
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Defining alternating sequences in polyurethanes: sequence-controlled photo-degradation in step polymerization
Xaver Kneidla,
Johannes Reeb-Begica,
Tongtong Cuia,
Patrick Theato ab and
Yosuke Akae†‡
*ac
ab and
Yosuke Akae†‡
*ac
aInstitute for Chemical Technology and Polymer Chemistry (ITCP), Karlsruhe Institute of Technology (KIT), 76131 Karlsruhe, Germany. E-mail: yosuke.akae@ac.rwth-aachen.de
bSoft Matter Synthesis Laboratory – Institute for Biological Interfaces III (IBG-3), Karlsruhe Institute of Technology (KIT), 76344 Eggenstein-Leopoldshafen, Germany
cResearch Fellow of Japan Society for the Promotion of Science, 102-0083 Tokyo, Japan
First published on 15th May 2026
Abstract
Programmable degradation is an important functionality for sustainable polymer materials; however, in conventional step polymerization of AA- and BB-type monomers, the polymer sequence itself has long lacked a clear and actionable definition. Polymers synthesized from AA- and BB-type monomers enable the incorporation of functional units directly into the polymer main chain, while inevitably featuring AA–BB connectivity; however, the concept of an “alternating” sequence has remained ambiguous. To address this fundamental limitation, we redefine sequence control in step polymerization by focusing on the connectivity unit rather than monomer composition. Using polyurethane as a representative model polymer, an AB-type monomer framework combined with dimeric species enables the explicit definition and practical implementation of (partially) alternating sequences. Incorporation of a photo-degradable monomer unit directly into the polymer backbone reveals pronounced sequence-dependent photo-degradability via main chain scission, which is drastically enhanced in alternating polymers, while thermal properties are likewise strongly influenced by polymer sequence. This work establishes the polymer sequence as an additional and actionable design parameter in polyurethanes as an example of step polymerization and provides a conceptual framework potentially applicable beyond polyurethanes to a broad range of AA/BB-based polymers. The utility of this framework for functional material development is demonstrated through sequence-controlled photo-degradation on the polymer main chain, offering a promising strategy toward sustainable polymer design.
Introduction
Controlling the polymer sequence is a fundamental challenge in polymer synthesis, essential both for understanding polymer primary structures and for enabling precise material design through tuning of properties such as degradability, recyclability, and mechanical performance.1–30 Accordingly, extensive efforts have been devoted to achieving increasingly sophisticated sequence control across diverse polymer classes. However, these developments have been concentrated on chain polymerization processes, such as vinyl polymerization, where the concept of sequence is inherently well-defined, and functional units are typically incorporated into polymer side chains.In contrast, step polymerization—despite its central importance in both academic and industrial polymer chemistry—has received limited attention from the perspective of sequence definition and control. Yet, many technologically relevant polymers, including polyesters, polyamides, polycarbonates, and polyurethanes (PUs), are synthesized via step-growth mechanisms. In these systems, typical polymers prepared from AA- and BB-type monomers inevitably exhibit AA–BB connectivity along the backbone. As a result, the concept of polymer sequence itself—and in particular, the meaning of an “alternating” sequence—remains ambiguous. This conceptual ambiguity has hindered systematic investigations of sequence–property relationships in polymers obtained by step polymerization, in sharp contrast to the well-established framework available for chain polymerization. While various advanced polymerization methods based on step polymerization have been developed, these approaches have largely focused on molecular weight control, leaving the polymer sequence inherently ambiguous.31–37 Because functional units are incorporated into the polymer main chain, sequence control in step polymerization provides a powerful tool for designing backbone-functional materials such as degradable polymers.
PU serves as a representative and industrially significant example of this issue.38–56 Conventionally, PUs are synthesized via highly efficient polyaddition reactions between diols and di-isocyanates (Fig. 1A), allowing broad compositional flexibility and facile tuning of material properties ranging from soft elastomers to rigid coatings. Owing to their practical utility, most PU research has focused on formulation-driven material development rather than on precise control of the polymer primary structure. Nevertheless, it is evident that the positioning of urethane bonds and comonomer units along the polymer backbone critically influences thermal, mechanical, and functional properties, underscoring the importance of sequence control. However, conventional AA/BB-type polyaddition intrinsically lacks a framework to define or implement distinct sequence motifs, such as alternating arrangements, within PU backbones (Fig. 1A).
 | ||
| Fig. 1 General concept of this work as illustrated using PU as a model system; (A) PU synthesis by the conventional method; (B) PU synthesis based on the AB-type monomer featuring both acyl azide and alcohol groups;69–73 (C) PU architectures accessible using the AB-type monomer. | ||
To address sequence control beyond conventional step polymerization, alternative PU production strategies have been explored. These include (i) ring-opening polymerization (ROP) of cyclic urethane monomers via chain-growth mechanisms and (ii) iterative one-by-one monomer addition to produce sequence-defined oligomers or polymers.57–68 While these approaches have successfully enabled advanced structural control, they are associated with substantial synthetic cost and limited practicality. ROP approaches require multistep cyclic monomer synthesis and offer restricted structural diversity, whereas iterative synthesis suffers from high workload and typically limits accessible molecular weights to a few thousand g per mol or less. Consequently, a practical, scalable method that allows systematic sequence definition and control within the step polymerization framework remains highly desirable.
Recently, we introduced a facile PU synthesis strategy based on AB-type monomers, which provides a new platform to address this long-standing issue.69–73 In this approach, monomers bearing both acyl azide and alcohol functionalities undergo heat-induced Curtius rearrangement to generate isocyanates in situ, triggering self-polyaddition between the isocyanate and alcohol groups (Fig. 1B). Because the acyl azide functionality is inert toward alcohols prior to the Curtius rearrangement, polymerization is effectively gated, enabling controlled step polymerization from AB-type building blocks. Importantly, this strategy establishes a clear definition of polymer sequence in PUs, enabling the construction of diverse polymer architectures, including block, statistical (random), grafted, and alternating polymers (Fig. 1C). The synthetic accessibility of AB-type monomers, readily derived from a diverse molecular pool of carboxylic acid species bearing alcohol groups, further allows flexible molecular design and broad monomer scope.
Using this framework, we have demonstrated that copolymerization behavior is strongly influenced by monomer reactivity, leading to statistical sequence distributions in certain systems. As expected, monomers featuring primary alcohol groups afford PUs more rapidly than those bearing secondary alcohol groups, resulting in statistical rather than completely random sequence distributions during copolymerization.69–73 Moreover, this approach has enabled the preparation of functional PU materials with advanced primary structure control, including photo-degradable polymers. While earlier studies have reported PU synthesis from acyl azide–alcohol monomers, these efforts largely focused on homopolymer formation for bio-based materials or dendrimer synthesis, rather than on sequence definition or copolymerization.74–77 Consequently, systematic implementation of alternating sequences within step polymerized PU backbones has remained unexplored.
Establishing a well-defined alternating sequence is particularly important for elucidating sequence–property relationships in step polymerization. To clearly evaluate the impact of sequence control, alternating monomer pairs should ideally exhibit distinct chemical or physical characteristics that enable unambiguous structural and property differentiation. In this context, we previously developed a photo-degradable PU monomer M1 featuring an o-nitrobenzyl alcohol (o-NBA) unit, which displayed a certain homo-diad preference during copolymerization (Fig. 2A).72 This uneven monomer distribution led to inefficient photo-degradation due to the formation of locally concentrated photo-cleavable segments. Introducing an alternating sequence represents a rational strategy to homogenize the distribution of photo-degradable units and thereby enhance degradation efficiency. Furthermore, alternating incorporation of rigid M1 units with flexible comonomers, such as M2 bearing a 2-ethylhexyl side chain, is expected to significantly influence the thermomechanical properties of the resulting polymers (Fig. 2B).73
 | ||
| Fig. 2 Conceptual overview of PU sequence control using photo-degradable monomer M1 in (A) previous work showing statistical sequence distribution;72 (B) this work enabling alternating sequence incorporation. | ||
In this study, we define and implement alternating sequences within a step polymerization framework using polyurethane as a representative model system to achieve advanced photo-degradability control. While our previous statistical sequence system demonstrated that the sequence could be discussed in AB-type polyurethane synthesis, it did not allow the explicit implementation of alternating connectivity; this limitation is addressed in this work through a dimer-based strategy. Such explicit alternating control is essential for examining sequence–property relationships without ambiguity arising from statistical sequence distributions. A newly designed dimeric monomer M3 is employed to introduce alternating connectivity in combination with AB-type monomers M1/M2 (Fig. 2B). The resulting polymers are systematically analyzed in terms of photo-degradability and thermal properties to elucidate the effects of the sequence control. As shown in this study, by redefining the connectivity unit through a combination of AB-type monomers and a dimeric species, step polymerization is transformed from a process with an ambiguous sequence definition into a sequence-definable one. This work therefore establishes a conceptual and synthetic framework for defining and implementing alternating sequences in step polymerization, while sequence-dependent properties are examined as validation of its chemical and functional significance.
