuO |
(002) |
35.59 |
62 |
| CuO |
(111) |
38.78 |
| CuO–A0.2 |
Cu |
(111) |
43.43 |
50.25 |
| Cu |
(200) |
50.60 |
| CuO |
(002) |
35.59 |
68.4 |
| CuO |
(111) |
38.78 |
| CuO–A0.5 |
Cu |
(111) |
43.43 |
55.54 |
| Cu |
(200) |
50.60 |
| CuO |
(002) |
35.59 |
43.86 |
| CuO |
(111) |
38.78 |
| Cu2O |
(111) |
36.4 |
53.74 |
| Cu2O |
(200) |
42.70 |
| CuO–A0.8 |
Cu |
(111) |
43.43 |
67.40 |
| Cu |
(200) |
50.60 |
| CuO |
(002) |
35.59 |
48.61 |
| CuO |
(111) |
38.78 |
| Cu2O |
(111) |
36.4 |
84.39 |
| Cu2O |
(200) |
42.70 |
| CuO–A1.1 |
Cu |
(111) |
43.33 |
70.55 |
| Cu |
(200) |
50.43 |
| CuO |
(002) |
35.54 |
37.09 |
| CuO |
(111) |
38.73 |
| Cu2O |
(111) |
36.47 |
39.34 |
| Cu2O |
(200) |
42.41 |
The crystallite size trends (Table 1) illustrate a stepwise CuO → Cu2O → Cu transformation. Pristine CuO exhibited large coherent domains (∼62 nm). Mild reduction (CuO–A0.2) slightly increased the CuO crystallite size (∼68 nm) and generated moderate-sized Cu (∼50 nm), consistent with surface oxygen loss and Cu nucleation.45 At CuO–A0.5, CuO crystallites shrank (∼44 nm) while fine Cu2O (∼54 nm) and larger Cu (∼56 nm) formed, indicating lattice disruption and nucleation-driven growth. Intermediate reduction (CuO–A0.8) promoted coalescence, with Cu2O (∼84 nm) and Cu (∼67 nm) growing significantly, and CuO partially regrowing (∼49 nm). In the most reduced sample (CuO–A1.1), Cu remained large (∼71 nm), Cu2O decreased (∼39 nm), and CuO further diminished (∼37 nm), suggesting consumption of Cu2O into metallic Cu. This evolution outlines a nucleation–growth–conversion sequence culminating in a Cu-dominant heterostructure. Overall, XRD analysis confirmed a controlled solid-state reduction of CuO into Cu2O and Cu phases, with phase composition systematically tuned by the ascorbic acid content. The progressive shift in peak intensities and variations in crystallite size evidenced a sequential CuO → Cu2O → Cu transformation, validating the successful formation of CuO–Cu2O–Cu heterostructures.
To verify that the CuO → Cu2O → Cu transformation originates solely from ascorbic acid, a control sample of pure CuO was subjected to the same calcination treatment (350 °C, 1 h, covered crucible) without any reducing agent. The XRD pattern (Fig. S1) revealed only diffraction peaks corresponding to monoclinic CuO, indicating no self-reduction or thermal decomposition under these conditions. In contrast, the ascorbic acid-treated samples progressively exhibited Cu2O and Cu reflections with increasing AA content (CuO–A0.2 → CuO–A1.1), confirming that the reduction arises from the ascorbic acid thermal treatment.
3.1.2 Reaction pathway and mechanism of CuO–Cu2O–Cu formation. At 350 °C, ascorbic acid (AA) acts not as a classical electron donor but as a solid precursor of reductive gases. The transient CO and H2 generated in situ extract oxygen from CuO, while the carbonaceous residue maintains a locally reducing atmosphere that suppresses re-oxidation of the reduced copper species.This temperature was deliberately chosen because it represents a balance between AA decomposition and CuO stability. Thermogravimetric and TG-FTIR studies show that AA begins to decompose at about 190–200 °C and continues up to ∼400 °C according to the literature,46 releasing CO, CO2, H2O, CH4, and carbonaceous fragments capable of reducing metal oxides. In contrast, CuO remains phase-stable when calcined alone at 350 °C for 1 h in air, as confirmed by XRD (Fig. S1). Therefore, any phase evolution in CuO–Ax (x = 0.2, 0.5, 0.8, and 1.1 g AA) mixtures must arise from redox interactions with the decomposition products of AA rather than from the intrinsic thermal instability of CuO. Upon heating, the generated gases react with CuO surfaces through heterogeneous reactions. These pathways lead to the stepwise reduction of CuO → Cu2O → Cu, driven by the spatially variable reducing strength of the locally formed gases.
The reduction strongly depends on the AA/CuO ratio. At low AA loading (CuO–A0.2), the total amount of reducing gases is small, but high local concentration at contact sites produces limited Cu nuclei alongside unreacted CuO. Increasing AA content (CuO–A0.5, CuO–A0.8) enlarges the reducing atmosphere, promoting the growth of Cu2O and Cu phases. At the highest AA dosage (CuO–A1.1), the environment becomes strongly reducing, yielding predominantly Cu and Cu2O with minimal CuO.
Overall, the results demonstrate that AA decomposition gases, not direct electron transfer, govern the reduction sequence and phase composition. The controlled partial reduction at 350 °C thus enables formation of a stable CuO–Cu2O–Cu heterostructure, providing abundant interfaces beneficial for charge transfer and photocatalytic activity. XRD analysis provides clear evidence of this transition. The progressive decreases the intensities of CuO peaks, and the emergence of Cu2O and Cu reflections in the XRD patterns indicates phase evolution with increasing ascorbic acid content. Scheme S1 illustrates this sequential reduction pathway of CuO depending on the AA/CuO ratio.
3.1.3 FTIR spectroscopy. The FTIR spectra (Fig. 1(b)) further support the structural evolution observed in XRD. Pure CuO displayed strong absorption bands between 700–400 cm−1, corresponding to Cu–O stretching vibrations in the monoclinic phase.47 With increasing ascorbic acid concentration, these Cu–O bands decreased in intensity, indicating progressive reduction of CuO. A characteristic absorption feature in the 1000–400 cm−1 region was assigned to Cu–O–Cu and O–Cu–O linkages involving Cu2+ and Cu+ in their lattice sites.48 The peak near 1630 cm−1 was attributed to adsorbed water on the nanostructured CuO surface. Interestingly, although XRD analysis of the CuO–A1.1 sample revealed metallic Cu and Cu2O as the dominant phases, its FTIR spectrum retained the signature of CuO. This apparent discrepancy can be explained by the infrared inactivity of metallic Cu49 and the relatively weak or overlapping Cu–O vibrational modes of Cu2O. Moreover, the stronger IR absorption of even small residual CuO fractions may dominate the spectrum. Together, the XRD and FTIR analyses confirm a controlled solid-state redox mechanism where ascorbic acid acts as the sole reducing agent, yielding hierarchical CuO–Cu2O–Cu heterostructures.
