
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZinc-based coordination exchange in epoxy networks cured via frontal polymerization
Christoph
Schmidleitner
 ab,
Stefan
Hirner
ab and
Elisabeth
Rossegger
*ab
ab,
Stefan
Hirner
ab and
Elisabeth
Rossegger
*ab
aPolymer Competence Center Leoben GmbH, Sauraugasse 1, 8700 Leoben, Austria. E-mail: elisabeth.rossegger@pccl.at
bGraz University of Technology, Institute of Chemistry and Technology of Materials, Stremayrgasse 9, 8010 Graz, Austria
First published on 1st April 2026
Abstract
Frontal polymerization (FP) provides high conversion, short reaction times, and excellent energy efficiency, making it an attractive method for the synthesis of polymeric materials. Epoxy resins represent a particularly relevant class of polymers due to their outstanding thermal and chemical resistance. However, their highly crosslinked nature hinders reprocessing, recycling, and repair, which poses significant challenges to sustainability. In this work, FP is combined with the concept of dynamic polymer networks to produce an epoxy-based material incorporating Zn2+ ions, enabling coordination-based bond exchange. The zinc content was systematically varied between 0, 2.5, 5, and 10 mol%. Characterization of the frontal parameters revealed consistently high propagation velocities, which decreased only at elevated zinc contents due to dark curing effects. Swelling experiments revealed a decrease in swelling degree with increasing Zn content, whereas the gel content remained nearly constant regardless of zinc concentration. Rheological measurements of a network with 5 mol% zinc salt showed a linear Arrhenius-type temperature dependence (R2 = 0.99), indicating thermally activated exchange dynamics. Variation of the zinc counter anion further revealed that relaxation behavior correlates with coordination strength of the anion rather than with cation acidity. These results suggest that coordination exchange between Zn2+ and the network's datively bound functional groups is the predominant exchange mechanism. Furthermore, the strength of these coordination-exchange interactions could be demonstrated through the successful thermal welding of two samples.
Introduction
Frontal polymerization describes self-perpetuating reactions, where a reaction zone, the front, propagates throughout the monomer in a wave like manner.1 The reaction is sustained by the high exothermicity generated by the polymerization and spreads throughout the monomer due to thermal diffusion. In general, the polymerization is locally initiated by an external stimulus, such as heat or light, and can be terminated through monomer depletion, dissipation of the exothermic heat, or external intervention.Major types of frontal polymerization are free radical, ring opening metathesis2,3 or cationic ring opening frontal polymerization.4 Additionally, FP can be classified according to the mode of initiation. While thermal stimulation represents the classical approach, photoinitiation5 is also widely employed. More advanced methods include the use of lasers,6 ultrasound7 or even magnetic fields.8 The present work focuses on the UV-light-induced cationic ring-opening frontal polymerization of epoxy resins, as these materials are of particular relevance for numerous industrial applications due to their mechanical performance, chemical resistance, and versatile processability.
Epoxy resins obtained via frontal polymerization form highly crosslinked networks, which are mechanically robust but difficult to repair once damaged. These limitations can be addressed by employing dynamic networks instead of conventional thermosets. In such systems, dynamically exchangeable bonds are incorporated into the polymer structure allowing for thermal welding or healing. Reported exchange mechanisms include the reversible cleavage and recombination of disulfides,9 thio-thioether exchange,10 the retro aza-Michael reaction11 and transesterification reactions.12 In addition, dynamic networks may rely on non-covalent, yet reversible, interactions. In the present work, the exchange mechanism is based on coordination dynamics involving zinc ions, as illustrated in Fig. 1. For epoxy systems, Zn2+ coordination is expected to occur predominantly with negatively charged alkoxide groups as well as with neutral oxygen atoms of carbonyl or ether functionalities serving as ligands.13
 | ||
| Fig. 1 Overview of the frontal polymerization and zinc-based coordination exchange mechanism together with the chemicals utilized. | ||
The combination of dynamic networks with frontal polymerization offers several advantages over conventional thermoset production. One key benefit is the rapid solidification of large material volumes. While conventional curing typically requires several hours, frontal polymerization can complete curing within minutes. For example, Tarafdar et al. reported a reduction in curing time from 15 h under conventional thermal oven curing to less than 2 min using UV-induced frontal polymerization (of a cationically cured epoxy resin).14 Moreover, the energy demand of frontal polymerization is substantially lower compared to conventional batch processes. For fiber-reinforced composite materials (also using a cationically cured epoxy resin as a polymer matrix), an energy reduction of up to 99.5% was reported in 2024.15 Beyond these process-related benefits, the use of dynamic networks provides superior repair- and reprocess-ability relative to traditional thermosets.16 Thus, we developed a frontally polymerizable epoxy resin that incorporates dynamic binding motifs through coordination exchange, offering a promising route towards more sustainable, more easily processable and repairable high-performance materials.
