DOI:
10.1039/D5RA08375C
(Review Article)
RSC Adv., 2026,
16, 2007-2029
Tetrahydropyridines: a recent update for their multicomponent synthesis
Received
31st October 2025
, Accepted 22nd December 2025
First published on 7th January 2026
Abstract
Tetrahydropyridines (THPs) are important nitrogen-based ring compounds found in many natural and synthetic molecules with wide biological importance. Their simple structure and chemical flexibility make them valuable in drug design and organic synthesis. Among different synthetic methods, multicomponent reactions (MCRs) have become a fast, efficient, and eco-friendly approach for preparing a large variety of THP derivatives in a single step. This review focuses on recent developments in the synthesis of tetrahydropyridines using MCRs with different nitrogen sources such as amines, enamines, imines, ammonia, ammonium acetate, etc. It also highlights green and sustainable methods like solvent-free, microwave-assisted, photoinduced, and catalyst-free reactions. Overall, this review explains how multicomponent reactions provide an easy, economical, and environmentally friendly route for building diverse tetrahydropyridine structures useful in chemical and medicinal research.
 Md Musawwer Khan | Dr Md. Musawwer Khan is an Associate Professor Organic Chemistry in the Department of Chemistry at Aligarh Muslim University, India. He completed his PhD at the Indian Institute of Technology (IIT) Guwahati, where he worked on developing new and simple methods for making important organic compounds. His main research areas include multicomponent reactions, heterocyclic chemistry, green chemistry, carbohydrate chemistry and the synthesis of small bioactive molecules. Dr Khan has published more than 45 research papers and 2 book chapters in reputed international journals. He is also co-editor of edited research topic “Five-Membered Heterocycles: Synthesis and Applications”. He has guided several PhD, M. Phil, M. Sc. and B. Sc. Project students in the field of organic synthesis and green methodologies. Dr Khan has received many awards, including the Star up Grant, Junio Research Fellowship, Senior Research Fellowship, DST Pre-Doctoral Fellowship, IAS Faculty Summer Research Fellowship, Royal Society of Chemistry Best Cited Author Award (2018) and the IAS, Summer Research Fellowship. His current research focuses on developing eco-friendly, greener novel synthetic methodologies and synthesis of valuable heterocyclic compounds with potential biological applications. |
 Faiz Ahmad Faiz | Faiz Ahmad Faiz is currently a fourth-year BSc Research student in the Department of Chemistry at Aligarh Muslim University, Aligarh, India. He was born in the district of Samastipur, Bihar. His research interests focus on the synthesis of organic compounds, especially nitrogen-containing heterocycles and drug-like molecules. He recently completed a research project titled “Efficient One-Pot Five-Component Synthesis of Piperidine Derivatives” under the Indo-US STEM APJ Abdul Kalam STEM Education and Research Program jointly conducted by The Ohio State University and Aligarh Muslim University under mentorship of Dr Md. Musawwer Khan. His long-term goal is to contribute to the development of new and efficient synthetic methods for medicinally important organic drugs. |
 Mohd Sufiyan Khan | Mr Mohd Sufiyan Khan, born on February 10, 2005, in Aligarh, India, is currently a fourth-year BSc Research student at Aligarh Muslim University. He recently completed a research project under supervision of Dr Md. Musawwer Khan on “Green One-Pot Synthesis of Pyranopyrazole Derivative Using Deep Eutectic Solvent” under the Indo–US STEM APJ Abdul Kalam STEM Education and Research Program, jointly conducted by The Ohio State University (USA) and Aligarh Muslim University (India). His research interests focus on green chemistry, heterocyclic synthesis, computational chemistry and sustainable organic transformations. He aims to develop environmentally friendly and efficient methodologies in synthetic organic chemistry to support sustainable scientific innovation. |
1 Introduction
Tetrahydropyridine (THP) scaffolds are a significant group of nitrogen-containing heterocycles because they are widely incorporated in natural bioactive compounds and are highly useful in synthetic chemistry. Their simple ring system, together with broad biological activities, makes THP derivatives valuable in drug discovery. Among the various synthetic approaches, multicomponent reactions (MCRs) are especially attractive since they are easy to carry out, save time, and allow the rapid preparation of many structurally diverse THP derivatives.1 The biological value of this scaffold is also seen in nature, as several alkaloids contain the THP ring. Some notable examples include bis-benzyl-isoquinoline alkaloids (BBIQAs, I) isolated from the Menispermaceae and Berberidaceae families,2 solacongestidine (II) isolated from Solanum congestiflorum which exhibits antifungal activity against Candida albicans, Trichophyton rubrum and Cryptococcus albidus3 and (+)-cannabisativine (III) from Cannabis sativa,4 and (–)-sedacrine (IV) from Sedum species,5 all of which show important biological effects as shown in Fig. 1. These natural products provide a strong base for designing synthetic analogues with potential pharmacological importance.
 |
| | Fig. 1 Natural alkaloids having tertrahydropyridine ring system. | |
Over the years, tetrahydropyridine derivatives have gained considerable attention in medicinal chemistry due to their broad spectrum of pharmacological activities.6 Numerous studies have reported that compounds containing the THP ring exhibit diverse bioactivities like -antimicrobial,7 anticonvulsant,8 antiproliferative,9 anti-inflammatory,10 antimalarial,11 antidiabetic12 etc. Some medicinally important compounds containing the tetrahydropyridine skeleton are shown in Fig. 2. In recent findings, tetrahydropyridine containing compound V has also emerged as a biologically relevant scaffold, demonstrating CDK2 kinase inhibition associated with anticancer activity, as well as DprE1 inhibition, which is important in antitubercular drug discovery.13 Additionally, thiazole-based tetrahydropyridines (VI) showed promising biological activity: several compounds exhibited strong insecticidal effects against Aphis laburni, and even resistant strains, thereby suggesting potential in resistance management.14 Furthermore, 4-aryl-1,2,3,6-tetrahydropyridines (VII) were studied as 5-HT2C receptor agonists, positioning them as potential agents for appetite suppression and obesity treatment.15
 |
| | Fig. 2 Some medicinally important compound containing tetrahydropyridine skeleton. | |
Tetrahydropyridine derivatives (VIII) also represent a class of muscarinic acetylcholine receptor ligands, with several compounds showing agonist activity at the M1 subtype involved in cognition and memory. The tetrahydropyridine ring contributes to effective receptor binding and functional activation. These compounds typically activate M1 receptors through Gq/11-mediated calcium signaling. Some tetrahydropyridine-based M1 agonists also stimulate ERK1/2 pathways and promote non-amyloidogenic amyloid precursor protein processing, supporting their relevance in Alzheimer's disease research.16 Tetrahydropyridine (THP) derivatives (IX) also represent a novel class of ligands with high affinity and selectivity for central nicotinic acetylcholine receptors (nAChRs), showing notable subtype selectivity, especially toward the α4β2 subtype of nAChRs, which is dominant in the brain. These compounds offer potential as central nervous system (CNS) therapeutics, with uses in studying nicotinic receptor function and possibly addressing neurological disorders like Alzheimer's or Parkinson's disease due to their selective cholinergic modulation.17
Moreover, THP derivatives (X) are particularly well recognized for their monoamine oxidase (MAO) inhibitory activity. Several substituted tetrahydropyridines effectively inhibit MAO-A and MAO-B enzymes, which are responsible for the metabolic degradation of key neurotransmitters such as dopamine, serotonin, and norepinephrine. MAO-A inhibition is mainly associated with antidepressant and anxiolytic effects, whereas MAO-B inhibition is relevant to the treatment of neurodegenerative disorders, especially Parkinson's disease. Subtle structural variations on the tetrahydropyridine ring strongly influence enzyme selectivity and inhibitory potency.18
Due to the above properties of THP, diverse synthetic approaches have been developed worldwide using multicomponent reactions (MCRs). MCRs serve as an efficient approach for rapidly assembling structurally diverse and complex molecules in a single synthetic operation. Compared to traditional stepwise methods, MCRs19 offer several advantages such as reduced cost, shorter reaction times, improved atom economy, and eco-friendly protocols. Additionally, they enable rapid exploration of structural diversity, making them highly suitable for combinatorial synthesis.
1.1. Recent progress for synthesis of tetrahydropyridines using multicomponent reaction
Tetrahydropyridine (THP) rings are widely studied N-heterocycles because of their valuable biological properties and practical applications, so chemists have focused on developing faster, cheaper, and greener synthetic method. One-pot multicomponent reactions (MCRs) have emerged as especially powerful tools for constructing diverse THP libraries by combining a few simple building blocks in a single operation; varying the amine component and other partners provides easy access to many substituted derivatives. In 2016, our group published a review summarized work up to 2015,20 and since then numerous research reports have extended and refined MCR strategies. In, this review therefore we cover a decade literature published after 2015 to the present and organizes the methods primarily by the type of amino building block employed in the multicomponent routes.