3.1.4 XPS analysis. XPS analysis provided deeper insight into the chemical states. The survey spectra of CuO–A0.5 and CuO–A1.1 confirmed Cu and O as the major elements, with only a minor C 1s peak from adventitious carbon used for charge calibration (Fig. 2(a and b)). The high-resolution Cu 2p spectra (Fig. 2(c)) showed clear differences between the samples. For CuO–A0.5, the Cu 2p3/2 region was dominated by a peak at ∼934.2 eV (Cu2+ in CuO) with a weaker feature at ∼932.2 eV (Cu+/Cu0). The corresponding Cu 2p1/2 components were observed at ∼954.3 eV (Cu2+) and ∼952.1 eV (Cu+/Cu0). Pronounced satellite shake-up peaks at ∼942.8 and ∼962.8 eV further confirmed the presence of Cu2+ species.50,51 A peak at 932.5 eV (Cu 2p3/2) was also observed, which may arise from either Cu0 or Cu+, since these states cannot be distinguished solely by binding energy.52 In contrast, CuO–A1.1 exhibited dominant low-binding-energy contributions (∼932.5 and ∼952.1 eV) characteristic of Cu+/Cu0, while Cu2+ peaks (∼934.1 and 954.2 eV) and their satellites were significantly diminished. This indicates that higher ascorbic acid loading led to extensive reduction of CuO into Cu2O and metallic Cu. The O 1s spectra (Fig. 2(d)) supported these assignments. In CuO–A0.5, two components were resolved: a lattice oxygen peak at ∼529.8 eV (O2− in Cu–O) and a higher-binding-energy feature at ∼531.4 eV, attributed to surface hydroxyls, chemisorbed oxygen, and vacancy-related species.51 For CuO–A1.1, the lattice oxygen peak (∼529.7 eV) dominated, with only a weak shoulder at ∼531.3 eV, suggesting a lower density of oxygen vacancies and a higher metallic Cu contribution. Overall, CuO–A0.5 exhibited a Cu2+-rich surface with abundant oxygen vacancies, while CuO–A1.1 was dominated by Cu+/Cu0 species with fewer surface oxygen states. This trend mirrors the XRD-determined reduction sequence (CuO → Cu2O → Cu) and correlates with the optical response: CuO–A1.1 leverages plasmonic Cu and multi-interface charge transfer to enhance photocatalytic performance, whereas CuO–A0.5 relies primarily on defect-assisted trapping.
 |
| | Fig. 2 XPS profile of CuO–A0.5 (a), and CuO–A1.1 (b), the core level XPS narrow scan of Cu 2p (c), and O 1s (d) of CuO–A0.5 and CuO–A1.1 heterostructures. | |
Along with XRD analysis, XPS analysis provides clear evidence of this transition. The progressive disappearance of CuO peaks and the emergence of Cu2O and Cu reflections in the XRD patterns indicate phase evolution with increasing ascorbic acid content. XPS Cu 2p spectra further confirm the reduction process through the decreasing intensity of the Cu2+ satellite peaks and the simultaneous growth of Cu+ and Cu0 components. The O 1s spectrum also reveals the formation of oxygen vacancies, which act as shallow electron traps, enhancing charge carrier separation and migration across interfaces. The combination of these structural and electronic transitions results in a robust CuO–Cu2O–Cu heterostructure, where the metallic Cu domains serve as electron mediators between CuO and Cu2O, facilitating efficient charge transfer and suppressing recombination. This synergistic interface coupling is key to the high photocatalytic performance observed for Cr(VI) reduction.
3.1.5 SEM and TEM analysis. SEM analysis of CuO–A0.5 and CuO–A1.1 (Fig. 3(a, b), S2(a and b)) highlights the strong influence of ascorbic acid concentration on the microstructure. The FE-SEM images of CuO–A0.5 (Fig. 3(a) and S2(a)) show broad, irregular flakes with thin, layered morphology. These loosely assembled sheets form a porous framework with rough textures and uneven edges, suggesting a high surface area favorable for photocatalytic reactions due to the abundance of exposed active sites.53 In contrast, CuO–A1.1 (Fig. 3(b) and S2(b)) exhibited larger, rounded cavities and interconnected channels, consistent with an expanded surface area that further enhances photocatalytic efficiency.54,55 The EDX spectra (Fig. 3(c and d)) confirmed Cu and O as the main elements in both samples. A noticeable increase in Cu peak intensity, accompanied by a decrease in O signal from CuO–A0.5 to CuO–A1.1, reflects the progressive reduction of CuO to Cu2O and Cu. Elemental mapping (Fig. S2(c and d)) further demonstrated the uniform distribution of Cu and O in both samples, while CuO–A1.1 exhibited localized Cu-rich domains, consistent with the coexistence of multiple Cu oxidation states in the heterostructure.
 |
| | Fig. 3 SEM images of CuO–A0.5 (a) and CuO–A1.1 (b) with EDX spectra of CuO–A0.5 (c) and CuO–A1.1 (d). | |
TEM analysis (Fig. 4 and S3) reveals that the ascorbic acid concentration dictates the architecture of the reduced particles. The CuO–A0.5 sample exhibits a distinct core–shell structure with a dark CuO-rich core and a lighter shell of reduced phases (Cu2O/Cu), as confirmed by SAED rings (Fig. 4(c and d)) corresponding to all three phases. In contrast, the CuO–A1.1 sample shows a more homogeneous contrast with SAED patterns dominated by Cu and Cu2O, indicating near-complete reduction of the original CuO framework. This structural evolution confirms that the ascorbic acid-mediated reduction proceeds from the surface inwards, creating cavity-like architectures at higher reductant loadings, which correlate with the enhanced photocatalytic activity.
 |
| | Fig. 4 TEM images of CuO–A0.5 (a) and CuO–A1.1 (b), along with SAED patterns of CuO–A0.5 (c) and CuO–A1.1 (d) samples. | |
3.1.6 BET surface area and pore structure analysis. The N2 adsorption–desorption isotherms (Fig. 5(a)) confirm the mesoporous nature of both pristine CuO and CuO–A1.1. Pristine CuO exhibits a BET surface area of 19.41 m2 g−1, while CuO–A1.1 shows a significantly higher value of 57.02 m2 g−1. The BJH pore size distribution (Fig. 5(b)) indicates that pristine CuO is dominated by small mesopores, whereas CuO–A1.1 displays a broader mesopore distribution with pore sizes mainly in the ranges of 1.9–3.2 nm and 3.2–9.5 nm. These textural features are consistent with the porous morphology observed in FESEM and HRTEM and, together with improved interfacial charge transfer in the CuO–Cu2O–Cu heterostructure, contribute to the enhanced Cr(VI) reduction performance.
 |
| | Fig. 5 N2 adsorption–desorption isotherms (a), and BJH pore size distribution curves (b) of pristine CuO and CuO–A1.1. | |
3.1.7 UV-vis diffuse reflectance spectroscopy (DRS). UV-Vis diffuse reflectance spectroscopy was used to evaluate changes in the electronic structure of the CuO–Cu2O–Cu heterostructures. Compared with pristine CuO, the reduced sample CuO–A1.1 shows a gradual shift of the absorption edge (Fig. 6(a)), indicating band-gap modification. The band gap energies were estimated from Tauc plots according to eqn (3):56,57| |
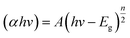 | (3) |
where α = absorption coefficient, h = Planck constant, A = constant, v = frequency, Eg = absorption band gap energy, and n = one or four for direct and indirect band gaps. Band gap values estimated from Tauc plots (Fig. 6(b) and S4) decrease from ∼1.70 eV for pristine CuO to ∼1.40 eV for CuO–A1.1. This band-gap narrowing is attributed specifically to the progressive reduction of Cu2+ in CuO to Cu+ (forming Cu2O) and Cu0 (metallic Cu), as confirmed by XRD and XPS (Fig. 1(a) and 2). The formation of CuO/Cu2O heterointerfaces creates mid-gap states, while plasmonic absorption from metallic Cu nanoparticles contributes to the extended visible-light absorption.
 |
| | Fig. 6 UV-Vis absorption spectra normalized to BET surface area of pristine CuO and reduced samples (a); Tauc plots (b) for band gap estimation of pristine CuO and CuO–A1.1. | |
3.2 Photocatalytic reduction of Cr(VI)
Photocatalysis is widely considered an environmentally sustainable approach for converting toxic Cr(VI) into the less harmful Cr(III) state. To verify the photocatalytic nature of the Cr(VI) reduction, control experiments were performed under three different conditions: (i) without a catalyst under visible light, (ii) with a catalyst in the dark, and (iii) with the CuO–A1.1 catalyst under visible-light irradiation. No change in the Cr(VI)–DPC absorption peak at 540 nm (Fig. S5) was observed in the light-only test, while a remarkable decrease was noted in the dark condition with catalyst, which stabilized after 30 min due to adsorption–desorption equilibrium. These results confirm that both light and the photocatalyst are necessary for effective Cr(VI) reduction. The CuO–Cu2O–Cu heterostructures exhibited markedly enhanced photocatalytic activity compared with pristine CuO (Fig. S6(a)). Complete reduction of Cr(VI) within 30 min was achieved using CuO–A1.1, whereas pristine CuO showed only minor activity under identical conditions. Samples with intermediate reductant levels (CuO–A0.2, CuO–A0.5, and CuO–A0.8) displayed progressively higher efficiencies, consistent with the effect of phase composition and heterointerface density. Kinetic analysis of photoreduction (ln(Ct/C0) vs. time, Fig. S6(b)) demonstrated that all samples followed pseudo-first-order behavior, with R2 values >0.95. The apparent rate constants (k) derived from the slopes (Fig. S6(c)) confirmed the superior activity of CuO–A1.1 (0.10359 min−1), nearly 50-fold greater than pristine CuO (0.0021 min−1). This enhancement arises from the synergistic interplay of Cu, Cu2O, and CuO domains, which improve charge separation, extend visible-light absorption, and promote electron transfer for Cr(VI) reduction. Although all reduced samples contain mixed phases, the optimized phase balance and high heterointerface density in CuO–A1.1 appear to be decisive for its enhanced photocatalytic performance.