Results and discussion
The frontal polymerization was conducted in a silicone mould and was initiated with UV light. Thermal infrared images of the curing process are provided in Fig. S1 (SI). The formulations consisted of the epoxides poly-(bisphenol A-co-epichlorhydrin)-glycidyl end capped (EPC) with 46 wt% and 3,4-epoxycyclohexylmethyl-3′,4′-epoxycyclohexane carboxylate (ECC) 54 wt%. EPC was selected because its hydroxyl groups provide additional coordination sites for the zinc cation. ECC was employed as a reactive diluent to supply the reactivity required for frontal polymerization. Moreover,1 mol% respective to the amount of ECC of p-(octyloxyphenyl)phenyl iodonium hexafluoro antimonate (IOC-8-SbF6), and 1,1,2,2-tetraphenyl-1,2-ethanediol (TPED) were used as initiators. Zinc triflate was employed to introduce dynamic coordination exchange and thereby enable the formation of a dynamic network; however, its slight acidity rendered the formulations susceptible to dark curing. With increasing zinc triflate content, the tendency for dark curing intensified, leading to the partial opening of epoxy groups prior to initiation. As a result, fewer reactive groups were available for the propagating front, reducing overall reactivity and thus increasing the required UV-initiation time. This can be seen in Fig. 2a. Between 2.5–5 mol%, there is no significant change, whereas from 5–10 mol% the illumination time more than doubled. Consequently 10 mol% of Zn2+ significantly promotes the dark reaction. A similar trend was observed for the frontal velocities of the formulations, as shown in Fig. 2b. The reference resin without Zn exhibited a velocity of 3.3 cm min−1, which is considered sufficiently high. In contrast, the formulation containing 2.5 mol% zinc displayed an increased velocity, likely due to accelerated catalysis by the zinc cation at elevated temperatures, which promotes polymerization in synergy with the photoacid generator. Incorporation of 5 mol% zinc salt led to only a minor reduction in front velocity, whereas 10 mol% resulted in a pronounced loss of reactivity and a corresponding decrease in velocity, consistent with the observed illumination times. This effect can be attributed to dark curing, which proceeds rapidly enough to compete with frontal polymerization. The maximum temperature, however, remained largely unaffected. Furthermore, Fig. 2c shows a FTIR-spectrum of a frontally polymerized sample before and after curing. The nearly complete conversion of the epoxide is evidenced by the disappearance of the characteristic epoxy band at 915 cm−1 as can be seen in Fig. 2c being emphasized by a circle. | ||
| Fig. 2 (a) Irradiation times to initiate frontal polymerization of samples with varying zinc content; (b) frontal velocity and maximum temperature in dependence to the zinc content; (c) FTIR spectrum of a frontally cured sample; (d) swelling degree and gel content related to the zinc content. | ||
To determine the influence of the Zn2+ ion content on the crosslinking density swelling tests have been conducted. The calculations for the degree of swelling as well as the gel content can be found in the experimental part of this article. As can be seen in Fig. 2d, the results show a clear trend from the neat system towards a higher zinc content. The swelling of the systems decreases from 115.1 ± 2.6% to 104.0 ± 0.4% owing to higher crosslinking density resulting from more coordinative bonds based on higher zinc loading. The observed gel contents exceeded 97% which suggests nearly complete conversion of the epoxides and is an indicator for a high crosslinking degree. This is expectable, though, since formulations used for frontal polymerization are characterized by their high reactivity, thus leading to a high conversion and followingly a high crosslinking density.17
To determine the glass transition temperature (Tg), dynamic mechanical analysis (DMA) was performed, and the results are shown in Fig. S2a (SI). The measurements reveal a decrease in Tg from 194 °C at 0 mol% Zn2+ to 128 °C at 5 mol% Zn2+. At first glance, this trend appears counterintuitive, as additional complexing cations would be expected to increase intermolecular interactions and potentially enhance crosslink density, consistent with the swelling experiments. However, Zn2+ not only participates in the formation of dynamic coordination bonds but also exerts a catalytic effect. Increasing zinc content promotes epoxy consumption prior to polymerization through dark reactions, thereby reducing the number of reactive groups available for subsequent network formation. Thus, the catalytic activity of Zn2+ can influence primary chain growth and consequently alter the resulting