
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel approach to POx–chelate conjugates and a comparison of their antibacterial activity with POx systems containing amino groups
Marcelina
Bochenek
 a,
Barbara
Mendrek
a,
Agnieszka
Kowalczuk
a,
Karolina
Olszowska
a,
Łukasz
Sieroń
b,
Katarzyna
Gawron
c and
Natalia
Oleszko-Torbus
*a
a,
Barbara
Mendrek
a,
Agnieszka
Kowalczuk
a,
Karolina
Olszowska
a,
Łukasz
Sieroń
b,
Katarzyna
Gawron
c and
Natalia
Oleszko-Torbus
*a
aCentre of Polymer and Carbon Materials, Polish Academy of Sciences, M. Curie-Skłodowskiej 34, 41-819 Zabrze, Poland. E-mail: noleszko@cmpw-pan.pl
bDepartment of Medical Genetics, Medical University of Silesia, Medyków 18, 40-752 Katowice, Poland
cDepartment of Medical Microbiology, Medical University of Silesia, Medyków 18, 40-752 Katowice, Poland
First published on 25th March 2026
Abstract
This study presents a novel synthetic strategy for the preparation of antibacterial poly(2-oxazoline)-based (POx) conjugates with chelating agents. A well-defined random copolymer of 2-ethyl-2-oxazoline and 2-[N-Boc-5-aminopentyl]-2-oxazoline was synthesized via microwave-assisted cationic ring-opening polymerization (CROP). Following deprotection, the copolymer was conjugated with diethylenetriaminepentaacetic acid (DTPA). This work demonstrates a simplified and efficient route to antibacterial POx conjugates, in which amino groups are obtained after polymerization and directly coupled to a chelator. No post-polymerization modification of the polymer is required to generate amino groups in the substituents. Comprehensive characterization of both the conjugated POx and its amino-modified precursor was carried out, including structural analysis and physicochemical properties. Preliminary antibacterial activity and cytotoxicity were assessed, including bacterial membrane assays and microscopic imaging. Both systems exhibited activity against E. coli, a commonly used representative Gram-negative strain; however, they act via different mechanisms: the conjugate likely acts through cation chelation and its amino-modified precursor through cationic polymer-mediated interactions. Importantly, both polymers were cytocompatible with human fibroblasts. Overall, this study establishes a versatile platform for designing polymeric antimicrobial agents, offering tunable ion-chelating functionality for future biomedical applications.
Introduction
Nowadays, infectious diseases constitute a global public health concern, mostly due to the ability of microorganisms to spread and reproduce efficiently.1,2 Their presence has an impact on healthcare systems, resulting in social and economic burdens, which demand the development and testing of novel antimicrobial systems.3 In recent years, there has been growing interest in polymers with antibacterial properties,4–7 among which poly(2-oxazoline)s (POx) have attracted significant attention.8–10 POx are synthesized via living cationic ring-opening polymerization (CROP) of 2-oxazolines, enabling precise control over the molar mass, architecture, and narrow dispersity of the polymer. This controlled ‘living’ polymerization behavior has been demonstrated under various conditions and monomer/initiator systems, yielding well-defined POx with low polydispersity.11–14 These polymers have shown great potential in the design of advanced bioactive materials due to their excellent biocompatibility, low immunogenicity, and ease of chemical modification, allowing precise tuning of their properties.15–24 Typically, the antibacterial activity of POx-based systems is associated with the presence of amino groups introduced into their structure, which can interact with bacterial cell membranes and lead to their disruption.25 The antibacterial activity of amine-functionalized POx arises primarily from their cationic nature, which disrupts bacterial membranes, much like antimicrobial peptides. Protonated amino or ammonium groups bind to negatively charged bacterial membranes, destabilizing them and increasing their permeability.8,10,26–29 While this mechanism is dominant for cationic POx, other POx-based systems exert antibacterial effects through alternative strategies, such as DNA targeting, interference with bacterial metabolism, or modulation of bacterial signalling pathways, providing additional approaches to combat resistant pathogens.30–33We previously developed a novel POx-based system containing a chelating moiety with demonstrated antibacterial activity.34–36 These findings indicated that the antibacterial effect may arise from the ability of the conjugated chelator to bind divalent cations (Ca2+ and Mg2+), which play a crucial role in stabilizing bacterial cell membranes. This revealed a new, alternative strategy for achieving antibacterial activity in POx systems through ion chelation. Two conjugation strategies were employed to obtain the POx–chelator system: coupling via N-hydroxysuccinimide (NHS) ester activation and a reaction using a triazine-based coupling agent. These approaches required the presence of amino groups in POx, which were introduced via post-polymerization thio-click chemistry. In the present study, we demonstrate a novel POx–chelator conjugate obtained via CROP of a Boc-protected monomer, followed by deprotection and subsequent direct coupling of the chelator, thereby eliminating the need for post-polymerization modification. A well-defined random copolymer of 2-ethyl-2-oxazoline (EtOx) and 2-[N-Boc-5-aminopentyl]-2-oxazoline (PentNHBocOx) was synthesized and designated as POx–Boc. Subsequent deprotection yielded the amino-functionalized copolymer, referred to as POx–NH2. The complete removal of Boc groups was confirmed, and the absolute molar mass was precisely determined. The selection of monomers for the copolymer design was based on both structural and functional considerations. EtOx was chosen to ensure good aqueous solubility of the resulting copolymer, while PentNHBocOx was incorporated to introduce protected amino groups positioned at a distance from the polymer backbone,37 facilitating their later function. A random copolymer microstructure was employed to distribute the functional monomer units throughout the polymer chain, reducing local density and steric hindrance, which enhances the efficiency of the conjugation reaction. Additionally, this choice of monomer ratio limits the overall content of amino groups in order to minimize potential cytotoxicity and maintain water solubility during the coupling step.34–36,38 Subsequently, the diethylenetriaminepentaacetic acid (DTPA) chelating agent was attached to the copolymer, resulting in a POx–chelator system (POx–DTPA). The selected chelator was chosen due to its superior metal ion binding efficiency.39,40 Complete conjugation (substitution of all amino groups with DTPA) and ion-binding capability were confirmed. Importantly, the antibacterial activity of the obtained POx–DTPA conjugate against a model E. coli strain was evaluated and compared with its amino-functionalized precursor, which had not been previously studied. This study demonstrates a simplified method for the synthesis of antibacterial POx–chelator systems and provides new insights into the design of metal-ion-scavenging polymeric antimicrobials.
