
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLiving polymerization of an amphiphilic, helical aramid diblock copolymer
Dinh Phuong Trinh
Nguyen
 ,
Meron
Debas
,
Rafael V. M.
Freire
,
Stefan
Salentinig
and
Andreas F. M.
Kilbinger
*
,
Meron
Debas
,
Rafael V. M.
Freire
,
Stefan
Salentinig
and
Andreas F. M.
Kilbinger
*
Department of Chemistry, University of Fribourg, Chemin du Musée 9, 1700 Fribourg, Switzerland. E-mail: andreas.kilbinger@unifr.ch
First published on 18th December 2025
Abstract
We report herein the living polymerization of an amphiphilic, helical aramid diblock copolymer made possible by the utilization of the reagent PHOS3. Two monomers, 2,4-bis((2,5,8,11-tetraoxapentadecan-15-yl)oxy)-5-aminobenzoic acid and 5-amino-2,4-difluorobenzoic acid, were used to build the hydrophilic and hydrophobic blocks, respectively. The diblock copolymer was characterized by NMR and GPC/SEC, and its aggregation behavior was investigated using atomic force microscopy (AFM), (depolarized) dynamic light scattering ((D)DLS), and small-angle X-ray scattering (SAXS). The analysis strongly suggests that the diblock copolymer self-assembles to form elongated, tube-like structures in water.
Introduction
Aromatic amides, also known as aramids, are an important class of polymers that possess exceptional mechanical strength and very high thermal resistance thanks to the presence of numerous hydrogen-bonding and aromatic–aromatic interactions.1 With their rigid backbone consisting of alternating aromatic and amide groups and shape-persistent nature, aramids are among the most studied systems for constructing folding oligomers and polymers (foldamers) that could ultimately mimic biomacromolecules such as proteins.2–4 In this effort, many groups, notably the groups of Huc and Gong, have developed families of aromatic oligoamide foldamers that have been clearly established to adopt well-defined, three-dimensional helical architectures in solution.5–10 Reports also demonstrated that aramid-based foldamers are capable of performing host–guest molecular recognition, molecular transport and crossing cell membranes.4,11–17Recently, our group developed a living polymerization method for aromatic amides that contributed to the expansion of the library of foldamers built on aramids. With a new coupling reagent, PHOS3, non-self-deactivating aromatic amino acid monomers and oligomers can be polymerized in a living fashion under slow monomer addition conditions, and block copolymers can be synthesized.18,19 With the desire to explore the new possibilities granted by this novel tool, we took interest in designing a non-charged, amphiphilic, helical aramid diblock copolymer. We were curious as to how such a polymer would behave in water, as, to our knowledge, no similar foldamer has been investigated before. We had previously synthesized a water-soluble oligomer utilizing 2,4-bis((2,5,8,11-tetraoxapentadecan-15-yl)oxy)-5-aminobenzoic acid 5 as the monomer, so we opted to exploit it again for the hydrophilic block. As for the hydrophobic block, our attention was drawn to 5-amino-2,4-difluorobenzoic acid 7 as the building block (see Scheme 2). Helical foldamers based on this monomer unit have been studied in molecular dynamics simulations, in the solid state and in solution.20–22 Reports on difluoro-substituted aromatic amide foldamers that form hydrogen bonds on the helical exterior are scarce. This is most likely due to their insolubility in most solvents. Monofluoro-substituted aromatic amides forming H-bonds on the concave side of the crescent shape have been shown to form five- and six-membered intramolecularly H-bonded rings and to arrange into well-defined secondary structures.21,23–26 Based on the simulations completed by the Pophristic group, dodecamers of difluoro-substituted arylamides generate helical structures 91%, 46% and 88% of the time in chloroform, methanol and water, respectively, due to the similar three-center hydrogen-bonding system they share with the well-investigated ortho-alkoxy-substituted aramids (Scheme 1).20 Although the hydrogen bond to the fluorine atom is weaker than that to the oxygen atom,21 the helical motif should still occur in solution due to additional solvophobic effects.