network architecture, giving rise to structural variations that affect the thermomechanical properties. In addition, at elevated temperatures, dynamic exchange processes become active and partial dissociation of coordination bonds may occur. This decreases the effective crosslink density under DMA conditions and further contributes to the observed reduction in Tg. Overall, the decrease in Tg can be attributed to the combined effects of Zn2+-induced catalytic influences on polymerization, modifications in network architecture, and the dynamic nature of the coordination bonds. At the same time, Fig. S2b (SI) shows that the storage modulus below Tg is higher in zinc-containing systems compared to the reference, indicating increased crosslinking due to coordination. A similar combination of reduced Tg and enhanced modulus has previously been reported for Zn2+-catalyzed epoxy–anhydride vitrimers.18
To further investigate the dynamic exchange and the underlying mechanism, different zinc salts with varying anions were incorporated. Subsequently, stress–relaxation experiments were performed using a rheometer in DMA mode at 200 °C. As shown in Fig. 3a, chloride displayed the slowest relaxation, perchlorate as well as tetrafluoroborate showed comparable behavior, while triflate enabled significantly faster relaxation than all other anions. This trend is consistent with coordination strengths reported in literature,19 where chloride exhibits the strongest coordination, BF4− and ClO4− fall within a similar intermediate range, and triflate represents the weakest coordinating anion (Fig. 3b). Furthermore, the observed relaxation behavior shows no significant correlation with the acidity of the corresponding zinc salts, as BF4− and ClO4− are more acidic than zinc triflate.20 These results indicate that relaxation is primarily governed by the availability of Zn2+ ions, which increases with weaker anion coordination. Consequently, weaker coordinating anions promote faster dynamic bond exchange and, in turn, faster stress relaxation. Since the triflate anion demonstrated the most favorable relaxation behavior, it was chosen for all subsequent experiments.
 | ||
| Fig. 3 (a) Stress relaxation measurements at 200 °C with various Zn2+ salts; (b) structures and coordination strengths of the anions used; (c) stress relaxation measurements of a sample containing 5 mol% zinc at different temperatures and (d) Arrhenius plot derived from these measurements. | ||
The resin containing 5 mol% zinc triflate was further analyzed at temperatures between 200 and 230 °C to evaluate the temperature dependence of the coordination exchange mechanism. As shown in Fig. 3c, relaxation times were determined using the Maxwell model at G/G0 = 1/e. The curves reveal efficient relaxation, reaching below 1/e within 13 min at the lowest temperature. With increasing temperature, relaxation times decreased and could be fitted in an Arrhenius plot (Fig. 3d), yielding an activation energy of 76.2 kJ mol−1 with excellent linearity (R2 = 0.99). Similar activation energies ranging from 68–90 kJ mol−1 were found for zinc catalysed transesterification reactions which are governed by similar coordination reactions.21
To qualitatively demonstrate the material's rapid stress–relaxation capability, a welding test was performed. Two DMA samples were placed in contact and welded in an oven at 175 °C for 45 minutes. The custom-made press used for welding is shown in Fig. 4a, while the resulting welded sample is shown in Fig. 4b. To demonstrate the load-bearing capacity of the welded sample, a 910 g bottle was attached using a double constrictor knot, as shown in Fig. 4c. The welded sample withstood the applied weight for several minutes without failure. Since slight discolouration was observed after welding, FT-IR spectra of the samples were recorded before and after (Fig. 4d). The spectra show no distinct changes, suggesting that the chemical structure of the network remained unchanged.
 | ||
| Fig. 4 (a) Custom-made press used for welding; (b) welded sample; (c) welded sample under load and (d) FTIR spectrum of the sample before and after welding. | ||
In summary, the measured material properties highlight the strong potential of this system for demanding applications. The combination of rapid curing, high glass transition temperatures, and reconfigurability owing to the incorporation of dynamic binding motifs, provides a unique property profile that is rarely achieved in conventional thermosets. These features make the material particularly attractive for use in repairable or weldable fibre-reinforced composites or reversible adhesives, where both mechanical robustness and dynamicity are required.