1.2. Synthesis of THP using amine as amino source
Using tert-butyl alcohol as a solvent, Ishmiyarov et al. described a one-pot, three-component synthesis of compound 4 by condensation of ethyl acetoacetate, methoxymethanol (formed in situ from paraformaldehyde), and primary amines or diamines in methanol.21 The reaction proceeds through an intermediate 2a, which then undergoes intramolecular cyclization to form the tetrahydropyridine core. High yields of mono and bicyclic tetrahydropyridine derivatives, up to 98%, were produced during the five-hour reaction at 65 °C. Depending on the amine's structure, the reaction produced compounds like diethyl 3-acetyl-1-isopropyl-6-methyl-1,2,3,4-tetrahydropyridine-3,5-dicarboxylate, diethyl 3-acetyl-1-benzyl-6-methyl-1,2,3,4-tetrahydropyridine-3,5-dicarboxylate, and diethyl 3-acetyl-1-(2-hydroxyethyl)-6-methyl-1,2,3,4-tetrahydropyridine-3,5-dicarboxylate. Tert-butyl alcohol was essential for the product selectivity. Intramolecular cyclization made possible by the use of diamines resulted in fused bicyclic systems. Under mild conditions, this multicomponent method offers a quick and easy way to obtain structurally varied tetrahydropyridine derivatives (Scheme 1).
 |
| | Scheme 1 Solvent selective multicomponent synthesis of monocyclic 1,2,3,4-tetrahydropyridine derivatives. | |
A one-pot method was established for the preparation of tetrahydropyridine-3-carboxylate derivatives 9 using a green and efficient approach. The reaction involves a four-component condensation of Meldrum's acid, ethyl cyanoacetate, aryl aldehydes, and aromatic amines in ethanol, promoted by nano nickel oxide (NiO) nanoparticles under ultrasound irradiation.22 This method provided high yields up to 95% in a short time and avoided the use of toxic reagents or harsh conditions. The process benefits from energy efficiency, catalyst reusability, and eco-friendly conditions. This innovative protocol was reported by Fathima shows the effective combination of nanocatalysis and sonochemistry in the green preparation of pharmacologically important tetrahydropyridine-3-carboxylates passing through an intermediate 8a (Scheme 2).
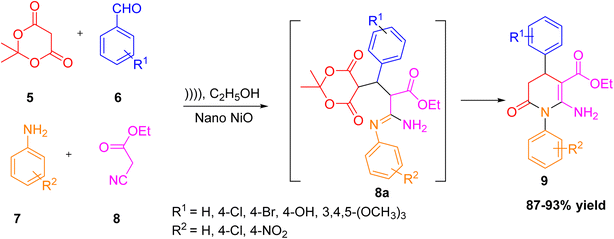 |
| | Scheme 2 Formation of tetrahydropyridine-3-carboxylates by using ultrasound irradiation. | |
Yang et al. developed an efficient Ru(II)-catalyzed one-pot synthesis of 1,2-dihydropyridines (DHPs) 13, which were subsequently converted into tetrahydropyridines (THPs) through an intermediate 12a.23 The reaction proceeds via a three-component strategy, employing cinnamaldehyde, p-anisidine, and diphenylacetylene as the starting materials. The catalytic system showed wide applicability to various substrates and was compatible with different functional groups, accommodating various aryl and heteroaryl aldehydes, amines, and internal alkynes. Notably, the use of RuCl2(COD) as a simple and accessible catalyst, in combination with additives such as Mg(OAc)2, TiO2, and BTBAC, significantly enhanced the reaction efficiency (Scheme 3).
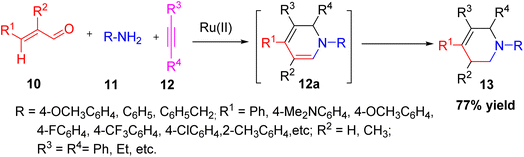 |
| | Scheme 3 One-pot three component formation of THPD using Ru(II). | |
Zanatta and co-workers reported a very fast, one-pot method to make 1-arylethyl-2-arylethylamino-5-trifluoroacetyl-1,2,3,4-tetrahydropyridines 16 in excellent yields.24 They started from 2-alkoxy-5-trifluoroacetyl-3,4-dihydro-2H-pyrans and simply added two equivalents of a 2-arylethanamine and related 2-ethanamines in methanol or ethanol at room temperature (Scheme 4). In just five minutes, the mixture cleanly converts into the target tetrahydropyridine products, each bearing a trifluoroacetyl ketone at C-5 and an arylethylamino group at C-2 in 90–98% isolated yield. The reactions proceeded rapidly, occurred at room temperature, afforded high yields, and were easy to purify and isolate. They have used many types of ethanamines, making this method useful for making many different compounds in the future. Furthermore, the same group also prepared these derivatives using an ionic liquid ([BMIM]BF4)25 in combination with microwave irradiation and achieved yield up to 97%.
 |
| | Scheme 4 One-pot formation of 1-arylethyl-2-arylethylamino-5-trifluoroacetyl-1,2,3,4 tetrahydropyridines. | |
Dudognon et al. reported a new and simple method to make enantioenriched polycyclic 1,2,3,4-tetrahydropyridines 20, which are important in drug research.26 They used a three-component reaction that combined β-ketoamides, (E)-cinnamaldehydes, and 2-aminophenols under the influence of a chiral iminium-based organocatalyst. This method forms the product 1,2,3,4-tetrahydropyridine derivatives via an intermediate 19a with three connected chiral centres and four new bonds formed in a single step. The reaction showed high regio-, diastereo-, and enantioselectivity. Although the yields were moderate, the stereocontrol and structural complexity obtained in one step show the synthetic value of this MCR approach (Scheme 5).
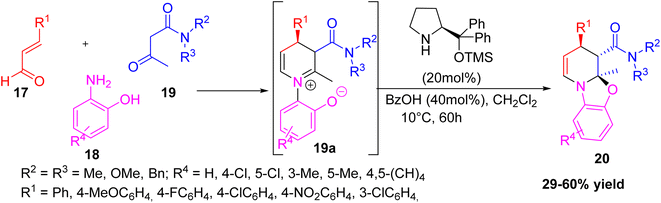 |
| | Scheme 5 Formation of polycyclic 1,2,3,4-tetrahydropyridines using organocatalyst. | |
Wan and co-workers developed a simple one-pot reaction using an enaminone (or nitroenamine), o-aminophenol, and cinnamaldehyde in the presence of lactic acid and an ethanol–water mixture to make fused tetrahydropyridines.27 The product, phenyl(3-phenyl-4,4a-dihydro-3H-benzo[4,5]oxazolo[3,2-a]pyridin-2-yl)methanone 22, was obtained in 76% yield as a single diastereomer (Scheme 6). The reaction goes through transamination of the enaminone, Michael addition to cinnamaldehyde, ring closure, and dehydration as shown in mechanism (Scheme 7). This method is efficient and sustainable, using a green solvent, a non-toxic bio-based catalyst, and air atmosphere at 90 °C to produce various tetrahydropyridine derivatives.
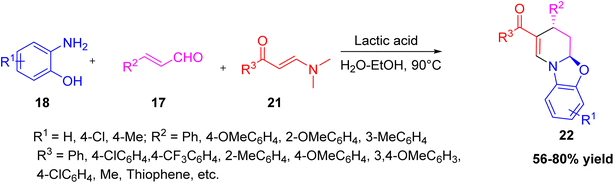 |
| | Scheme 6 One-pot three component formation of fused tetrahydropyridine by lactic acid. | |
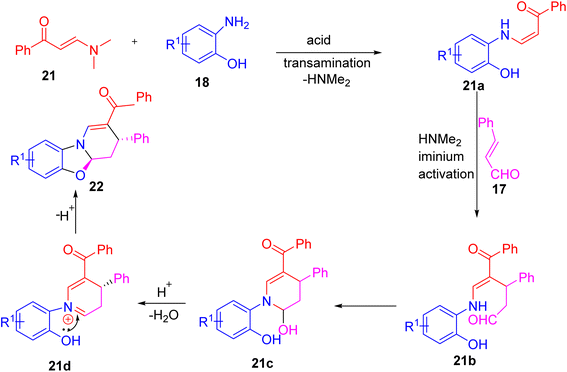 |
| | Scheme 7 Mechanistic approach to explain for synthesis of fused tetrahydropyridine. | |
Mohammadi et al. reported a simple and efficient one-pot, four-component reaction to synthesize new tetrahydropyridine derivatives.28 The target product, 10b-hydroxy-4-nitro-5-phenyl-2,3,5,5a-tetrahydro-1H-imidazo[1,2-a]indeno[2,1-e]pyridin-6(10bH)-one 27, was synthesized using ethylenediamine, 1,1-bis(methylthio)-2-nitroethylene, various aldehydes, and 1,3-indandione in ethanol without using any catalyst (Scheme 8). The reaction involves a Knoevenagel condensation, Michael addition, tautomerization, and cyclization steps. The reaction provides high yields and exhibits excellent stereoselectivity in the formation of the final products. Additionally, molecular docking analysis revealed that several of these compounds exhibit strong binding affinity toward the active site of the HIV protease enzyme with binding energies comparable to the standard drug saquinavir. The synthesized tetrahydropyridine compounds exhibit potential for further development as HIV protease inhibitors.
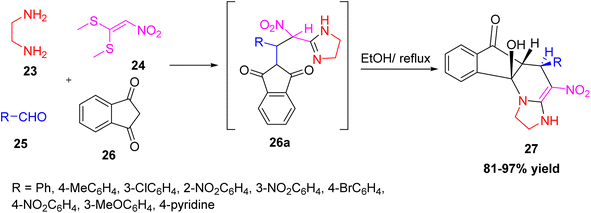 |
| | Scheme 8 Synthesis of 10b-hydroxy-4-nitro-5-phenyl-2,3,5,5a-tetrahydro-1H-imidazo[1,2-a]indeno[2,1-e]pyridin-6(10bH)-one via MCR. | |
Gibadullina et al. reported a convenient one-pot multicomponent synthesis of 1,1′-(α,ω-alkanediyl)bis(1,2,3,4-tetrahydropyridines), 30 recognized for exhibiting diverse biological activities.29 The method involves 1,3-dicarbonyl compounds (acetylacetone or acetoacetic ester), aqueous 33% formaldehyde, and various α,ω-diamines such as ethylenediamine, 1,3-diaminopropane, and 1,5-diaminopentane. The reaction proceeded efficiently in DMF at 50 °C over 7 hours, yielding the bis-tetrahydropyridine derivatives in 28–69% yields. Notably, the process follows a multicomponent cyclocondensation mechanism, where the intermediate 29a formed from formaldehyde and 1,3-diketones undergoes nucleophilic addition with imines and intramolecular cyclization (Scheme 9).