Results and discussion
M1–M3 were all synthesized by following previously reported standard synthesis including the final step to convert a carboxylic acid group into an acyl azide group using diphenyl phosphoryl azide (DPPA).69–73,78–83 Newly synthesized M3 was obtained as a solid at room temperature, the same as M1, while M2 is liquid. According to previous results, liquid monomers typically show good polymerizability under bulk conditions, while solid ones often need some solvent during the polymerization for supplying enough fluidity to the reaction mixture, but this issue is affected by the Tg/Tm of the corresponding polymers. Since the polymerization behaviors of M1 and M2 were already investigated in previous studies, that of M3 is first evaluated.The kinetic analysis of the Curtius rearrangement of M3 was studied by following previous reports (Scheme 1).69–73,83–86 Namely, M3 was dissolved in PhCl-d5 at 0.020 M concentration and heated at 100 °C to induce the Curtius rearrangement. 1H NMR measurements were conducted after 5, 10, and 15 min of heating to estimate the time-course of the spectral integration for calculating the kinetic constant. The results are summarized in the SI, and the obtained kinetic values are consistent with previous reports.69–73 Namely, the Curtius rearrangement reaches a quantitative conversion within 60 min at 100 °C, and 30 min at 110 °C, which will be used for the setup of the polymerization conditions.
 | ||
| Scheme 1 Curtius rearrangement of M3. | ||
Next, to gain an insight into the polymerization behavior of M3, its homopolymerization was conducted under four standard reaction conditions (Scheme 2 and Table 1). Namely, the polymerization was carried out at 110 °C for 30 min in bulk or DMF (1.0 M) solvent, in the absence or presence of the dibutyltin dilaurate (DBTDL) catalyst, which was subsequently purified by precipitation to cold MeOH. The SEC profiles of products showed unimodal signals, while their 1H NMR spectra clearly indicated the successful formation of polyM3 (Fig. 3, 4A, and B).83 Namely, 1H NMR signals of the polymer became broader than the respective monomer signals, and each peak shift before and after the polymerization was consistent with previous reports,69–73 while the generation of a new urethane signal e1 at 10.4 ppm clearly indicated the occurrence of polymerization. Comparing the investigated four conditions (Table 1), the polymerization in bulk with the catalyst afforded the highest yield and the highest molecular weight (polyM3a, 59%, Mn = 11 kDa, and Đ = 1.6), while other reaction conditions also successfully provided the desired polymer. Since the effective copolymerization conditions for M1/M2/M3 would depend on the comonomer combination, the observed applicability of M3 on various polymerization conditions is useful. Although the polymerization under DMF solution conditions without the catalyst resulted in a low yield (27%), the corresponding homopolymerization of M2 only afforded a trace amount of polyM2,73 suggesting the higher reactivity of M3 than M2 and/or the lower solubility in the precipitation solvent (cold MeOH) of the polyM3 framework than that of polyM2. Since polyM3 has the equivalent structure to poly(M1-alt-M2), PU featuring the alternating sequence was successfully synthesized by step polymerization. For clarification, “alternating sequence”, in this manuscript, refers to the strict alternating connection of M1 and M2 units provided by the dimeric monomer M3, distinguishing it from the inherent repeating unit alternation in traditional AA/BB polymerization.
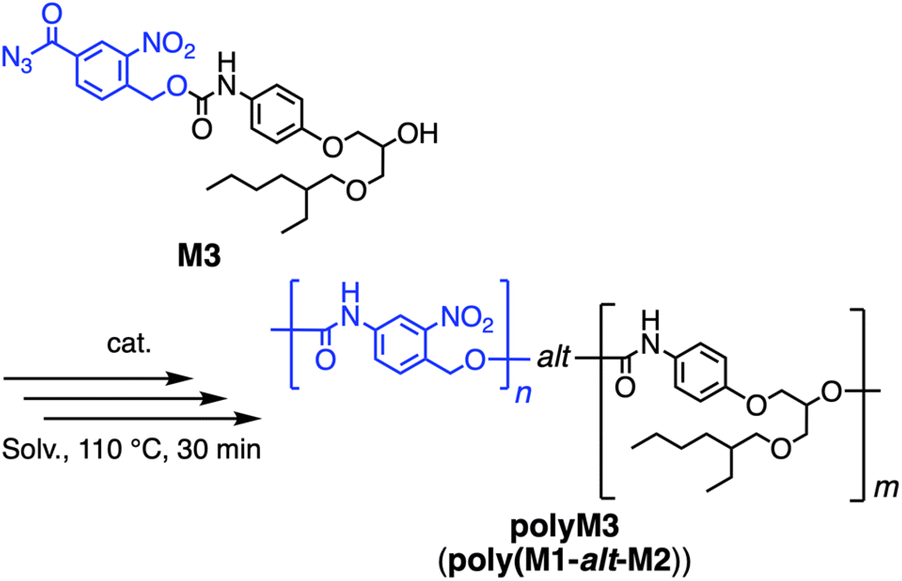 | ||
| Scheme 2 Homopolymerization of M3. | ||
 | ||
| Fig. 3 SEC profile of polyM3a (eluent: DMAc, flow rate: 0.5 mL min−1, and 50 °C). | ||
 | ||
| Fig. 4 1H NMR spectra of (A) M3; (B) polyM3a (poly(M1-alt-M2)); (C) poly(M1-stat-M2)a (400 MHz, 298 K, DMSO-d6). | ||
To study the sequence-property control on various M1/M2 feed ratios, copolymerization of M1, M2, and M3 was conducted (Scheme 3 and Table 2). Here, the M1/M2 feed ratio is set as 1/1, 4/1, and 1/4, and the alternating sequence was partially integrated by copolymerization with M3 (Schemes 3B and C). In the case of high M1 content, the DMF solution conditions were applied to promote effective polymerization, while the bulk conditions were applied for high M2 content, considering the preferable polymerization conditions of M1 and M2.69–73 When M1/M2 is 1/1, both the solution and the bulk conditions were applied (poly(M1-stat-M2)a and poly(M1-stat-M2)d). As a result, all the copolymers were successfully synthesized in moderate to high yields as confirmed by each unimodal SEC signal with Mn values of around 6–13 kDa.83 All the 1H NMR signals of the copolymer were clearly assigned in a similar manner to polyM3, which also indicated the successful polymer synthesis (Fig. 4C).83 Interestingly, certain differences were observed in 1H NMR signals between the alternating polymer polyM3 and the M1/M2 = 1/1 statistical copolymer poly(M1-stat-M2)a (Fig. 4B and C). In general, the statistical copolymer showed broader peaks than the alternating copolymer, which indicated a more diverse chemical status of the corresponding signals on the former. This broadening effect is consistent considering the existence of various sequence patterns on the statistical copolymer, while the alternating one has only a single pattern. Moreover, multiple aliphatic signals near urethane bonds showed two signals on the statistical polymer, while only single signals were observed for the alternating copolymer, e.g. the benzyl proton of the M1 unit d at 5.4 ppm and alkyl signals h at 4.2 ppm, i at 5.2 ppm, and j at 3.7 ppm of the M2 unit. These equally split signals in the statistical copolymer are attributed to two different local diad environments, depending on whether the corresponding repeating unit is next to M1 or M2, which is also consistent with the previous observation on other PUs from AB-type monomers.69–73 In contrast, the alternating copolymer provides only a single local sequence environment, resulting in one corresponding signal for each resonance. Thus, the sequence effect, statistical vs. alternating, was clearly observed in NMR measurement. The compositions of the obtained copolymers were calculated using 1H NMR analyses and approximately followed the feed ratio as summarized in Table 2.83 Meanwhile, further detailed analysis of diad or triad patterns based on NMR was difficult because the difference between them could not be clearly detected in our current system. Although the homopolymerization of M1 did not proceed effectively under bulk conditions, its copolymerization with M2 certainly worked at the M1/M2 ratio = 1/1 or 1/4, suggesting that the M2 unit provides sufficient fluidity for the effective reaction progress during the polymerization. Including homopolymer polyM3 (= poly(M1-alt-M2)) in addition to these copolymers, PUs with multiple M1/M2 compositions were successfully synthesized featuring a certain degree of the alternating sequence, which has been difficult to achieve by other existing synthetic methods.