Conclusion
In this work, a dynamic epoxy-based network curable by frontal cationic polymerization was presented for the first time. The resulting material combines high crosslink density and elevated glass transition temperature with excellent thermomechanical performance. Despite its high Tg, the network exhibits efficient relaxation at elevated temperatures, with stress relaxation times as short as 240 s at 230 °C. Furthermore, a novel coordination-based exchange mechanism involving zinc ions, their counterions, and the polymer backbone was systematically demonstrated. These findings highlight that fast-curing, high-Tg dynamic networks can be readily obtained by frontal polymerization, offering a promising route towards energy-efficient manufacturing of repairable and re-processable materials.Experimental
Materials
Poly-(bisphenol A-co-epichlorhydrin)-glycidyl end capped (EPC), 1,1,2,2-tetraphenyl-1,2-ethanediol (TPED), zinc chloride, zinc tetrafluoroborate hydrate, zinc perchlorate hexahydrate as well as zinc trifluoromethanesulfonate were purchased from Sigma Aldrich. The photoinitiator p-(octyloxyphenyl)phenyl iodonium hexafluoro antimonate (IOC-8 SbF6) was bought from BLD Pharmatech Inc. and 3,4-epoxycyclohexylmethyl-3′,4′-epoxycyclohexane carboxylate (ECC) was acquired from IGM Resins.Resin preparation
For preparation of the resin EPC and ECC were mixed in a ratio of 46![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :54 in weight percent. To this mixture 1 mol%, with regard to the amount of ECC used, of IOC-8 SbF6 as well as TPED were added. Zinc salts were then added to the resin after dissolution in acetone. This was done in concentrations of 2.5, 5 and 10 mol% in regard to the amount of ECC. The solvent was removed under reduced pressure within some minutes to avoid ring opening polymerization of the epoxides.
:54 in weight percent. To this mixture 1 mol%, with regard to the amount of ECC used, of IOC-8 SbF6 as well as TPED were added. Zinc salts were then added to the resin after dissolution in acetone. This was done in concentrations of 2.5, 5 and 10 mol% in regard to the amount of ECC. The solvent was removed under reduced pressure within some minutes to avoid ring opening polymerization of the epoxides.
Frontal polymerization
Frontal polymerization was conducted in a silicone mould (50 × 10 × 5 mm). Irradiation of the samples was done with a an Omnicure Series 2000 lamp at an intensity of 2 W cm−2. The lightguide of the UV-lamp was placed ca. 2 mm over the resin to induce the reaction. The frontal velocity was measured using indicating elevations of the mould with one being placed every centimeter. The reaction was recorded using an IR-camera (FLIR E54). This also enabled the measurement of maximum reaction temperatures. The temperature and velocity were measured in three different defined zones and the mean value as well as the standard deviation were calculated. The initiation time was measured using the timer of the UV-lamp in combination with the IR-camera to see whether the reaction had been intiated.Characterization techniques
To determine the Tg of the networks, DMA measurements were conducted using a Mettler Toledo DMA/STDA in a temperature range from 30 to 250 °C at a frequency of 1 Hz. Samples were casted in a silicone mould with the dimensions 25 × 5 × 1 mm with a glass slide on top and polymerized at a 140 °C in a drying oven.To study the stress relaxation kinetics of the developed material, rheometer measurements were carried out. The rheometer samples were made using a silicone mould with the dimensions 25 × 5 × 1 mm. The measurements were conducted on an Anton Paar Physica MCR501 in DMA mode with a deformation of 1% and a normal force of 0.05 N at temperatures ranging from 200–240 °C.
To evaluate the degree of swelling and the gel content, samples were prepared by drop casting on a glass slide and subsequently cured at 140 °C. Afterwards the samples were weighed and emerged in dichloromethane for 48 h. After weighing, the samples were dried in a vacuum oven at 100 °C for one week and subsequently weighed again. The degree of swelling was calculated according to formula (1) from the wet weight of the sample (mwet) divided by the original sample weight (m0); the gel content was calculated by division of the dried sample weight (mdry) by the original sample weight (formula (2)).