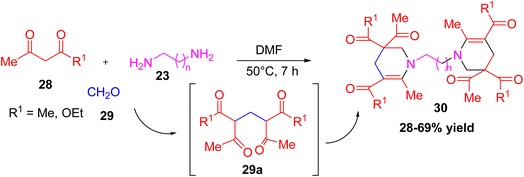 |
| | Scheme 9 Multicomponent preparation of polyfunctionalized tetrahydropyridine. | |
Govindaraju et al. reported the ultrasound-assisted, silica iodide (SiO2-I)-catalyzed one-pot four-component synthesis of novel 1,4,5,6-tetrahydropyridine-3-carboxylate derivatives 31, passes through an intermediate 7a.30 The reaction involved Meldrum's acid, substituted aryl aldehydes, aromatic amines, and ethyl acetoacetate, carried out under solvent-free conditions and ultrasonic irradiation. This green and efficient methodology afforded different products with excellent yields, notably ethyl-1-(4′-chlorophenyl)-2-methyl-6-oxo-4-(3′,4′,5′-trimethoxyphenyl)-1,4,5,6-tetrahydropyridine-3-carboxylate in 95% yield under optimized conditions. Compared to conventional methods, this approach offers a shorter reaction time, mild reaction conditions, reusability of the catalyst, and high product purity without the need for chromatographic purification (Scheme 10).
 |
| | Scheme 10 Ultrasound assisted formation of 1,4,5,6-tetrahydropyridine-3-carboxylate derivatives. | |
Vinoth and co-workers developed a green and efficient approach for the synthesis of tetrahydropyridine derivatives, specifically fused tetrahydropyridines like oxazolo[3,2-a]pyridines (Scheme 11).31 The method involves a three-component reaction of amino alcohols, 1,3-dicarbonyl compounds, and α,β-unsaturated aldehydes in water, without using any catalyst. This simple, one-pot procedure allows the construction of the tetrahydropyridine core in good yields through a sequence of enamine formation, Michael addition, and intramolecular cyclization steps (Scheme 12). The reaction showed excellent control over stereochemistry, mainly producing the trans isomers in most cases. It also followed principles of atom and step economy, as it generated only water as a by-product. Impressively, the process formed two fused rings and four new bonds, including one carbon–carbon, two carbon–nitrogen, and one carbon–oxygen all in a single step.
 |
| | Scheme 11 A catalyst free one-pot synthesis of tetrahydropyridine derivative. | |
 |
| | Scheme 12 Probable mechanism to explain synthesis of tetrahydropyridine derivative. | |
Wei et al. reported an efficient one-pot, three-component formation of 4-aryl-1,2,3,4-tetrahydropyridines 38 using Morita–Baylis–Hillman (MBH) carbonates 36, 1,3-ketoesters, and primary amines as starting materials.32 The reaction is catalyzed by DABCO, a Lewis base, and follows a formal [3 + 2 + 1] cyclization mechanism. This organocatalytic method provides a variety of tetrahydropyridine derivatives under mild conditions and in excellent yields. The reaction is compatible with various aromatic groups, amines, and esters, offering a simple and rapid route to construct complex molecular frameworks (Scheme 13).
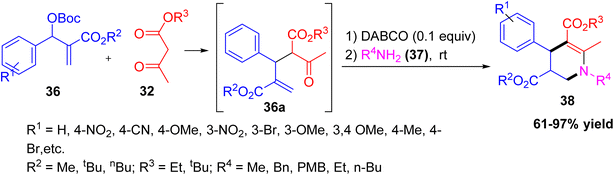 |
| | Scheme 13 Multicomponent synthesis of 4-aryl-1,2,3,4-tetrahydropyridines catalysed by DABCO. | |
Li et al. reported an easy and effective approach for the synthesis of multisubstituted tetrahydropyridine derivatives 41 using a photoinduced copper-catalyzed multicomponent reaction.33 The reaction uses readily available starting materials like alkyl amines, alkynes, and aldehydes and proceeds under mild conditions using visible light. The product was formed through an interesting [3 + 2 + 1] radical cyclization, which creates three carbon–carbon (C–C) bonds and one carbon–nitrogen (C–N) bond in a single step. The method shows excellent regio- and diastereoselectivity, and the reaction occurs through an α-aminoalkyl radical intermediate. This new approach offers a green and economical way to prepare complex bicyclic and spirocyclic tetrahydropyridines, which are important in pharmaceutical and chemical research (Scheme 14).
 |
| | Scheme 14 Photoinduced multicomponent synthesis of highly substituted tetrahydropyridine. | |
Rossetti et al. reported a simple one-pot three-component Gewald reaction to synthesize a series of novel chiral derivatives of 2-amino-4,5,6,7-tetrahydrothieno[2,3-c]pyridine, a type of fused tetrahydropyridine compound 45.34 The main starting materials used in the reaction were N-substituted piperidones, ethyl cyanoacetate, and sulfur, followed by reaction with 4-nitrobenzoyl chloride. The main aim was to introduce chiral amine substituents on the nitrogen atom of the tetrahydropyridine ring to study their impact on antimicrobial activity. The synthesized compounds were tested against two bacterial strains: Sarcina lutea (Gram-positive) and Escherichia coli (Gram-negative). The results showed that only Gram-positive bacteria were inhibited, and notably, the (R)-enantiomers had stronger activity than their (S)-counterparts. This shows the significance of chirality in biological activity and supports the potential of these tetrahydropyridine derivatives as new antibacterial agents (Scheme 15).
 |
| | Scheme 15 Three component synthesis of 2-amino-4,5,6,7-tetrahydrothieno[2,3-c]pyridine. | |
A novel and highly stereoselective multicomponent reaction (MCR) for the synthesis of penta substituted tetrahydropyridines was developed by Echemendia and co-workers.35 This one-pot, organocatalytic procedure involved the combination of benzoylacetonitrile, cinnamaldehyde, a primary or secondary amine, and an isocyanide, catalyzed by the Jørgensen–Hayashi catalyst and 3,5-dinitrobenzoic acid. The reaction proceeded under microwave irradiation in trifluoroethanol. This strategy also allowed the incorporation of peptide, sugar, and steroid moieties, resulting in complex chimeric tetrahydropyridine hybrids (Scheme 16).
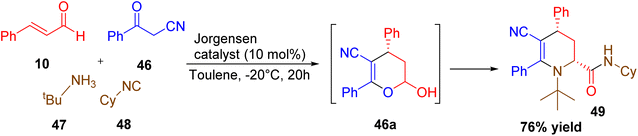 |
| | Scheme 16 Organocatalytic multicomponent synthesis of pentasubstitued tetrahydropyridine. | |
Tang and co-workers reported the diastereoselective synthesis of two fused tetrahydropyridine derivatives 51 and 52 via a one-pot multicomponent reaction.36 The synthesis was achieved by using cinnamaldehyde, o-aminophenol, with starting materials 32 and 50 in refluxing ethanol using acetic acid (AcOH) as a catalyst proceeds via an intermediate 32a and 50a respectively. The reaction proceeded efficiently to deliver the trans isomer in high yield and with excellent stereoselectivity (Scheme 17).
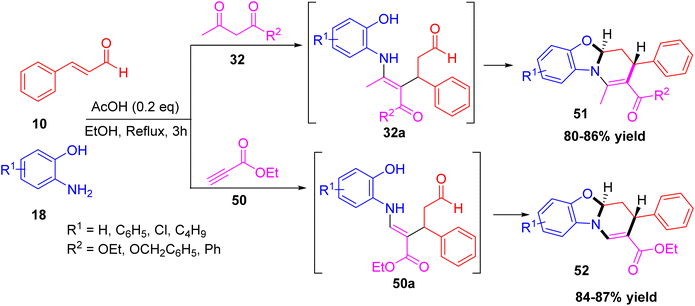 |
| | Scheme 17 Diastereoselective synthesis of ethyl 3-phenyl-4,4a-dihydro-3H-benzo[4,5]oxazolo[3,2-a]pyridine-2-carboxylate. | |
Zhu et al. developed a simple and effective method to prepare indol-3-yl-substituted tetrahydropyridines using a three-component reaction.37 In this reaction, 1-amino-3,3-diethoxypropane, diethyl acetylenedicarboxylate, and indole derivatives were combined in acetonitrile at 60 °C with Al(OTf)3 as the catalyst. The reaction proceeds through an intermediate 55a, which then cyclizes to form the tetrahydropyridine core. The process gave a wide range of products in moderate to good yields. Different indoles, including 5-methoxyindole, 6-nitroindole, 2-methylindole, and N-substituted indoles, worked well, showing the broad scope of the method. This one-pot reaction is attractive because it uses easily available starting materials and a low-cost catalyst to make biologically valuable tetrahydropyridine derivatives in a straightforward way (Scheme 18).