Experimental
Reagents
Methyl 4-nitrobenzenesulfonate (99%, Sigma-Aldrich, St Louis, MO, USA), n-propylamine (>99%, Sigma-Aldrich, St Louis, MO, USA), trifluoroacetic acid (TFA, 99%, Alfa Aesar, Haverhill, MA, USA), dichloromethane (DCM, 99.5%, Pure Land Chemical, Shanghai, China), methanol (gradient grade for HPLC, ChemSolve, Tewksbury, MA, USA), ethanol (99.8% for HPLC, POCH S.A., Gliwice, Poland), CaH2 (95%, Sigma-Aldrich, St Louis, MO, USA), diethylenetriaminepentaacetic acid (DTPA, >99%, Carl Roth GmbH, Karlsruhe, Germany), 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methyl morpholinium chloride (DMTMM, 97%, Acros Organics, Geel, Belgium), NaOH (pure for analysis, Stanlab Sp. z o.o., Lublin, Poland), MgCl2·6H2O (pure, POCH S.A., Gliwice, Poland), eriochrome black T (Loba Chemie, Mumbai, India), LiBr (99%, Alfa Aesar, Haverhill, MA, USA), NaNO3 (99%, Thermo Scientific, Waltham, MA, USA), ammonium buffer (pH 10, TarChem Sp. z o.o., Tarnowskie Góry, Poland), CaCl2 (95%, Sigma-Aldrich, St Louis, MO, USA) and ammonium purpurate (murexide, extra pure, Loba Chemie, Mumbai, India) were used as received. PentNHBocOx (>97%, Polymer Source, Dorval, QC, Canada) was dried under reduced pressure (1 × 10−4 mbar) using a high-vacuum pump set (nEXT Turbomolecular Pumping Station equipped with an nEXT85H turbomolecular pump and an nXDS6i backing pump, Edwards Ltd, Crawley, UK) for 8 h and stored under an argon atmosphere. EtOx (>99%, Sigma-Aldrich, St Louis, MO, USA) was distilled under reduced pressure, dried over CaH2, and distilled again. The purity of EtOx was analyzed by gas chromatography (Shimadzu Nexis GC 2030, Shimadzu Corporation, Kyoto, Japan) equipped with two chromatographic lines with FID and BID-2030 detectors and TR-5 (30 m × 0.32 mm ID; 0.25 µm, Thermo Scientific, Waltham, MA, USA) and SH-I-17 (30 m × 0.32 mm ID; 0.50 μm, Shimadzu Corporation, Kyoto, Japan) columns. Acetonitrile (ACN, >99%, Merck KGaA, Darmstadt, Germany) was prepared as described previously.41N,N-Dimethylformamide (DMF) (for HPLC, POCH S.A., Gliwice, Poland) was distilled before use.Biological reagents
Luria–Bertani (LB) broth (Sigma-Aldrich, St Louis, MO, USA), Mueller–Hinton (MH) broth (Sigma-Aldrich), Dulbecco's modified Eagle's medium (DMEM, Sigma-Aldrich, St Louis, MO, USA), 1-N-phenylnaphthylamine (NPN, Sigma-Aldrich, St Louis, MO, USA), trypan blue (PAN Biotech, Aidenbach, Germany) and Triton X-100 (Sigma-Aldrich, St Louis, MO, USA) were used as received. The Escherichia coli TOP10 strain was used. Primary dermal fibroblasts (PDFs) were purchased from the American Type Culture Collection (ATCC, Cat. No. PCS-201-010™).Synthesis of POx–Boc
CROP was carried out in a microwave reactor (Anton Paar Monowave 400, Graz, Austria) at 140 °C. The progress of copolymerization was monitored by nuclear magnetic resonance (1H NMR). Methyl 4-nitrobenzenesulfonate, dissolved in acetonitrile, was placed in a microwave vial equipped with a magnetic stirrer. The copolymer was synthesized via a one-pot reaction, in which EtOx and PentNHBocOx were added simultaneously to the vial under an argon atmosphere and polymerized. After 2 hours, the reaction was quenched with n-propylamine. Following confirmation of total conversion by 1H NMR, the copolymer was purified by evaporation of acetonitrile, dissolution in methanol, dialysis (1 kDa Spectra/Por® membranes), and drying under reduced pressure. After purification, the copolymer was named POx–Boc and analyzed by 1H NMR to determine the chemical composition.Deprotection of POx–Boc
The Boc-protected copolymer was dissolved in DCM, followed by the addition of TFA in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) ratio, using a 1.5 mol% excess relative to the Boc groups in the polymer. The reaction was carried out for 1 h at room temperature (RT) under stirring, after which TFA and DCM were evaporated under reduced pressure. The polymer in its salt form, containing trifluoroacetate as the counterion (named POx–NH3+TFA−), was used for the conjugation reaction with DTPA. To remove the TFA anion, POx–NH3+TFA− was dissolved in methanol and treated with Amberlyst A-21 ion-exchange resin (Thermo Scientific, Waltham, MA, USA). Then, methanol was evaporated, and the copolymer was purified by dialysis against water (1 kDa Spectra/Por® membranes) and lyophilization. The copolymer containing amino groups (POx–NH2) was used for cytotoxicity and antibacterial activity studies.
:1 (v/v) ratio, using a 1.5 mol% excess relative to the Boc groups in the polymer. The reaction was carried out for 1 h at room temperature (RT) under stirring, after which TFA and DCM were evaporated under reduced pressure. The polymer in its salt form, containing trifluoroacetate as the counterion (named POx–NH3+TFA−), was used for the conjugation reaction with DTPA. To remove the TFA anion, POx–NH3+TFA− was dissolved in methanol and treated with Amberlyst A-21 ion-exchange resin (Thermo Scientific, Waltham, MA, USA). Then, methanol was evaporated, and the copolymer was purified by dialysis against water (1 kDa Spectra/Por® membranes) and lyophilization. The copolymer containing amino groups (POx–NH2) was used for cytotoxicity and antibacterial activity studies.