 | (1) |
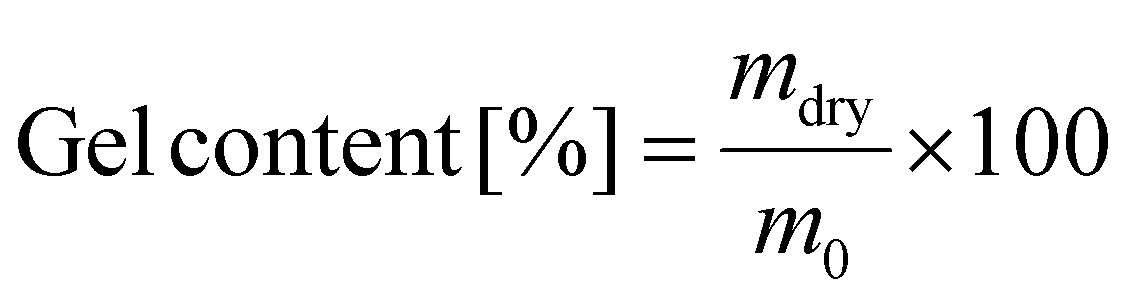 | (2) |
Fourier transformed infrared spectroscopy was performed using a Bruker Avance P spectrometer at a resolution of 4 cm−1 with 24 scans per measurement.
Welding
Samples were prepared the same way as described for DMA measurements. The formulation used had a concentration of 5 mol% of Zn2+. The samples were then placed in a Teflon press, secured with screws, and heated in an oven at 175 °C for 45 min. To apply a load to the welded samples, they were connected with a thread—tied using a double constrictor knot—to a water-filled glass bottle weighing 910 g.Conflicts of interest
The authors declare no conflict of interest.Data availability
The data supporting this article have been included as part of the supplementary information (SI). See DOI: https://doi.org/10.1039/d5py01170a.Acknowledgements
The research work was performed within the COMET-project “Photostructurable Encapsulation Molds and Magnetic Composites” (project-no.: VII-S2) at the Polymer Competence Center Leoben GmbH (PCCL, Austria) within the framework of the COMET-program of the Federal Ministry for Climate Action, Environment, Energy, Mobility, Innovation and Technology and the Federal Ministry for Digital and Economic Affairs with contributions by the Graz University of Technology. The PCCL is funded by the Austrian Governments and the State Governments of Styria, Lower Austria and Upper Austria. The authors thank Sebastian Maar from PCCL for conducting the DMA measurements and David Reisinger for conducting Rheometer measurements.References
- Q. Li, H.-X. Shen, C. Liu, C.-F. Wang, L. Zhu and S. Chen, Advances in frontal polymerization strategy: From fundamentals to applications, Prog. Polym. Sci., 2022, 127, 101514, DOI:10.1016/j.progpolymsci.2022.101514.
- S. Sutthasupa, M. Shiotsuki and F. Sanda, Recent advances in ring-opening metathesis polymerization, and application to synthesis of functional materials, Polym. J., 2010, 42, 905–915, DOI:10.1038/pj.2010.94.
- J. C. Cooper, J. E. Paul, N. Ramlawi, C. Saengow, A. Sharma, B. A. Suslick, R. H. Ewoldt, N. R. Sottos and J. S. Moore, Reprocessability in Engineering Thermosets Achieved Through Frontal Ring-Opening Metathesis Polymerization, Adv. Mater., 2024, 36, e2402627, DOI:10.1002/adma.202402627.
- D. Bomze, P. Knaack, T. Koch, H. Jin and R. Liska, Radical induced cationic frontal polymerization as a versatile tool for epoxy curing and composite production, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3751–3759, DOI:10.1002/pola.28274.
- C. Schmidleitner, M. U. Kriehuber, R. Korotkov, S. Schlögl and E. Rossegger, Frontal Polymerization of Thiol-Acrylate Covalent Adaptable Networks, Polym. Chem., 2025, 16, 963–971, 10.1039/D4PY01106F.
- Z.-F. Zhou, C. Yu, X.-Q. Wang, W.-Q. Tang, C.-F. Wang and S. Chen, Facile access to poly(NMA-co-VCL) hydrogels via long range laser ignited frontal polymerization, J. Mater. Chem. A, 2013, 1, 7326, 10.1039/c3ta11409k.
- J. Rigolini, F. Bombled, F. Ehrenfeld, K. E. Omari, Y. L. Guer and B. Grassl, 2D-Infrared Thermography Monitoring of Ultrasound-Assisted Polymerization of Water-Soluble Monomer in a Gel Process, Macromolecules, 2011, 44, 4462–4469, DOI:10.1021/ma200706j.