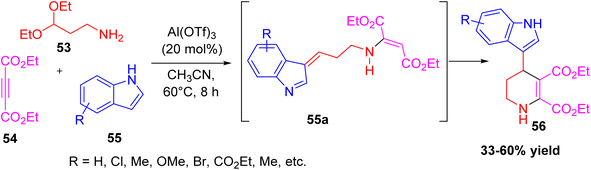 |
| | Scheme 18 Al(OTf)3 catalysed three component synthesis of indol-3-yl-substituted tetrahydropyridines | |
Ramaraju et al. reported an efficient enantioselective multicomponent synthesis of 1,2,5,6-tetrahydropyridines (THPs) through a one-pot domino sequence. The reaction involved the proline-catalyzed Mannich reaction and cyclization of glutaraldehyde with in situ generated imines, which were prepared from aromatic aldehydes and p-anisidine, followed by site-selective oxidation using IBX (2-iodoxybenzoic acid) and NaBH4 reduction under mild conditions.38 The reaction was carried out in DMSO as the solvent, giving chiral THPs in good yields (up to 80%) and excellent enantioselectivity. The method was also applied for the synthesis of several medicinally important nitrogen heterocycles, including hexahydrochromeno[4,3-b]pyridine, polyhydroxylated piperidines, guvacine, and nipecotic acid derivatives. This simple, environmentally friendly, and economical one-pot strategy demonstrates the efficiency of L-proline catalysis in constructing biologically significant tetrahydropyridine derivatives (Scheme 19).
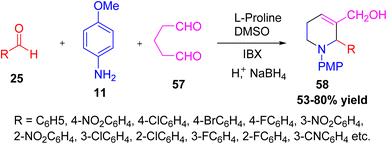 |
| | Scheme 19 Enantioselective formation of 1,2,5,6-tetrahydropyridines by L-proline catalyzed. | |
Khan et al. reported a green and efficient multicomponent synthesis of functionalized tetrahydropyridine (THPD) derivatives using lemon juice as a natural, biodegradable, and eco-friendly catalyst. The reaction involved β-ketoesters, aromatic aldehydes and its substituted derivatives, and aromatic amines in ethanol at room temperature. Using only 0.25 mL of lemon juice as a catalyst, the reaction proceeded via intermediate 32a to give the desired tetrahydropyridine products in good to excellent yields (76–86%) within a few hours.39a This green protocol eliminates the need for toxic reagents, harsh conditions, and column chromatography, offering a simple, cost-effective, and sustainable approach for synthesizing biologically significant tetrahydropyridine derivatives with high atom economy and operational ease (Scheme 20). Several methods have also been reported by L-proline,39b Ni(Salen),39c Fe3O4@S–TiO2,39d N-methyl pyridinium tosylate (NMPyTs) ionic liquid,39e ammonium trifluoroacetate,39f phosphomolybdic acid,39g nano-Fe3O4@walnut shell,39h, [Cu(2-pic)2]2H2O}n],39i etc.
 |
| | Scheme 20 Five-component synthesis of tetrahydropyridine using lemon juice as a green catalyst. | |
1.3. Synthesis of THP using enamine as amino source
Dhinakaran et al. developed an efficient and eco-friendly method for the multicomponent synthesis of ethyl 5-cyano-1-(4-ethylphenyl)-6-imino-4-(4-methoxybenzoyl)-1,4,5,6-tetrahydropyridine-3-carboxylate 63 (Scheme 21).40 The reaction proceeds via an intermediate 62a formed during the domino Knoevenagel condensation and Michael addition, which then undergoes intramolecular cyclization to give the tetrahydropyridine product. The process uses ethyl (E)-3-(arylamino) acrylates 62, 2,2-dihydroxy-1-arylethan-1-ones 60, and malononitrile in a single step under solvent-free and catalyst-free grinding conditions, offering high yields and excellent selectivity. Importantly, the selective formation of tetrahydropyridine depends on the presence of electron-donating groups on the N-aryl ring of the acrylate. This green synthesis protocol provides a practical and sustainable method for preparing bioactive tetrahydropyridine compounds.
 |
| | Scheme 21 A catalyst free three component synthesis of tetrahydropyridine-3-carboxylate. | |
Zhu et al. introduced a BTFPBA-catalyzed three-component reaction to make C6-(indol-3-yl)-substituted tetrahydropyridines 67.37 This reaction used ethyl 3-aminocrotonate 64, acrolein 65, and 2-methylindole derivatives 66 in dichloromethane at 40 °C, with 2,4-bis(trifluoromethyl)phenylboronic acid (BTFPBA) as the catalyst. The reaction proceeds via an intermediate 66a, which then cyclizes to give the desired products in moderate to good yields. It worked well with different indoles such as 5-methylindole, 5-methoxyindole, 2-phenylindole, and N-substituted indoles. This approach is notable because it provides an easy one-pot way to build diverse indole-tetrahydropyridine hybrids, which are valuable scaffolds in medicinal chemistry (Scheme 22).
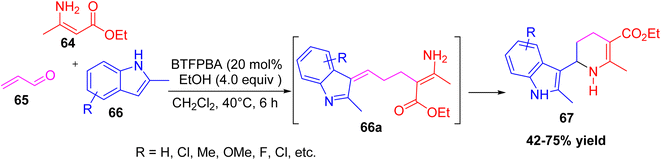 |
| | Scheme 22 One pot three component synthesis of indol-3-yl-substituted tetrahydropyridines using BTFPBA as a catalyst. | |
Darakshan and Parvin developed a simple and eco-friendly method to synthesize benzophenazine tethered tetrahydropyridopyrimidine derivatives using a one-pot four-component reaction (Scheme 23).41 The reaction involves cinnamaldehyde or crotonaldehyde 17, 2-hydroxy-1,4-naphthoquinone 68, 6-amino-1,3-dimethyl uracil 69, and o-phenylenediamine 70 in ethanol with p-toluenesulfonic acid (p-TSA) as a catalyst. The reaction proceeds via a Knoevenagel condensation, followed by nucleophilic attack, tautomerization, and intramolecular cyclization, ultimately leading to the selective formation of product 71 through sequential addition of the reactants (Scheme 24). The final products combine three bioactive structures: benzophenazine, tetrahydropyridine, and pyrimidine, which makes them promising for medicinal use. The advantages of this method include simple procedure, metal-free conditions, availability of reactants, and easy purification.
 |
| | Scheme 23 p-TSA catalysed synthesis of benzophenazine containing THPD via MCR. | |
 |
| | Scheme 24 Probable mechanism to explain synthesis of benzophenazine containing THPD via MCR. | |
The product 73 was successfully synthesized through a three-component reaction using cinnamaldehyde (α,β-unsaturated aldehyde), 2-hydroxy-1,4-naphthaquinone (a cyclic 1,3-dicarbonyl compound), and 1,3-dimethyl-6-aminouracil. Kumari et al. demonstrated a microwave-assisted, regioselective one-pot method in ethanol using FeCl3·6H2O as a green and inexpensive catalyst.42 The reaction proceeds via an intermediate 72a, which then cyclizes to give the desired product in high yield (up to 85%) under mild conditions within 30 minutes. The developed method is simple, environmentally friendly, and useful for synthesizing pyrimidine-fused tetrahydropyridine derivatives, which show biological and medicinal applications (Scheme 25).
 |
| | Scheme 25 Regioselective microwave irradiation multicomponent synthesis of tetrahydropyridine derivative. | |
1.4. Synthesis of THP using ammonia and ammonium acetate as amino source
Khumalo et al. presented a clean and eco-friendly method for synthesizing tetrahydropyridine-3-carboxamide derivatives through a microwave-assisted, catalyst-free one-pot reaction.43a The reaction involves a combination of aromatic aldehydes, ethyl cyanoacetate, acetoacetanilide, and ammonium acetate, using ethanol as a green solvent (Scheme 26). Under microwave irradiation, the products were formed rapidly within 10 minutes, with high yields ranging from 91% to 97%. This protocol avoids the use of toxic catalysts and lengthy purification, as the products can be easily isolated without column chromatography. The method also benefits from short reaction times, energy efficiency, and operational simplicity. Furthermore, another method using a CeO2/MWCNT nanocomposite catalyst, also reported for achieving the same product up to 97% yield in 15 minutes.43b
 |
| | Scheme 26 Microwave irradiation one-pot catalysed formation of tetrahydropyridine-3-carboxamide. | |
A novel and efficient four-component synthesis of 1,4,5,6-tetrahydropyridine derivatives was developed by Vereshchagin et al., involving benzylidenemalononitriles, aromatic aldehydes, esters of 3-oxocarboxylic acids, and ammonium acetate.44 The reaction proceeds through a cascade sequence passing through an intermediate 77a formed during the Michael addition and Mannich reaction, followed by cyclization and dehydration in methanol, to afford highly diastereoselective 2-substituted alkyl (4SR,6RS)-4,6-diaryl-5,5-dicyano-1,4,5,6-tetrahydropyridine-3-carboxylates 78. This method achieves excellent yields (66–92%) in a single step and eliminates the need for column chromatography. Ammonium acetate plays a dual role as a base and a nitrogen source (Scheme 27).
 |
| | Scheme 27 Multicomponent synthesis of 1,4,5,6-tetrahydropyridine derivatives by Mannich reaction. | |
Iliyasov et al. reported the stereoselective one-pot five-component synthesis of substituted 1,4,5,6-tetrahydropyridines, particularly 2-substituted alkyl (4SR,6RS)-4,6-diaryl-5,5-dicyano-1,4,5,6-tetrahydropyridine-3-carboxylates and 3,5-dialkyl (4RS,5SR,6RS)-5-cyano-2,4,6-triaryl-1,4,5,6-tetrahydropyridine-5,3-carboxylates (Scheme 28).45 The method involves a series of Knoevenagel condensation, Michael addition, Mannich reaction, cyclization, and dehydration using aromatic aldehydes, malononitrile or ethyl cyanoacetate, esters of 3-oxocarboxylic acids, and ammonium acetate as the key reactants (Scheme 29). Ammonium acetate plays a dual role as a base and a nitrogen source. The reaction proceeds under mild reflux conditions in methanol and leads to high yields (57–84%).