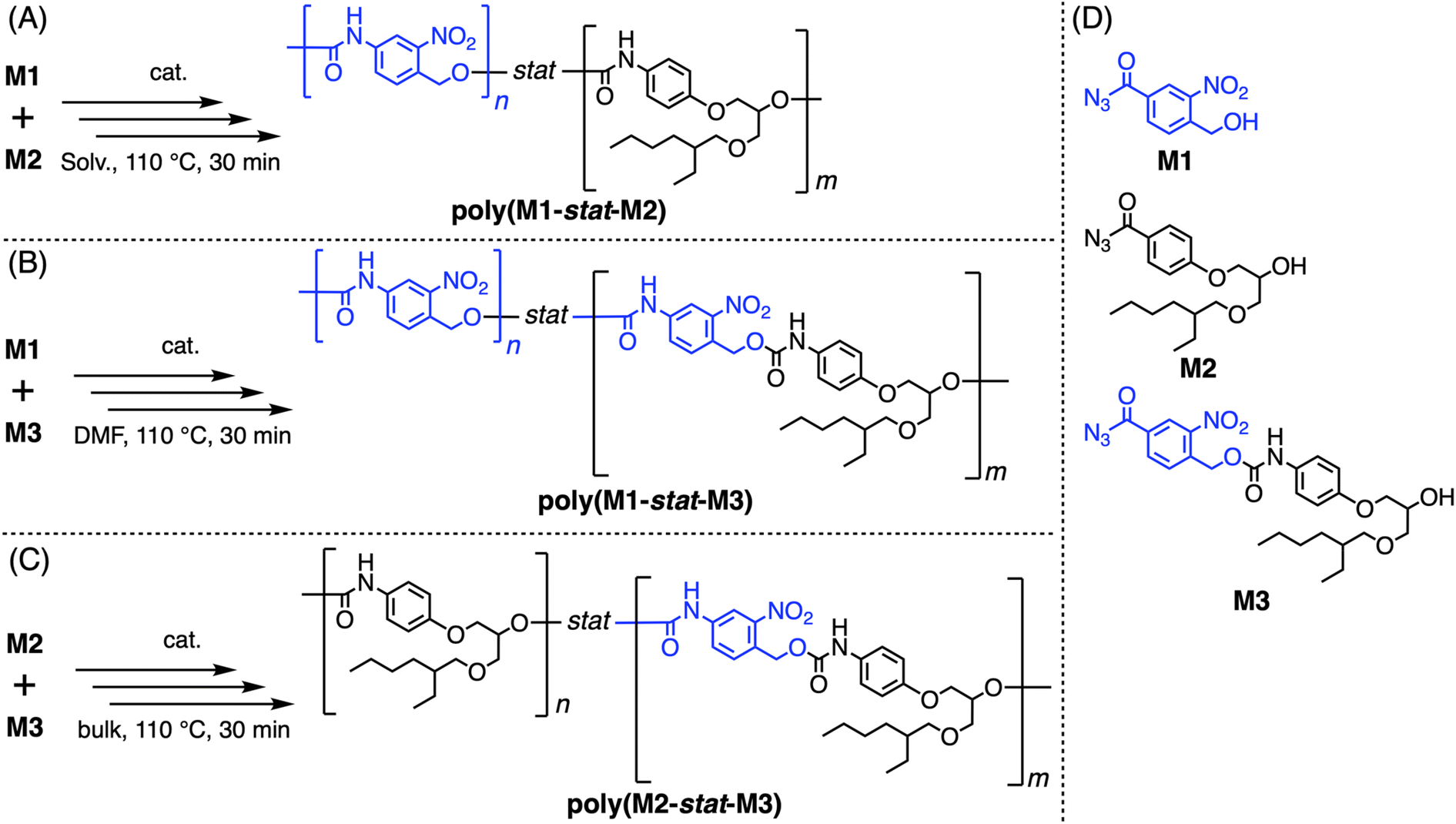 | ||
| Scheme 3 Copolymerization of (A) M1/M2; (B) M1/M3; (C) M2/M3. (D) Chemical structures of M1–M3. | ||
| Polymer | Monomer feed ratio | M1/M2 unit feed ratio | Solv. | Compositiona M1/M2 | Yield [%] | Mn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b [kDa] b [kDa] |
Mwb [kDa] |
Đb |
|---|---|---|---|---|---|---|---|---|
| a Calculated from 1H NMR spectra.b Calculated by SEC (eluent: DMAc, flow rate: 0.5 mL min−1, and 50 °C). | ||||||||
| poly(M1-stat-M2)a | M1/M2 = 1/1 | 1/1 | DMF | 49/51 | 65 | 8.6 | 12 | 1.3 |
| poly(M1-stat-M2)b | M1/M2 = 4/1 | 4/1 | DMF | 70/30 | 77 | 8.0 | 15 | 1.9 |
| poly(M1-stat-M2)c | M1/M2 = 1/4 | 1/4 | bulk | 22/78 | 29 | 6.0 | 10 | 1.7 |
| poly(M1-stat-M2)d | M1/M2 = 1/1 | 1/1 | bulk | 49/51 | 67 | 13 | 17 | 1.4 |
| poly(M1-stat-M3) | M1/M3 = 3/1 | 4/1 | DMF | 72/28 | 97 | 8.2 | 15 | 1.8 |
| poly(M2-stat-M3) | M2/M3 = 3/1 | 1/4 | bulk | 24/76 | 55 | 9.3 | 13 | 1.4 |
Next, thermal properties of the obtained polymers were analyzed using DSC and TGA measurements (Table 3).83 As previously observed, polyM1 has a thermal self-immolative nature by the conjugative depolymerization path, including the decarboxylation step (Scheme 4),70,72,87–89 leading to the lower Td5 values of copolymers containing a high M1 content (around 145 °C) than the ones with a high M2 content (around 240 °C). Considering high Td5 values of M1/M2 = 1/1 copolymers, the thermal degradation of the polyM1 segment does not proceed effectively in these copolymers, probably because the M2 unit stops the conjugative degradation path. Owing to the higher rigidity of the M1 unit than M2, copolymers bearing the higher M1 content generally showed higher Tg values, while certain differences were observed according to the sequence. A clear Tg difference was observed in M1/M2 = 4/1 samples, poly(M1-stat-M2)b and poly(M1-stat-M3) (107 °C and 87 °C). In the latter, the soft M2 unit is always located next to the M1 unit through the alternating sequence, which effectively reduces the packing of the local homo-M1 segment, while the generation of a certain amount of homo-M1 or M2 segments is inevitable in the former, which could induce easier homo-segment packing to increase the Tg value (Fig. 5). A similar Tg difference was observed in M1/M2 = 1/4 samples, poly(M1-stat-M2)c and poly(M2-stat-M3) (56 °C and 40 °C), which can be explained in the same manner. Namely, the dimer integration induced the distribution of the hard M1 unit always next to the soft M2 unit, while the generation of a certain content of homo-M1/M2 was inevitable in the copolymerization without the dimer M3, which caused the trend of easier packing by local homo-polymer segments to increase the Tg value. In these cases, the partial alternating sequence (M3 unit) introduction significantly reduced the packing nature of the local polymer segments compared with the corresponding statistical copolymers, resulting in the decrease of Tg values. Meanwhile, the reason for the difference in Tg values between M1/M2 = 1/1 statistical polymers (poly(M1-stat-M2)a and poly(M1-stat-M2)d) was not completely clear, but could have been influenced by the sequence formation process during the polymerization. Namely, the bulk conditions used for poly(M1-stat-M2)d reduced the fluidity of the polymerization mixture during the copolymerization, which would have caused the local homo-M1/M2 segment formation, while the DMF solution conditions used for poly(M1-stat-M2)a induced a more homogeneous distribution of M1/M2 units in the polymer framework. The resulting sequence difference would have caused the different Tg values. Interestingly, poly(M1-stat-M3) having a 20% hard M1 content showed a slightly lower Tg value than the 100% soft segment homopolymer polyM2 (40 °C and 42 °C respectively). This suggested that the small content of M1 on the former disturbed the effective packing of the aliphatic side chain moieties of the entire polyM2 framework to reduce Tg, and this effect would work more dominantly than the increase of the rigidity by the introduction of the hard M1 unit. Considering the above discussion about the polymer sequence, the implementation of the alternating sequence was clearly proved to affect the thermal properties of the polymer derived from step polymerization (PU in this case), underscoring the importance of the “sequence” as a parameter to control the basic properties of the polymer obtained by step polymerization. Moreover, the pronounced sequence dependence observed in thermal properties indicates that the sequence effect is not limited to photo-cleavable systems, although photo-degradability was chosen in this work as a particularly clear model response.