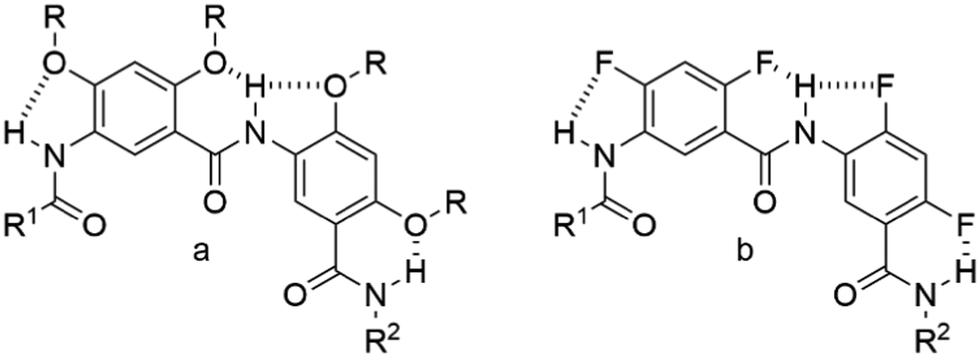 | ||
| Scheme 1 (a) Scheme of the intramolecular three-center hydrogen bonding system involving the amide bond and the ortho-OR group. (b) Scheme of the intramolecular three-center hydrogen bonding system involving the amide bond and the ortho-F atom. | ||
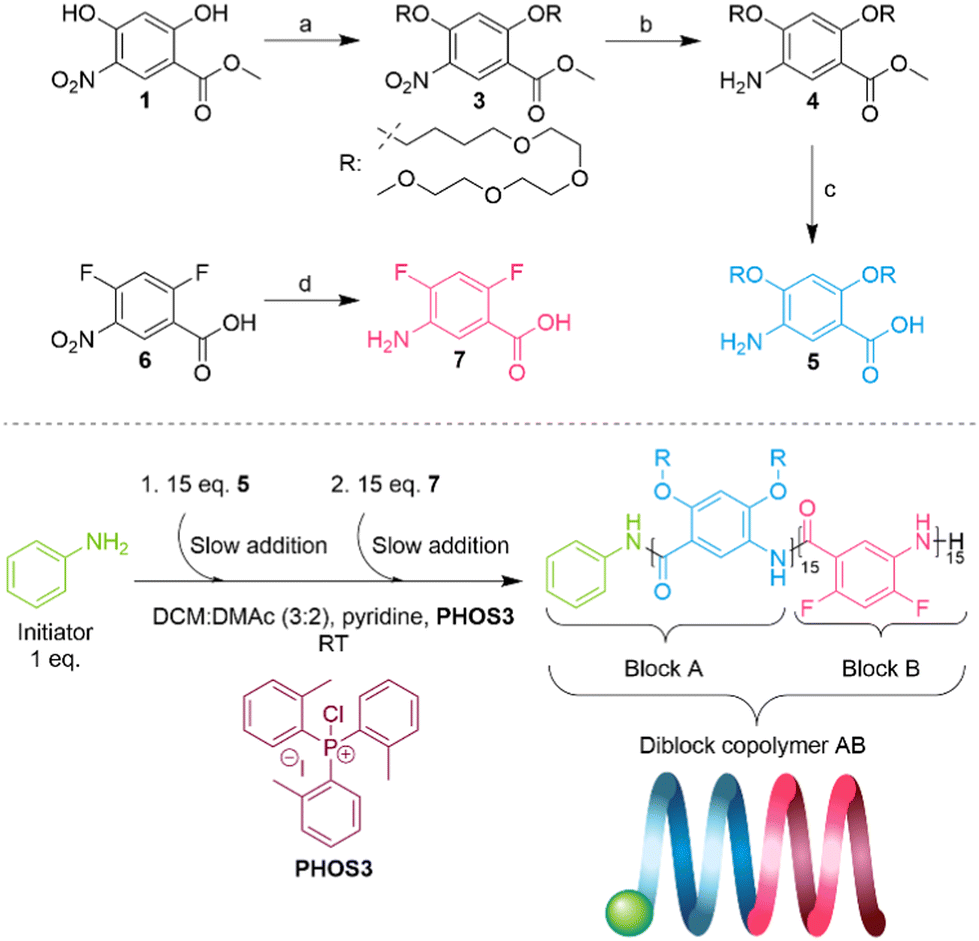 | ||
| Scheme 2 Top: Synthetic paths for monomers 5 and 7. (a) K2CO3, DMF, 15-bromo-2,5,8,11-tetraoxapentadecane 2, 51%. (b) H2/Pd/C, MeOH, EtOAc, quant. (c) KOH, EtOH, 60%. (d) H2/Pd/C, MeOH, EtOAc, quant. Bottom: Polymerization conditions of diblock copolymer AB. | ||
Several reports have determined that the number of aromatic units (residues) per turn required to form a substituted helix for monomers similar to 5 and 7 in Scheme 2 is 7.2,20 With that in mind, we decided to target a four-turn amphiphilic diblock copolymer helix, meaning that the hydrophilic and hydrophobic moieties would each be comprised of 15 aromatic residues, for a total of 30 aromatic residues.