Conjugation of POx–NH3+TFA− with the chelating agent
DTPA (ca. 80 eq. per amino group in the polymer) was dissolved in H2O (c ∼1 g mL−1) after the addition of NaOH (5 mol L−1) until the solution reached pH 8.5. DMTMM (ca. 5 eq. per polymeric amino group) was dissolved in water (c ∼0.05 g mL−1) and added to the aqueous DTPA solution under stirring. Then, an aqueous solution of POx–NH3+TFA− (c ∼0.05 g mL−1) was added to the mixture. The reaction solution was stirred overnight at RT, then transferred to a 1 kDa Spectra/Por® membrane and dialyzed against water. The polymer was recovered by freeze-drying and named POx–DTPA.Measurements
where wPEtOx is the weight ratio of 2-ethyl-2-oxazoline in the copolymer and wPBocOx is the weight ratio of 2-[N-Boc-5-aminopentyl]-2-oxazoline;
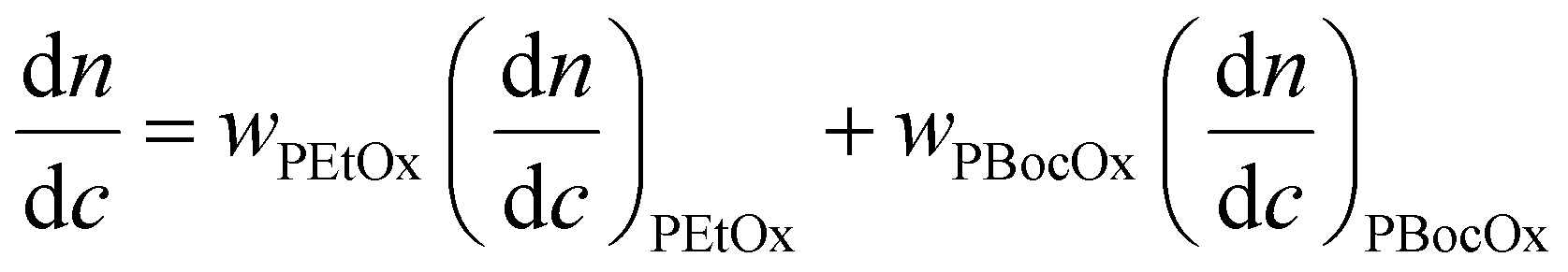
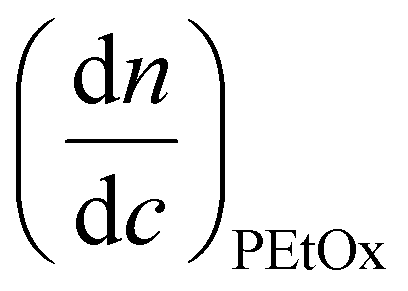 is the refractive index increment of the homopolymer of 2-ethyl-2-oxazoline and is equal to 0.083 mL g−1, and
is the refractive index increment of the homopolymer of 2-ethyl-2-oxazoline and is equal to 0.083 mL g−1, and 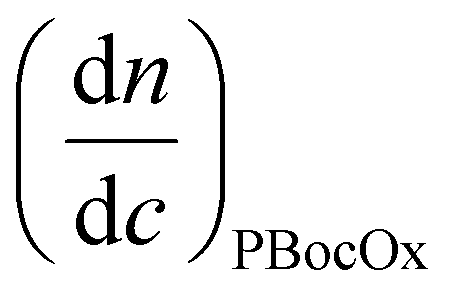 is the refractive index increment of the homopolymer of 2-[N-Boc-5-aminopentyl]-2-oxazoline (measured independently) and is equal to 0.068 mL g−1. The refractive index increment (dn/dc) of the copolymer after the deprotection was calculated from the equation
is the refractive index increment of the homopolymer of 2-[N-Boc-5-aminopentyl]-2-oxazoline (measured independently) and is equal to 0.068 mL g−1. The refractive index increment (dn/dc) of the copolymer after the deprotection was calculated from the equationwhere wPEtOx is the weight ratio of 2-ethyl-2-oxazoline in the copolymer and wPNH2Ox is the weight ratio of 2-[5-aminopentyl]-2-oxazoline;
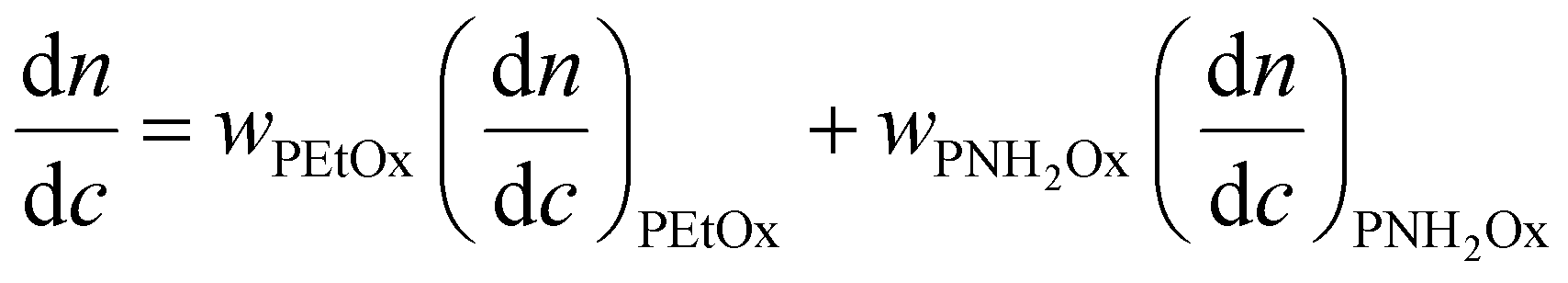
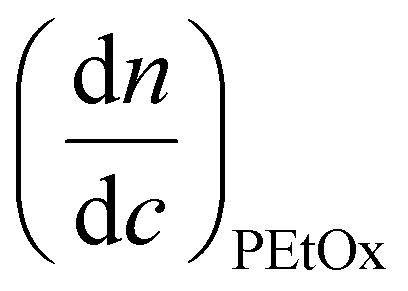 is the refractive index increment of the homopolymer of 2-ethyl-2-oxazoline and is equal to 0.083 mL g−1, and
is the refractive index increment of the homopolymer of 2-ethyl-2-oxazoline and is equal to 0.083 mL g−1, and 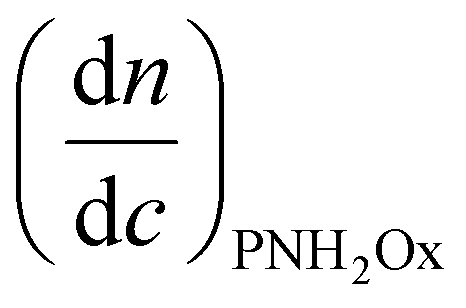 is the refractive index increment of the homopolymer of 2-[5-aminopentyl]-2-oxazoline and is equal to 0.049 mL g−1. The refractive index increments of all homopolymers were determined independently using an SEC-3010 dn/dc refractometer (λ = 620 nm, WGE Dr Bures, Düren, Germany). Five concentrations in DMF were prepared, with a minimum of three measurements per concentration. Data were analyzed using BI-DNDCW software.