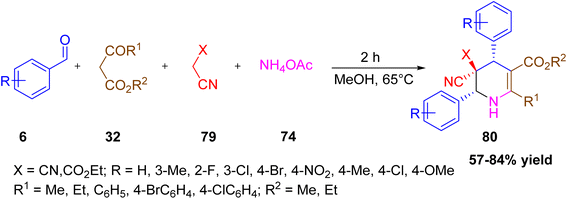 |
| | Scheme 28 Stereoselective five-component synthesis of highly substituted tetrahydropyridine. | |
 |
| | Scheme 29 Plausible mechanism to explain synthesis of highly substituted tetrahydropyridine. | |
Spirooxindole derivatives containing 2-piperidinone and 1,2,3,4-tetrahydropyridine46 rings were synthesized via a one-pot asymmetric multicomponent reaction developed by Huang et al. This four-component cascade proceeds through an intermediate 82a formed during the Michael/Mannich sequence, followed by cyclization, and is catalyzed by a recyclable fluorous Cinchona alkaloid/thiourea-based organocatalyst. The key reactants include electron-deficient olefinic oxindoles, diethyl malonate or 1,3-diketones, aromatic or heteroaromatic aldehydes, and ammonium acetate or amines. The optimized method yielded products with up to 99% enantiomeric excess and excellent diastereoselectivity. The catalyst could be easily recovered and reused without loss of efficiency. This environmentally friendly synthesis provides valuable spirooxindole–tetrahydropyridine hybrids, which are of great interest in drug discovery due to their structural similarity to biologically active molecules like MDM2 inhibitors (Scheme 30).
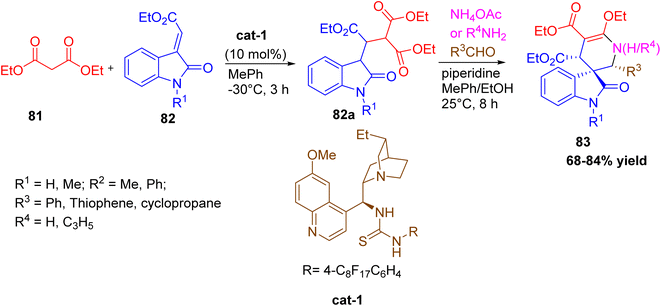 |
| | Scheme 30 Four component synthesis of 1,2,3,4-tetrahydropyridine derivative using organocatalyst. | |
Vereshchagin et al. reported a simple and efficient method for synthesizing (4RS,6SR)-4,6-diaryl-5,5-dicyano-2-methyl-1,4,5,6-tetrahydropyridine-3-carboxylates, a type of tetrahydropyridine derivative with a wide range of biological activity.47 Method is multicomponent reaction involving three key reactants: benzylidenemalononitrile, 2-acetyl-3-arylacrylate, and aqueous ammonia in methanol at room temperature. The reaction proceeds through an intermediate 85a, leading to high diastereoselectivity and yields ranging from 55% to 87%. Ammonia plays a dual role, acting as both a nitrogen source and a base. This method offers a mild, clean, and simple synthetic route to complex nitrogen-containing heterocycles (Scheme 31).
 |
| | Scheme 31 Diastereoselective multicomponent synthesis of 1,4,5,6-tetrahydropyridine-3-carboxylates. | |
1.5. Synthesis of THP using imine as amino source
Zhang et al. describe a very simple three-component reaction was developed using α,β-unsaturated N-arylaldimines, dialkyl acetylenedicarboxylates, and arylidenemalononitriles in dry acetonitrile at room temperature (Scheme 32).48 This reaction leads to the formation of 6-styryl-1,4,5,6-tetrahydropyridines with different cis/trans forms in good yields. Similar results were obtained when using other reactants like ethyl arylidenecyanoacetates or arylidene pivaloylacetonitriles. The reaction works through a domino [2 + 2 + 2] cycloaddition involving 1,4-dipolar intermediates (Scheme 33). These intermediates form when α,β-unsaturated imines react with electron-poor alkynes. This method allows easy access to functionalized tetrahydropyridines under mild conditions. It offers advantages such as simple setup, good yields, and broad substrate scope. The process is useful for making nitrogen-containing heterocycles and shows promise for applications in organic and medicinal chemistry.
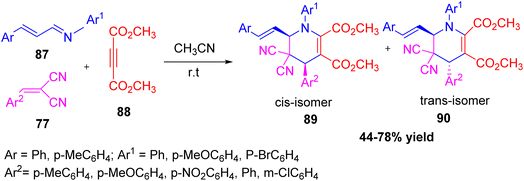 |
| | Scheme 32 Three component synthesis of 6-styryl-1,4,5,6-tetrahydropyridines via cycloaddition reaction. | |
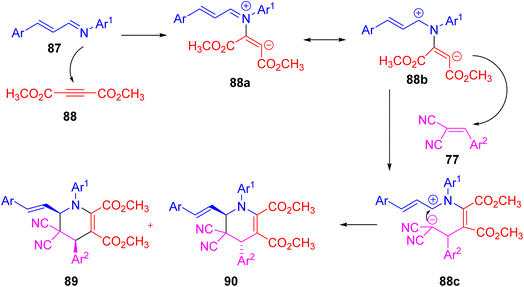 |
| | Scheme 33 Probable mechanism to represent the synthesis of 6-styryl-1,4,5,6-tetrahydropyridines. | |
Rai et al. reported a novel one-pot three-component synthesis of highly substituted tetrahydropyridines (THPs) using a Morita–Baylis–Hillman (MBH) enal-based triple cascade strategy (Scheme 34).49 The reaction involves [E]-α-cyano/nitro cinnamaldehyde, malononitrile (an active methylene compound), and aldimines or phenyl-N-tosyl-methanimines as reactants. This method uses iminium-enamine catalysis facilitated by a secondary amine catalyst and proceeds through a sequential cascade forming C–C and C–N bonds in one pot (Scheme 35). The reaction provides the THP products in high yields (83–94%) and with excellent diastereoselectivity (95–99%) favouring the anti-isomer. This method is a useful and selective way to make complex THP compounds and shows the role of MBH adducts in making complicated heterocycles.
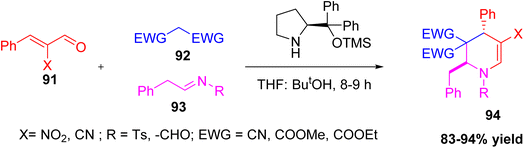 |
| | Scheme 34 Three component synthesis of tetrahydropyridine derivative via cascade reaction. | |
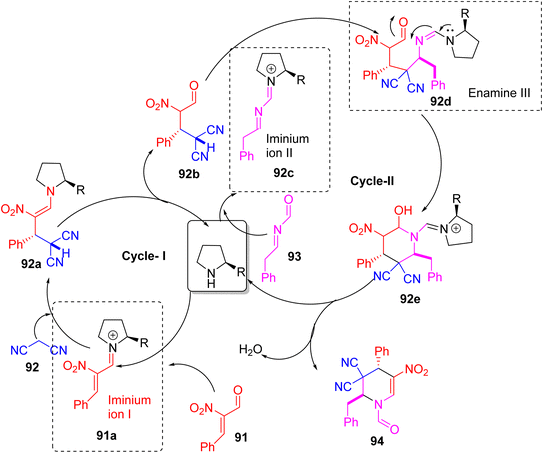 |
| | Scheme 35 Mechanistic way to show the formation of highly substituted tetrahydropyridines | |
Mehrabi and Mohebbi reported a simple and efficient one-pot, two-step synthesis of highly substituted tetrahydropyridines.50 The method involves the initial formation of N-benzylidenemethanamines from benzylamines and benzaldehydes in the presence of acetic acid, followed by cyclization with dialkyl acetylenedicarboxylates and benzylidenemalononitriles in acetonitrile at 50 °C under catalyst-free condition. This multicomponent process efficiently yields products like dimethyl 1-benzyl-5,5-dicyano-1,4,5,6-tetrahydro-4,6-diphenylpyridine-2,3-dicarboxylate in good yields (up to 85%). This method is easy to use, uses inexpensive starting materials, works well under mild conditions, and gives good yields (Scheme 36).
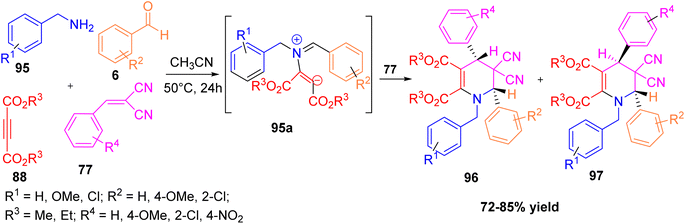 |
| | Scheme 36 One-pot synthesis of highly substituted tetrahydropyridine without using catalyst. | |
1.6. Synthesis of THP using miscellaneous reactions
Zangouei et al. developed a new and efficient one-pot, four-component method for the synthesis of alkyl 6-amino-5-cyano-1-(4,6-dimethylpyrimidin-2-yl)-2-oxo-4-phenyl-1,2,3,4-tetrahydropyridine-3-carboxylates 99 (Scheme 37).51 The reaction combines aromatic aldehydes 6, malononitrile 61, 6,8-dimethyl-2-hydroxy-4H-pyrimido[1,2-a]pyrimidine-4-one 98, and alcohols in the presence of diisopropylethylamine (DIPEA) under reflux (Scheme 38). This unexpected transformation produced the target compounds 99 with excellent diastereoselectivity and in high yields ranging from 85% to 95%. The stereochemistry of the products was confirmed by single-crystal X-ray analysis. The advantages of this method include mild reaction conditions, short reaction times, and simple purification. Optimization experiments showed that ethanol and DIPEA were essential for achieving good results, while other solvents or solvent-free systems failed to produce the desired product. The mechanism of product formation is given in Scheme 38, which revealed that the key intermediate 98a obtained by reaction of alcohol with compound 98.