In addition to synthesizing and characterizing the amphiphilic helical aramid diblock copolymer, an investigation of its aggregation behavior with the help of atomic force microscopy (AFM), multi-angle (depolarized) dynamic light scattering ((D)DLS), and small-angle X-ray scattering (SAXS) is presented in this study.
Results and discussion
Synthesis of the monomers and polymerization
The first monomer, 2,4-bis((2,5,8,11-tetraoxapentadecan-15-yl)oxy)-5-aminobenzoic acid 5, was synthesized following our previously established protocol.18 To obtain 5, methyl 2,4-dihydroxy-5-nitrobenzoate27 was first alkylated with 15-bromo-2,5,8,11-tetraoxapentadecane 2 (see the SI), followed by the reduction of the nitro group. Then, hydrolysis of methyl ester with an excess of KOH in ethanol afforded amino acid 5 (Scheme 2).We opted for 5-amino-2,4-difluorobenzoic acid 7 as the hydrophobic monomer because of its straightforward synthesis. 7 can be obtained through the reduction of the nitro group of the commercially available 2,4-difluoro-5-nitrobenzoic acid (Scheme 2, see the SI). When we attempted to make homopolymers of 7 with our polymerization method using chloroform as the solvent, we saw that they precipitated in solution when they reached around a 15-mer in size.
The polymerization was carried out following a modified protocol.197 was not soluble in chloroform unless pyridine was added to the monomer solution; therefore, we used a DCM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMAc (3:2) mixture instead as the solvent. To avoid solubility problems related to the homopolymer of 7, we decided to start the copolymerization with monomer 5 for the first block and monomer 7 for the second block.
:DMAc (3:2) mixture instead as the solvent. To avoid solubility problems related to the homopolymer of 7, we decided to start the copolymerization with monomer 5 for the first block and monomer 7 for the second block.
First, 15 eq. of monomer 5 were added slowly (0.1 mL h−1) to the reaction mixture containing pyridine, reagent PHOS3 and 1 eq. of initiator aniline; this constituted the formation of block A with a molecular weight of 7.7 kDa (Đ = 1.1, theoretical Mn = 8.9 kDa) (Fig. S1). The second block was synthesized by slow addition (0.1 mL h−1) of 15 eq. of monomer 7, and diblock copolymer AB was obtained with a molecular weight of 12 kDa (Đ = 1.16, theoretical Mn = 11 kDa) (Fig. S1). The SEC elugrams show excellent control of the oligomer and polymer sizes with narrow dispersity. The crude SEC trace of diblock AB in DMF shows a slight shoulder at lower molecular weights but at a higher molecular weight than that of the first block A. We speculate that this is caused by aggregation processes in DMF rather than poor block transfer. A DOSY NMR spectrum of the purified block copolymer AB showed no evidence of residual homopolymer A present in the sample, which was used for all subsequent analytical measurements. The polymerization conditions are summarized in Scheme 2 and detailed in the SI.
After the removal of pyridine from the crude polymer solution by acidic extraction, the concentrated residue was dissolved in chloroform, and the pure polymer was obtained through recycling GPC (chloroform). Diblock copolymer AB was soluble in chloroform, methanol, dimethyl formamide and water.