3.2.1 Effect of solution pH. The effect of solution pH on photocatalytic performance was further investigated using CuO–A1.1 over a pH range of 2.5–8 (Fig. 7(a)). All experimental runs were conducted in triplicate (n = 3). At pH > 3.5, the reduction efficiency decreased markedly. This pronounced pH dependence is governed by two interconnected factors: Cr(VI) speciation/surface precipitation and catalyst surface charge. Under acidic conditions (pH < ∼4), Cr(VI) predominantly exists as the highly reducible dichromate ion (Cr2O72−, eqn (6)), while the reduced product Cr3+ remains soluble. At higher pH, Cr(VI) shifts to CrO42−, and Cr3+ precipitates as Cr(OH)3 (eqn (5)), forming surface deposits that block active sites and hinder light absorption.58–61 In addition, the catalyst surface charge plays a decisive role in regulating Cr(VI) reduction. The point of zero charge (pHpzc) of CuO–A1.1, determined by the pH drift method (Fig. S7), is approximately 5.4. At the optimal reaction pH of 2.5 (well below the pHpzc), the catalyst surface is strongly positively charged, electrostatically attracting the anionic Cr(VI) species (HCrO4−/Cr2O72−) and thereby enhancing adsorption and interfacial electron transfer. As the pH increases toward and beyond the pHpzc, the surface charge becomes neutral and then negative, weakening this crucial electrostatic interaction. Consequently, the reduction proceeded most efficiently at pH 2.5, achieving nearly complete detoxification within 30 min. Kinetic fitting confirmed pseudo-first-order behavior (Fig. 7(b), R2 > 0.94), with the rate constant at pH 2.5 (0.1178 min−1) being orders of magnitude higher than at pH 8 (1.53 × 10−18 min−1), unequivocally demonstrating the critical role of proton concentration and surface electrostatics in the photoreduction process.| | |
CuO–Cu2O–Cu + hv → h+ + e−
| (4) |
| | |
CrO42− + 4H2O + 3e− → Cr(OH)3 + 5OH−
| (5) |
| | |
Cr2O72− + 14H+ + 6e− →2Cr3+ + 7H2O
| (6) |
| | |
4h+ + 2H2O → O2 + 4H+
| (7) |
 |
| | Fig. 7 (a) Variation of Cr(VI) concentration (Ct/C0) with time at different pH values. (b) Pseudo-first-order kinetic plots of ln(Ct/C0) versus time. (c) Apparent rate constants (k) at each pH. (d) Mean photoreduction efficiency (%) with standard deviations. (Cr(VI) concentration: 25 mg L−1; catalyst dosage: 20 mg). | |
Descriptive statistics for Cr(VI) photoreduction at different pH levels are presented in Table S4, summarizing the mean, standard deviation, and standard error of the mean (n = 3 for each condition). To statistically confirm the effect of pH, one-way ANOVA was performed (Tables S5 and S6). The analysis revealed a highly significant influence of pH on Cr(VI) photoreduction efficiency (F = 762.06, p < 0.0001), with an R2 = 0.9965, confirming that pH accounted for over 99% of the total variation in removal performance. Post hoc Tukey's HSD test showed that the mean photoreduction efficiencies at pH 2.5 and 3.5 were significantly lower (p < 0.001) than those at pH 3.5 and 8. Pairwise comparisons indicated that differences between adjacent levels (e.g., pH 2.5 vs. 3.5, pH 3.5 vs. 5.8, and pH 5.8 vs. 8) were all statistically significant at α = 0.05, confirming the steep decline in reduction efficiency as pH increased, as shown in Table S7. The mean photoreduction efficiency (%) with standard deviations for different concentration levels is shown in Fig. 7(d). Levene's test verified the homogeneity of variance (F = 1.20, p = 0.369), confirming that the ANOVA assumptions were satisfied (Table S8). The small coefficient of variation (3.9%) and low root MSE (2.31) also demonstrate the precision and reproducibility of the data, as shown in Table S6.
Overall, both experimental and statistical results highlight that acidic conditions (pH ≈ 2.5) are optimal for achieving maximum Cr(VI) photoreduction efficiency, while alkalinity strongly suppresses the process due to reduced Cr(VI) speciation reactivity and surface site deactivation.
3.2.2 Effect of initial Cr(VI) concentration. The influence of initial Cr(VI) concentration on photocatalytic reduction efficiency was investigated in the range of 15–45 mg L−1 under identical experimental conditions using CuO–A1.1 (Fig. 8(a)). The photoreduction efficiency decreased gradually with increasing Cr(VI) concentration. At lower concentrations (15–25 mg L−1), nearly complete reduction was achieved within 20–30 min, whereas at higher levels (≥35 mg L−1), the reaction rate and photoreduction efficiency declined markedly. This trend is attributed to the higher number of Cr(VI) ions competing for the same number of active sites, as well as increased light scattering and reduced photon penetration in concentrated solutions. Moreover, the limited generation of photoexcited charge carriers relative to the excess Cr(VI) at higher concentrations can hinder complete reduction.62 Kinetic fitting confirmed pseudo-first-order behavior (Fig. 8(b)) across all concentrations, with apparent rate constants (k) decreasing from 0.1246 min−1 at 25 mg L−1 to 0.0335 min−1 at 45 mg L−1 (Fig. 8(c)). However, at elevated concentrations (≥35 mg L−1), pseudo-first-order kinetics were maintained (R2 > 0.95), confirming that the reduction mechanism remains dominated by surface-mediated electron transfer rather than mass transport limitations. The apparent rate constant decreased from 0.0872 min−1 at 35 mg L−1 to 0.0335 min−1 at 45 mg L−1, indicating active site saturation and competitive surface adsorption effects. Statistical analysis further validated these trends. One-way ANOVA (Table S5) revealed a highly significant effect of concentration on photoreduction efficiency (F = 92.46, p < 0.001, R2 = 0.972). Tukey's post hoc test (Table S7) indicated that the 45 mg L−1 treatment was significantly different (p < 0.05) from the other concentrations, while no significant difference was observed among 15–35 mg L−1. The mean photoreduction efficiency (%) with standard deviations for different pH levels is shown in Fig. 8(d). The Levene's test confirmed the homogeneity of variance (F = 2.64, p = 0.121), validating the use of ANOVA (Table S8). The low coefficient of variation (2.13%) and small root MSE (1.99) (Table S6) further support the precision and reproducibility of the data. These results confirm that lower initial Cr(VI) concentrations favor faster kinetics and higher overall photoreduction efficiency due to more efficient light utilization and greater availability of reactive sites per Cr(VI) ion.