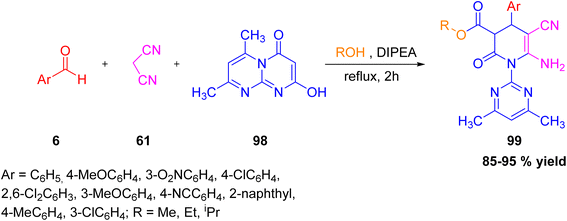 |
| | Scheme 37 Four-component synthesis of tetrahydropyridine derivative using DIPEA as a catalyst. | |
 |
| | Scheme 38 Mechanism proposed to explain the synthesis of tetrahydropyridine derivative via MCR. | |
A simple and efficient multicomponent method was developed for synthesizing tetrahydropyridine derivatives through a domino ring-opening and cyclization reaction.52 Kermani used aziridines, isocyanides, and malononitrile as reactants, with tetrabutylphosphonium acetate (TBPAc) as the catalyst in hexafluoroisopropanol (HFIP) under mild conditions. The reaction proceeds via an intermediate 101a, which subsequently undergoes cyclization to afford products 102 in good to excellent yields with high atom economy and regioselectivity. This approach allows rapid construction of structurally diverse tetrahydropyridines, which are known for their biological and pharmaceutical relevance. This approach demonstrates the effectiveness of TBPAc as an organocatalyst and provides a useful strategy for building nitrogen-containing six-membered heterocycles (Scheme 39).
 |
| | Scheme 39 Multicomponent synthesis of diverse tetrahydropyridine using TBPAc catalyst through domino ring opening. | |
A novel and efficient one-pot multicomponent synthesis of tetrahydropyridine phosphonate derivatives has been reported using diethylphosphoramidate, aromatic aldehydes, and ethyl acetoacetate as starting materials in the presence of ceric ammonium nitrate (CAN) as a catalyst in acetonitrile at room temperature.53 The reaction passes via an intermediate 104a to afford the tetrahydropyridine products under mild conditions with good yields and high atom economy. The main product synthesized was ethyl 1-(diethoxyphosphoryl)-4-((diethoxyphosphoryl)amino)-2,6-diaryl-1,2,5,6-tetrahydropyridine-3-carboxylate. Molecular docking studies were carried out for the synthesized tetrahydropyridine phosphonate derivatives to evaluate their interaction with the α-glucosidase enzyme, a key target in the treatment of type II diabetes mellitus. Since this enzyme plays a crucial role in carbohydrate digestion, its inhibition can help regulate blood sugar levels. The docking analysis helped to understand the probable binding mechanisms between the synthesized compounds and the enzyme's active site. Reddy et al., demonstrated an environmentally friendly and effective approach for the synthesis of bioactive tetrahydropyridines (Scheme 40).
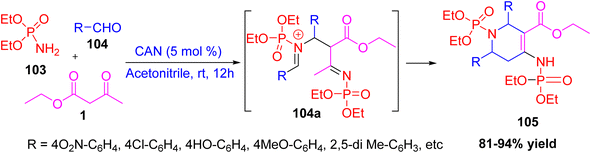 |
| | Scheme 40 Multicomponent synthesis of tetrahydropyridine phosphonate derivatives using CAN as a catalyst. | |
Kang et al. reported a simple one-pot method to make 3-(halomethyl)-4-aryl-5-(aryl or alkylsulfonyl)-1-tosyl-1,2,3,6-tetrahydropyridine derivatives 109 using a three-component reaction.54 The starting materials were N-centered 1,6-enynes, sodium sulfinates, and tetrabutylammonium halides 108 (TBAX). In the presence of a copper catalyst, sulfonyl radicals are generated, which add to the triple bond to form an intermediate 108a that subsequently undergoes 6-endo-dig cyclization and halogenation to afford the desired products. This method works well with many types of reactants, including aromatic and aliphatic ones, and gives good yields (Scheme 41).
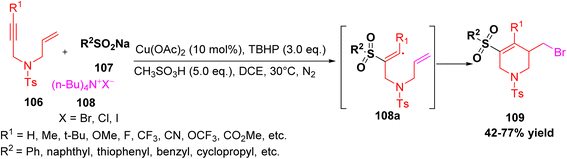 |
| | Scheme 41 Three-component synthesis of tetrahydropyridine derivative via ring formation. | |
Zhang et al. developed a new and easy one-pot method to make useful fluorinated compounds called mono fluorocyclohexenes and 6-fluoro-1,2,3,4-tetrahydropyridines 113 which are important building blocks in synthetic chemistry and the pharmaceutical industry.55 They used three starting materials: α-CF3 alkenes, electron-rich alkenes and dimethyl esters or sulfonamides. These react together in the presence of light and a base to break two C–F bonds and form a ring structure. When dimethyl malonate is used, the product is monofluorocyclohexene. When sulfonamides are used, the product is 6-fluoro-1,2,3,4-tetrahydropyridine. These functional products can be used for synthetic applications such as decarboxylation, cyclization, reduction, and oxidation (Scheme 42).
 |
| | Scheme 42 Photoinduced multicomponent synthesis of 6-fluoro-1,2,3,4-tetrahydropyridines | |
Ershov et al. developed a simple and efficient multicomponent method to synthesize 6-hydroxy-2-chloro-1,4,5,6-tetrahydropyridine-3,4,4-tricarbonitrile derivatives 116 through a three-component reaction involving tetracyanoethylene, various ketones, and hydrogen chloride (HCl) in 1,4-dioxane at 30–40 °C.56 The reaction follows a domino mechanism, where an intermediate 115a is formed and then converted to the final product. The yields of the final compounds ranged from 43% to 72%. These tetrahydropyridine derivatives are significant because they are part of many biologically active compounds (Scheme 43).
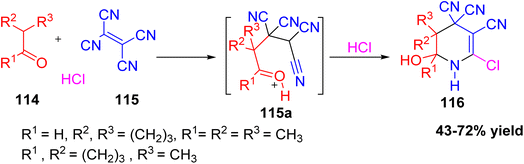 |
| | Scheme 43 One-pot synthesis of 6-hydroxy-2-chloro-1,4,5,6-tetrahydropyridine-3,4,4-tricarbonitrile derivatives. | |
An and Wu reported a mild and catalyst-free multicomponent method to make sulfonated tetrahydropyridine derivatives 120.57 The reaction uses 1,6-enynes, sulfur dioxide (from DABCO·(SO2)2), and aryldiazonium tetrafluoroborates as the main starting materials (Scheme 44). It works through a radical cyclization process started by arylsulfonyl radicals passing through an intermediate 119a formed during the reaction. Two molecules of the aryldiazonium salt are involved in this transformation. The best results were obtained by heating the mixture in dichloroethane at 50 °C under nitrogen, without any catalyst, giving moderate to good yields. This metal-free method is an efficient way to make functional tetrahydropyridine compounds, which are useful in medicinal and synthetic chemistry.
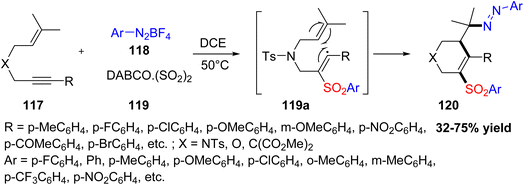 |
| | Scheme 44 Multicomponent synthesis of sulfonated tetrahydropyridine derivatives via cyclization process. | |
Rigi and Shaterian reported a straightforward, green four-component synthesis of 1,6-diamino-2-oxo-1,2,3,4-tetrahydropyridine-3,5-dicarbonitriles 123 by reacting hydrazine hydrate, malononitrile, ethyl cyanoacetate, and ketones at 80 °C under solvent-free conditions (Scheme 45).58a The reaction proceeds via an intermediate 122a, which subsequently undergoes cyclization to afford the desired tetrahydropyridine products. Using a magnetic nanoparticle-supported ionic liquid catalyst (MNPs@DABCO·Cl) enables easy product separation with an external magnet, high isolated yields, and catalyst reuse for at least four cycles without loss of activity. Overall, this work gives a high yield, short reaction time, efficient, and recyclable protocol for making tetrahydropyridine derivatives in an environmentally friendly manner. Some other methods, such as ultrasonication and the use of 4-(dimethylamino)pyridine (DMAP) and nano-Fe2O3 as catalysts, have also been reported, offering advantages such as high yields, a broader substrate scope, and short reaction times.58b–d
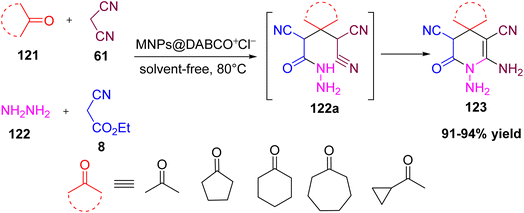 |
| | Scheme 45 A four-component synthesis of 1,6-diamino-2-oxo-1,2,3,4-tetrahydropyridine-3,5-dicarbonitriles. | |
Nazeri et al. reported an environmentally friendly and simple one-pot multicomponent approach has been developed for producing pyrazolo-tetrahydropyridine derivatives 128 through a chemo- and diastereoselective intramolecular Aza–Diels–Alder reaction (ADAR). In this approach, four key starting materials; benzoylacetonitrile derivatives, phenylhydrazine, salicylaldehyde derivatives, and styrenesulfonyl or cinnamoyl chloride 126 react in water as an environmentally friendly solvent (Scheme 46).59 The reaction proceeds smoothly with good yields and avoids traditional purification steps such as recrystallization and column chromatography methods by employing the Group-Assisted Purification (GAP) chemistry technique. This approach enables the quick formation of complex heterocyclic structures that incorporate pharmacologically significant components.