NMR analysis
A sample was taken after the complete addition of monomer 5 and analyzed by 1H-NMR spectroscopy in CDCl3 (Fig. 1a and Fig. S7). This confirmed the total consumption of 5, as the sharp signals corresponding to the protons of the butyl-TEG side chains between 3.10 and 4.20 ppm in the 1H-NMR spectrum of monomer 5 (Fig. 1b and Fig. S2) all became broadened in the 1H-NMR spectrum of the crude polymer solution. This broadening is a strong indicator of the self-aggregation of sufficiently long aromatic oligoamides in chloroform.28 The presence of amide protons was also confirmed by new peaks in the downfield region (7.5–9.00 ppm) (Fig. S7). Next, the total consumption of 7 was verified by taking a sample after the complete addition of monomer 7. 19F-NMR spectroscopy of the crude AB diblock copolymer solution in DMSO-d6 (Fig. S9) revealed major chemical shifts of the fluorine signals, moving from −122.86 ppm and −124.03 ppm in the spectrum of monomer 7 (Fig. S4) to −112.39 ppm and −112.68 ppm for the block copolymer, respectively. Once diblock copolymer AB was purified by recycling GPC, 19F-NMR spectroscopy in CDCl3 (Fig. 1d and Fig. S11) also showed two weak signals at −117.12 ppm and −123.77 ppm, which were also significantly shifted from −118.11 ppm and −119.28 ppm observed in the 19F-NMR spectrum of monomer 7 (Fig. 1d and Fig. S5). The broadening of the fluorine peaks in CDCl3 is likely due to the aggregation of polymer AB in this solvent, similar to the broadening of the proton signals of the butyl-TEG side chains. | ||
| Fig. 1 Partial 1H-NMR spectra of 5 (a) and crude block A polymer solution (b) in CDCl3. 19F-NMR spectra of 7 in CDCl3 (c) and polymer AB in CDCl3 (d). Shifting and broadening of signals are clear indications of polymer formation. | ||
The shifting of the fluorine signals before and after polymerization can only be attributed to the successful formation of block B onto block A and not to the formation of an independent oligomer, as SEC analysis in DMF also shows a clear shift in molecular weight from block A to diblock copolymer AB. A DOSY experiment in CDCl3 also supports the formation of the diblock copolymer (a single diffusing species) and the total consumption of monomers (Fig. S12).
Self-assembly structure study
Several Atomic Force Microscopy (AFM) images of block copolymer AB at different concentrations in water were recorded (see the SI); in all of them, extended, micrometer-long, fiber-like structures can be observed. At 2 mg L−1 and 4 mg L−1, the elongated self-assemblies are jagged and break into different directions with hard angles. Individual or smaller aggregates can also be seen as light-colored spots all around the long filaments. At these concentrations, polymer AB appears to be self-assembling into individual strands; some of these strands start to align side by side, but do not fully develop into uniform fibers. At 6 mg L−1, however, we observe longer, more ordered, and neater fiber arrangements. This suggests that the concentration of polymer AB has an influence on the formation of the fibers.Multi-angle depolarized dynamic light scattering (DDLS) and small-angle X-ray scattering (SAXS) were used to provide insights into the nanostructure of block copolymer AB in water. The DDLS signal is highly dependent on particle shape anisotropy, with isotropic spherical particles giving no signal in the DDLS configuration.19 For block copolymer AB in water, DDLS autocorrelation curves could be obtained at different scattering angles (see Fig. 2a), which is initial evidence of particle anisotropy. From the linear fit of the decay rates calculated from the DDLS autocorrelation curves as a function of q2, it is possible to extract the translational diffusion coefficient Dt from the slope and the rotational diffusion coefficient Dr from the intercept of the linear fit (see the SI for details).29,30 For polymer AB, a Dt value of 1.23 × 106 nm2 s−1 and a Dr value of 26.3 s−1 (1/6th of the intercept value) were obtained (Fig. 2b). The cylinder length and diameter can be obtained from empirical mathematical expressions such as the Broersma relations.31 The best possible solution of the Broersma system of equations using the experimental values of Dt and Dr resulted in the estimation of a cylinder length of 515.1 nm and a diameter of 135.5 nm. The Broersma relations are not ideal for length/diameter ratios smaller than 5 (as in the present case), and therefore the length and diameter estimated here may deviate from the real values.