 |
| | Fig. 8 (a) Variation of Cr(VI) concentration (Ct/C0) with time at different solution concentration values. (b) Pseudo-first-order kinetic plots of ln(Ct/C0) versus time. (c) Apparent rate constants (k) at each solution concentration. (d) Mean photoreduction efficiency (%) with standard deviations (catalyst dosage: 20 mg; pH: 2.5). | |
3.2.3 Effect of catalyst dosage. The effect of catalyst dosage on Cr(VI) photoreduction efficiency was examined using CuO–A1.1 at loadings of 20–50 mg (Fig. 9(a)). Increasing the dosage from 20 to 30 mg slightly enhanced the photoreduction efficiency from 99.0% to 99.8%, while a minor decrease to 99.0% was observed at 50 mg. The reduction in efficiency at higher loadings can be attributed to excess particle concentration, which leads to light scattering and reduced photon penetration through the suspension.62 Nevertheless, pseudo-first-order kinetics (Fig. 9(b)) are maintained under all dosage conditions (R2 > 0.95), indicating that the reaction mechanism remained unchanged. The apparent rate constant (k) from pseudo-first-order kinetics increased from 0.1178 min−1 at 20 mg to 0.1509 min−1 at 50 mg (Fig. 9(c)), suggesting that a moderate increase in catalyst amount improves active site availability up to an optimal level. Despite these variations, pseudo-first-order kinetics (Fig. 9(b)) were maintained under all conditions (R2 > 0.95). This indicated that moderate catalyst dosages provide a favorable balance between active-site density and light absorption. Compared with other reported CuO and Cu2O-based photocatalysts, the cavity-engineered Cu–Cu2O–CuO heterostructure (CuO–A1.1) displayed relatively higher activity for Cr(VI) reduction (Table 2), attributable to its optimized phase composition and heterointerface density.
 |
| | Fig. 9 (a) Variation of Cr(VI) concentration (Ct/C0) with time at different catalyst dosage values. (b) Pseudo-first-order kinetic plots of ln(Ct/C0) versus time. (c) Apparent rate constants (k) at each catalyst dosage value. (d) Mean photoreduction efficiency (%) with standard deviations (solution concentration: 25 mg L−1; pH: 2.5). | |
Table 2 Performance of different catalysts for chromium photoreduction
| Catalyst |
Usage (mg) |
Cr conc. (mg L−1) |
Photoreduction time (min) |
Photoreduction efficiency (%) |
Ref. |
| CuO |
20 |
25 |
60 |
6 |
This study |
| Cu2O nanocrystals |
50 |
40 |
180 |
40 |
63 |
| Ag/Cu2O/CuO |
Not reported |
1.5 |
240 |
57 |
64 |
| Cu–Cu2O/SiO2 |
125 |
50 |
180 |
57.34 |
65 |
| 3D/2D Cu2O/BiOBr S |
40 |
15 |
100 |
65.46 |
66 |
| 2D CuO/rGO |
50 |
10 |
60 |
75 |
67 |
| Nb2O5/CuO |
10 |
50 |
210 |
84 |
68 |
| CuO–Cu2O–Cu (CuO–A1.1) |
20 |
25 |
30 |
100 |
This study |
Statistical evaluation supported these findings. One-way ANOVA indicated that catalyst dosage had no statistically significant effect on the overall reduction efficiency (F = 0.565, p = 0.596, R2 = 0.158) as shown in Table S5. Tukey's post hoc comparison confirmed that the differences among 20, 30, and 50 mg loadings were not statistically significant (p > 0.05) (Table S7). However, Levene's analysis (F = 7.52, p = 0.023) revealed that variation in dosage moderately influenced the apparent rate constant, reflecting changes in surface active-site density (Table S8). These results confirm that increasing the catalyst dosage beyond the optimal 30 mg offers no substantial improvement in Cr(VI) removal efficiency due to light attenuation effects. The mean photoreduction percentages for each dosage (n = 3) are presented in Fig. 9(d). Overall, the optimal conditions for maximum efficiency were highly acidic (pH 2.5), with moderate Cr(VI) concentration (25 mg L−1) and a catalyst dosage of 30 mg, resulting in nearly complete photoreduction of Cr(VI) within 30 minutes. The heterostructure maintained high performance under real tap-water conditions, confirming its structural stability and strong interfacial charge transport, making it suitable for wastewater detoxification.
3.3 Photocatalytic performance in real tap water
Although the primary experiments were conducted in a controlled Cr(VI) aqueous solution to isolate the intrinsic activity of the CuO–Cu2O–Cu heterostructure, to assess the practical applicability of the CuO–Cu2O–Cu heterostructure, photocatalytic tests were carried out in Cr(VI)-spiked tap water (conductivity = 290–350 µS cm−1, TDS = 191–231 ppm) containing common ions e.g., (Cl−, SO42−, HCO3−). Under identical experimental conditions to the deionized-water system (Cr(VI) = 35 mg L−1, catalyst dose = 20 mg, pH = 2.5, 40 mL), the reduction efficiency decreased slightly from 95.7% to 82.5% within 30 min, as revealed in Fig. S8. This small drop in performance is attributed to ionic competition and minor light scattering effects, confirming the high stability and efficiency of the heterostructure under realistic water conditions. In conclusion, the study found that Cr(VI) reduction is a photocatalytic process that requires visible light and the CuO–Cu2O–Cu heterostructure. CuO–A1.1 showed the highest photocatalytic activity, achieving complete Cr(VI) reduction within 30 minutes.
3.4 Verification of Cr(VI) reduction by complexation and UV-vis spectroscopy
To confirm the chemical transformation of Cr(VI) during photocatalysis, complementary complexation and UV-Vis spectroscopic analyses were performed. As shown in Scheme 2(a), residual Cr(VI) species in the treated solution were detected via complexation with 1,5-DPC, forming a violet Cr(VI)–DPC complex characterized by a strong absorption band at 540 nm.69,70 The disappearance or pronounced decrease of this absorption feature after illumination directly confirms the reduction of Cr(VI) to Cr(III). For qualitative visualization of the resulting trivalent chromium species, the filtrate was treated with Na2CO3, yielding a bluish-green hydroxo complex typical of Cr(III) under mildly basic conditions, Scheme 2(b).71 This carbonate-based colorimetric change was used only as supportive evidence, while the primary verification relied on the diminished Cr(VI)–DPC absorption band. To further strengthen the identification, comparative UV-Vis spectra of standard Cr(VI)–DPC and Cr(III) solutions were recorded (Fig. S9). The spectra confirmed the disappearance of the 540 nm Cr(VI)–DPC peak in the photocatalyzed sample, providing strong evidence for the complete reduction of Cr(VI) to Cr(III) during the process.
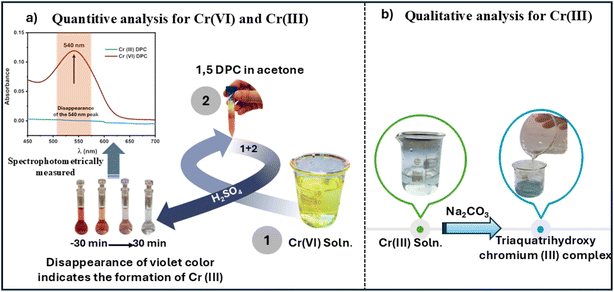 |
| | Scheme 2 Chromium analysis using 1,5-diphenylcarbazide (DPC). (a) Quantitative analysis of Cr(VI) and Cr(III). The inset shows the corresponding absorbance spectra (presented separately in Fig. S9). (b) Qualitative analysis of Cr(III). Photographs shown are original and were taken by the corresponding author. | |
3.5 Recyclability test
Cyclic photoreduction experiments were conducted to evaluate the long-term stability and reusability of the CuO–Al.1 catalyst. Fig. 10(a) presents the stability trend over seven consecutive cycles, while the corresponding full kinetic profiles (Ct/C0 vs. time) for each cycle are provided in Fig. S10. The plots the photocatalytic reduction efficiency (%) for each cycle, providing a dynamic view of the stability trend. The efficiency remained above 97% throughout all seven runs (99.4% in Cycle 1 to 97.3% in Cycle 7), confirming outstanding operational stability with only a gradual, minimal decay. No abrupt activity loss was observed at any specific cycle, highlighting the structural robustness of the heterostructure. This stability and durability arise from its compositional gradient and self-regulating redox equilibrium. During illumination, photogenerated electrons are transferred through the Cu2O–CuO cascade and accumulate at the metallic Cu interface, which serves as the primary site of reduction. In the acidic chloride medium, dissolved Cu species exist mainly as Cu–Cl complexes (CuCl, CuCl2−),72 that remain concentrated near the Cu surface. These complexes readily capture the accumulated electrons and are reduced back to Cu0 or Cu2O, leading to in situ photo-redeposition of copper73,74 and effective self-healing of the catalyst surface. Thermodynamically, the outer Cu and Cu2O layers, with lower oxidation potentials (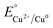 (+0.34 V) and
(+0.34 V) and 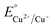 (+0.16 V)), preferentially donate electrons to Cr(VI)
(+0.16 V)), preferentially donate electrons to Cr(VI) 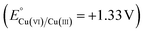 , thus preventing self-corrosion. This thermodynamic hierarchy, combined with rapid interfacial electron transfer and physical encapsulation of the CuO core, ensures long-term catalytic stability, as verified by post-cycling XRD, SEM, and XPS analyses. Post-reaction XRD (Fig S11(a)) and XPS analyses (Fig. 10(b and c)) confirmed that the Cu2+, Cu+, and Cu0 phases remained structurally stable even after multiple photocatalytic runs. Only minor variations in relative peak intensities were detected, suggesting limited redistribution among Cu2+, Cu+, and Cu0 species under reaction conditions (discussed in detail in the Subsection no. 3.8). As revealed in SEM images (Fig. S11(b)), after multiple cycles, the overall morphology was largely retained, showing only minor surface smoothing and partial aggregation, without any evidence of severe degradation or particle sintering. Thus, maintaining >97% activity retention after seven cycles.