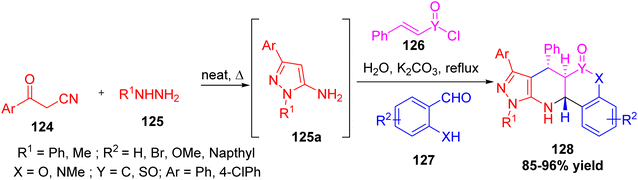 |
| | Scheme 46 One-pot synthesis of pyrazolo-tetrahydropyridines derivatives via Aza–Diels–Alder reaction. | |
Ravindernath and Reddy reported an efficient one-pot multicomponent synthesis of benzo[d]imidazolyl tetrahydropyridine carboxylates using (E)-5-(benzylidene amino)-1H-benzo[d]imidazole-2-thiol, 5-amino-2-mercaptobenzimidazole, aromatic aldehydes, and ethyl acetoacetate as reactants in acetonitrile solvent with ceric ammonium nitrate (CAN) as a Lewis acid catalyst.60 The reaction proceeds smoothly at room temperature to afford the desired tetrahydropyridine derivatives in good yields. The synthesized compounds were evaluated for anti-inflammatory, antioxidant, antibacterial, and antifungal activities. Most of the synthesized derivatives showed significant biological properties, with several compounds exhibiting comparable or better activity than standard drugs such as diclofenac sodium, ciprofloxacin, and fluconazole. This study highlights that combining benzimidazole and tetrahydropyridine frameworks within a single molecule enhances pharmacological potential. Overall, this multicomponent approach offers a simple, economical, and environmentally friendly route to biologically active benzo[d]imidazolyl tetrahydropyridine carboxylates (Scheme 47).
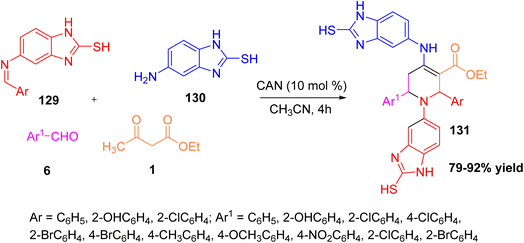 |
| | Scheme 47 Formation of benzo[d]imidazolyl tetrahydropyridine carboxylates using CAN as a catalyst. | |
Gibadullina et al. reported an efficient one-pot multicomponent synthesis of difluoromethyl-containing 1,2,3,4-tetrahydropyridine derivatives. The reaction involved ethyl 4,4-difluoroacetoacetate, formaldehyde, and various primary amine hydrochlorides or α-amino acid ester hydrochlorides as reactants.61 The process proceeds through an intermediate 133a formed during the Mannich-type condensation, which subsequently cyclizes to afford the tetrahydropyridine framework. The optimized conditions employed an acetate buffer (pH 4) as the reaction medium in the presence of sodium chloride (NaCl) as an electrolyte additive, which significantly enhanced the selectivity and yield of the desired products. Under these conditions, diethyl 3-(difluoroacetyl)-6-(difluoromethyl)-1,2,3,4-tetrahydropyridine-3,5-dicarboxylate was obtained in good to excellent yield (Scheme 48).
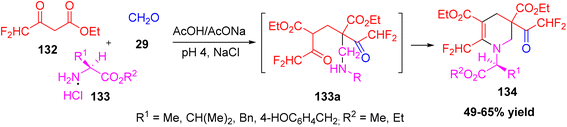 |
| | Scheme 48 One pot synthesis of 1,2,3,4-tetrahydropyridine-3,5-dicarboxylate. | |
2 Conclusion
In conclusion, multicomponent reactions (MCRs) have proven to be a powerful, efficient, and eco-friendly approach for the synthesis of tetrahydropyridine (THP) derivatives. Over the past decade, various MCR strategies using different nitrogen sources such as amines, enamines, imines, ammonia, and ammonium acetate, etc. have enabled the rapid and diverse construction of THP frameworks with wide biological and pharmaceutical importance. These methods often proceed under mild, solvent-free, or green conditions, with high yields and excellent selectivity. The development of catalyst-free, microwave-assisted, photoinduced, and nanocatalytic systems further highlights the progress toward sustainable and atom-economical chemistry. Overall, the advances summarized in this review show that MCR-based synthesis provides a simple, versatile, and environmentally responsible route for designing structurally complex THP derivatives, paving the way for their future applications in medicinal and organic chemistry.
Conflicts of interest
There are no conflicts to declare.
Data availability
This is a review article, therefore no additional data is available.
References
- P. Vinoth, P. S. R. Prasad, T. Vivekanand, C. U. Maheswari, S. Nagarajan, J. C. Menéndez and V. Sridharan, RSC Adv., 2015, 5, 81881–81888 RSC.
- C. Weber and T. Opatz, Alkaloids, 2019, 81, 1–114 CAS.
- L. Dai, M. R. Jacob, S. I. Khan, I. A. Khan, A. M. Clark and X.-C. Li, J. Nat. Prod., 2011, 74, 2023–2026 CrossRef CAS PubMed.
- C. E. Turner, M. F. H Hsu, J. E Knapp, P. L S. Jr and D. J. Slatkin, J. Pharm. Sci., 1976, 65, 1084–1085 CrossRef CAS PubMed.
- F.-X. Felpin and J. Lebreton, Curr. Org. Synth., 2004, 1, 83–109 CrossRef CAS.
- N. A. Mohsin and M. Ahmad, Turk. J. Chem., 2018, 42, 1191–1216 CrossRef CAS.
- G. Aridoss, L. Amirthaganesan, K. Senthamaraikannan, Y. T. Jeong, A. Kumar and A. Nanjundan, Bioorg. Med. Chem. Lett., 2008, 18, 6542–6548 CrossRef CAS PubMed.
- H. Bin, A. M. Crider and J. P. Stables, Eur. J. Med. Chem., 2001, 36, 265–286 CrossRef PubMed.
- R. Romagnoli, P. G. Baraldi, M. D. Carrion, O. Cruz-Lopez, C. Lopez Cara, M. Tolomeo, S. Grimaudo, A. Di Cristina, M. R. Pipitone, J. Balzarini, S. Kandil, A. Brancale, T. Sarkar and E. Hamel, Bioorg. Med. Chem. Lett., 2008, 18, 5041–5045 CrossRef CAS PubMed; M. Misra, S. K. Pandey, V. P. Pandey, J. Pandey, R. Tripathi and R. P. Tripathi, Bioorg. Med. Chem., 2009, 17, 625–633 CrossRef PubMed.
- S. A. Khanum, V. Girish, S. S. Suparshwa and N. F. Khanum, Bioorg. Med. Chem. Lett., 2009, 19, 1887–1891 CrossRef CAS PubMed.
- M. Misra, S. K. Pandey, V. P. Pandey, J. Pandey, R. Tripathi and R. P. Tripathi, Bioorg. Med. Chem., 2009, 17, 625–633 CrossRef CAS PubMed.
- Y. Fang, S. Zhang, W. Wu, Y. Liu, J. Yang, Y. Li, M. Li, H. Dong, Y. Jin, R. Liu and Z. Yang, Bioorg. Med. Chem., 2020, 30, 103390 CrossRef PubMed.
- P. R. Kharade, U. B. Chougale, D. S. Gaikwad, S. S. Kadam and S. S. Rathod, Res. Chem. Intermed., 2024, 50, 1777–1808 CrossRef CAS.
- Y. J. Zhu, X. F. Guo, Z. J. Fan, L. Chen, L. Y. Ma, H. X. Wang, Y. Wei, X. M. Xu, J. P. Lin and V. A. Bakulev, RSC Adv., 2016, 6, 112704–112711 RSC.
- R. J. Conway, C. Valant, A. Christopoulos, A. D. Robertson, B. Capuano and I. T. Crosby, Bioorg. Med. Chem. Lett., 2012, 22, 2560–2564 CrossRef CAS PubMed.
- M. Huang, D.-H. Suk, N.-C. Cho, D. Bhattarai, S. B. Kang, Y. Kim, A. N. Pae, H. Rhim and G. Keum, Bioorg. Med. Chem. Lett., 2015, 25, 1546–1551 CrossRef CAS PubMed.
- P. H. Olesen, M. D. B. Swedberg and K. Rimvall, Bioorg. Med. Chem., 1998, 6, 1623–1629 CrossRef CAS.
- O. U. R. Khan, B. A. Khan, S. S. Hamdani, S. Jalil, P. A. Sidhom, K. E. Ibrahim, T. Abalkhail, J. Iqbal, H. Tallima, T. Shoeib and M. A. A. Ibrahim, ChemistryOpen, 2025, 14, e202400516 Search PubMed.
- H. J. Wang, L. P. Mo and Z. H. Zhang, ACS Comb. Sci., 2011, 13, 181–185 CrossRef CAS PubMed.
- M. M. Khan, S. Khan, Saigal and S. Iqbal, RSC Adv., 2016, 6, 42045–42061 Search PubMed.
- E. R. Ishmiyarov, N. T. Rakhimova, D. R. Latypova, L. V. Spirikhin, M. I. Abdullin and V. A. Dokichev, Russ. J. Org. Chem., 2015, 51, 1770–1773 CrossRef CAS.