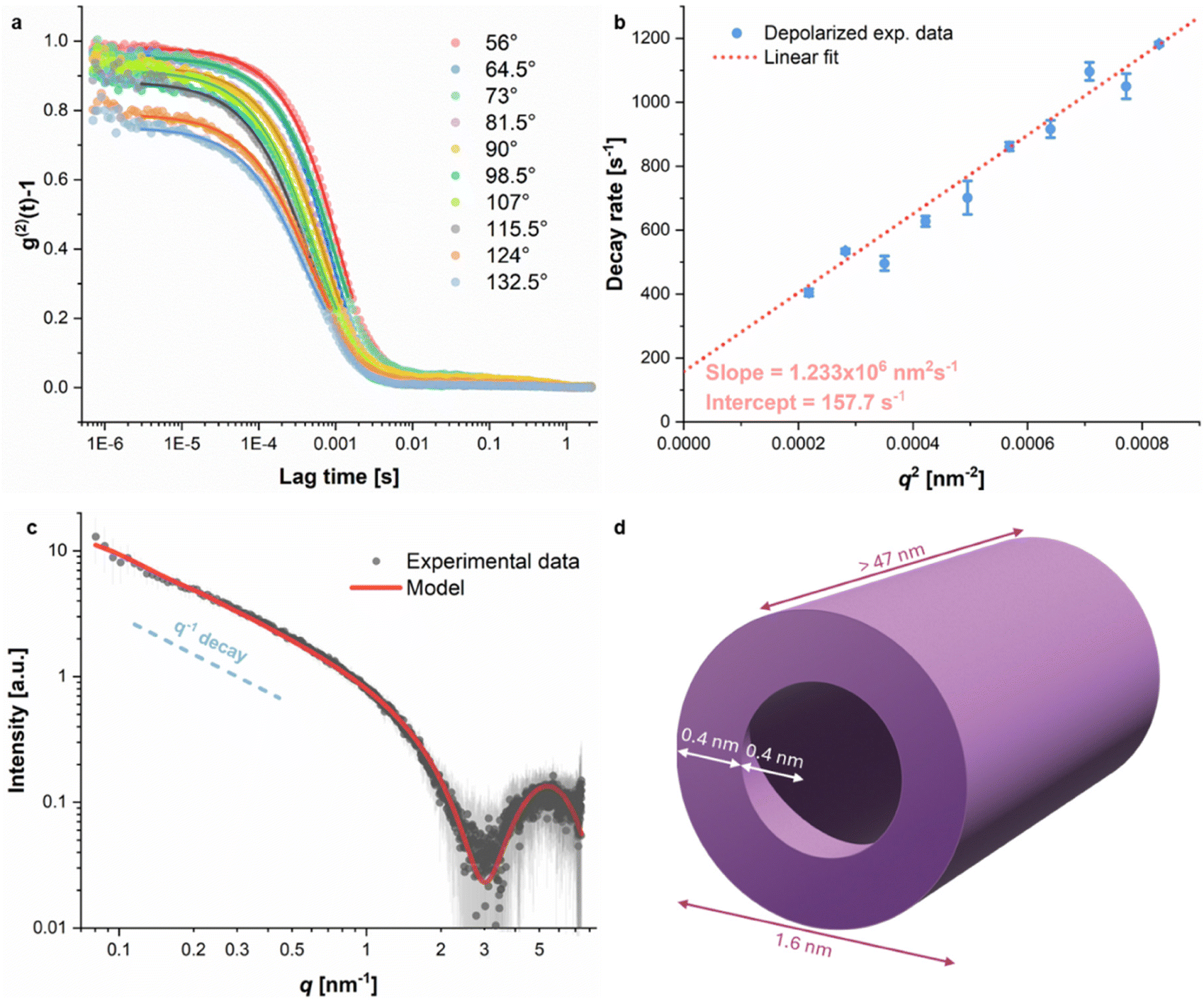 | ||
| Fig. 2 (a) DDLS autocorrelation curves obtained at different angles for diblock copolymer AB in water at 25 °C. Solid lines correspond to 2nd order cumulant fits of the experimental data. (b) Decay rates as a function of q2 for polymer AB in water, calculated from the cumulant fits of the DDLS autocorrelation curves at different angles. The dashed line corresponds to the linear fit of the data, from which the slope and intercept are used to estimate Dt and Dr, respectively. (c) SAXS curve of block copolymer AB in water along with the fitted core–shell cylinder model. The dashed blue line is a visual guide for a q−1 decay slope, typical of elongated structures. (d) Core–shell cylinder visualization of aggregated block copolymer AB. | ||
The SAXS curve (Fig. 2c) for our diblock copolymer exhibited a decay proportional to q−1 at lower q values (q < 0.5 nm−1) and also a bump at higher q values (q > 3 nm−1). The observed curve shape is typical of elongated structures such as cylinders, with the reported q−1 power-law decay characteristic of such structures.32 The sample's scattering data could not be fitted with a homogeneous cylinder model due to large variations in the electronic density of its cross-section, which are reflected in the strong bump seen at high q values. A core–shell cylinder model33 was therefore employed to accommodate such changes in the cross-sectional electronic density, with an estimated core radius of 0.4 nm and a shell thickness of 0.4 nm, resulting in a total diameter of 1.6 nm (Fig. 2d). The cylinder length of 47 nm results from a mathematical cut-off due to an insufficient q-range at lower q values from the experimental setup, and the real length is larger than the fitted value.