, thus preventing self-corrosion. This thermodynamic hierarchy, combined with rapid interfacial electron transfer and physical encapsulation of the CuO core, ensures long-term catalytic stability, as verified by post-cycling XRD, SEM, and XPS analyses. Post-reaction XRD (Fig S11(a)) and XPS analyses (Fig. 10(b and c)) confirmed that the Cu2+, Cu+, and Cu0 phases remained structurally stable even after multiple photocatalytic runs. Only minor variations in relative peak intensities were detected, suggesting limited redistribution among Cu2+, Cu+, and Cu0 species under reaction conditions (discussed in detail in the Subsection no. 3.8). As revealed in SEM images (Fig. S11(b)), after multiple cycles, the overall morphology was largely retained, showing only minor surface smoothing and partial aggregation, without any evidence of severe degradation or particle sintering. Thus, maintaining >97% activity retention after seven cycles.
 |
| | Fig. 10 Recyclability photocatalytic Cr(VI) reduction over CuO–A1.1 under visible light for 7 successive cycles (a). Post XPS of CuO–A1.1: Cu 2p spectra (b), and O 1s spectra (c). | |
3.6 Scavenger experiments
The role of reactive species in the Cr(VI) reduction process was further investigated through scavenger experiments (Fig. 11(a)). Methanol was used as a hole scavenger, isopropanol (IPA) as a hydroxyl radical scavenger, benzoquinone (BQ) as a superoxide radical (˙O2−) scavenger, and AgNO3 as an electron scavenger. The addition of IPA caused only a minor decrease in activity (97.1%), suggesting that hydroxyl radicals contribute little to the overall process. In contrast, BQ led to a significant reduction in activity (74.6%), highlighting the important role of superoxide radicals. The strongest inhibition was observed with AgNO3, where activity decreased to 42.3%, demonstrating that photogenerated electrons are the dominant reactive species driving Cr(VI) photoreduction. Electron paramagnetic resonance (EPR) spectroscopy was employed to directly identify the reactive species generated during the photocatalytic process using DMPO as a spin-trapping agent. As shown in Fig. 11(b and c), no EPR signals were detected in the dark for either DMPO-˙O2− or DMPO-˙OH, indicating negligible radical formation in the absence of light. Under visible-light irradiation, a strong characteristic quartet signal corresponding to DMPO-˙O2− was clearly observed, confirming the generation of superoxide radicals. In contrast, no detectable DMPO-˙OH signal was observed under identical conditions, demonstrating that hydroxyl radicals are not involved in the photocatalytic Cr(VI) reduction process. These results are consistent with the scavenger experiments, where isopropanol (˙OH scavenger) exhibited only a weak inhibitory effect. Combined with the pronounced suppression observed in the presence of electron and ˙O2− scavengers, the EPR results confirm that Cr(VI) reduction proceeds predominantly via a direct electron transfer and ˙O2− mediated pathway, rather than through ˙OH radicals.
 |
| | Fig. 11 Effect of scavengers (IPA, BQ, AgNO3) (a), EPR spectra of the DMPO-˙O2− adducts (b) and DMPO-˙OH adducts (c) obtained with the CuO–A1.1 and Nyquist plots obtained from EIS measurements for pristine CuO and CuO–A1.1 under identical conditions (d). | |
3.7 Band structure and charge separation
Upon exposure to light, photogenerated electron–hole (e and h+) pairs are produced in the conduction band (CB) and valence band (VB) of semiconductor materials. The electronic band structure of the CuO–Cu2O–Cu heterostructure can be assessed by estimating the positions of the conduction and valence band edges using the empirical formulas stated in the literature.75,76 The valence and conduction band edges were estimated for CuO and Cu2O. For CuO, EVB = 2.162 eV and ECB = 0.462 eV; for Cu2O, EVB = 2.064 eV and ECB = −0.406 eV. A plausible mechanism is suggested to elucidate the augmented photocatalytic activity of the Cu–Cu2O–CuO heterostructure (Scheme 3). As seen in Scheme 3 CuO and Cu2O exhibit a staggered band alignment that promotes charge separation at their interface. Both CuO and Cu2O produce electron–hole pairs when exposed to visible-light irradiation. The photogenerated electrons in the conduction band (CB) of Cu2O (−0.41 eV) are transported to the CB of CuO (+0.46 eV), whereas the photogenerated holes in the valence band (VB) of CuO (+2.16 eV) migrate toward the VB of Cu2O (+2.06 eV).77 This bidirectional transfer inhibits charge recombination and leads to spatially distinct redox sites. The electrons accumulated in the CuO conduction band, facilitated by metallic Cu as an electron mediator, convert O2 into superoxide radicals (˙O2−). These radicals possess a moderately reducing potential (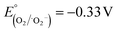 vs. NHE) that enables them to transfer electrons to Cr(VI) species, facilitating the stepwise conversion of Cr2O72− → Cr3+ (
vs. NHE) that enables them to transfer electrons to Cr(VI) species, facilitating the stepwise conversion of Cr2O72− → Cr3+ (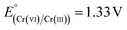 vs. NHE). The band structure analysis shows that the valence band positions of CuO and Cu2O lie above the H2O/˙OH oxidation potential (+2.40 eV),78 indicating that photogenerated holes cannot generate ˙OH. The added methanol served as a sacrificial hole scavenger, since it was reported that the redox level of ˙CH2OH/CH3OH is 1.45 V,79 which is well above the valence band potential of the catalyst under study (for CuO (+2.16 eV) and Cu2O (+2.06 eV)), so positive holes can easily oxidize the methanol, enabling efficient hole consumption.
vs. NHE). The band structure analysis shows that the valence band positions of CuO and Cu2O lie above the H2O/˙OH oxidation potential (+2.40 eV),78 indicating that photogenerated holes cannot generate ˙OH. The added methanol served as a sacrificial hole scavenger, since it was reported that the redox level of ˙CH2OH/CH3OH is 1.45 V,79 which is well above the valence band potential of the catalyst under study (for CuO (+2.16 eV) and Cu2O (+2.06 eV)), so positive holes can easily oxidize the methanol, enabling efficient hole consumption.