- C. Yu, C.-F. Wang and S. Chen, Facile access to versatile hydrogels via interface-directed frontal polymerization derived from the magnetocaloric effect, J. Mater. Chem. A, 2015, 3, 17351–17358, 10.1039/C5TA03811A.
- B. Sölle, M. Schmallegger, S. Schlögl and E. Rossegger, Wavelength-Dependent Dynamic Behavior in Thiol-Ene Networks Based on Disulfide Exchange, J. Am. Chem. Soc., 2024, 146, 34152–34157, DOI:10.1021/jacs.4c13735.
- N. J. Bongiardina, K. F. Long, M. Podgórski and C. N. Bowman, Substituted Thiols in Dynamic Thiol–Thioester Reactions, Macromolecules, 2021, 54, 8341–8351, DOI:10.1021/acs.macromol.1c00649.
- C. Ivaldi, E. Laguzzi, V. M. Ospina, D. Antonioli, R. Chiarcos, F. Campo, N. Cuminetti, J. de Buck and M. Laus, Exploiting β-amino ester chemistry to obtain methacrylate-based covalent adaptable networks, Polymer, 2024, 293, 126636, DOI:10.1016/j.polymer.2023.126636.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Silica-Like Malleable Materials from Permanent Organic Networks, Science, 2011, 334, 965–968, DOI:10.1126/science.1211649.
- A. Demongeot, S. J. Mougnier, S. Okada, C. Soulié-Ziakovic and F. Tournilhac, Coordination and catalysis of Zn 2+ in epoxy-based vitrimers, Polym. Chem., 2016, 7, 4486–4493, 10.1039/C6PY00752J.
- A. Tarafdar, W. Lin, A. Naderi, X. Wang, K. Fu, I. D. Hosein and Y. Wang, UV-induced frontal polymerization for optimized in-situ curing of epoxy resin for excellent tensile and flexural properties, Compos. Commun., 2024, 46, 101832, DOI:10.1016/j.coco.2024.101832.
- J. Staal, B. Caglar and V. Michaud, Self-catalysed frontal polymerisation enables fast and low-energy processing of fibre reinforced polymer composites, Compos. Sci. Technol., 2024, 251, 110584, DOI:10.1016/j.compscitech.2024.110584.
- A. E. Gerdroodbar, V. Karimkhani, E. Dashtimoghadam and M. Salami-Kalajahi, Vitrimerization as a bridge of chemical and mechanical recycling, J. Environ. Chem. Eng., 2024, 12, 112897, DOI:10.1016/j.jece.2024.112897.
- B. A. Suslick, J. Hemmer, B. R. Groce, K. J. Stawiasz, P. H. Geubelle, G. Malucelli, A. Mariani, J. S. Moore, J. A. Pojman and N. R. Sottos, Frontal Polymerizations: From Chemical Perspectives to Macroscopic Properties and Applications, Chem. Rev., 2023, 123, 3237–3298, DOI:10.1021/acs.chemrev.2c00686.
- L. Yue, H. Guo, A. Kennedy, A. Patel, X. Gong, T. Ju, T. Gray and I. Manas-Zloczower, Vitrimerization: Converting Thermoset Polymers into Vitrimers, ACS Macro Lett., 2020, 9, 836–842, DOI:10.1021/acsmacrolett.0c00299.
- I. M. Riddlestone, A. Kraft, J. Schaefer and I. Krossing, Taming the Cationic Beast: Novel Developments in the Synthesis and Application of Weakly Coordinating Anions, Angew. Chem., Int. Ed., 2018, 57, 13982–14024, DOI:10.1002/anie.201710782.
- Shivani, B. Pujala and A. K. Chakraborti, Zinc(II) perchlorate hexahydrate catalyzed opening of epoxide ring by amines: applications to synthesis of (RS)/(R)-propranolols and (RS)/(R)/(S)-naftopidils, J. Org. Chem., 2007, 72, 3713–3722, DOI:10.1021/jo062674j.
- X. Niu, F. Wang, X. Li, R. Zhang, Q. Wu and P. Sun, Using Zn 2+ Ionomer To Catalyze Transesterification Reaction in Epoxy Vitrimer, Ind. Eng. Chem. Res., 2019, 58, 5698–5706, DOI:10.1021/acs.iecr.9b00090.
| This journal is © The Royal Society of Chemistry 2026 |