The core–shell cylinder model fits incredibly well with block copolymer AB being a hollow, tubular helix in solution, as the inner diameter (0.8 nm) shown by SAXS measurements is consistent with the inner cavity diameter found in crystal structures of previous helices with an identical backbone (0.9–1 nm).2,10 Simulations by the Pophristic group of dodecamers of dimethoxy- and difluoro-substituted arylamides present pore diameters of 1.39 nm and 1.43 nm, respectively,20 which are both larger than what is observed experimentally. The length of the 4-turn polymer, however, should only be ∼1.5 nm if we consider its helical pitch to be ∼0.35 nm.10 A cylinder length of more than 47 nm is very indicative of the stacking of individual, helical block copolymers AB onto themselves to create a long, tube-like structure in water.
DDLS and SAXS provide evidence that the formation of elongated self-assemblies by diblock copolymer AB occurs not only in the dried state, as observed in AFM, but also in solution. The diameter of the structures estimated by DDLS analysis (135.5 nm) is considerable, similar to the assemblies observed by AFM. On the other hand, SAXS data suggested much smaller structures, with an estimated diameter of 1.6 nm. Given the smaller dimensions probed by the SAXS technique, we believe that the rod-like structures observed by SAXS may correspond to the individual polymer molecules, which would assume an elongated shape in water and act as sub-fibers. Our hypothesis is that these sub-fibers would aggregate into much larger one-dimensional structures (fibers) with diameters over 100 nm. The additional upturn observed in the SAXS curve at q < 0.1 nm−1 could also be a sign of the aggregation of the polymers. These large hierarchical fiber-like structures are likely those observed by both DDLS and AFM.
To probe the impact of the hydrophobic moiety on the aggregation behavior of diblock copolymer AB, we polymerized 15-mer P1 (Mn, theo = 8.9 kDa, Mn, DMF-GPC = 7.6 kDa, Đ = 1.20) (Fig. S2) and 30-mer P2 (Mn, theo = 18 kDa, Mn, DMF-GPC = 17 kDa, Đ = 1.34) (Fig. S3) homopolymers using 5 (see the SI) as reference polymers. The scattering of P1 and P2 hydrophilic homopolymers was analyzed in water with SAXS (Fig. S28). For both polymer solutions, an upturn at low q indicates the presence of aggregated particles, which was not observed for the diblock copolymer solution. This is in agreement with DLS results (measured at an angle of 90°), indicating the presence of large structures with an estimated diameter of 380 nm (PDI = 0.4) and 322 nm (PDI = 0.75) for the 15- and 30-mer homopolymers, respectively (Fig. S29). The SAXS pattern in the intermediate power-law region presents similar decay to that of the AB diblock copolymer, while the strong bump at higher q values is absent for the homopolymers. The SAXS patterns of the homopolymers in water were therefore best fitted with a homogeneous cylinder model, and an additional power-law model was implemented to account for the additional low-q upturn. The fitted diameter of the cylindrical assemblies converged around 2.2 nm for both 15- and 30-mer, while the estimated intermediate length increased from 3.5 nm to 6.8 nm, which is expected with the increase in monomer units in the homopolymers.
The fittings of the SAXS data for the AB block copolymer and the homopolymers strongly indicate their ability to form cylindrical structures of different lengths in water. The presence of hydrophobic segments in the AB polymer appears to be critical for the formation of supramolecular, elongated fibers, as supported by the DDLS and AFM measurements. The homopolymers tend to form smaller cylindrical structures compared to the block copolymer, as indicated by SAXS, which can further aggregate into more random structures.
We hypothesize that the density of TEG side chains in homopolymers P1 and P2 is extremely high, leading to partial coverage of the cross-sectional aromatic pi-faces of the helical ends. This renders the overall shape of the fully TEG-substituted homopolymers P1 and P2 more like solid cylinders. We believe that exposed pi-faces are not favored in water and lead to helical end-to-end stacking. In the case of the diblock copolymer, the fluorine-containing block B allows for some steric relaxation of the TEG chains in block A. Therefore, the TEG chains are not sterically forced to cover the pi-faces of the TEG-containing A block and can possibly not reach the pi-face of the end of the B block. Overall, the diblock copolymer resembles the shape of a hollow cylinder.