:ethanol 1:1 v/v (2.5 mg mL−1) were added to the solution. A change in the color of the solution from transparent to light purple was observed.
000×.
is the refractive index increment of the homopolymer of 2-[5-aminopentyl]-2-oxazoline and is equal to 0.049 mL g−1. The refractive index increments of all homopolymers were determined independently using an SEC-3010 dn/dc refractometer (λ = 620 nm, WGE Dr Bures, Düren, Germany). Five concentrations in DMF were prepared, with a minimum of three measurements per concentration. Data were analyzed using BI-DNDCW software.
:ethanol 1:1 v/v (2.5 mg mL−1) were added to the solution. A change in the color of the solution from transparent to light purple was observed.
000×.
Antibacterial activity assays
Outer membrane (OM) permeability was assessed using NPN (20 μM in PBS) with E. coli (OD600 = 0.5) incubated with POx–NH2, POx–DTPA-Ca or POx–DTPA (1 mg mL−1) prior to fluorescence measurement (Ex/Em 355/460 nm) using a Victor X5 plate reader (PerkinElmer, Waltham, MA, USA). POx–DTPA-Ca was prepared as follows: POx–DTPA was dissolved in water, followed by the addition of CaCl2 in equimolar amounts relative to DTPA units in the conjugate. The mixture was stirred for 24 h, dialyzed against water (1 kDa Spectra/Por® membrane) and lyophilized. For inner membrane (IM) permeability assays, E. coli suspensions were treated with POx–NH2 or POx–DTPA (1 mg mL−1), followed by staining with trypan blue (0.4%) and analysis by flow cytometry using a BD FACSAria instrument (BD Biosciences, San Jose, CA, USA; PE channel). Triton X-100 (0.01%) served as a positive control. Results from three independent biological replicates confirmed the trends observed in the NPN and flow cytometry assays.Determination of minimum inhibitory concentration (MIC)
MICs for POx–DTPA, POx–DTPA-Ca and POx–NH2 were determined using E. coli, applying the microdilution method. A fresh MH broth was prepared, and the bacterial inoculum was standardized to an optical density (OD600) of 0.5, corresponding to approximately 1 × 108 CFU mL−1, and then diluted to 1 × 106 CFU mL−1. Serial two-fold dilutions of the polymers were prepared in the broth and added to sterile 96-well microtiter plates. Each well contained 100 μL of the polymer solution and 100 μL of the bacterial inoculum, resulting in a final volume of 200 μL per well. Growth controls (broth and inoculum) and sterility controls (broth only) were included. Plates were incubated at 37 °C for 24 h. The MIC was determined as the lowest conjugate concentration that completely inhibited visible bacterial growth, measured at OD600 or by visual inspection for turbidity. All tests were performed in three independent biological experiments, each with three technical replicates. MIC values are reported as the average of the three experiments.Cytotoxicity studies
PDFs were seeded in 24-well plates at a density of 5 × 104 cells per well and maintained in DMEM supplemented with 10% FBS, 1% L-glutamine, 1000 U mL−1 penicillin, 100 μg mL−1 streptomycin, and 250 μg mL−1 amphotericin B at 37 °C under a 5% CO2 atmosphere for 24 h. Subsequently, POx–NH2 or POx–DTPA (0.1 and 1 mg mL−1) were added to the cultures and incubated for 24, 48, and 72 h. Cell viability was evaluated at each time point using a 10% Alamar Blue solution. Untreated cells served as controls. At each time point, the culture medium was replaced with 200 μL of Alamar Blue solution, followed by incubation at 37 °C for 1 h. After incubation, 100 μL of medium was transferred to a 96-well plate, and cell viability was measured with a VICTOR™ Multilabel Plate Reader (PerkinElmer, Waltham, MA, USA) using a 1 s exposure time.Results and discussion
Synthesis and characterization of the copolymer precursor (POx–Boc) and its deprotection
Microwave-assisted CROP of PentNHBocOx and EtOx, initiated with methyl 4-nitrobenzenesulfonate and terminated with n-propylamine, was performed according to the scheme (Fig. 1). The obtained copolymer was named POx–Boc. The characterization of POx–Boc is shown in Table 1. | ||
| Fig. 1 Scheme for the synthesis of the random copolymer of PentNHBocOx and EtOx (POx–Boc), its deprotection (POx–NH3+TFA−) and ion exchange (POx–NH2). | ||
| POx–Boc | POx–NH2 | ||||||
|---|---|---|---|---|---|---|---|
| DP(t) EtOx:PentNHBocOx |
M n(t) (g mol−1) | Conv. (%) | DP(NMR) EtOx:PentNHBocOx |
M n(SEC-MALLS) (g mol−1) | Đ | M n(SEC-MALLS) (g mol−1) | Đ |
| DP(t) – targeted DP; Mn(t) – theoretical Mn; Conv. – total conversion of monomers; Mn(SEC-MALLS) – absolute molar masses; Đ – dispersity. | |||||||
| 70:30 |
14610 |
100 | 70:30 |
15000 |
1.45 | 13000 |
1.15 |
The total conversion of monomers of POx–Boc was calculated using 1H NMR spectra based on the integration ratio of signals corresponding to protons of methylene groups in the main chain, originating from the monomer and polymer at δ = 4.20–4.24 ppm, δ = 3.79–3.85 ppm and δ = 3.25–3.60 ppm, respectively. Mn shown is the absolute molar mass, determined by SEC-MALLS and calculated using the refractive index increment derived from the equations provided in the experimental section. The absolute molar mass of POx–Boc was in good agreement with the theoretical value, and the peak was monomodal (Fig. 2).
 | ||
| Fig. 2 SEC traces of POx–Boc and POx–NH2 (DMF with 5 mmol LiBr as the eluent, 1 mL min−1, RI signal). | ||
The Boc-protecting groups were removed from the copolymer substituents (DCM/TFA, RT), following established procedures,30,43 yielding POx–NH3+TFA−, as depicted in the scheme in Fig. 1. Subsequently, in order to remove the resulting TFA anion, the deprotected copolymer was treated with ion-exchange resins, according to ref. 44, yielding POx with pendant amino functions (POx–NH2). Quantitative deprotection was confirmed by NMR spectroscopy. Fig. 3 presents the 1H NMR spectra of POx–Boc and POx–NH2.
 | ||
| Fig. 3 1H NMR of POx–Boc (10 mg mL−1 solution in CDCl3) and POx–NH2 (10 mg mL−1 solution in D2O). | ||
The signal corresponding to the protons of the tert-butyl group at δ = 1.45 ppm (signal k) has vanished, thus indicating the total removal of the Boc groups.