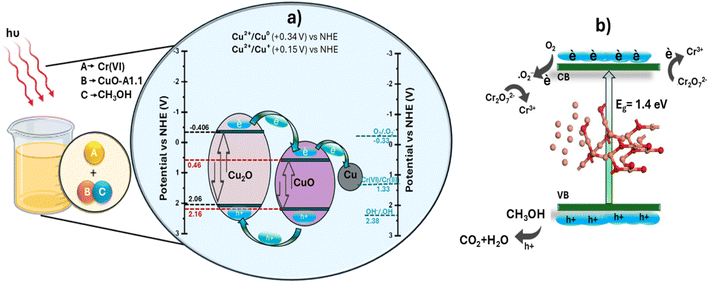 |
| | Scheme 3 Energy band structures of CuO–A1.1 (a), mechanism of the photocatalytic reaction with CuO–A1.1 as photocatalyst (b). | |
3.8 Experimental verification: electronic redistribution and charge transfer
To further verify the proposed photocatalytic mechanism, post-reaction XPS analyses were performed. The high-resolution Cu 2p spectrum (Fig. 10(b)) revealed a slight red shift and increased intensity of the Cu+ component, consistent with electron accumulation and partial reduction of Cu2+. Conversely, the Cu2+ peak exhibited a slight blue shift and decreased intensity, indicating partial transformation toward lower oxidation states. The O 1s spectrum (Fig. 10(c)) showed an increase in the low-binding-energy component associated with oxygen vacancies, confirming the generation of surface defects during the reaction. Consistently, EPR measurements (Fig. 11(b and c)) directly confirm the generation of superoxide radicals (˙O2−) under visible-light irradiation, with no detectable ˙OH signals. Together, these results demonstrate that dynamic electronic redistribution and defect formation in the CuO–Cu2O–Cu heterostructure promote efficient electron transfer and ˙O2− mediated Cr(VI) reduction.
The charge transfer kinetics were evaluated using electrochemical impedance spectroscopy (EIS) Nyquist plots (Fig. 11(d)), where the arc diameter directly represents charge transfer resistance (Rct).80 While pristine CuO exhibits a large semicircle indicating poor carrier mobility, the semicircle for CuO–A1.1 effectively vanishes. This transition to a diagonal line signifies negligible charge Rct, reduced resistance, and more efficient electron transport.81 Together, the XPS, EPR, and EIS results demonstrate that dynamic electronic redistribution, defect formation, and enhanced interfacial charge mobility in the CuO–Cu2O–Cu heterostructure work synergistically to promote rapid electron transfer and efficient ˙O2−-mediated Cr(VI) reduction, fully accounting for the superior photocatalytic performance of CuO–A1.1.
4 Conclusions
CuO–Cu2O–Cu heterostructures were successfully synthesized via an ascorbic acid-mediated solid-state route, achieving the study's objective of developing an efficient, stable, and environmentally benign photocatalyst for Cr(VI) reduction under visible light. Structural investigation revealed the presence of Cu, Cu2O, and CuO phases, whilst FESEM and HRTEM images displayed cavity-like morphologies with dense heterointerfaces that improved charge transfer. The optimized CuO–A1.1 catalyst exhibited a band gap of 1.42 eV (decreased from 1.67 eV for pristine CuO) and an enhanced Cu+/Cu2+ ratio with oxygen vacancies, as validated by the XPS study. Under visible light, CuO–A1.1 achieved complete Cr(VI) reduction (100%) within 30 minutes, corresponding to an apparent rate constant (k = 0.1036 min−1) nearly 50 times higher than that of CuO (0.0021 min−1). One-way ANOVA followed by Tukey's post hoc test statistically verified the significance of pH (F = 762.06, p < 0.0001, R2 = 0.9965) and initial Cr(VI) concentration (F = 92.46, p < 0.001), while catalyst dosage showed no significant effect on overall efficiency (p > 0.05). Levene's test confirmed variance homogeneity across all experimental sets, supporting the reliability of the ANOVA assumptions. The catalyst maintained over 97% efficiency after seven reuse cycles, demonstrating excellent structural and electronic stability verified by post-reaction XRD and XPS. Scavenger experiments identified electrons and superoxide radicals as the primary active species, validating the proposed Z-scheme charge transfer mechanism. Overall, the CuO–Cu2O–Cu heterostructure showed efficient charge separation, strong visible-light response, and high stability, enabling superior Cr(VI) reduction. The simple ascorbic acid synthesis makes it a promising and scalable photocatalyst for wastewater treatment. Future research will explore their integration into continuous-flow photoreactors, performance under solar irradiation, and potential applications in persistent contaminants like dyes and pharmaceutical residues.
Ethical approval
Institutional review board statement: this study does not involve human participants, animal subjects, or any ethically regulated biological materials. General approval for conducting laboratory-based chemical and environmental experiments was granted by the Ethical Committee of the Faculty of Science, Benha University (Approval No. BUFS-REC-2025-467Chm) in accordance with institutional safety and research guidelines.
Author contributions
Mona S. NourEldien: writing – original draft, visualization, methodology, formal analysis, data curation. Mostafa Y. Nassar: writing – review & editing, validation, supervision, investigation, conceptualization, project administration.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability
This article incorporates all the data produced or examined during this study.
Supplementary information (SI): additional experimental details, characterization techniques, statistical analyses, figures, and tables that further support the results presented in the main text. See DOI: https://doi.org/10.1039/d5ra09131d.
Acknowledgements
This work was supported by the Deanship of Scientific Research, Vice Presidency for Graduate Studies and Scientific Research, King Faisal University, Saudi Arabia [Grant No. KFU253221]. During the preparation of this work, the author(s) used QuillBot to assist with paraphrasing and improving the clarity of sentences. After using this tool, the author(s) reviewed and edited the content as needed and take(s) full responsibility for the content of the published article.
References
- I. Y. El-Sherif, S. Tolani, K. Ofosu, O. A. Mohamed and A. K. Wanekaya, J. Environ. Manage., 2013, 129, 410–413 CrossRef CAS PubMed.
- Y. Zou, X. Wang, A. Khan, P. Wang, Y. Liu, A. Alsaedi, T. Hayat and X. Wang, Environ. Sci. Technol., 2016, 50, 7290–7304 CrossRef CAS PubMed.
- G. Tjandraatmadja, C. Diaper, Y. Gozukara, L. Burch, C. Sheedy and G. Price, Sources of Critical Contaminants in Domestic Wastewater: Contaminant Contribution from Household Products, Australia, 2008 Search PubMed.
- M. Taseidifar, F. Makavipour, R. M. Pashley and A. M. Rahman, Environ. Technol. Innovation, 2017, 8, 182–190 CrossRef.
- M. El-Sharkawy, M. O. Alotaibi, J. Li, D. Du and E. Mahmoud, Sustainability, 2025, 17, 701 CrossRef CAS.
- W. R. García-Niño and J. Pedraza-Chaverrí, Food Chem. Toxicol., 2014, 69, 182–201 CrossRef PubMed.
- K. Duan, Y. Li, W. Yang, Y. Lin, L. Rao and C. Han, Sustainability, 2025, 17, 5343 CrossRef CAS.
- E. Matei, M. Râpă, I. M. Mateş, A.-F. Popescu, A. Bădiceanu, A. I. Balint and C. I. Covaliu-Mierlă, Molecules, 2025, 30, 1455 CrossRef CAS PubMed.
- P. Sahu, A. R. Patel, A. Pandey, M. Hait and G. K. Patra, Inorg. Chim. Acta, 2025, 585, 122751 CrossRef CAS.
- R. Singh, S. Chakma and V. Birke, Environ. Sci. Pollut. Res. Int., 2025, 1–19 Search PubMed.
- Y. Zhigalenok, A. Tazhibayeva, S. Kokhmetova, A. Starodubtseva, T. Kan, D. Isbergenova and F. Malchik, RSC Adv., 2025, 15, 21439–21464 RSC.
- R. Saha, R. Nandi and B. Saha, J. Coord. Chem., 2011, 64, 1782–1806 CrossRef CAS.
- C.-C. Wang, X.-D. Du, J. Li, X.-X. Guo, P. Wang and J. Zhang, Appl. Catal., B, 2016, 193, 198–216 CrossRef CAS.
- H.-L. Ma, Y. Zhang, Q.-H. Hu, D. Yan, Z.-Z. Yu and M. Zhai, J. Mater. Chem., 2012, 22, 5914–5916 RSC.