The exposed cross-sectional pi-faces of the helical diblock copolymer are, therefore, more likely to be exposed to the solvent, water, and hence form extended linear stacks in aqueous solution, in contrast to the more densely covered TEG-substituted homopolymers.
Conclusions
In conclusion, an amphiphilic helical aramid diblock copolymer was successfully synthesized for the first time. SEC analysis of polymer block A and diblock copolymer AB further proves the livingness of the polymerization method using reagent PHOS3, as the measured Mn is consistent with the target Mn and the dispersity Đ is very narrow. Despite its very insoluble nature, the second block, composed of difluoro-substituted aromatic amides, was effectively dissolved in water thanks to the highly water-soluble butyl-TEG-substituted aromatic amide block. Analysis of block copolymer AB in the solid state by AFM revealed the formation of long, multi-stranded fibrils after the evaporation of water. A DDLS study also showed that block copolymer AB aggregates in solution to form very large, elongated structures, comparable to those observed in AFM. Finally, SAXS data corroborated the tube-like nature of block copolymer AB self-assemblies in water as well as the formation of supra-structures. The investigation showed that block copolymer AB develops into very robust hollow architectures in both the solid state and in water compared to hydrophilic homopolymers of the same size. This neutrally charged block copolymer could have interesting applications in membrane transport thanks to its solubility in both aqueous and organic media.Author contributions
D. P. T. N. and A. F. M. K. designed the experiments. D. P. T. N. synthesized the phosphorous reagent and the monomers and conducted all polymerizations as well as the polymer and molecular analysis. M. D. and S. S. conducted the DLS and SAXS measurements of the homopolymers and their analyses. R. V. M. F. and S. S. conducted the DLS and SAXS measurements of the diblock copolymer and their analyses. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: syntheses of monomers and polymers, NMR, HRMS, SEC, AFM, SAXS and DDLS characterization data. See DOI: https://doi.org/10.1039/d5py01096a.Acknowledgements
We thank Dr J. Adamcik for taking and providing the AFM images. A. F. M. K., D. P. T. N., M. D., R. V. M. F. and S. S. thank the National Center of Competence in Research (NCCR Bioinspired Materials) for their support.References
- J. M. García, F. C. García, F. Serna and J. L. de la Peña, Prog. Polym. Sci., 2010, 35, 623–686 CrossRef.
- B. Gong, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11583–11588 CrossRef CAS PubMed.
- L. Yuan, H. Zeng, K. Yamato, A. R. Sanford, W. Feng, H. S. Atreya, D. K. Sukumaran, T. Szyperski and B. Gong, J. Am. Chem. Soc., 2004, 126, 16528–16537 Search PubMed.
- K. Ziach, C. Chollet, V. Parissi, P. Prabhakaran, M. Marchivie, V. Corvaglia, P. P. Bose, K. Laxmi-Reddy, F. Godde, J.-M. Schmitter, S. Chaignepain, P. Pourquier and I. Huc, Nat. Chem., 2018, 10, 511–518 CrossRef CAS PubMed.
- Y.-X. Lu, Z.-M. Shi, Z.-T. Li and Z. Guan, Chem. Commun., 2010, 46, 9019–9021 RSC.
- B. Gong, Acc. Chem. Res., 2008, 41, 1376–1386 CrossRef CAS PubMed.
- C.-F. Wu, Z.-M. Li, X.-N. Xu, Z.-X. Zhao, X. Zhao, R.-X. Wang and Z.-T. Li, Chem. – Eur. J., 2014, 20, 1418–1426 CrossRef CAS PubMed.
- Y. Huo and H. Zeng, Acc. Chem. Res., 2016, 49, 922–930 CrossRef CAS.
- Y. Ferrand and I. Huc, Acc. Chem. Res., 2018, 51, 970–977 CrossRef CAS.
- Y. Zhong, B. Kauffmann, W. Xu, Z.-L. Lu, Y. Ferrand, I. Huc, X. C. Zeng, R. Liu and B. Gong, Org. Lett., 2020, 22, 6938–6942 CrossRef CAS PubMed.
- K. Yamato, L. Yuan, W. Feng, A. J. Helsel, A. R. Sanford, J. Zhu, J. Deng, X. C. Zeng and B. Gong, Org. Biomol. Chem., 2009, 7, 3643–3647 RSC.