 | ||
| Scheme 4 Thermal degradation of the M1 segment. | ||
 | ||
| Fig. 5 Schematics of the plausible local polymer packing structure of (A) the homo-M1 segment; (B) homo-M2 segment; (C) M1/M2 hetero-segment. | ||
| Polymer | Monomer feed ratio | M1/M2 unit feed ratio | Tgb [°C] |
Td5c [°C] |
|---|---|---|---|---|
| a Not detected.b Estimated by DSC (2nd heating).c Estimated by TGA. | ||||
| polyM1 | — | — | NDa | 141 |
| polyM2 | — | — | 42 | 272 |
| polyM3a | — | 1/1 | 78 | 245 |
| poly(M1-stat-M2)a | M1/M2 = 1/1 | 1/1 | 66 | 210 |
| poly(M1-stat-M2)b | M1/M2 = 4/1 | 4/1 | 107 | 144 |
| poly(M1-stat-M2)c | M1/M2 = 1/4 | 1/4 | 56 | 237 |
| poly(M1-stat-M2)d | M1/M2 = 1/1 | 1/1 | 78 | 246 |
| poly(M1-stat-M3) | M1/M3 = 3/1 | 4/1 | 87 | 147 |
| poly(M1-stat-M3) | M2/M3 = 3/1 | 1/4 | 40 | 238 |
Next, the photo-degradability of copolymers was investigated to evaluate the effect of their sequences on it by following the previous report (Fig. 6 and 7 and Scheme 5).72 Namely, each copolymer was dissolved in THF or DMAc under diluted conditions (1 mg mL−1), and 365 nm light was irradiated to induce the photo-cleavage reaction of the o-NBA framework on the M1 unit, which was traced by SEC measurements after 0, 5, 10, 15, 30, 60, and 120 min of light irradiation and summarized as a time-course Mn plot (Fig. 7). Here, the result of the THF system is discussed, because the result of DMAc indicated a similar trend, and the former showed sharper SEC signals that could be clearly detected.83 First, all the samples showed a clear decrease in the molecular weight even after 5 min of irradiation, and the continuous irradiation induced a further decrease that was mostly completed within one hour, while the control test of UV irradiation on polyM2 did not change its SEC profile at all even after several hours, clearly indicating the occurrence of photo-degradation in the M1 unit.83 The general trend of the photo-degradability is consistent with the previous report,72 indicating that copolymers bearing a high M1 content are likely to show more intensive photo-degradability, while certain effects of the sequence were clearly observed as discussed in the following.
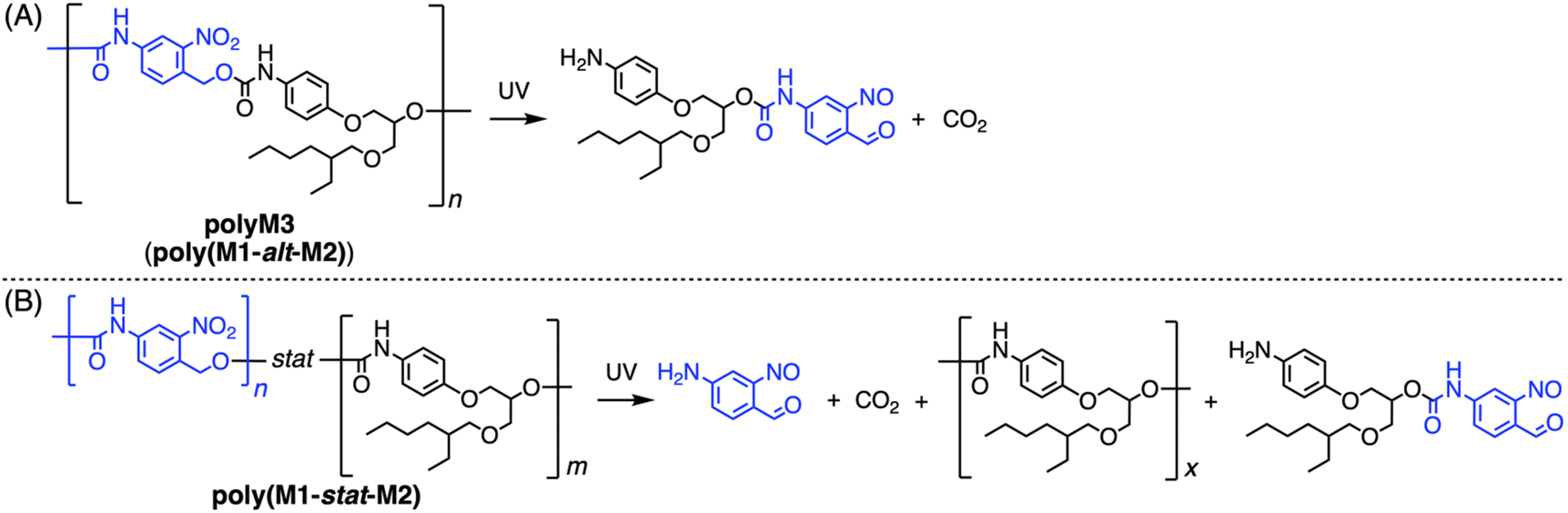 | ||
| Scheme 5 Photo-degradation of (A) polyM3 (poly(M1-alt-M2)) and (B) poly(M1-stat-M2). | ||
 | ||
| Fig. 6 Time course of SEC profiles of (A) polyM3a; (B) poly(M1-stat-M2)d; (C) poly(M1-stat-M2)a; (D) poly(M1-stat-M3); (E) poly(M1-stat-M2)b; (F) poly(M2-stat-M3); (G) poly(M1-stat-M2)c (UV irradiation for 0, 5, 10, 15, 30, 60, and 120 min; eluent: THF, flow rate: 1.0 mL min−1, and 25 °C). The red dotted line indicates the dimeric fragment generation, while the blue one indicates the monomeric fragment. | ||
 | ||
| Fig. 7 Plot of Mn [%] vs. UV irradiation time of copolymers. The column of Mn shows the initial Mn before UV irradiation (eluent: THF, flow rate: 1.0 mL min−1, and 25 °C). | ||
Comparing polyM3a and polyM3c, both of which feature the alternating sequence poly(M1-alt-M2), the former showed more intensive degradation than the latter. Since both polymers have the same sequence/composition, this difference would be caused by the molecular weight difference. In this method, the minimum unit after the photo-degradation is the monomeric unit from M1 (Mw ∼ 150 Da) or the dimeric unit from the M2–M1 diad (Mw ∼ 470 Da), indicating that the size of the initial polymer certainly affects the percentage of the Mn decrease when it is around 10 kDa or less. Thus, the initially bigger polyM3a (Mn = 8.4 kDa) showed a greater decrease in the percentage of Mn from the beginning to the complete degradation than polyM3c (Mn = 5.2 kDa). It also affects the more intensive degradation of polyM3a than the copolymers bearing an 80% M1 content (poly(M1-stat-M3) and poly(M1-stat-M2)b). In this case, the high M1 content increases the photo-degradability of polymers, but it also causes a lower molecular weight in the resulting polymer, possibly because of the high rigidity of the M1 unit,72 which could cause low fluidity during the polymerization, hindering the propagation reaction. The effect of the molecular weight would be more dominant than the high content of the M1 unit and would have induced the highest photo-degradability of polyM3a in this measurement. Except for polyM3a, copolymers bearing more than 50% M1 content showed a similar trend in the time-course of the Mn decrease regardless of their sequence or the M1 content, suggesting that these parameters do not significantly influence their photo-degradability, possibly because they are already highly photo-degradable. Meanwhile, a large photo-degradability difference was observed in copolymers bearing a 20% M1 content (poly(M2-stat-M3) and poly(M1-stat-M2)c), the smallest M1 content in this work. Both degraded less than the others, but poly(M2-stat-M3) (∼45% decrease of Mn) degraded substantially less than poly(M1-stat-M2)c (∼70% decrease of Mn), which was associated with the sequence difference. Namely, the alternating sequence from M3 induces more homogeneous distribution of photo-degradable M1 units in the entire polymer chain, while the M1/M2 statistical polymer without M3 includes inevitable local homo-M1 and M2 polymer segments, which decrease the photo-degradability owing to the non-photo-degradable homo-M2 segment. When the M1 content is higher than 50%, this effect would not be dominant, but it has a significant influence on the low M1 content copolymers, resulting in a drastic photo-degradability difference between the partially alternating polymer and the complete statistical polymer. This “alternating vs. statistical” sequence-dependent photo-degradability was first observed by the implementation of sequence definition/control on the polymer derived from step polymerization (PU in this case), supporting its importance.