A monomodal molar mass distribution was observed for POx–NH2 (Fig. 2), confirming the successful removal of the protecting groups without polymer degradation. The decrease in the polymer's molar mass, resulting from the detachment of the protecting moieties, was also observed (Table 1).
To briefly summarize, it was possible to quantitatively remove the protective groups from the macromolecular substituents and remove the TFA anion from the copolymer to yield POx with pendant amino groups.
Synthesis of POx–DTPA and the ion-trapping study
The chelating agent DTPA was conjugated to POx–NH3+TFA− using a method based on the triazine-derived coupling agent DMTMM.45 The reaction scheme is presented in Fig. 4. | ||
| Fig. 4 Scheme of POx–NH3+TFA− and DTPA conjugation leading to POx–DTPA. | ||
After the reaction was completed, the conjugate was designated as POx–DTPA and characterized by 13C NMR (Fig. 5).
 | ||
| Fig. 5 13C NMR spectra of POx–NH3+TFA− and POx–DTPA (15 mg mL−1 solution in D2O). | ||
The complete substitution with DTPA was confirmed by the shift of the peak originating from the carbon atom of the methylene group at the nitrogen atom from 42.08 ppm to 42.03 ppm. In addition, the shift of the peak in the POx–DTPA spectrum at 177.03 ppm, originating from the carbon atoms of the amide group, and also the shifts of the peaks at 173.26 ppm and 179.57 ppm, derived from the carbon atoms of the carboxyl groups of DTPA, demonstrated the reaction between POx–NH3+TFA− and DTPA. All amino groups in the copolymer (30 mol%) were substituted with the chelating agent DTPA. This design ensures that the density of functional groups is identical in both POx–NH2 and POx–DTPA, allowing a direct comparison of their activities.
This experiment showed that a chelating agent can be efficiently attached to copolymers obtained via the CROP of 2-oxazolines with a protected amino group, followed by deprotection.
The obtained POx–DTPA conjugate was able to entrap model Mg2+ and Ca2+ ions with 100% efficiency, as confirmed by the eriochrome black T and ammonium purpurate probes, respectively (Table 2). After adding eriochrome black T to the conjugate solution in water, to which an equimolar amount of magnesium ions relative to DTPA in the copolymer had been previously added, the solution immediately changed color from transparent to purple. This color change indicates the absence of free magnesium ions in the solution. Similarly, immediately after the addition of murexide to the solution of POx–DTPA in water, to which an equimolar amount of calcium ions relative to DTPA in the copolymer had been previously added, the mixture turned light purple, indicating the absence of free calcium ions in the solution. The highly efficient trapping of metal ions from solution highlights the strong potential of these polymeric systems.
| Ions | Mol of DTPA in the sample (×10−5) | Mol of ions added to the sample (×10−5) | Mol of chelated ions (×10−5) | Chelation efficiency (%) |
|---|---|---|---|---|
| Ca2+ | 6.23 | 6.22 | 6.22 | 100 |
| Mg2+ | 6.18 | 6.08 | 6.08 | 100 |
Following dialysis to remove indicators from the conjugate solutions, the resulting conjugates complexed with calcium and magnesium ions were designated as POx–DTPA-Ca and POx–DTPA-Mg, correspondingly. The pH of the aqueous solution of POx–DTPA with complexed ions increased to 6–7, indicating that carboxyl groups of the chelating agent were involved in the formation of chemical bonds with Ca2+ and Mg2+.
Elemental analysis of the samples was carried out using EDX to confirm the presence of ions complexed by the conjugate. The conjugate samples containing complexed calcium or magnesium ions were prepared by drop-casting onto a silicon substrate. Additionally, the spatial distribution of the complexed ions across the substrate surface was analyzed in order to assess their localization within the sample. The EDX spectrum, displaying X-ray energy versus intensity, along with the corresponding EDX mapping analysis, is presented in Fig. 6.
 | ||
| Fig. 6 The EDX curves of POx–DTPA-Ca, POx–DTPA-Mg and POx–DTPA (A); EDX mapping analysis of POx–DTPA-Ca and POx–DTPA-Mg. Elements at the same spot correspond to Ca and Mg, respectively; the scale bar is 20 μm (B). Samples were prepared on silicon wafers from POx–DTPA (10 mg mL−1 solution in water) complexed with Ca2+ and Mg2+ ions in equimolar amounts relative to DTPA units, incubated for 24 h, and air-dried, followed by drying under reduced pressure. | ||
A significant increase in signal intensity was observed in the carbon region (0.27–0.28 keV) of the spectra, as well as in the regions corresponding to oxygen (0.53 keV) and nitrogen (0.39–0.40 keV) present in the structure of the tested conjugates. The signal from Si (1.75 keV) was ascribed to the silicon wafer used for sample preparation. Our results confirmed the complexation of Ca and Mg ions with the conjugates, as evidenced by the presence of peaks at energy values characteristic of these ions: 3.69–3.80 keV for calcium and 1.19–1.38 keV for magnesium. These peaks were not observed in the EDX curves of the control POx–DTPA, which was not complexed with metal ions.
The EDX mapping results, shown in Fig. 6B, illustrate the surface coverage of Ca and Mg ions through their characteristic X-ray signals. The uniform distribution of calcium and magnesium ions across the surface of the analyzed sample was observed. Elemental maps were acquired to visualize the spatial localization of these ions, providing insight into how the complexed species are arranged within the conjugate layer. This type of analysis enables qualitative assessment of elemental dispersion and may serve as a basis for further quantitative evaluation.