- N. Fathima, Mater. Today: Proc., 2021, 45, 4063–4066 CAS.
- J. Yang, B. Liu and J. Chang, Org. Lett., 2023, 25, 1476–1480 CrossRef CAS PubMed.
- N. Zanatta, M. M. Lobo, J. M. dos Santos, L. de A. Souza, V. P. de Andrade, H. G. Bonacorso, M. A. P. Martins and J. Heterocyclic, Chem, 2015, 52, 1776–1781 CAS.
- V. P. Andrade, M. Mittersteiner, M. M. Lobo, C. P. Frizzo, H. G. Bonacorso, M. A. P. Martins and N. Zanatta, Tetrahedron Lett., 2018, 59, 891–894 CrossRef CAS.
- Y. Dudognon, H. Du, J. Rodriguez, X. Bugaut and T. Constantieux, Chem. Commun., 2015, 51, 1980–1982 RSC.
- J. P. Wan, S. Zhong and Y. Liu, Synth., 2015, 47, 3611–3617 CrossRef CAS.
- A. A. Mohammadi, S. Taheri, A. Amouzegar, R. Ahdenov, M. R. Halvagar and A. S. Sadr, J. Mol. Struct., 2017, 1139, 166–174 CrossRef CAS.
- N. N. Gibadullina, D. R. Latypova, T. R. Nugumanov, L. V. Spirikhin and V. A. Dokichev, Chem. Heterocycl. Compd., 2017, 53, 1098–1102 CrossRef CAS.
- S. Govindaraju, S. Tabassum and M. A. Pasha, Phosphorus, Sulfur Silicon Relat. Elem., 2017, 192, 292–299 Search PubMed.
- P. Vinoth, P. S. R. Prasad, T. Vivekanand, C. U. Maheswari, S. Nagarajan, J. C. Menéndez and V. Sridharan, RSC Adv., 2015, 5, 81881–81888 Search PubMed.
- J. Wei, Y. Li, C. Tao, H. Wang, B. Cheng, H. Zhai and Y. Li, J. Org. Chem., 2018, 83, 835–842 CrossRef CAS.
- S. Li, J. Li, H. Zhang, G. Zhang and R. Guo, Org. Lett., 2025, 27, 5057–5062 CrossRef CAS.
- A. Rossetti, N. Bono, G. Candiani, F. Meneghetti, G. Roda and A. Sacchetti, Chem. Biodiversity, 2019, 16, e1900097 CrossRef.
- R. Echemendia, G. P. da Silva, M. Y. Kawamura, A. F. de la Torre, A. G. Correa, M. A. B. Ferreira, D. G. Rivera and M. W. Paixao, Chem. Commun., 2018, 54, 6412–6415 RSC.
- Y. Tang, Z. S. Ma, R. Wang, W. Zhang, Z. H. Qi, Y. H. Yu and H. B. Tan, J. Mol. Struct., 2022, 1251, 131935 CrossRef CAS.
- H. Zhu, L. Chen, R. Bai and Y. Gu, Adv. Synth. Catal., 2022, 364, 2883–2888 Search PubMed.
- P. Ramaraju, N. A. Mir, D. Singh and I. Kumar, RSC Adv., 2016, 6, 60422–60432 RSC.
-
(a) M. M. Khan, S. Khan and S. C. Sahoo, ChemistrySelect, 2018, 3, 1371–1380 CrossRef CAS;
(b) R. M. Muhiebes, L. Fatolahi and S. Sajjadifar, Asian J. Green Chem., 2023, 7, 121–131 CAS;
(c) A. N. Yankin and M. V. Dmitriev, Synth. Commun., 2020, 50, 3481–3489 Search PubMed;
(d) Z. Nezami and H. Eshghi, J. Iran. Chem. Soc., 2021, 18, 1997–2008 Search PubMed;
(e) A. V. Sapkal, J. M. Dinore, A. A. Yelwande, M. P. Palve and B. R. Madje, Polycyclic Aromat. Compd., 2024, 44, 3964–3974 CrossRef CAS;
(f) R. Duraisamy, P. Nagaraja, A. Nizam, N. V. Katharigatta, H. Nagarajaiah, M. B. M. Reddy and D. Thirumalai, ChemistrySelect, 2023, 8, e202302533 CrossRef CAS;
(g) V. Palermo, J.-C. Castillo, M. A. Macías, J. J. Martínez, P. Langer and G. P. Romanelli, ChemistrySelect, 2024, 9, e202403739 Search PubMed;
(h) M. A. K. Zarchi, K. Behboodi, B. B. F. Mirjalili and A. Bamoniri, J. Polym. Res., 2022, 29, 260 CrossRef CAS;
(i) M. Aier, F. R. Gayen and A. Puzari, J. Organomet. Chem., 2023, 993, 122712 CrossRef CAS.
- I. Dhinakaran, V. Padmini and N. Bhuvanesh, ACS Comb. Sci., 2016, 18, 236–242 Search PubMed.
- Darakshan and T. Parvin, Mol. Diversity, 2023, 27, 313–322 CrossRef CAS.
- P. Kumari, R. Yadav, R. Bharti and T. Parvin, Mol. Diversity, 2020, 24, 107–117 CrossRef CAS PubMed.
-
(a) M. Khumalo, S. N. Maddila, S. Maddila and S. B. Jonnalagadda, J. Chem. Sci., 2020, 132, 36 CrossRef;
(b) S. Harikrishna, A. R. Robert, H. Ganja, S. Maddila and S. B. Jonnalagadda, Appl. Organomet. Chem., 2020, 34, e5796 CrossRef CAS.
- A. N. Vereshchagin, T. M. Iliyasov, K. A. Karpenko, V. A. Smirnov, I. E. Ushakov and M. N. Elinson, Chem. Heterocycl. Compd., 2021, 57, 929–933 CrossRef CAS.
- T. M. Iliyasov, K. A. Karpenko, A. S. Akulinin, A. N. Fakhrutdinov, V. A. Korolev, M. N. Elinson and A. N. Vereshchagin, Arkivoc, 2022, 232–244 Search PubMed.
- X. Huang, K. Pham, W. Yi, X. Zhang, C. Clamens, J. H. Hyatt, J. P. Jasinsk, U. Tayvah and W. Zhang, Adv. Synth. Catal., 2015, 357, 3820–3824 CrossRef CAS.
- A. N. Vereshchagin, K. A. Karpenko, T. M. Iliyasov, M. N. Elinson, E. O. Dorofeeva, A. N. Fakhrutdinov and M. P. Egorov, Russ. Chem. Bull., 2018, 67, 2049–2053 CrossRef CAS.
- J. Zhang, W. J. Yang, J. Sun and C. G. Yan, Eur. J. Org Chem., 2015, 34, 7571–7582 CrossRef.
- V. K. Rai, F. Verma, M. Satnami, M. Singh and A. Rai, Tetrahedron Lett., 2018, 59, 1783–1786 CrossRef CAS.
- H. Mehrabi and A. Mohebbi, Arkivoc, 2016, 89–100 Search PubMed.
- M. Zangouei, A. A. Esmaeili and J. T. Mague, Synlett, 2016, 27, 1669–1673 CrossRef CAS.
- A. S. Kermani, Monatshefte fur Cheme, 2019, 150, 1495–1501 CrossRef.
- K. M. K. Reddy, K. Peddanna, M. Varalakshmi, N. B. Reddy, G. Sravya, G. V. Zyryanov and C. S. Reddy, Phosphorus, Sulfur Silicon Relat. Elem., 2019, 194, 812–819 CrossRef.
- T. M. Kang, Y. W. Wu, W. S. Zheng, X. H. Zhang and X. G. Zhang, Tetrahedron, 2023, 135, 133339 CrossRef CAS.
- S. Zhang, M. Gan, F. Zhang, L. Zhou and W. Li, Org. Lett., 2025, 27, 3013–3018 CrossRef CAS PubMed.
- O. V. Ershov, K. V. Lipin, A. V. Eremkin, O. E. Nasakin, V. P. Sheverdov, P. I. Fedorov and V. A. Tafeenko, Russ. J. Org. Chem., 2017, 53, 215–221 CrossRef CAS.
- Y. An and J. Wu, Org. Lett., 2017, 19, 6028–6031 CrossRef CAS PubMed.
-
(a) F. Rigi and H. R. Shaterian, J. Chin. Chem. Soc., 2016, 63, 557–561 Search PubMed;
(b) M. Shokoohian, N. Hazeri, M. T. Maghsoodlou and M. Lashkari, Chem. J. Mold., 2019, 14, 97–104 Search PubMed;
(c) J. Yang, Q. Li, J. Zhang, W. Lin, J. Wang, Y. Wang, Z. Huang and D. Shi, Molecules, 2013, 18, 14519–14528 CrossRef PubMed;
(d) F. Rigi and H. R. Shaterian, Polycyclic Aromat. Compd., 2017, 37, 314–326 CrossRef CAS.
- M. T. Nazeri, S. Javanbakht, A. Shaabani and H. R. Khavasi, ChemistrySelect, 2019, 4, 14271–14275 CrossRef CAS.
- A. Ravindernath and M. S. Reddy, Arab. J. Chem., 2017, 10, 1172–1179 CrossRef.
- N. N. Gibadullina, D. R. Kireeva, A. N. Lobov and V. A. Dokichev, J. Fluorine Chem., 2023, 266, 110088 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Faiz Ahmad Faiz
and
Mohd Sufiyan Khan
*,
Faiz Ahmad Faiz
and
Mohd Sufiyan Khan




















