Meanwhile, SEC profiles showed further dependence of the resulting residues from the photo-degradation on the sequence (Fig. 6). According to the photo-degradation of polyM3a, the SEC intensity around 21.5 min (red dotted line in Fig. 6A) increased, suggesting the formation of dimeric fragments after the alternating polymer framework cleavage. The peaks at the same retention time were also observed on other polymers containing an alternating sequence (Fig. 6D and F). On the other hand, copolymers made from M1 show an SEC peak increase around 22.5 min in proportion to the UV irradiation time (blue dotted lines in Fig. 6), which would be derived from the monomeric fragment after the photo-degradation of the homo-M1 segment. Statistical polymers without M3 integration also showed peaks from dimeric fragments around 21.5 min, suggesting the existence of various sequence patterns there, including the homo-M1 segment and other hetero-segment types. Comparing two copolymers featuring 20% M1 content, the one with the alternating sequence showed an intense dimeric fragment peak on the residue (Fig. 6F), while the one without the alternating sequence exhibited an intense monomeric fragment peak (Fig. 6G), highlighting the sequential difference and the homo-M1 segment formation in poly(M1-stat-M2)c, which induced its more decreased photo-degradability and higher Tg than its partially alternating sequenced counterpart poly(M2-stat-M3). Finally, comparing two statistical copolymers featuring M1/M2 = 1/1 content, the one made in DMF solution showed a more intensive dimeric peak (Fig. 6C) than the one made in the bulk (Fig. 6B), while both showed monomeric fragment peaks. This difference is probably because the solvent increased the fluidity of the reaction mixture to induce a more homogeneous polymerization system, while the bulk conditions could cause the local aggregation of the homo-polymer segments, resulting in monomeric fragment generation through photo-degradation and higher Tg due to their higher packing preference. Further spectroscopic analysis of the degradation products would provide deeper mechanistic insight into this system, but such analysis was experimentally difficult for the present degraded samples owing to the low product amount in diluted solution. Therefore, the degradation mechanism proposed here remains partly speculative and should be interpreted with appropriate caution. A more detailed structural elucidation of the degradation products will be the subject of future study.
Considering the above, the thermal properties and photo-degradability of M1/M2 copolymers were consistently associated with their polymer sequence, which was clearly controlled by the (partial) integration of the alternating sequence. The detailed analysis indicated a certain level of sequence-property control through the polymerization conditions (bulk vs. solution), which could supportively contribute to the polymer design. In the end, this work successfully demonstrated the definition of an alternating sequence in the polymer derived from step polymerization conventionally featuring only AA–BB-type connectivity by the combination of an AB-type monomer framework and the dimeric species, and the property analysis of redefined polymers suggested that the polymer sequence acts as an independent design parameter in step polymerization, giving rise to pronounced sequence-property divergence (Fig. 8).
 | ||
| Fig. 8 Sequence definition of the polymer derived from step polymerization demonstrated in this work, highlighting the resulting sequence–property divergence for photo-degradation control. | ||
Conclusion
In this work, we addressed a fundamental yet ambiguous question in step polymerization by defining what constitutes an alternating sequence in polymers synthesized from AA- and BB-type monomers. An AB-type monomer framework enabled explicit sequence definition, while a dimer-based strategy allowed the practical implementation of alternating connectivity. Using polyurethane as a model system, we demonstrated that defined alternating sequences give rise to pronounced sequence–property relationships, as reflected in thermal properties and photo-degradability, enabling programmable material degradation. The main contribution of this study is the establishment of a conceptual and synthetic basis for alternating sequences in step polymerization, while the observed sequence-dependent properties confirm the practical relevance of this framework. This study establishes polymer sequence as an additional and actionable design parameter in polyurethane as a model system for step polymerization, beyond conventional monomer and composition design. The framework demonstrated in this work would potentially be applicable to other step polymerization systems. By enabling the controlled incorporation of the functional unit into the polymer main chain rather than the side chain, this method provides a complementary tool for contributing to functional/sustainable material development other than well-studied chain polymerization.Author contributions
X. K.: data curation, formal analysis, investigation, and visualization; J. R.-B.: data curation and investigation; T. C.: data curation; P. T.: resources and writing – review & editing; Y. A. conceptualization, formal analysis, funding acquisition, methodology, project administration, resources, supervision, validation, visualization, writing – original draft, and writing – review & editing.Conflicts of interest
The authors declare no conflicts of interest.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information (SI): general methods, chemical synthesis, property analysis of the synthesized compounds, kinetic analysis, and spectra of the synthesized compounds. See DOI: https://doi.org/10.1039/d6py00409a.Acknowledgements
This work was supported by JSPS KAKENHI (Japan) grant numbers JP20J00360 (Y. A.), JP21K14672 (Y. A.), and JP22KJ1489 (Y. A.). The authors acknowledge financial support from the Helmholtz Association.References
- J. F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Sequence- Controlled Polymers, Science, 2013, 341, 1238149 CrossRef PubMed.
- S. L. Perry and C. E. Sing, 100th Anniversary of Macromolecular Science Viewpoint: Opportunities in the Physics of Sequence-Defined Polymers, ACS Macro Lett., 2020, 9, 216–225 CrossRef CAS PubMed.
- Q. Shia, Z. Denga, M. Houa, X. Hua and S. Liu, Engineering precise sequence-defined polymers for advanced functions, Prog. Polym. Sci., 2023, 141, 101677 CrossRef.
- M. A. R. Meier and C. Barner-Kowollik, A New Class of Materials: Sequence-Defined Macromolecules and Their Emerging Applications, Adv. Mater., 2019, 31, 1806027 CrossRef PubMed.
- Z. Gan, J. Liu, Z. Xu, S. Jia and X.-H. Dong, Precision polymers: advances in synthesis, structural engineering, and functional optimization, Prog. Polym. Sci., 2025, 170, 102030 CrossRef CAS.
- L. D. Thai, J. A. Kammerer, D. Golberg, H. Mutlu and C. Barner-Kowollik, Sequence-defined main-chain photoswitching macromolecules with odd-even effect-controlled properties, Chem, 2025, 11, 102341 CAS.