- H. Zhao, S. Sheng, Y. Hong and H. Zeng, J. Am. Chem. Soc., 2014, 136, 14270–14276 CrossRef CAS.
- X. Li, X. Yuan, P. Deng, L. Chen, Y. Ren, C. Wang, L. Wu, W. Feng, B. Gong and L. Yuan, Chem. Sci., 2017, 8, 2091–2100 RSC.
- Y. Zhong, T. A. Sobiech, B. Kauffmann, B. Song, X. Li, Y. Ferrand, Y. Huc and B. Gong, Chem. Sci., 2023, 14, 4759–4768 Search PubMed.
- Q. Gan, Y. Ferrand, C. Bao, B. Kauffmann, A. Grélard, H. Jiang and I. Huc, Science, 2011, 331, 1172–1175 CrossRef CAS PubMed.
- E. R. Gillies, F. Deiss, C. Staedel, J.-M. Schmitter and I. Huc, Angew. Chem., Int. Ed., 2007, 46, 4081–4084 Search PubMed.
- S. Farooq, J. A. Malla, M. Nedyalkova, R. V. M. Freire, I. Mandal, A. Crochet, S. Salentinig, M. Lattuada, C. T. McTernan and A. F. M. Kilbinger, Angew. Chem., Int. Ed., 2025, 64, e202504170 CrossRef CAS.
- S. Pal, D. P. T. Nguyen, A. Molliet, M. Alizadeh, A. Crochet, R. D. Ortuso, A. Petri-Fink and A. F. M. Kilbinger, Nat. Chem., 2021, 13, 705–713 CrossRef CAS PubMed.
- S. Pal, L. Hong, R. V. M. Freire, S. Farooq, S. Salentinig and A. F. M. Kilbinger, Macromolecules, 2023, 56, 7984–7992 CrossRef CAS.
- Z. Liu, A. M. Abramyana and V. Pophristic, New J. Chem., 2015, 39, 3229–3240 RSC.
- Y.-Y. Zhu, J. Wu, C. Li, J. Zhu, J.-L. Hou, C.-Z. Li, X.-K. Jiang and Z.-T. Li, Cryst. Growth Des., 2007, 7, 1490–1496 Search PubMed.
- S. Farooq, M. Nedyalkova, S. Pal, A. Crochet, M. Lattuada and A. F. M. Kilbinger, Macromolecules, 2025, 58, 5990–5994 CrossRef CAS.
- X. Zhao, X.-Z. Wang, X.-K. Jiang, Y.-Q. Chen, Z.-T. Li and G.-J. Chen, J. Am. Chem. Soc., 2003, 125, 15128–15139 CrossRef CAS.
- C. Li, S.-F. Ren, J.-L. Hou, H.-P. Yi, S.-Z. Zhu, X.-K. Jiang and Z.-T. Li, Angew. Chem., Int. Ed., 2005, 44, 5725–5729 Search PubMed.
- J.-L. Hou, C. Li, H.-P. Yi and Z.-T. Li, Chem. – Asian J., 2006, 1, 766–778 CrossRef.
- J.-L. Hou, C. Li and Z.-T. Li, Acc. Chem. Res., 2008, 41, 1343–1353 CrossRef PubMed.
- L. Yuan, A. R. Sanford, W. Feng, A. Zhang, J. Zhu, H. Zeng, K. Yamato, M. Li, J. S. Ferguson and B. Gong, J. Org. Chem., 2005, 70, 10660–10669 CrossRef CAS PubMed.
- Y. Zhao, A. L. Connor, T. A. Sobiech and B. Gong, Org. Lett., 2018, 20, 5486–5489 CrossRef CAS.
- A. Molliet, S. Doninelli, L. Hong, B. Tran, M. Debas, S. Salentinig, A. F. M. Kilbinger and T. Casalini, J. Am. Chem. Soc., 2023, 145, 27830–27837 CrossRef CAS PubMed.
- R. Nixon-Luke and G. Bryant, Part. Part. Syst. Charact., 2018, 36 Search PubMed.
- S. Broersma, J. Chem. Phys., 1960, 32, 1626–1631 CrossRef CAS.
- D. McDowall, D. J. Adams and A. M. Seddon, Soft Matter, 2022, 18, 1577–1590 RSC.
- I. Livsey, J. Chem. Soc., Faraday Trans. 2, 1987, 83 Search PubMed.
| This journal is © The Royal Society of Chemistry 2026 |