- W. Zheng, J. Hu, Z. Han, Z. Wang, Z. Zheng, J. Langer and J. Economy, Chem. Commun., 2015, 51, 9853–9856 RSC.
- J. Fang, Z. Gu, D. Gang, C. Liu, E. S. Ilton and B. Deng, Environ. Sci. Technol., 2007, 41, 4748–4753 CrossRef CAS PubMed.
- Z. Yang, X. Jin, H. Lu, S. Tong, J. Chen, M. Deng and F. Chen, Mater. Sci. Semicond. Process., 2025, 193, 109510 CrossRef CAS.
- H. Luo, S. Guo, Y. Bao and Z. Jia, Appl. Surf. Sci., 2023, 618, 156596 CrossRef CAS.
- M. Sakar, S. Balakumar, P. Saravanan and S. Bharathkumar, Nanoscale, 2016, 8, 1147–1160 RSC.
- E. Zhengyang, J. Liang, Y. Dong, Q. Chao, P. Li and Q. Fan, Environ. Res., 2023, 236, 116819 CrossRef PubMed.
- Z. Ding, W. Zhang, J. Liang, S. Qiang, W. Wang, P. Li and Q. Fan, Chemosphere, 2023, 311, 137136 CrossRef CAS PubMed.
- M. S. S. Danish, L. L. Estrella, I. M. A. Alemaida, A. Lisin, N. Moiseev, M. Ahmadi, M. Nazari, M. Wali, H. Zaheb and T. Senjyu, Metals, 2021, 11, 80 CrossRef CAS.
- M. A Sliem, A. Youssef Salim, R. M Abdelnaby, G. Genidy Mohamed and A. S. Amin, Benha J. Appl. Sci., 2022, 7, 41–47 CrossRef.
- V. Kumar, S. Masudy-Panah, C. Tan, T. Wong, D. Chi and G. Dalapati, Copper oxide based low cost thin film solar cells, Singapore, 2013, DOI:10.1109/INEC.2013.6466072.
- M. A. Khalifa, Phys. Scr., 2024, 99, 075934 CrossRef CAS.
- S. Bonthula, S. R. Bonthula, R. Pothu, R. K. Srivastava, R. Boddula, A. B. Radwan and N. Al-Qahtani, Sustainable Chem., 2023, 4, 246–271 CrossRef CAS.
- N. Manjula, G. Sarma, B. M. Shilpa and K. Suresh Kumar, in Phytonanotechnology, Springer, 2022, pp. 255–276 Search PubMed.
- M. Kumar, B. Meena, P. Subramanyam, D. Suryakala and C. Subrahmanyam, Catalysts, 2022, 12, 1198 Search PubMed.
- A. M. Mohammed, S. S. Mohtar, F. Aziz, S. A. Mhamad and M. Aziz, J. Environ. Chem. Eng., 2021, 9, 105138 CrossRef CAS.
- S. Bandaranayake, A. Patnaik, E. Hruska, Q. Zhu, S. Das and L. R. Baker, ACS Appl. Mater. Interfaces, 2024, 16, 41616–41625 CrossRef CAS PubMed.
- A. Enesca, L. Isac and A. Duta, Thin Solid Films, 2013, 542, 31–37 CrossRef CAS.
- R. Ran, X. Meng and Z. Zhang, Appl. Catal., B, 2016, 196, 1–15 CrossRef CAS.
- M. Abd Elkodous, G. Kawamura, W. K. Tan and A. Matsuda, Mater. Lett., 2022, 323, 132606 CrossRef CAS.
- P. Wang, Z. Liu, C. Han, X. Ma, Z. Tong and B. Tan, J. Nanopart. Res., 2021, 23, 268 CrossRef CAS.
- P. Uthirakumar, M. Devendiran, T. H. Kim, S. Kalaiarasan and I.-H. Lee, Mater. Sci. Eng. B, 2020, 260, 114652 CrossRef CAS.
- H. Li, Z. Su, S. Hu and Y. Yan, Appl. Catal., B, 2017, 207, 134–142 CrossRef CAS.
- K. Yang, B. Zhou, K. Dong, Y. Wu, Q. Huang, J. Lv, Z. Wan and M. Yang, Catal. Lett., 2025, 155, 199 CrossRef CAS.
- C.-J. Chang, C.-W. Kang and A. Pundi, Catalysts, 2024, 14, 926 CrossRef CAS.
- S. Kumar, Bhawna, A. Gupta, R. Kumar, A. Bharti, A. Kumar and V. Kumar, J. Phys. Chem. C, 2023, 127, 7095–7106 CrossRef CAS.
- R. Subagyo, G. R. Anindika, A. A. Aufkani, L. Zhang, H. Ardhyananta, R. P. Burhan, Z. V. Nugraheni, S. Akhlus, H. Bahruji and D. Prasetyoko, Mater. Sci. Energy Technol., 2025, 8, 131–142 CAS.
- Z. Martínez-Corona, D. García-Zaleta, R. López-González, C. García-Mendoza, M. Alvarez-Lemus, C. Encarnacion-Gomez and S. Gómez-Cornelio, J. Mater. Sci., 2025, 20, 70 Search PubMed.
- X. Zhang, D. Zheng and A. Tian, J. Mol. Struct., 2025, 1337, 142237 CrossRef CAS.
- N. Wang, Y. Xu, L. Zhu, X. Shen and H. Tang, J. Photochem. Photobiol., A, 2009, 201, 121–127 CrossRef CAS.
- T. Jiang, Y. Wang, D. Meng, X. Wu, J. Wang and J. Chen, Appl. Surf. Sci., 2014, 311, 602–608 CrossRef CAS.
- L. D. L. S. Valladares, D. H. Salinas, A. B. Dominguez, D. A. Najarro, S. Khondaker, T. Mitrelias, C. Barnes, J. A. Aguiar and Y. Majima, Thin Solid Films, 2012, 520, 6368–6374 CrossRef CAS.
- S. Jingyan, L. Yuwen, W. Zhiyong and W. Cunxin, J. Pharm. Biomed. Anal., 2013, 77, 116–119 CrossRef PubMed.
- K. Karthik, N. V. Jaya, M. Kanagaraj and S. Arumugam, Solid State Commun., 2011, 151, 564–568 CrossRef CAS.
- R. Subagyo, G. R. Anindika, A. A. Aufkani, L. Zhang, H. Ardhyananta, R. P. Burhan, Z. V. Nugraheni, S. Akhlus, H. Bahruji and D. Prasetyoko, Mater. Sci. Energy Technol., 2025, 8, 131–142 CAS.
- E. Kociołek-Balawejder, K. Winiarska, J. Winiarski and I. Mucha, Materials, 2024, 17, 3893 CrossRef PubMed.
- J. Choi, D. A. Reddy, M. J. Islam, B. Seo, S. H. Joo and T. K. Kim, Appl. Surf. Sci., 2015, 358, 159–167 CrossRef CAS.
- I. Platzman, R. Brener, H. Haick and R. Tannenbaum, J. Phys. Chem. C, 2008, 112, 1101–1108 CrossRef CAS.
- P. Uthirakumar, M. Devendiran, D. Van Dao, H. Son, Y. H. Cho and I.-H. Lee, J. Environ. Chem. Eng., 2021, 9, 106396 CrossRef CAS.
- O. Gohar, M. Z. Khan, I. Bibi, N. Bashir, U. Tariq, M. Bakhtiar, M. R. A. Karim, F. Ali, M. B. Hanif and M. Motola, Mater. Des., 2024, 241, 112930 CrossRef CAS.
- M.-R. Pallavolu, A.-N. Banerjee and S.-W. Joo, Coatings, 2023, 13, 1337 CrossRef CAS.
- S. Li, C. You, K. Rong, C. Zhuang, X. Chen and B. Zhang, Adv. Powder Mater., 2024, 3, 100183 CrossRef.
- A. Al-Keisy, L. Ren, D. Cui, Z. Xu, X. Xu, X. Su, W. Hao, S. X. Dou and Y. Du, J. Mater. Chem. A, 2016, 4, 10992–10999 RSC.