- H. S. Wang, M. Agrachev, H. Kim, N. P. Truong, T.-L. Choi, G. Jeschke and A. Anastasaki, Visible Light–Triggered Depolymerization of Commercial Polymethacrylates, Science, 2025, 387, 874–880 CrossRef CAS PubMed.
- L. Cederholm, P. Olsen, M. Hakkarainen and K. Odelius, Design for Recycling: Polyester- and Polycarbonate-Based A–B–A Block Copolymers and Their Recyclability Back to Monomers, Macromolecules, 2023, 56, 3641–3649 CrossRef CAS.
- Y. Takahashi, A. Kanazawa and S. Aoshima, 3-Alkoxyphthalides as Nonhomopolymerizable, Highly Reactive Comonomers for ABC Pseudo-Periodic Terpolymers and Degradable Polymers via Cationic co- and Terpolymerizations With Oxiranes and/or Vinyl Ethers, Macromolecules, 2023, 56, 4198–4207 CrossRef CAS.
- M. Uchiyama, Y. Murakami, K. Satoh and M. Kamigaito, Synthesis and Degradation of Vinyl Polymers with Evenly Distributed Thioacetal Bonds in Main Chains: Cationic DT Copolymerization of Vinyl Ethers and Cyclic Thioacetals, Angew. Chem., Int. Ed., 2023, 62, e202215021 CrossRef CAS PubMed.
- Y. Xia, T. Shao, Y. Sin, J. Wang, C. Gu, C. Zhang and X. Zhang, Precise Placement of Thioester Bonds Into Sequence-Controlled Polymers Containing ABAC-type Units, Nat. Commun., 2025, 16, 1974 CrossRef CAS PubMed.
- Y. Akae and P. Theato, Aggregation Behavior of Cyclodextrin- Based [3]Rotaxanes, Chem. – Eur. J., 2023, 29, e202301582 CrossRef CAS PubMed.
- M. Uchiyama, M. Imai and M. Kamigaito, Synthesis of Degradable Polymers via 1,5-Shift Radical Isomerization Polymerization of Vinyl Ether Derivatives With a Cleavable Bond, Polym. J., 2024, 56, 359–368 CrossRef CAS.
- Y. Akae, Cyclodextrin-Based Rotaxanes for Polymer Materials: Challenge on Simultaneous Realization of Inexpensive Production and Defined Structures, Beilstein J. Org. Chem., 2024, 20, 3026–3049 CrossRef CAS PubMed.
- H. G. Hester, Z. B. A. Abel and G. W. Coates, Ultra-High-Molecular- Weight Poly(Dioxolane): Enhancing the Mechanical Performance of a Chemically Recyclable Polymer, J. Am. Chem. Soc., 2023, 145, 8800–8804 CrossRef CAS PubMed.
- Y. Akae and T. Takata, O-Acylative Vinylation of Cyclodextrin- Based [3]Rotaxane Towards Rotaxane Crosslinkers, Eur. J. Org. Chem., 2023, 26, e202300009 CrossRef CAS.
- A. Tardy, J. Nicolas, D. Gigmes, C. Lefay and Y. Guillaneuf, Radi- cal Ring-Opening Polymerization: Scope, Limitations, and Application to (Bio)degradable Materials, Chem. Rev., 2017, 117, 1319–1406 CrossRef CAS PubMed.
- H. Sogawa, C.-G. Wang, S. Monjiyama, Y. Akae and T. Takata, Aliphatic Ditopic Nitrile N-Oxide Crosslinker: Synthesis, Chemical Stability, and Catalyst-Free Crosslinking Reactions, Polymer, 2021, 213, 123291 CrossRef CAS.
- C. Weng, X. Li and X. Tang, Solvent-Dependent Sequence-Controlled Copolymerization of Lactones: Tailoring Material Properties from Robust Plastics to Tough Elastomers, Angew. Chem., Int. Ed., 2025, 64, e202415388 CrossRef CAS PubMed.
- R. D. Rittinghaus, J. Zenner, A. Pich, M. Kol and S. Herres-Pawlis, Master of Chaos and Order: Opposite Microstructures of PCL-co-PGA-co-PLA Accessible by a Single Catalyst, Angew. Chem., Int. Ed., 2022, 61, e202112853 CrossRef CAS PubMed.
- M. Uchiyama, Y. Murakami, K. Satoh and M. Kamigaito, Synthesis and Degradation of Vinyl Polymers with Evenly Distributed Thioacetal Bonds in Main Chains: Cationic DT Copolymerization of Vinyl Ethers and Cyclic Thioacetals, Angew. Chem., Int. Ed., 2023, 62, e202215021 CrossRef CAS PubMed.
- T. Kimura, K. Kuroda, H. Kubota and M. Ouchi, Metal-Catalyzed Switching Degradation of Vinyl Polymers via Introduction of an “In-Chain” Carbon−Halogen Bond as the Trigger, ACS Macro Lett., 2021, 10, 1535–1539 CrossRef CAS PubMed.
- Y. Akae, H. Sogawa and T. Takata, Cyclodextrin Based [3]Rotaxane Crosslinked Fluorescent Polymer: Synthesis and De Crosslinking Using Size Complementarity, Angew. Chem., Int. Ed., 2018, 57, 14832–14836 CrossRef CAS PubMed.
- K. Watanabe, N. Kaizawa, B. J. Ree, T. Yamamoto, K. Tajima, T. Isono and T. Satoh, One-Shot Intrablock Cross-Linking of Linear Diblock Copolymer to Realize Janus-Shaped Single-Chain Nanoparticles, Angew. Chem., Int. Ed., 2021, 60, 18122–18128 CrossRef CAS PubMed.
- J. J. Lessard, G. M. Scheutz, S. H. Sung, K. A. Lantz, T. H. Epps III and B. S. Sumerlin, Block Copolymer Vitrimers, J. Am. Chem. Soc., 2020, 142, 283–289 CrossRef CAS PubMed.
- M. Appold, C. Mari, C. Lederle, J. Elbert, C. Schmidt, I. Ott, B. Stühn, G. Gasser and M. Gallei, Multi-stimuli responsive block copolymers as a smart release platform for a polypyridyl ruthenium complex, Polym. Chem., 2017, 8, 890–900 RSC.
- Y. Akae, Y. Koyama, H. Sogawa, Y. Hayashi, S. Kawauchi, S. Kuwata and T. Takata, Structural Analysis and Inclusion Mechanism of Native and Permethylated α-Cyclodextrin-Based Rotaxanes Containing Alkylene Axles, Chem. – Eur. J., 2016, 22, 5335–5341 CrossRef CAS PubMed.
- K. Satoh, D.-H. Lee, K. Nagai and M. Kamigaito, Precision Synthesis of Bio-Based Acrylic Thermoplastic Elastomer by RAFT Polymerization of Itaconic Acid Derivatives, Macromol. Rapid Commun., 2014, 35, 161–167 CrossRef CAS PubMed.
- Y. Akae, T. Arai, Y. Koyama, H. Okamura, K. Johmoto, H. Uekusa, S. Kuwata and T. Takata, One-Pot Synthesis of Permethylated α-CD-Based Rotaxanes Having Alkylene Chain Axles and Their Structural Characteristics, Chem. Lett., 2012, 41, 806–808 CrossRef CAS.
- J. Li, R. M. Stayshich and T. Y. Meyer, Exploiting Sequence to Control the Hydrolysis Behavior of Biodegradable PLGA Copolymers, J. Am. Chem. Soc., 2011, 133, 6910–6913 CrossRef CAS PubMed.
- A. Ravve, Step-Growth Polymerization and Step-Growth Polymers, in Principles of Polymer Chemistry, Springer, New York, 2012, pp. 403–535 Search PubMed.
- M. Ueda, K. Kimura and T. Yokozawa, Polycondensation, in Polymer Synthesis, ed. T. Endo, Kodansha, Tokyo, 2010, pp. 569–744 Search PubMed.
- T. Yokozawa and Y. Ohta, Transformation of Step-Growth Polymerization into Living Chain-Growth Polymerization, Chem. Rev., 2016, 116, 1950–1968 CrossRef CAS PubMed.