Comparison of antibacterial activity and in vitro cytotoxicity of POx–DTPA and POx–NH2
To compare the antibacterial activity of POx–DTPA and POx–NH2, a series of microbiological assays were conducted to evaluate their capacity to disrupt both the IM and OM of a model E. coli strain commonly used for preliminary antibacterial screening. These assays included assessments of membrane permeability and integrity, allowing for the investigation of potential differences in the mechanisms of action between the two conjugates. This dual-membrane approach provides a more comprehensive understanding of the antibacterial effects and membrane-targeting capabilities of the tested compounds. Since the number of functional groups is identical in both POx–NH2 and POx–DTPA, this allows a direct comparison of their activities. Additionally, the activity of POx–DTPA saturated with model calcium ions (POx–DTPA-Ca) against E. coli was tested.In the first stage, the minimum inhibitory concentration (MIC) was determined for POx–DTPA, POx–DTPA-Ca and POx–NH2, using the broth microdilution method against E. coli. The MIC is defined as the lowest concentration of an antimicrobial agent that completely inhibits the visible growth of the tested microorganism. POx–DTPA exhibited a MIC value of 0.15 mg mL−1, whereas POx–DTPA-Ca showed a MIC of 1.25 mg mL−1. This indicates a significant reduction in antibacterial activity upon calcium saturation, supporting the role of metal ion chelation in the mechanism of action of POx–DTPA. Literature reports indicate that MIC values of cationic POx-based antibacterial systems against E. coli typically range from a few hundredths of mg mL−1 to a few mg mL−1, depending on the polymer architecture and mode of action.46–48 In this context, the MIC value of 0.15 mg mL−1 observed for our new chelating system POx–DTPA against E. coli places it well within the activity range reported for POx-based antimicrobial systems. The MIC value observed for POx–NH2 was 1 mg mL−1, indicating that a higher concentration was required to exert a lethal effect compared to POx–DTPA.
To assess the ability of POx–DTPA and POx–NH2 to damage the integrity of the IM, a trypan blue exclusion assay was conducted. E. coli cultures were incubated with the tested polymers (1 mg mL−1 of each), followed by staining with the dye. The uptake of trypan blue, indicative of membrane damage, was quantified using flow cytometry. Untreated cells served as a negative control, while Triton X-100, a known membrane-disrupting agent, was used as a positive control for IM disruption.49 Results are depicted in the fluorescence histograms (Fig. 7).
 | ||
| Fig. 7 Flow cytometry analysis of trypan blue staining of E. coli treated with POx–DTPA and POx–NH2. For membrane permeability assays, E. coli suspensions were treated with POx–NH2 and POx–DTPA (1 mg mL−1), followed by staining with trypan blue (0.4%) (Triton X-100 (0.01%) served as a positive control). | ||
The fluorescence intensity of E. coli cells treated with POx–DTPA and POx–NH2 and subsequently stained with trypan blue exhibited a noticeable shift toward the fluorescence profile characteristic of Triton X-100-treated bacteria, which is commonly used as a positive control for membrane disruption. This shift indicates a loss of membrane integrity, specifically suggesting an increase in IM permeability. The comparable fluorescence patterns observed for both polymer-treated samples imply that POx–DTPA and POx–NH2 induce IM permeabilization to a similar extent. These findings are consistent with the ability of both conjugates to facilitate the entry of the otherwise membrane-impermeant trypan blue dye into the cytoplasm, reflecting compromised membrane barrier function.
To evaluate potential damage to the bacterial OM, E. coli cultures were incubated with the tested polymers in LB medium (1 mg mL−1) in the presence of NPN dye, followed by fluorescence intensity measurements. NPN is a hydrophobic fluorescent probe that exhibits minimal fluorescence in aqueous environments or in intact bacterial cells. However, upon disruption of the OM, NPN is able to partition into the phospholipid-rich periplasmic space, resulting in a significant increase in fluorescence.50 This method is widely used to assess the integrity of the bacterial OM and to detect membrane-permeabilizing agents. The fluorescence intensities of NPN observed after treatment with POx–DTPA and POx–NH2 are presented in Fig. 8. Additionally, this test was also performed for POx–DTPA-Ca (saturated with calcium ions).
 | ||
| Fig. 8 Relative fluorescence units (RFUs) for E. coli cultures treated for 1 h with NPN (20 μM in PBS) in the presence of POx–DTPA, POx–DTPA-Ca and POx–NH2 (1 mg mL−1) (untreated E. coli used as a control). | ||
An increase in fluorescence intensity was observed in E. coli cells treated with either POx–DTPA or POx–NH2 compared to the untreated control, which indicates disruption of the OM. In contrast, the sample of POx–DTPA pre-saturated with calcium ions exhibited fluorescence intensity at a level comparable to that of the control, indicating that it did not induce outer membrane disruption. Notably, the fluorescence intensity for POx–DTPA was slightly higher than that observed for POx–NH2.
These findings suggest that both POx–DTPA and POx–NH2 have the capacity to damage the integrity of bacterial membranes, thereby facilitating the increased uptake of hydrophobic molecules such as hydrophobic dyes. Differences in fluorescence intensity in both copolymer-treated cells may indicate distinct mechanisms of action, possibly related to the chelating properties of the DTPA moiety. Specifically, the POx–DTPA conjugate may exert its disruptive effect through the complexation of divalent cations (Ca2+ and Mg2+), which are known to play a crucial role in stabilizing the lipopolysaccharide (LPS) layer of the OM of Gram-negative bacteria. This mechanism was indirectly confirmed by the NPN assay, in which calcium-saturated POx–DTPA exhibited no antibacterial activity. In contrast, POx–NH2, which lacks chelating groups, may interact with the membrane primarily through electrostatic interactions or hydrogen bonding with membrane components, leading to membrane perturbation via a different mechanism. Understanding these differences is crucial for tailoring polymer conjugates for targeted antibacterial applications, as the mode of membrane interaction influences both efficacy and specificity. Further studies, including membrane biophysical analyses, would allow the elucidation of the precise molecular interactions that contribute to the enhanced permeabilization induced by POx–DTPA.