- V. Štengl and T. M. Grygar, Int. J. Photoenergy, 2011, 2011, 685935 Search PubMed.
- Q. Wang, W. Ma, J. Qian, N. Li, C. Zhang, M. Deng and H. Du, Environ. Pollut., 2024, 347, 123707 CrossRef CAS PubMed.
- C. Jin, J. Liu, Y. Yin and Z. Li, RSC Adv., 2024, 14, 5142–5148 Search PubMed.
- N. Hassan, A. Jalil, E. S. Aldeen, M. Bahari, L. Teh, S. Rajendran, N. Jusoh and N. Ainirazali, Water Process Eng., 2024, 59, 105050 Search PubMed.
- L.-W. Wei, M.-W. Zheng, S.-H. Liu, H. P. Wang, Y.-C. Pu and V.-C. Nguyen, Chem. Eng. J., 2024, 487, 150712 Search PubMed.
- M. Rhaya, H. Ighnih, H. Abou Oualid, B. Ennasraoui, R. Haounati, H. Ouachtak, A. Jada and A. Ait Addi, J. Mol. Struct., 2025, 1338, 142333 Search PubMed.
- B. Qin, Y. Zhao, H. Li, L. Qiu and Z. Fan, Chin. J. Catal., 2015, 36, 1321–1325 CrossRef CAS.
- M. Zhu, L. Hao, Q. Zhao, T. Hu, S. Guan and Y. Lu, J. Alloys Compd., 2025, 1010, 177822 Search PubMed.
- P. Zhu, Y. Li, Y. Ma, X. Ruan and Q. Zhang, Ceram. Int., 2023, 49, 12518–12528 CrossRef CAS.
- X. Dou, C. Zhang and H. Shi, Sep. Purif. Technol., 2022, 282, 120023 CrossRef CAS.
- I. Gomaa, A. Helal, M. A. Ibrahim and N. M. Hosny, BMC Chem., 2025, 19, 174 CrossRef CAS PubMed.
- A. E. Nogueira, O. F. Lopes, A. B. Neto and C. Ribeiro, Chem. Eng. J., 2017, 312, 220–227 Search PubMed.
- A. Wiryawan, R. Retnowati, P. Burhan and S. Syekhfani, J. Environ. Eng. Sustainable Technol., 2018, 5, 37–46 Search PubMed.
- L. Suryati, H. Sulistyarti and A. Atikah, J. Pure Appl. Chem. Res., 2015, 4, 34 CrossRef CAS.
- A. Ashar, I. A. Bhatti, A. Jilani, M. Mohsin, S. Rasul, J. Iqbal, M. B. Shakoor, A. G. Al-Sehemi, S. Wageh and A. A. Al-Ghamdi, Polymers, 2021, 13, 4047 Search PubMed.
- G. Lacarbonara, L. Faggiano, S. Porcu, P. C. Ricci, S. Rapino, D. P. Casey, J. F. Rohan and C. Arbizzani, Batteries, 2021, 7, 83 CrossRef CAS.
- L. Lu, S. Hu, H.-I. Lee, C. Wöll and R. A. Fischer, J. Nanopart. Res., 2007, 9, 491–496 CrossRef CAS.
- S. Cao, C.-J. Wang, G.-Q. Wang, Y. Chen, X.-J. Lv and W.-F. Fu, RSC Adv., 2020, 10, 5930–5937 RSC.
- S. Mihai, D. Cursaru, D. Matei, A. Manta, R. Somoghi and G. Branoiu, Sci. Rep., 2019, 9, 18798 CrossRef CAS PubMed.
- Y. Wu, J. Zhang, B. Long and H. Zhang, ACS Omega, 2021, 6, 15057–15067 Search PubMed.
- S. Mosleh, M. R. Rahimi, M. Ghaedi, K. Dashtian and S. Hajati, Ultrason. Sonochem., 2018, 40, 601–610 CrossRef CAS PubMed.
- T. Cao, J. Xu and M. Chen, Sep. Purif. Technol., 2022, 291, 120896 CrossRef CAS.
- A. Samad, S. Ahsan, I. Tateishi, M. Furukawa, H. Katsumata, T. Suzuki and S. Kaneco, Chin. J. Chem. Eng., 2018, 26, 529–533 Search PubMed.
- M. S. NourEldien, M. Y. Nassar, I. M. Ibrahim and H. M. Aly, Mater. Adv., 2024, 5, 9107–9123 Search PubMed.
- L. Yuan, B. Weng, J. C. Colmenares, Y. Sun and Y. J. Xu, Small, 2017, 13, 1702253 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Mostafa Y. Nassar
*a and
Mostafa Y. Nassar













 (+0.34 V) and
(+0.34 V) and  (+0.16 V)), preferentially donate electrons to Cr(VI)
(+0.16 V)), preferentially donate electrons to Cr(VI)  , thus preventing self-corrosion. This thermodynamic hierarchy, combined with rapid interfacial electron transfer and physical encapsulation of the CuO core, ensures long-term catalytic stability, as verified by post-cycling XRD, SEM, and XPS analyses. Post-reaction XRD (Fig S11(a)) and XPS analyses (Fig. 10(b and c)) confirmed that the Cu2+, Cu+, and Cu0 phases remained structurally stable even after multiple photocatalytic runs. Only minor variations in relative peak intensities were detected, suggesting limited redistribution among Cu2+, Cu+, and Cu0 species under reaction conditions (discussed in detail in the Subsection no. 3.8). As revealed in SEM images (Fig. S11(b)), after multiple cycles, the overall morphology was largely retained, showing only minor surface smoothing and partial aggregation, without any evidence of severe degradation or particle sintering. Thus, maintaining >97% activity retention after seven cycles.
, thus preventing self-corrosion. This thermodynamic hierarchy, combined with rapid interfacial electron transfer and physical encapsulation of the CuO core, ensures long-term catalytic stability, as verified by post-cycling XRD, SEM, and XPS analyses. Post-reaction XRD (Fig S11(a)) and XPS analyses (Fig. 10(b and c)) confirmed that the Cu2+, Cu+, and Cu0 phases remained structurally stable even after multiple photocatalytic runs. Only minor variations in relative peak intensities were detected, suggesting limited redistribution among Cu2+, Cu+, and Cu0 species under reaction conditions (discussed in detail in the Subsection no. 3.8). As revealed in SEM images (Fig. S11(b)), after multiple cycles, the overall morphology was largely retained, showing only minor surface smoothing and partial aggregation, without any evidence of severe degradation or particle sintering. Thus, maintaining >97% activity retention after seven cycles.


 vs. NHE) that enables them to transfer electrons to Cr(VI) species, facilitating the stepwise conversion of Cr2O72− → Cr3+ (
vs. NHE) that enables them to transfer electrons to Cr(VI) species, facilitating the stepwise conversion of Cr2O72− → Cr3+ ( vs. NHE). The band structure analysis shows that the valence band positions of CuO and Cu2O lie above the H2O/˙OH oxidation potential (+2.40 eV),78 indicating that photogenerated holes cannot generate ˙OH. The added methanol served as a sacrificial hole scavenger, since it was reported that the redox level of ˙CH2OH/CH3OH is 1.45 V,79 which is well above the valence band potential of the catalyst under study (for CuO (+2.16 eV) and Cu2O (+2.06 eV)), so positive holes can easily oxidize the methanol, enabling efficient hole consumption.
vs. NHE). The band structure analysis shows that the valence band positions of CuO and Cu2O lie above the H2O/˙OH oxidation potential (+2.40 eV),78 indicating that photogenerated holes cannot generate ˙OH. The added methanol served as a sacrificial hole scavenger, since it was reported that the redox level of ˙CH2OH/CH3OH is 1.45 V,79 which is well above the valence band potential of the catalyst under study (for CuO (+2.16 eV) and Cu2O (+2.06 eV)), so positive holes can easily oxidize the methanol, enabling efficient hole consumption.