- A. Yokoyama and T. Yokozawa, Converting Step-Growth to Chain-Growth Condensation Polymerization, Macromolecules, 2007, 40, 4093–4101 CrossRef CAS.
- S. Sakai, T. Nakamura, S. Uchida, Y. Nakauchi, M. Uchiyama, T. Kubo, M. Kamigaito and K. Satoh, Controlled/Living Click Polymerization with Possible Bidirectional Chain-Growth Propagation during Polyaddition, J. Am. Chem. Soc., 2025, 147, 21459–21467 CrossRef CAS PubMed.
- J. Yang, L. Chen, M. Zhu, M. W. Ishaq, S. Chen and L. Li, Investigation of the Multimer Cyclization Effect during Click Step- Growth Polymerization of AB-Type Macromonomers, Macromolecules, 2022, 55, 6830–6840 CrossRef CAS.
- R. Ejima, M. Nakahata, Y. Kamon and A. Hashidzume, Synthesis and Metal Ion Adsorption Properties of a Dense Triazole Polymer Carrying Cysteine Residues, J. Polym. Sci., 2025, 63, 1570–1579 CrossRef CAS.
- E. Delebecq, J. P. Pascault, B. Boutevin and F. Ganachaud, On the Versatility of Urethane/Urea Bonds: Reversibility, Blocked Isocyanate, and Non-Isocyanate Polyurethane, Chem. Rev., 2013, 113, 80–118 CrossRef CAS PubMed.
- D. Liu, C. Huyan, H. Li, J. Ge, F. Chen and L. Zhang, Recycling and Upcycling of Polyurethane Thermosets: The Second Life of Polymers, Adv. Mater., 2026, e15809, DOI:10.1002/adma.202515809.
- E. Balla, D. N. Bikiaris, N. Pardalis and N. D. Bikiaris, Toward Sustainable Polyurethane Alternatives: A Review of the Synthesis, Applications, and Lifecycle of Non-Isocyanate Polyurethanes (NIPUs), Polymers, 2025, 17, 1364 CrossRef CAS PubMed.
- A. Gomez-Lopez, F. Elizalde, I. Calvob and H. Sardon, Trends in non-isocyanate polyurethane (NIPU) development, Chem. Commun., 2021, 57, 12254 RSC.
- F. Orabona, F. Recupido, G. C. Lama, K. Polaczek, F. Taddeo, T. Salmi, M. Di Serio, L. Verdolotti and V. Russo, Cutting-edge development of non-isocyanate polyurethane (NIPU) foams: from sustainable precursors to environmental impact evaluation, Green Chem., 2025, 27, 7403 RSC.
- C. Liang, U. R. Gracida-Alvarez, E. T. Gallant, P. A. Gillis, Y. A. Marques, G. P. Abramo, T. R. Hawkins and J. B. Dunn, Material Flows of Polyurethane in the United States, Environ. Sci. Technol., 2021, 55, 14215–14224 CrossRef CAS PubMed.
- N. G. Jaques, A. Llevot, É. Grau, T. Vidil, M. A. R. Meier and H. Cramail, High Molar Mass Non-Isocyanate Polyurethanes by Transurethanization of Diols with Isophorone-Based Bismethylcarbamate, Macromol. Chem. Phys., 2025, 226, 2500068 CrossRef CAS.
- F. C. Destaso, C. Libretti, C. L. Coz, E. Grau, H. Cramail and M. A. R. Meier, Optimized synthesis of a high oleic sunflower oil derived polyamine and its lignin-based NIPUs, Green Chem., 2025, 27, 1440 RSC.
- J. Sawada, H. Sogawa, H. Marubayashi, S. Nojima, H. Otsuka, K. Nakajima, Y. Akae and T. Takata, Segmented Polyurethanes Containing Movable Rotaxane Units on the Main Chain: Synthesis, Structure, and Mechanical Properties, Polymer, 2020, 193, 122358 CrossRef CAS.
- Y. Akae and P. Theato, Polyurethane Type Poly[3]rotaxanes Synthesized from Cyclodextrin Based [3]Rotaxane Diol and Diisocyanate, Macromol. Rapid Commun., 2024, 45, 2400441 CrossRef CAS PubMed.
- H. Sogawa, M. Abe, R. Shintani, T. Sotani, K. Tabaru, T. Watanabe, Y. Obora and F. Sanda, Polyurethanes containing platinum in the main chain: synthesis, structure and mechanofluorochromism, Polym. J., 2023, 55, 1119–1128 CrossRef CAS.
- P. Schara, A. M. Cristadoro, R. P. Sijbesma and Z. Tomovic, Solvent- Free Synthesis of Acetal-Containing Polyols for Use in Recyclable Polyurethanes, Macromolecules, 2023, 56, 8866–8877 CrossRef CAS.
- Y. Akae, K. Iijima, M. Tanaka, T. Tarao and T. Takata, Main Chain-Type Polyrotaxanes Derived From Cyclodextrin-Based Pseudo[3]Rotaxane Diamine and Macromolecular Diisocyanate: Synthesis, Modification, and Characterization, Macromolecules, 2020, 53, 2169–2176 CrossRef CAS.
- J. Futter, M. Holzer and B. Rieger, Formic Acid as Feedstock in the Phosgene-Free Dehydrogenative Coupling of Formamides and Alcohols to Polyurethanes, Macromolecules, 2025, 58, 1817–1826 CrossRef CAS.
- Q. Jaussaud, I. M. Ogbu, G. G. Pawar, E. Grau, F. Robert, T. Vidil, Y. Landais and H. Cramail, Synthesis of polyurethanes through the oxidative decarboxylation of oxamic acids: a new gateway toward self-blown foams, Chem. Sci., 2024, 15, 13475 RSC.
- T. Noda, A. Tanaka, Y. Akae and Y. Kohsaka, Unsaturated Polyurethanes Degradable by Conjugate Substitution Reactions With Amines and Carboxylate Anions, RSC Adv., 2023, 13, 20336–20341 RSC.
- J. Lee, S. Baek, H. H. Moon, S. U. Son and C. Song, Furandiacylazide: A Biomass-Derived Versatile Polymer Platform toward Photodegradable and Nonflammable Polyurethanes, ACS Appl. Polym. Mater., 2021, 3, 5767–5777 CrossRef CAS.
- H. Tomita, F. Sanda and T. Endo, Model Reaction for the Synthesis of Polyhydroxyurethanes from Cyclic Carbonates with Amines: Substituent Effect on the Reactivity and Selectivity of Ring-Opening Direction in the Reaction of Five-Membered Cyclic Carbonates with Amine, J. Polym. Sci., Part A:Polym. Chem., 2001, 39, 3678–3685 CrossRef CAS.
- S.-D. Lee, F. Sanda and T. Endo, A Novel Synthetic Method of a Polyurethane from an Aminimide as a “Latent Monomer”, J. Polym. Sci., Part A:Polym. Chem., 1997, 35, 1333–1335 CrossRef CAS.
- S. Neffgen, H. Keul and H. Höcker, Cationic Ring-Opening Polymerization of Trimethylene Urethane: A Mechanistic Study, Macromolecules, 1997, 30, 1289–1297 CrossRef CAS.
- L. Rubino, V. Patamia, A. Rescifina, M. Galimberti and V. Barbera, Cyclic carbamates based on (R)-(+)-limonene oxide for ring-opening polymerization, Sci. Rep., 2024, 14, 23521–21297 CrossRef CAS PubMed.
- D. Zhang, Y. Zhang, Y. Fan, M.-N. Rager, V. Gueŕineau, L. Bouteiller, M.-H. Li and C. M. Thomas, Polymerization of Cyclic Carbamates: A Practical Route to Aliphatic Polyurethanes, Macromolecules, 2019, 52, 2719–2724 CrossRef CAS.
- E. A. Hoff, G. X. de Hoe, C. M. Mulvaney, M. A. Hillmyer and C. A. Alabi, Thiol–Ene Networks From Sequence-Defined Polyurethane Macromers, J. Am. Chem. Soc., 2020, 142, 6729–6736 CrossRef CAS PubMed.