To further investigate the antibacterial activity of the tested polymers, SEM analysis was performed. The E. coli cultures were incubated for 2 h in the presence of POx–DTPA or POx–NH2 (1 mg mL−1 of each). Next, samples were collected, applied onto solid substrates, air-dried, and subsequently imaged using SEM. As shown in Fig. 9A, dense bacterial colonies are visible prior to exposure to the polymer solutions. After incubation with POx–DTPA or POx–NH2, no intact bacterial cells were detected (Fig. 9B and C). Instead, only residual debris or fragments of disrupted bacterial cells can be observed, which together indicate effective bactericidal activity and severe membrane damage induced by both tested polymers.
 | ||
| Fig. 9 SEM images of E. coli colonies (108 CFU mL−1) (A) and polymers incubated with bacteria for 2 h: POx–DTPA (1 mg mL−1 in water with E. coli (108 CFU mL−1)) (B) and POx–NH2 (1 mg mL−1 in water with E. coli (108 CFU mL−1)) (C). Samples were prepared by depositing 3 μL of the samples onto silicon wafers, incubating them for 2 h at RT and air-drying. Prior to imaging, the samples were sputter-coated with a 5 nm gold layer. | ||
In addition to the studies of the antibacterial activity of the polymer systems, their cytotoxicity toward model human skin fibroblasts was assessed (Fig. 10). The results demonstrated that both POx–DTPA and POx–NH2 exhibit negligible cytotoxicity within the concentration range corresponding to their antibacterial activity, even after 72 h of exposure. This observation indicates biocompatibility and suggests that the polymer conjugates are safe for potential biomedical applications.
 | ||
| Fig. 10 The cytotoxicity assays of POx–DTPA and POx–NH2 at 1 and 0.1 mg mL−1, employing model human skin fibroblasts. The results are presented as a percentage of untreated (control) cells, which constituted 100%. | ||
Conclusions
This study demonstrates an efficient route to POx–chelator conjugates via direct CROP of functionalized monomers, followed by deprotection and DTPA conjugation, providing a simplified bioactive polymeric platform. Structural characterization confirmed polymer integrity and successful conjugation, while the resulting POx–DTPA effectively chelated divalent cations essential for bacterial membrane stability. Microbiological assays using a model E. coli strain revealed efficient inner membrane disruption by both the conjugate and its amino-functionalized precursor, with POx–DTPA causing slightly greater OM damage. In future studies, a broader panel of bacterial strains, including both Gram-negative and Gram-positive species, merits assessment. Both polymers exhibited minimal cytotoxicity, highlighting their potential as versatile antimicrobial materials. The obtained POx system combines structural versatility with tunable monomer composition and functionalization, enabling precise control over physicochemical and biological properties. In this context, the platform presented here serves as a robust foundation for future optimization rather than representing definitive endpoint structures. Taken together, these findings establish POx-based conjugates as a versatile and tunable platform for antimicrobial materials.Author contributions
All authors contributed to this study. M. B., B. M., and A. K.: synthesis and characterization of the conjugate, analysis of the obtained results, and preparation of the manuscript; K. O.: EDX and SEM analyses; Ł. S. and K. G.: bacteriological studies and cytotoxicity tests; N. O. T.: concept, synthesis and characterization of the conjugate, analysis of the obtained results, and preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
Repository for open data: https://doi.org/10.18150/PNTKPM.Acknowledgements
This work was supported by the National Science Centre, project 2021/43/B/ST4/01493.References
- D. E. Bloom, S. Black and R. Rappuoli, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 4055–4059 CrossRef CAS PubMed.
- Q. Liu, J. Deng, W. Yan, C. Qin, M. Du, Y. Wang, S. Zhang, M. Liu and J. Liu, Glob. Health Res. Policy, 2024, 9, 23 CrossRef PubMed.
- B. Punz, C. Christ, A. Waldl, S. Li, Y. Liu, L. Johnson, V. Auer, O. Cardozo, P. M. A. Farias, A. C. D. S. Andrade, A. Stingl, G. Wang, Y. Li and M. Himly, Environ. Sci. Nano, 2025, 12, 1710–1739 RSC.
- D. Olmos and J. González-benito, Polymers, 2021, 13, 613 CrossRef CAS PubMed.
- S. Alkarri, H. Bin Saad and M. Soliman, Polymers, 2024, 16, 771 CrossRef CAS PubMed.
- S. C. Williams, M. B. Chosy, C. K. Jons, C. Dong, A. N. Prossnitz, X. Liu, H. L. Lopez Hernandez, L. Cegelski and E. A. Appel, ACS Cent. Sci., 2025, 11, 486–496 CrossRef CAS PubMed.
- R. Bryaskova, N. Philipova, V. Bakov and N. Georgiev, Appl. Sci., 2025, 15, 1780 CrossRef CAS.
- M. Zhou, W. Jiang, J. Xie, W. Zhang, Z. Ji, J. Zou, Z. Cong, X. Xiao, J. Gu and R. Liu, ChemMedChem, 2021, 16, 309–315 CrossRef CAS PubMed.
- W. Jiang, M. Zhou, S. Chen, J. Xie, M. Chen, H. Zhang, Y. Wu, X. Chen and R. Liu, J. Am. Chem. Soc., 2023, 145, 25753–25765 CrossRef CAS PubMed.
- M. Concilio, R. Garcia Maset, L. P. Lemonche, V. Kontrimas, J. I. Song, S. K. Rajendrakumar, F. Harrison, C. R. Becer and S. Perrier, Adv. Healthcare Mater., 2023, 12, 2301961 CrossRef CAS PubMed.
- S. Borova and R. Luxenhofer, Beilstein J. Org. Chem., 2023, 19, 217–230 CrossRef CAS PubMed.
- Y. C. M. Wu and T. M. Swager, J. Am. Chem. Soc., 2019, 141, 12498–12501 CrossRef CAS PubMed.
- C. Petit, B. Grassl, E. Mignard, K. P. Luef, F. Wiesbrock and S. Reynaud, Polym. Chem., 2017, 8, 5910–5917 RSC.
- C. Guerrero-Sanchez, R. Hoogenboom and U. S. Schubert, Chem. Commun., 2006, 3797–3799 RSC.
- R. Luxenhofer, Y. Han, A. Schulz, J. Tong, Z. He, A. V. Kabanov and R. Jordan, Macromol. Rapid Commun., 2012, 33, 1613–1631 CrossRef CAS PubMed.
- B. Guillerm, S. Monge, V. Lapinte and J. J. Robin, Macromol. Rapid Commun., 2012, 33, 1600–1612 CrossRef CAS PubMed.
- W. Wałach, N. Oleszko-Torbus, A. Utrata-Wesolek, M. Bochenek, E. Kijeńska-Gawrońska, Z. Górecka, W. Święszkowski and A. Dworak, Polymers, 2020, 12, 295 CrossRef PubMed.
- S. Jana and M. Uchman, Prog. Polym. Sci., 2020, 106, 101252 CrossRef CAS.
- N. Oleszko-Torbus, Polym. Rev., 2022, 62, 529–548 CrossRef CAS.