- T. Mondal, L. Charles and J.-F. Lutz, Damage and Repair in Infor- mational Poly(N-Substituted Urethane)s, Angew. Chem., Int. Ed., 2020, 59, 20390–20393 CrossRef CAS PubMed.
- J. V. Hoorde, N. Badi and F. E. DuPrez, Scalable Design of Uni- form Oligourethanes for Impact Study of Chain Length, Sequence and End Groups on Thermal Properties, Polym. Chem., 2024, 15, 4319 RSC.
- H. Yamauchi, S. Inayama, M. Nakabayashi and S. Hayashi, Sys- tematic Order-Made Synthesis of Sequence-Defined Polyurethanes With Length, Types, and Topologies, ACS Macro Lett., 2023, 12, 1264–1271 CrossRef CAS PubMed.
- W. Forysiak, A. Lizak and R. Szweda, Sequence of Monomers and Position of Stereocenters Matter for Thermal Properties of Stereocon- trolled Oligourethanes, ChemPhysChem, 2024, 25, e202400366 CrossRef CAS PubMed.
- J. R. Sculuk, H. A. Fargher and E. V. Anslyn, The Utility of Chain-End Degradation for De Novo Sequencing of Sequence-Defined Oligourethanes, Acc. Chem. Res., 2026, 59, 958–968 CrossRef PubMed.
- S. D. Dahlhauser, P. R. Escamilla, A. N. VandeWalle, J. T. York, R. M. Rapagnani, J. S. Shei, S. A. Glass, J. N. Coronado, S. R. Moor, D. P. Saunders and E. V. Anslyn, Sequencing of Sequence-Defined Oligourethanes via Controlled Self-Immolation, J. Am. Chem. Soc., 2020, 142, 2744–2749 CrossRef CAS PubMed.
- W. Forysiak, S. Kozub, Ł. John and R. Szweda, “Discrete oligourethanes of sequence-regulated properties – impact of stereocontrol, Polym. Chem., 2022, 13, 2980–2987 RSC.
- A. Sharma, P. Cwynar, A. Jose, V. Gupta and R. Szweda, Step-economy approach for scalable synthesis of stereocontrolled and sequence-defined polyurethanes, Eur. Polym. J., 2025, 236, 114120 CrossRef CAS.
- Y. Akae, M. Sakurai and T. Takata, Alcohol Moiety-Tethering Acylazides as Versatile Isocyanate Precursors for Polymerization Initiators and Monomers, Macromolecules, 2021, 54, 8488–8494 CrossRef CAS.
- Y. Akae and P. Theato, Polyurethanes by Ring-Opening Polymerization Initiated From Alcohol Moiety-Tethering Acylazide, J. Polym. Sci., 2024, 62, 4127–4135 CrossRef CAS.
- Y. Shen, P. Theato and Y. Akae, Tailored Polyurethane Synthesis from AB-Type Monomers, Macromolecules, 2024, 57, 8162–8175 CAS.
- J. Reeb, Y. Shen, P. Theato and Y. Akae, Controllable Photo- Degradability on Polyurethane From AB-Type Monomers, Macromolecules, 2025, 58, 2701–2708 CrossRef CAS.
- X. Kneidl, L. Forkel, T. Cui, P. Theato and Y. Akae, The Effect of Aliphatic or Aromatic Substituents on the Synthesis and Properties of Polyurethanes From AB-Type Monomers, J. Polym. Sci., 2026, 64, 366–375 CrossRef CAS.
- D. V. Palaskar, A. Boyer, E. Cloutet, C. Alfos and H. Cramail, Synthesis of Biobased Polyurethane From Oleic and Ricinoleic Acids as the Renewable Resources via the AB-Type Self-Condensation Approach, Biomacromolecules, 2010, 11, 1202–1211 CrossRef CAS PubMed.
- A. S. More, B. Gadenne, C. Alfos and H. Cramail, AB Type Polyaddition Route to Thermoplastic Polyurethanes From Fatty Acid Deriva- tives, Polym. Chem., 2012, 3, 1594 RSC.
- A. S. More, L. Maisonneuve, T. Lebarbe, B. Gadenne, C. Alfos and H. Cramail, Vegetable-Based Building-Blocks for the Synthesis of Thermoplastic Renewable Polyurethanes and Polyesters, Eur. J. Lipid Sci. Technol., 2013, 115, 61–75 CrossRef CAS.
- C. Sivakumar and A. S. Nasar, Hydroxyl- and Amine-Terminated Hyperbranched Polyurethanes Using AB2-Type Azide Monomers: Synthesis, Characterization, Fluorescence, and Charge-Transfer Complexation Studies, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3337–3351 CrossRef CAS.
- T. Shioiri, K. Ishihara and M. Matsui, Cutting Edge of Diphenyl Phosphorazidate (DPPA) as a Synthetic Reagent – A Fifty-Year Odyssey, Org. Chem. Front., 2022, 9, 3360 RSC.
- Y. Akae, J. Sawada, K. Nakajima and T. Takata, The Effect of the Axle End Structure and Number of Through-Space Bonds on the Properties of Rotaxane Crosslinked Polymers, Angew. Chem., Int. Ed., 2023, 62, e202303341 CrossRef CAS PubMed.
- Y. Akae, H. Sogawa and T. Takata, Effective Synthesis and Modification of α-Cyclodextrin Based [3]Rotaxanes Enabling Versatile Molecular Design, Eur. J. Org. Chem., 2019, 22, 3605–3613 CrossRef.
- Y. Akae, H. Sogawa and T. Takata, Evaluation of Induced Circular Dichroism via Through Space Chirality Transfer in alpha-Cyclodextrin-Based Rotaxanes Directed toward Fine Tuning, Bull. Chem. Soc. Jpn., 2019, 92, 1413–1418 CrossRef CAS.
- T. Shioiri, K. Ninomiya and S. Yamada, Diphenylphosphoryl Azide. New Convenient Reagent for a Modified Curtius Reaction and for Peptide Synthesis, J. Am. Chem. Soc., 1972, 94, 6203–6205 CrossRef CAS PubMed.
- See ESI.
- Y. Akae, H. Okamura, Y. Koyama, T. Arai and T. Takata, Selective Synthesis of a [3]Rotaxane Consisting of Size-Complementary Components and Its Stepwise Deslippage, Org. Lett., 2012, 14, 2226–2229 CrossRef CAS PubMed.
- Y. Akae, Y. Koyama, S. Kuwata and T. Takata, Cyclodextrin-Based Size-Complementary [3]Rotaxanes: Selective Synthesis and Specific Dissociation, Chem. – Eur. J., 2014, 20, 17132–17136 CrossRef CAS PubMed.
- Y. Akae, H. Sogawa and T. Takata, Synthesis of a Structure Definite α-Cyclodextrin Based Macromolecular [3]Rotaxane Using a Size-Complementary Method, Angew. Chem., Int. Ed., 2018, 57, 11742–11746 (Angew. Chem., 2018, 130, 11916–11920) CrossRef CAS PubMed.
- O. Shelef, S. Gnaim and D. Shabat, Self-Immolative Polymers: An Emerging Class of Degradable Materials With Distinct Disassem- bly Profiles, J. Am. Chem. Soc., 2021, 143, 21177–21188 CrossRef CAS PubMed.
- R. E. Yardley, A. R. Kenaree and E. R. Gilles, Triggering Depolym- erization: Progress and Opportunities for Self-Immolative Polymers, Macromolecules, 2019, 52, 6342–6360 CrossRef CAS.
- G. I. Peterson, M. B. Larsen and A. J. Boydston, Controlled Depolymerization: Stimuli-Responsive Self-Immolative Polymers, Macromolecules, 2012, 45, 7317–7328 CrossRef CAS.
Footnotes |
| † Present address: Department of Applied Chemistry, Graduate School of Engineering, The University of Osaka, 2-1 Yamadaoka, Suita, 565-0871 Osaka, Japan. |
| ‡ Present address: Institute of Inorganic Chemistry, RWTH Aachen University, Landoltweg 1a, 52074 Aachen, Germany. |
| This journal is © The Royal Society of Chemistry 2026 |