- T. Lorson, M. M. Lübtow, E. Wegener, M. S. Haider, S. Borova, D. Nahm, R. Jordan, M. Sokolski-Papkov, A. V. Kabanov and R. Luxenhofer, Biomaterials, 2018, 178, 204–280 CrossRef CAS PubMed.
- N. Oleszko-Torbus, B. Mendrek, A. Kowalczuk, A. Utrata-Wesołek, A. Dworak and W. Wałach, Materials, 2020, 13, 3403 CrossRef CAS PubMed.
- N. Oleszko-Torbus, M. Bochenek, A. Utrata-Wesołek, A. Kowalczuk, A. Marcinkowski, A. Dworak, A. Fus-Kujawa, A. L. Sieroń and W. Wałach, Materials, 2020, 13, 2702 CrossRef CAS PubMed.
- M. N. Leiske, Eur. Polym. J., 2023, 185, 111832 CrossRef CAS.
- A. Lusina, T. Nazim and M. Cegłowski, Polymers, 2022, 14, 4176 CrossRef CAS PubMed.
- N. Oleszko-Torbus, M. Bochenek and A. Kowalczuk, Polym. Chem., 2025, 16, 2574–2579 RSC.
- W. Jiang, M. Zhou, Z. Cong, J. Xie, W. Zhang, S. Chen, J. Zou, Z. Ji, N. Shao, X. Chen, M. Li and R. Liu, Angew. Chem., Int. Ed., 2022, 61, e202200778 CrossRef CAS PubMed.
- M. Lin and J. Sun, Biosaf. Health, 2022, 4, 269–279 CrossRef.
- C. J. Waschinski and J. C. Tiller, Biomacromolecules, 2005, 6, 235–243 CrossRef CAS PubMed.
- M. Zhou, Y. Qian, J. Xie, W. Zhang, W. Jiang, X. Xiao, S. Chen, C. Dai, Z. Cong, Z. Ji, N. Shao, L. Liu, Y. Wu and R. Liu, Angew. Chem., Int. Ed., 2020, 59, 6412–6419 CrossRef CAS PubMed.
- C. Dai, M. Zhou, W. Jiang, X. Xiao, J. Zou, Y. Qian, Z. Cong, Z. Ji, L. Liu, J. Xie, Z. Qiao and R. Liu, J. Mater. Sci. Technol., 2020, 59, 220–226 CrossRef CAS.
- S. Buyuksungur, T. E. Tanir, V. Hasirci and N. Hasirci, Biomacromolecules, 2025, 26, 3139–3154 CrossRef CAS PubMed.
- L. Benski and J. C. Tiller, Eur. Polym. J., 2019, 120, 109233 CrossRef.
- A. M. Kelly, V. Kaltenhauser, I. Mühlbacher, K. Rametsteiner, H. Kren, C. Slugovc, F. Stelzer and F. Wiesbrock, Macromol. Biosci., 2013, 13, 116–125 CrossRef CAS PubMed.
- M. Bochenek, B. Mendrek, W. Wałach, A. Foryś, J. Kubacki, Ł. Jałowiecki, J. Borgulat, G. Płaza, A. Klama-Baryła, A. Sitkowska, A. Kowalczuk and N. Oleszko-Torbus, Polym. Chem., 2024, 15, 2387–2396 RSC.
- B. Mendrek, A. Kowalczuk, W. Wałach, M. Bochenek and N. Oleszko-Torbus, J. Mol. Liq., 2025, 428, 127540 CrossRef CAS.
- M. Bochenek, B. Mendrek, A. Kowalczuk, W. Wałach, Ł. Jałowiecki, J. Borgulat, G. Płaza, J. Kubacki, M. Sikora, A. Fus-Kujawa, Ł. Sieroń, K. Gawron and N. Oleszko-Torbus, Biomater. Sci., 2025, 13, 3876–3886 RSC.
- S. Cesana, J. Auernheimer, R. Jordan, H. Kessler and O. Nuyken, Macromol. Chem. Phys., 2006, 207, 183–192 CrossRef CAS.
- M. Bochenek, B. Mendrek, W. Wałach, A. Kowalczuk, A. Utrata-Wesołek, A. Fus-Kujawa and N. Oleszko-Torbus, eXPRESS Polym. Lett., 2025, 19, 1226–1237 CrossRef CAS.
- G. Chauhan, K. K. Pant and K. D. P. Nigam, Environ. Sci.: Processes Impacts, 2015, 17, 12–40 RSC.
- E. Zeini Jahromi and J. Gailer, Metallomics, 2012, 4, 995–1003 Search PubMed.
- N. Oleszko-Torbus, B. Mendrek, W. Wałach, A. Fus-Kujawa, V. Mitova, N. Koseva and A. Kowalczuk, Polym. Chem., 2024, 156, 742–753 Search PubMed.
- O. Gjems, Analyst, 1960, 85, 738–744 Search PubMed.
- Z. He, L. Miao, R. Jordan, D. S-Manickam, R. Luxenhofer and A. V. Kabanov, Macromol. Biosci., 2015, 15, 1004–1020 Search PubMed.
- M. Hartlieb, D. Pretzel, K. Kempe, C. Fritzsche, R. M. Paulus, M. Gottschaldt and U. S. Schubert, Soft Matter, 2013, 9, 4693–4704 Search PubMed.
- D. Majonis, I. Herrera, O. Ornatsky, M. Schulze, X. Lou, M. Soleimani, M. Nitz and M. A. Winnik, Anal. Chem., 2010, 82, 8961–8969 Search PubMed.
- D. Kozon, J. Mierzejewska, T. Kobiela, A. Grochowska, K. Dudnyk, A. Głogowska, A. Sobiepanek, A. Kuźmińska, T. Ciach, E. Augustynowicz-Kopeć and D. Jańczewski, Macromol. Biosci., 2019, 19, 1–10 Search PubMed.
- C. Krumm, M. Hijazi, S. Trump, S. Saal, L. Richter, G. Georg, G. G. F. K. Noschmann, T.-D. Nguyen, K. Preslikoska, T. Moll and J. C. Tiller, Polymers, 2017, 118, 107–115 Search PubMed.
- C. J. Waschinski, V. Herdes, F. Schueler and J. C. Tiller, Macromol. Biosci., 2005, 5, 149–156 Search PubMed.
- B. Mattei, R. B. Lira, K. R. Perez and K. A. Riske, Chem. Phys. Lipids, 2017, 202, 28–37 Search PubMed.
- I. M. Helander and T. Mattila-Sandholm, J. Appl. Microbiol., 2000, 88, 213–219 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |