
Preparation and performance characterization of block cross-linked copolymers PCaPE(Br) and PCaPE(I) via RAFT polymerization and quaternization reaction
Chaoyue
Shan
 *,
Haohao
Zhang
,
Yan
Gao
,
Hui
Wang
,
Li
Han
,
Yuanyuan
Du
,
Wen
He
,
Mingyuan
Pang
,
Xing
Gao
and
Yen Leng
Pak
*
*,
Haohao
Zhang
,
Yan
Gao
,
Hui
Wang
,
Li
Han
,
Yuanyuan
Du
,
Wen
He
,
Mingyuan
Pang
,
Xing
Gao
and
Yen Leng
Pak
*
College of Biological and Chemical Engineering, Qilu Institute of Technology, Jinan 250200, China. E-mail: QITscy@163.com
First published on 22nd November 2025
Abstract
Reversible addition–fragmentation chain transfer (RAFT) polymerization enables precise regulation of polymer chain growth and active center distribution. This study adopted a two-step approach with RAFT polymerization followed by quaternization to synthesize antibacterial materials, specifically tunable porous polymers PCaPE(Br) and PCaPE(I). The work focused on the structure–activity relationship between RAFT-derived block structures and the antibacterial properties of these materials. RAFT-mediated polymerization precisely controlled the block copolymer precursor PE-PDE, which is PEMA-b-P(DMAEA-co-EGDMA), and this control facilitated the subsequent formation of ordered porous structures. When the molar ratio of poly(ethyl methacrylate) (PEMA) to ethylene glycol dimethacrylate (EGDMA) was set to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6, the final quaternized products PCaPE2(Br) and PCaPE2(I) exhibited porous and fluffy microstructures. These microstructures were characterized by large specific surface area and high porosity. This structure, combined with uniformly distributed quaternary ammonium salt groups from the quaternization of N,N-dimethylaminoethyl acrylate (DMAEA) tertiary amine groups, enhanced the contact between antibacterial active sites and bacteria as well as antibacterial efficacy. PCaPE(Br) and PCaPE(I) showed excellent broad-spectrum antibacterial activity against E. coli and S. aureus. They also maintained good reusability with over 95% activity retention after five cycles. Such advantages support their prospects in biomedicine, agricultural antibacterials and wastewater treatment.
:6, the final quaternized products PCaPE2(Br) and PCaPE2(I) exhibited porous and fluffy microstructures. These microstructures were characterized by large specific surface area and high porosity. This structure, combined with uniformly distributed quaternary ammonium salt groups from the quaternization of N,N-dimethylaminoethyl acrylate (DMAEA) tertiary amine groups, enhanced the contact between antibacterial active sites and bacteria as well as antibacterial efficacy. PCaPE(Br) and PCaPE(I) showed excellent broad-spectrum antibacterial activity against E. coli and S. aureus. They also maintained good reusability with over 95% activity retention after five cycles. Such advantages support their prospects in biomedicine, agricultural antibacterials and wastewater treatment.
Introduction
Over recent decades, unprecedented global population growth and widespread antibiotic misuse have significantly exacerbated environmental microbial contamination.1,2 Gram-negative and Gram-positive bacteria are ubiquitous on medical devices, food packaging, and frequently touched surfaces, driving a sharp rise in microbial infections and posing a serious public health threat. Addressing this challenge urgently requires the development of novel antibacterial materials with high efficiency, long durability, and low risk of inducing bacterial resistance. Quaternary ammonium salts (QAS) are effective antibacterial agents: their positively charged (R4N+)3 cations interact electrostatically with negatively charged bacterial membrane components,4 disrupting integrity, inducing intracellular leakage, and ultimately inhibiting bacterial proliferation or causing lysis.5–7 Although quaternary ammonium salts can inhibit a wide range of bacteria, the common low-molecular-weight ones have several limitations. They exhibit poor thermal stability, lack long-lasting bacteriostatic activity, have poor solubility in solvents, and are prone to inducing bacterial resistance. These limitations severely restrict their practical application, especially in scenarios where sustained and stable antibacterial effects are imperative.To overcome these limitations, polymer-based antibacterial agents have emerged as a promising alternative. Unlike low-molecular-weight QAS, polymeric antibacterial materials act through direct contact with bacteria without releasing toxic active substances, offering advantages of stable antibacterial effects, prolonged durability, and enhanced biocompatibility.8,9 Among polymeric systems, cross-linked copolymers have attracted particular attention due to their tunable mechanical properties and stability,10,11 which further extend their service life in practical applications. However, most of the currently available cross-linked antibacterial polymers feature a random cross-linking structure. This results in an uneven distribution of antibacterial functional groups, a low specific surface area, and inadequate contact with bacteria, all of which serve to constrain their antibacterial efficiency. Block cross-linked copolymers, a special class of cross-linked polymers, are constructed by covalently linking polymer segments with distinct chemical properties.12–14 Their unique self-assembly behavior enables the formation of well-defined microstructures (e.g., porous) when functional groups are introduced, which can significantly optimize the exposure of antibacterial moieties and interaction with bacteria.15,16 Integrating quaternary ammonium groups into block cross-linked copolymers offers great potential for the development of high-performance antibacterial materials with strong antibacterial activity, stability, and controllable morphology.17–21 For instance, Guo et al. fabricated copolymers with excellent antibacterial activity and good stability by modifying polymer side chains through nucleophilic substitution reactions.22 However, these studies primarily focused on random cross-linked structures or linear block copolymers, and the design of block cross-linked architectures specifically tailored for enhancing QAS-based antibacterial performance remains underexplored.
Reversible addition–fragmentation chain transfer (RAFT) polymerization is a reactive radical polymerization technique.23–25 By adding chain transfer agents (CTAs),26,27 it can precisely control the growth of polymer chains based on the reversible reactions between CTAs and free radicals. This leads to polymers with narrow molecular weight distributions and predetermined structures and molecular weights.28,29 Previous studies have demonstrated the potential of RAFT polymerization in preparing functional polymers. Yang et al. synthesized a bioactive polymer based on baicalin for the treatment of osteoarthritis by combining RAFT polymerization with esterification.29 Zeng et al. prepared an ABC triblock copolymer assembly with reversible “breathing” behavior through RAFT.30
In this study, the block cross-linked structure was precisely constructed through RAFT polymerization, and then the PCaPE series was prepared by quaternization. Compared with traditional random cross-linked polymers, this structure increases the specific surface area, promotes the quaternization reaction, and optimizes the antibacterial mechanism through the synergistic effect of hydrophilic and hydrophobic blocks, aiming to solve the problems of antibacterial efficiency and persistence of existing materials.
Experimental
Materials
Ethyl methacrylate (EMA) and 1,4-dioxane were obtained from Jinan Xilu Biotechnology Co., Ltd (China). Azobisisobutyronitrile (AIBN) and ethylene glycol dimethacrylate (EGDMA) were obtained from Beijing Inokai Biochemical Technology Co., Ltd (China). 2-(Dodecyl trithiocarbonate)-2-methylpropionic acid (DMP), n-hexane, methyl iodide (CH3I), 1-bromobutane (C4H9Br) and dimethylaminoethyl acrylate (DMAEA) were obtained from Shanghai Aladdin Biochemical Technology Co., Ltd (China). All reagents were of analytical grade (≥99.9%) and were used as received without further purification.Synthesis of macromolecular chain transfer agent PEMA
17.7 mL of EMA, 0.13 g of DMP, 0.06 g of AIBN, and 20 mL of 1,4-dioxane (as solvent) were added to the reaction flask. The mixture was then stirred at a constant temperature of 70 °C for 6 hours. Poly(ethyl methacrylate) (PEMA) was obtained after three washes with n-hexane and vacuum drying. This PEMA, functionalized as a chain transfer agent (CTA), provided an important building block for the subsequent synthesis of block cross-linked copolymers.31Synthesis of block cross-linked copolymer precursor (PE-PDE)
1.27 g of PEMA was dissolved in 10 mL of 1,4-dioxane, followed by the addition of 6.25 mL of DMAEA, different volumes of EGDMA (0 mL, 0.72 mL, 1.44 mL), and 12.8 mg of AIBN; the reaction was then conducted at 65 °C for 5 h with constant stirring. The reaction was quenched by exposure to air, the products were washed and dried under vacuum to obtain PEMA-b-P(DMAEA-co-EGDMA) (denoted as PE-PDE). The products were designated as PE-PDE1, PE-PDE2, and PE-PDE3 corresponding to the addition amounts of EGDMA (0 mL, 0.72 mL, and 1.44 mL, respectively).Synthesis of quaternized block cross-linked polymers (PCaPE(Br) and PCaPE(I))
20 mL of 1,4-dioxane and 0.9 g of PE-PDE were added, and the mixture was stirred until fully dissolved. For quaternization, 1.56 mL of C4H9Br was added for the synthesis of PCaPE(Br), while 1.25 mL of CH3I was used for PCaPE(I). The reaction was carried out at 55 °C for 24 h. After centrifugation, washing with n-hexane, and vacuum drying, quaternary ammonium bromide PCaPE(Br) and iodide PCaPE(I) were obtained. The quaternization products were named PCaPE1(Br), PCaPE2(Br), PCaPE3(Br), and PCaPE1(I), PCaPE2(I), PCaPE3(I), corresponding to their parent polymers PE-PDE1, PE-PDE2, and PE-PDE3, respectively. The synthetic routes of PCaPE(Br) and PCaPE(I) are shown in Scheme 1.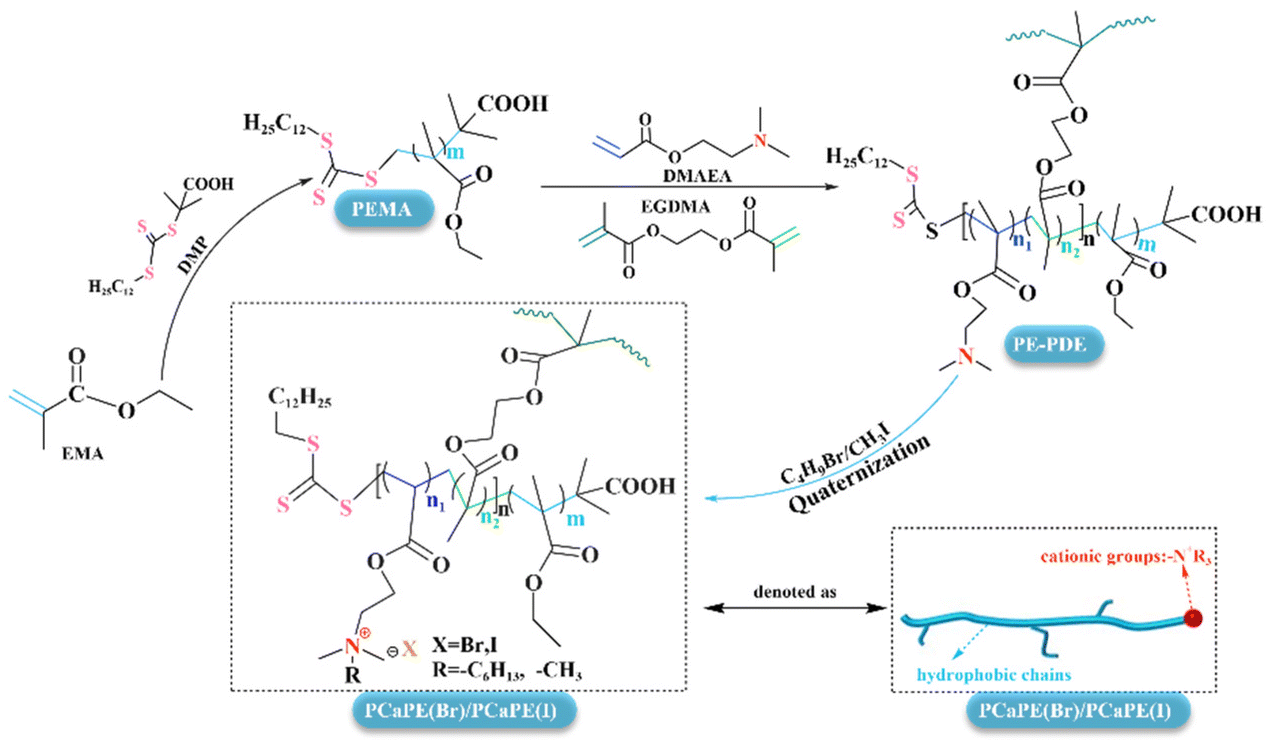 | ||
| Scheme 1 Synthesis of PCaPE(Br) and PCaPE(I). | ||
Synthesis of control samples (random cross-linked quaternary ammonium polymer, RCP(Br) and RCP(I))
To verify the antibacterial enhancement effect of the block cross-linked structure, control samples were prepared via conventional free-radical copolymerization (without employing the RAFT process or PEMA), specifically designated as randomly cross-linked quaternary ammonium salt cationic polymers (abbreviated as RCP(Br) and RCP(I), respectively).The synthesis conditions for the random cross-linked precursor were identical to those for PE-PDE2 (the precursor of PCaPE2). Specifically, 6.25 mL of DMAEA, 0.72 mL of EGDMA, 12.8 mg of AIBN, and 10 mL of 1,4-dioxane were added to a 50 mL three-necked flask. The flask was purged with N2 for 30 min to remove O2, then sealed and placed in a 65 °C constant-temperature water bath for a 5-hour reaction under magnetic stirring. The product was obtained through washing and vacuum drying. The resulting solid powder, defined as random cross-linked poly(DMAEA-co-EGDMA), was designated as RC-PDE.
The quaternization conditions for RC-PDE were identical to those for PE-PDE. Briefly, 0.9 g of prepared RC-PDE and 20 mL of 1,4-dioxane were added to a round-bottomed flask and magnetically stirred at room temperature until complete dissolution. For RCP(Br) preparation, 1.56 mL of C4H9Br was added to the solution; for RCP(I), 1.25 mL of CH3I was added instead. The flask was then placed in a 55 °C constant-temperature water bath for 24 h under magnetic stirring to proceed with quaternization. After reaction completion, the mixture was washed with n-hexane and vacuum drying yielded the random cross-linked quaternary ammonium polymers RCP(Br) and RCP(I), respectively.
Characterization
The reaction conversion rates of three key polymerization steps were quantified by the gravimetric method. These steps included RAFT polymerization for synthesizing PEMA, RAFT block copolymerization for synthesizing PE-PDE, and conventional free-radical copolymerization for synthesizing the control sample RC-PDE. For each polymerization system, first, the weight of an empty 5 mL centrifuge tube was measured and denoted as m0; then, 1.00 mL of the uniform prepolymerization mixture was transferred into the tube, immediately weighed and recorded as m1, so as to calculate the total mass of monomers in the 1.00 mL mixture. Upon completion of the polymerization under the corresponding conditions, 1.00 mL of the post-polymerization solution was sampled and placed into a new pre-weighed 5 mL centrifuge tube. The tube was then weighed and recorded as m2. Subsequently, the centrifuge tube was placed in a vacuum oven at 60 °C for 24 h until a constant weight was achieved, at which point the weight was recorded as m3. The reaction conversion rate was calculated using the following equation: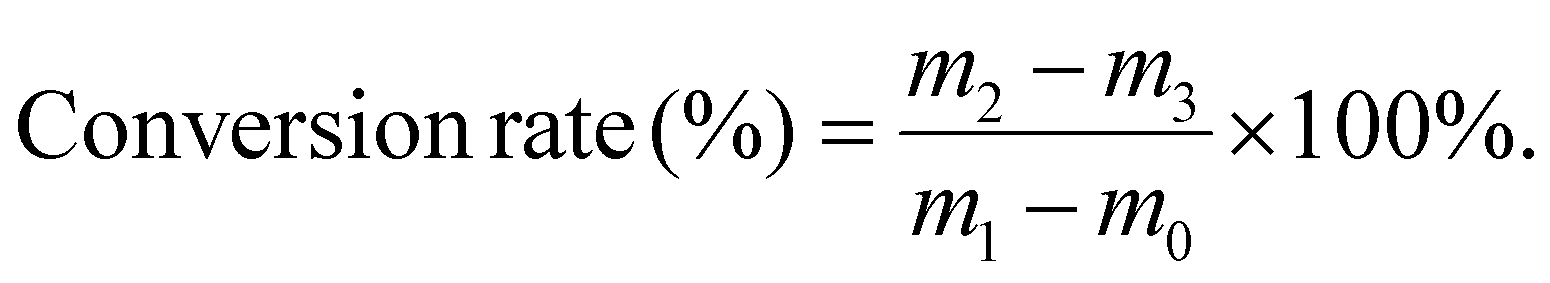
Each polymerization experiment was repeated three times, and the conversion rates were expressed as mean ± standard deviation to ensure data repeatability.
The content of quaternary ammonium groups in PCaPE(Br), PCaPE(I), and reference samples RCP(Br)/RCP(I) was quantified via the Mohr method to determine their quaternization degrees, with the focus on measuring counterions (Br− or I−) bound to quaternary ammonium groups. A 0.1000 g portion of dried polymer sample was dissolved in 50 mL of a 1:1 (v/v) n-butanol/deionized water mixture to form a homogeneous solution, ensuring complete dissolution of the polymer and full exposure of counterions. Approximately 3 drops of 5 wt% potassium chromate (K2CrO4) solution were added as the indicator. The sample solution was then titrated with a standard 0.0100 mol L−1 silver nitrate (AgNO3) solution at room temperature under constant magnetic stirring. The titration endpoint was identified by the persistent formation of a brick-red silver chromate (Ag2CrO4) precipitate. A blank titration was performed simultaneously using the 1:1 (v/v) n-butanol/deionized water mixture (without polymer) to correct for potential interference from trace halide ions in the solvent. The quaternization degree of each sample was calculated based on the stoichiometric relationship between quaternary ammonium groups and counterions (1:1 molar ratio) using the concentration and volume of AgNO3 consumed at the endpoint.
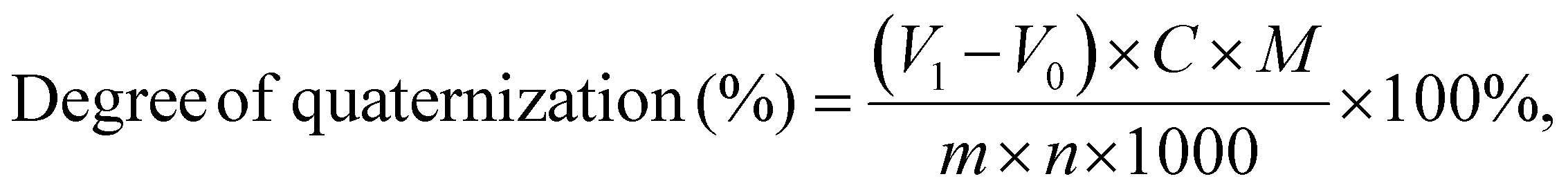
To demonstrate the precise control of RAFT polymerization over molecular weight, conventional free-radical copolymerization was used as a reference due to its broader molecular weight distribution. The number-average molecular weight (Mn), weight-average molecular weight (Mw), and polydispersity index (PDI) of PEMA, PE-PDE2, and RC-PDE were determined via gel permeation chromatography (GPC, Agilent 1260 Infinity II, USA). The GPC analysis was performed using chromatographic grade DMF (>99.9%, containing 0.1% lithium bromide) as the mobile phase at 45 °C, with a flow rate of 1.0 mL min−1. The system was equipped with a refractive index detector and two serially connected PL-MIXED-C columns (7.5 × 50 mm, 5 μm; 7.5 × 300 mm, 5 μm), and apparent molecular weights were calibrated against polystyrene (PS) standards. For samples preparation, each sample was dissolved in DMF to a concentration of 3.0 mg mL−1, followed by sonication at room temperature for 5 h to ensure complete dissolution. The solution was then filtered through a 0.22 μm organic syringe filter to remove insoluble impurities prior to injection.
To analyze the microstructure, the samples were sputter-coated with gold to form a metallic layer, and the micrographs of the samples were obtained using a field emission scanning electron microscope (FESEM, SIGMA 300, ZEISS, Germany). Additionally, the elemental composition and distribution of the prepared polymer samples were analyzed using an attached energy dispersive X-ray spectroscopy (EDS) module.
The specific surface area and pore structure of PE-PDE2, PCaPE2(Br), and the control sample (RCP(Br)) were determinedvia nitrogen adsorption–desorption isotherms at 77 K using a surface area and porosity analyzer (Micromeritics ASAP 2460, U.S.A). Prior to testing, all samples were degassed under vacuum at 105 °C for 6 hours to remove adsorbed water and residual solvents, ensuring the accuracy of adsorption data. The specific surface area was calculated using the Brunauer–Emmett–Teller (BET) model in the relative pressure range of P/P0 = 0.05–0.99. The pore size distribution was derived from the adsorption branch of the isotherm using the Barrett–Joyner–Halenda (BJH) method, and the total pore volume was determined at a relative pressure of P/P0 = 0.99.
To verify the successful synthesis of the macromolecular CTA PEMA, 1H NMR spectra of the products were obtained using a nuclear magnetic resonance spectrometer (Ascend Evo 400, Bruker, Germany).
Fourier transform infrared spectroscopy (FT-IR) was used to characterize the chemical structures of PEMA, PE-PDE2, PCaPE(Br), PCaPE(I) and RCP(Br). The samples were mixed with dry KBr at a mass ratio of 1:100 (w/w), ground to a particle size of <2 μm, and then pressed into a thin sheet. Spectra were collected using an FT-IR spectrometer (Nicolet iS5, Thermo Fisher Scientific, U.S.A) with a resolution of 4 cm−1, 32 scans, and a wavenumber range of 4000–500 cm−1.
The elemental composition and valence states of I, Br, C, O, and N on the surface of the samples were determined by X-ray photoelectron spectroscopy (XPS, ESCALAB 250XI, Thermo Fisher, U.S.A).
The contact angle data of PE-PDE, RCP(Br), RCP(I), PCaPE(Br), and PCaPE(I) were determined using a contact angle meter (OCA20, DataPhysics, Germany). The test method followed the procedure described in ref. 32.
The testing method for emulsifying performance can be found in ref. 33. Briefly, 3 g each of PE-PDE, PCaPE(Br), and PCaPE(I) was dispersed respectively in 3 mL of deionized water, mixed thoroughly, allowed to stand for 10 minutes, and photographed for documentation. For further assessment, 3 mL of rhodamine B-labeled n-butanol solution was added to a glass sample bottle, dispersed in 3 mL of deionized water, thoroughly mixed, and photographed after standing for 10 minutes. Additionally, 3 g of each sample (PE-PDE, PCaPE(Br), and PCaPE(I)) was slowly added to 3 mL of rhodamine B-labeled n-butanol solution, followed by dispersion in 3 mL of deionized water; the mixtures were thoroughly mixed and photographed after standing for 10 minutes. The mixing behavior of the samples with n-butanol and deionized water was observed and recorded, enabling visual assessment of their emulsification performance.
The minimum inhibitory concentration (MIC)34,35 of PCaPE(Br) and PCaPE(I) was determined by the dilution plating method. Escherichia coli (E. coli, representative of Gram-negative bacteria) and Staphylococcus aureus (S. aureus, representative of Gram-positive bacteria) were used as test organisms, and bacterial suspensions were prepared by incubating the bacteria to the logarithmic growth phase. The samples were dissolved in 1,4-dioxane to prepare stock solutions. A series of solutions were then prepared, with concentrations ranging from 0 to 1.6 mg mL−1 in 0.1 mg mL−1 increments. These solutions were mixed with an equal volume of the bacterial suspension and incubated at 37 °C for 24 h. The treated bacterial suspension was spread on the agar plates, which were then incubated inverted at 37 °C for 24 h to observe colony growth.
Bacterial growth curves were generated by monitoring the absorbance of bacterial suspensions at different time points after adding solutions of RCP(Br), RCP(I), PCaPE(Br), and PCaPE(I). This indirectly reflects the inhibitory effects of these compounds on microbial growth. Solutions of RCP(Br), RCP(I), PCaPE(Br) and PCaPE(I) (at a concentration of 0.3 mg mL−1) were prepared using 1,4-dioxane as the solvent. Bacterial suspensions of E. coli and S. aureus were diluted to a concentration of 1 × 106 CFU mL−1. The above solutions were mixed with an equal volume of bacterial suspensions, respectively. Meanwhile, a blank control group was set up, in which only 1,4-dioxane was added. Subsequently, the absorbance of each group at different time points was measured using a UV spectrophotometer (UV752, Qingdao Jingcheng Instrumentation Co., Ltd, China) with a 1 cm quartz cuvette at 600 nm. The inhibition rate (IR) was then calculated using the following equation based on the absorbance (A) values:36
| IR(\percnt) = [(A0 − Aa)/A0] × 100% |
Under simulated cyclic antibacterial conditions, the reusability stability of quaternized block cross-linked polymers (i.e., PCaPE2(Br) and PCaPE2(I)) was evaluated by assessing their antibacterial activity retention across multiple “antibacterial-recovery-re-antibacterial” cycles. First, PCaPE2(Br), PCaPE2(I), and the control samples (RCP(Br)/RCP(I)) were fabricated into uniform films of identical dimensions (10 mm in diameter, 50 μm in thickness), which were then sterilized for subsequent use. Each sterilized film was immersed in 10 mL of S. aureus suspension (1 × 106 CFU mL−1), while a blank control group containing only the bacterial suspension but no membrane was simultaneously established. The mixture was incubated with shaking at 37 °C for 24 h. After incubation, the inhibition rate was determined via the optical density method. Thereafter, the film was retrieved and gently rinsed three times with sterile normal saline (10 s per rinse) to remove surface-adhered bacteria, with gentle handling to avoid mechanical damage. The film was then dried in a vacuum oven at 60 °C for 2 h to restore its initial state. The above procedure was repeated to conduct the 2nd to 5th antibacterial cycles, and fresh bacterial suspension was used in each cycle to ensure consistent experimental conditions.
The antibacterial activity of PCaPE(Br) was evaluated using the paper diffusion method with E. coli as the test organism, to eliminate interference of solvents and raw materials with the antibacterial properties of the final products. Four filter paper discs were immersed in 0.6 mg mL−1 PCaPE2(Br) solution, 0.6 mg mL−1 PEMA solution, 0.6 mg mL−1 PE-PDE solution, and 1,4-dioxane, labeled as a, b, c, and d, respectively. The bacterial suspension was adjusted to a turbidity of 0.5 McFarland (approximately 1.5 × 108 CFU mL−1). This E. coli suspension was uniformly spread over the surface of the nutrient agar medium, and sterile pre-soaked filter discs were attached to the agar surface. The plates were then incubated inverted at 37 °C for 24 h. After incubation, the agar plates were removed, and the diameter of the inhibition zone was measured to determine the antibacterial activity of the samples.
Results and discussion
Analysis of the macroscopic morphologies of PEMA, PE-PDE, RCP(Br), RCP(I), PCaPE(Br), and PCaPE(I)
The macroscopic morphologies of the intermediates PEMA and PE-PDE are shown in Fig. 1a and b, respectively, and both are beige solid particles. The macroscopic morphology of the control samples (RCP(Br)/RCP(I)) are shown in Fig. 1c and d. PEMA block segments were not incorporated during synthesis, so these samples could not form the “hydrophobic–hydrophilic” segment microphase separation effect, which is a key requirement for generating a fluffy porous structure. Moreover, the samples only feature a simple cross-linked network, with polymer chains distributed randomly. As a result, they exhibit macroscopic properties of relatively high hardness and brittleness. In contrast to the control samples, PCaPE products with different quaternization degrees (i.e., PCaPE(Br) and PCaPE(I), including PCaPE1–3(Br) and PCaPE1–3(I)) exhibit spongy solid morphologies. However, in contrast to the spongy morphology of other PCaPE products, cross-linker-free PCaPE1(Br) (Fig. 1e) and PCaPE1(I) (Fig. 1h) show a dense blocky state. PCaPE2(Br) (Fig. 1f) and PCaPE2(I) (Fig. 1i), which contain a small amount of cross-linker, display a fluffy structure with numerous pores. In contrast, PCaPE3(Br) (Fig. 1g) and PCaPE3(I) (Fig. 1j) with an increased cross-linker dosage appear as fine particles and no longer exhibit a porous morphology. Therefore, when the ratio of PEMA and EGDMA is 1:6 (corresponding to PCaPE2(Br) and PCaPE2(I)), the products show a fluffy appearance with high porosity and a high specific surface area. This high porosity can increase the contact area with bacteria, thereby improving bacteriostatic activity.37,38 Hence, PCaPE2(Br) and PCaPE2(I) are expected to exhibit the optimal bacteriostatic effect.
 | ||
| Fig. 1 The macroscopic morphology of the product: (a) PEMA, (b) PE-PDE, (c) RCP(Br), (d) RCP(I), (e) PCaPE1(Br), (f) PCaPE2(Br), (g) PCaPE3(Br), (h) PCaPE1(I), (i) PCaPE2(I), (j) PCaPE3(I). | ||
The reaction conversion rates of RAFT polymerization, RAFT block copolymerization and conventional free-radical copolymerization
The reaction conversion rates of RAFT polymerization for preparing PEMA, RAFT block copolymerization for preparing PE-PDE, and conventional free-radical copolymerization for preparing the control sample precursor RC-PDE are summarized in Table 1. Conversion rates of the RAFT-mediated systems were significantly higher than those of the conventional free-radical polymerization system. The conversion rate for PEMA synthesis was the highest, reaching 87.5 ± 1.2%. This was ascribed to the reversible chain-transfer capability of the chain transfer agent DMP in RAFT polymerization, which could inhibit the premature termination of free radicals, prolong the chain growth period, and facilitate the full conversion of EMA. The synthesis conversion rate of PE-PDE slightly decreased to 82.3 ± 1.0%, mainly due to the steric hindrance effect of the crosslinking agent EGDMA that hindered the combination of some free radicals with monomers. Nevertheless, the precise regulation of RAFT polymerization still maintained a relatively high conversion rate with a small standard deviation, demonstrating the controllability of the reaction. In contrast, the synthesis conversion rate of RC-PDE without RAFT regulation was the lowest with a larger standard deviation. This is because free radicals are prone to uncontrollable termination in conventional free-radical polymerization, leading to insufficient monomer conversion and inferior reaction reproducibility. These differences in conversion rates confirm that RAFT polymerization not only enhances monomer conversion efficiency but also ensures the controllability and repeatability of the reaction, thereby laying a foundation for the precise construction and performance optimization of the subsequent PCaPE(Br)/PCaPE(I) block cross-linked structures.| Polymerization step | Polymerization method | Monomer composition | Conversion rate (%) (mean ± SD, n = 3) |
|---|---|---|---|
| PEMA synthesis | RAFT homopolymerization | EMA | 87.5 ± 1.2 |
| PE-PDE2 synthesis | RAFT block copolymerization | DMAEA + EGDMA | 82.3 ± 1.0 |
| RC-PDE synthesis | Conventional free-radical copolymerization | DMAEA + EGDMA | 77.8 ± 1.5 |
The quaternization degrees of PCaPE(Br)/PCaPE(I), and the control sample RCP(Br)/RCP(I)
The quaternization degrees of PCaPE and RCP samples were quantified, with results summarized in Fig. 2a. PCaPE2(Br) and PCaPE2(I) exhibited higher quaternization degrees at 92.3% ± 0.6% and 89.1% ± 0.6% respectively, compared to their reference counterparts RCP(Br) at 73.0% ± 1.3% and RCP(I) at 70.8% ± 1.1%. This indicates that the modified synthesis route for PCaPE effectively promotes the quaternization reaction, likely due to the enhanced accessibility of reactive sites in the polymer, which facilitates the binding of counterions such as Br− or I−. The slightly lower quaternization degree of I−-containing samples, as seen in PCaPE(I) relative to PCaPE(Br), may be attributed to the larger ionic radius of I−. This could induce steric hindrance during the ion-binding process. | ||
| Fig. 2 (a) The quaternization degrees of PCaPE and RCP samples, (b) the molecular weight and polydispersity index of PEMA, PE-PDE2, and RC-PDE. | ||
Molecular weight and polydispersity indices analysis
The molecular weight and polydispersity index of PEMA, PE-PDE2, and RC-PDE are illustrated in Fig. 2b. For the samples synthesized via RAFT polymerization, PEMA exhibited a well-defined Mn of 9.0 kg mol−1 and Mw of 13.3 kg mol−1 with a narrow PDI of 1.5. Its derived copolymer PE-PDE2 showed a slightly increased Mn of 10.7 kg mol−1 and Mw of 26.7 kg mol−1 with a PDI of 2.5. This mild PDI expansion is controllability of RAFT polymerization over molecular weight and distribution. Such control is enabled by its reversible chain transfer mechanism that mitigates chain length disparity.39 In contrast, free radical polymerization lacks such regulation, leading to random chain growth and extreme molecular weight heterogeneity.40 This is particularly evident in RC-PDE, where Mn is 28.7 kg mol−1, Mw is 118.2 kg mol−1, and the PDI is wide, at 4.1. It is worth noting that the conversion rate of PE-PDE2 is higher than that of RC-PDE, but its molecular weight is lower than that of RC-PDE. This result confirms the advantages of RAFT polymerization in regulating the conversion rate and molecular weight, as it can maintain a narrow distribution of the target molecular weight even at high conversion rates. The well-defined molecular weights of RAft-derived PEMA and PE-PDE2 are pivotal for ensuring uniform cross-linking density and ordered amphiphilic block assembly in subsequent reactions. This lays a structural foundation for the formation of porous morphologies and stable antibacterial performance of the final quaternized copolymers.Micro-morphological analysis of PE-PDE, PCaPE(Br) and PCaPE(I)
SEM images showed that the surface of PE-PDE2 (Fig. 3a) was dense and lumpy, while the surface of PCaPE2(Br) (Fig. 3b) showed a loose and porous aerogel-like structure. This structural change implies that the quaternization treatment not only increases the specific surface area of the product, but also enhances the hydrophilicity of the polymer surface, which is more conducive to the full contact of the product surface with bacteria. The elemental composition and distribution of the PCaPE2(Br) polymer were analyzed using EDS, as shown in Fig. 3c–e. Br and N were clearly detected in the selected regions, which confirms the successful preparation of PCaPE2(Br). | ||
| Fig. 3 SEM images: (a) PE-PDE2, (b) PCaPE2(Br); (c) scanning area of PCaPE2(Br), EDS surface distribution maps: (d) Br element, (e) N element. | ||
Similarly, we compared the SEM results of different PCaPE(I) products obtained via quaternization. PCaPE2(I) (Fig. 4b) exhibited a porous gel structure, while the other products, PCaPE1(I) (Fig. 4a) and PCaPE3(I) (Fig. 4c), showed lamellar and porous wafer morphologies, respectively. These SEM observations are consistent with the macroscopic morphological characteristics of the iodinated block cross-linked copolymers (PCaPE1(I), PCaPE2(I), and PCaPE3(I)). Uniform distribution of N and I was observed in the EDS mapping of PCaPE2(I) (Fig. 4d–f), which confirms the successful preparation of PCaPE2(I).
 | ||
| Fig. 4 SEM images: (a) PCaPE1(I), (b) PCaPE2(I), (c) PCaPE3(I); EDS surface distribution maps: (d) scanning position of PCaPE2(I), (e) I element, (f) N element. | ||
1H NMR spectra analysis
1H NMR spectra were used to examine the molecular structure of the samples. The 1H NMR spectrum of DMP, one of the reactants used in synthesizing the macromolecular CTA, is shown in Fig. 5a. It has the following chemical shifts: δ = 3.28 ppm (–CH2–(S)3–), δ = 1.90 ppm (–CH2–S3–C(CH3)2–COOH), δ = 1.73 ppm (–CH2CH2–S3–), δ = 1.37 ppm (–CH2CH2CH2–S3–), δ = 1.25 ppm (–(CH2)8CH2CH2CH2CH2–S3–), and δ = 0.86 ppm (CH3(CH2)11–S3–). The 1H NMR spectrum of the macromolecular chain transfer agent PEMA is shown in Fig. 5b. Its chemical shifts were: δ = 1.83 ppm (–S3–CH2C–), δ = 1.26 ppm (–CH2–S3–C(CH3)2–COOH), δ = 4.02 ppm (–CH2CH2CH2–S3–), δ = 3.71 ppm (CH3OCO–), and δ = 0.86–1.01 ppm (CH3C-, –(CH2)10CH2–S3–). Compared with the DMP CTA (where the CH3(CH2)11–S3– group appears at δ = 0.86 ppm), the presence of these specific characteristic shifts in PEMA confirms the formation of its block structure and indicates that the S element in DMP has been successfully introduced into the macromolecular chain during the reaction. This confirms the successful preparation of the macromolecular chain transfer agent PEMA. | ||
| Fig. 5 1H NMR spectra: (a) DMP (CDCl3); (b) PEMA (CDCl3). | ||
BET specific surface area and porosity analysis
The Fig. 6 presents N2 adsorption–desorption isotherms and pore size distribution. PCaPE2(Br) exhibits a type IV isotherm, with a BET surface area of 80.9 m2 g−1, a BJH adsorption cumulative pore volume of 0.2 cm3 g−1 for pores ranging from 1.70 nm to 200.00 nm, and an average pore diameter of 8.9 nm, alongside abundant mesopores centered at 40–80 nm. In contrast, RCP(Br) and PE-PDE2 show negligible N2 adsorption, indicating their non-porous nature. This porous characteristic of PCaPE2(Br) is consistent with its FESEM results, which also reveal a well-developed mesoporous morphology. The disparity in porosity originates from structural differences. PCaPE2(Br) benefits from its RAFT-synthesized block cross-linked architecture, where amphiphilic self-assembly of hydrophobic PEMA segments and hydrophilic quaternized segments induces the formation of interconnected mesopores. RCP(Br), prepared via uncontrolled free radical polymerization, undergoes random cross-linking and chain aggregation, leading to a dense structure without ordered pores. PE-PDE2, as a precursor, lacks porosity before quaternization and structural rearrangement, which are necessary to drive pore formation in PCaPE2(Br). The differences in the porosity of the three samples highlight that both RAFT polymerization and quaternization reactions play a promoting role in constructing mesoporous structures that are conducive to antibacterial properties. | ||
| Fig. 6 N2 adsorption–desorption isotherms and pore size distribution. | ||
FT-IR analysis
The FT-IR spectra of PEMA, PE-PDE2, RCP(Br), and PCaPE2(Br) are displayed in Fig. 7a, while those of PCaPE(Br) and PCaPE(I) are shown in Fig. 7b. The absorption peak at 1732 cm−1, assigned to the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration within the polymer chain, confirms the presence of carbonyl groups. Notably, RCP(Br) lacks this ester group characteristic peak at 1732 cm−1, which is attributed to the absence of structural regulation via RAFT polymerization during synthesis or the specific cross-linking mechanism employed; additionally, its spectrum exhibits slight peak broadening. The peak at 1450 cm−1 corresponds to C–H bending vibrations of alkyl groups, specifically –CH3 and –CH2– moieties. A key feature is the thiocarbonyl (CS) stretching vibration at 1042 cm−1, which serves as definitive evidence for the successful incorporation of the PEMA chain transfer agent into the polymer backbone. For the cationic porous polymer PCaPE2(Br), which contains –N+(CH3)3 groups, the absorption peak at approximately 987 cm−1 is assigned to C–N+ stretching vibrations. This peak is characteristic of quaternary ammonium groups and thus confirms the cationic nature of the polymer. The distinct distinction between the peaks at 2982 cm−1 and 2925 cm−1 arises from the differing C–H stretching vibrations in –CH2–N+(CH3)3versus –CH2–N(CH3)2 moieties. Furthermore, the absorption peak at 1149 cm−1 is attributed to the C–N bending vibration of the amino groups in DMAEA, confirming the presence of this functionality, which may influence the physical and chemical properties of the synthesized polymers, such as solubility and reactivity.41–43 Collectively, these results verify the successful preparation of PEMA, PE-PDE2, RCP, PCaPE with varying cross-linker ratios, as well as validate the polymerization reactions, block structure formation, and quaternization processes.
O stretching vibration within the polymer chain, confirms the presence of carbonyl groups. Notably, RCP(Br) lacks this ester group characteristic peak at 1732 cm−1, which is attributed to the absence of structural regulation via RAFT polymerization during synthesis or the specific cross-linking mechanism employed; additionally, its spectrum exhibits slight peak broadening. The peak at 1450 cm−1 corresponds to C–H bending vibrations of alkyl groups, specifically –CH3 and –CH2– moieties. A key feature is the thiocarbonyl (CS) stretching vibration at 1042 cm−1, which serves as definitive evidence for the successful incorporation of the PEMA chain transfer agent into the polymer backbone. For the cationic porous polymer PCaPE2(Br), which contains –N+(CH3)3 groups, the absorption peak at approximately 987 cm−1 is assigned to C–N+ stretching vibrations. This peak is characteristic of quaternary ammonium groups and thus confirms the cationic nature of the polymer. The distinct distinction between the peaks at 2982 cm−1 and 2925 cm−1 arises from the differing C–H stretching vibrations in –CH2–N+(CH3)3versus –CH2–N(CH3)2 moieties. Furthermore, the absorption peak at 1149 cm−1 is attributed to the C–N bending vibration of the amino groups in DMAEA, confirming the presence of this functionality, which may influence the physical and chemical properties of the synthesized polymers, such as solubility and reactivity.41–43 Collectively, these results verify the successful preparation of PEMA, PE-PDE2, RCP, PCaPE with varying cross-linker ratios, as well as validate the polymerization reactions, block structure formation, and quaternization processes.
 | ||
| Fig. 7 FT-IR spectra (KBr pellet): (a) PEMA, PE-PDE2, RCP(Br), PCaPE2(Br); (b) PCaPE(Br) and PCaPE(I). | ||
XPS analysis
The XPS spectrum of PCaPE2(Br) (Fig. 8a) clearly confirms the presence of C, O, N, and Br elements. In the C 1s spectrum (Fig. 8b), the characteristic peak at 284.3 eV is assigned to the C–H, C–C, and CC bonds within the alkyl chains. The peaks at 285.6 eV and 288.2 eV correspond to C–O/C–N and CO bonds, respectively, confirming the presence of ester (–COO–) in the polymer chain. In the N 1s spectrum (Fig. 8c), the main peak at 401.6 eV is assigned to the quaternary ammonium groups (–N+(CH3)3) on the surface of PCaPE2(Br), indicating successful quaternization. In the Br 3d spectrum (Fig. 8d), two prominent peaks are observed at 67.4 eV and 68.7 eV, which correspond to Br 3d5/2 and Br 3d3/2, respectively, providing further confirmation of the presence of bromine in the polymer. In the O 1s spectrum (Fig. 8e), the peaks at 531.5 eV and 533.1 eV are attributed to O–CO and CO bonds, consistent with the oxygen environments of ester (–COO–) and amide (–CONH–) groups. These results collectively confirm the successful preparation of quaternized PCaPE2(Br). Following the analysis of PCaPE2(Br) XPS spectra, we next characterized PCaPE2(I). The full spectrum of PCaPE2(I) (Fig. 8a) confirms the presence of C, N, O, and I elements. The I 3d spectrum (Fig. 8h) exhibits two characteristic peaks at 618 eV and 629 eV, assigned to I 3d5/2 and I 3d3/2, respectively, confirming the successful preparation of PCaPE2(I).
 | ||
| Fig. 8 XPS spectra of PCaPE2(Br) and PCaPE2(I): (a) survey spectra of PCaPE2(Br) and PCaPE2(I), (b) C 1s spectrum of PCaPE2(Br), (c) N 1s spectrum of PCaPE2(Br), (d) Br 3d spectrum of PCaPE2(Br), (e) O 1s spectrum of PCaPE2(Br); (f) C 1s spectrum of PCaPE2(I), (g) N 1s spectrum of PCaPE2(I), (h) I 3d spectrum of PCaPE2(I), (i) O 1s spectrum of PCaPE2(I). | ||
Contact angle analysis
When the material is hydrophilic, it can rapidly adsorb water and form a water film, which promotes adequate contact between quaternary ammonium cations and the negative charges on the bacterial surface, thereby accelerating the bacteriostatic activity.44 Thus, the hydrophilicity of the samples contributes to an enhanced bacteriostatic efficacy.45 The hydrophilicity of PE-PDE2, RCP(Br), RCP(I), PCaPE2(Br), and PCaPE2(I) was evaluated via contact angle measurements showed in Fig. 9. After quaternization, all cationic samples exhibited lower contact angles, yet there were distinct differences between the systems. The RAFT-synthesized PCaPE2 series possessed well-developed mesopores, as indicated by a BET surface area of 80.9 m2 g−1, and demonstrated superior hydrophilicity. Specifically, PCaPE2(Br) had a contact angle of 27.50°, while PCaPE2(I) had a contact angle of 35.71°. The mesopores in these samples offered channels for water infiltration and storage, and the uniformly distributed quaternary ammonium groups enhanced their affinity for water, which contributed to their better hydrophilicity. In contrast, the RCP series featured a random cross-linked structure and showed significantly higher contact angles. RCP(Br) had a contact angle of 60.50°, and RCP(I) had a contact angle of 70.15°. The absence of pores in these samples meant there was no capacity to retain water, and the uneven distribution of hydrophilic groups reduced their wetting ability, resulting in higher contact angles. | ||
| Fig. 9 Contact angle test results of PE-PDE2, RCP(Br), RCP(I), PCaPE2(Br) and PCaPE2(I). | ||
Within each series, samples containing Br− had smaller contact angles compared to those containing I−. This was because the smaller ionic radius of Br− reduced the steric hindrance for water interaction. Given its mesoporous structure and strong hydrophilicity, PCaPE2(Br) is anticipated to have the best bacteriostatic activity, as it can facilitate more effective contact between cations and bacteria.
Emulsification performance tests analysis
Many bacteriostatic substances with amphiphilic structures exhibit surfactant-like properties.46–49 An excellent emulsifying capacity enables bacteriostatic ingredients to disperse more uniformly in stains or biofilms, thereby disrupting the environment for bacterial attachment and improving bacteriostatic efficiency.50 Moreover, testing emulsification can evaluate the compatibility of bacteriostatic substances with other components; superior emulsification of a sample prevents interactions between components that may cause precipitation, delamination, or loss of activity.51 The precursor PE-PDE2 and its quaternized derivatives PCaPE2(Br) and PCaPE2(I) were dispersed in deionized water. PE-PDE2, which was not quaternized, was insoluble in water, whereas after quaternization, PCaPE2(Br) and PCaPE2(I) became water-dispersible due to the generation of quaternary ammonium cations, forming homogeneous milky white and yellow emulsions, respectively (Fig. 10a). When rhodamine B-labeled n-butanol was mixed with deionized water, distinct delamination occurred, indicating immiscibility between the oil and water phases (Fig. 10b). Subsequently, the three samples were added to mixtures of deionized water and rhodamine B-labeled n-butanol, thoroughly shaken, and then left to stand. The unquaternized PE-PDE2 exhibited no emulsifying activity and still showed delamination, whereas the quaternized PCaPE2(Br) and PCaPE2(I) exhibited excellent emulsifying capacity for both n-butanol and water, enabling the formation of stable n-butanol–water emulsions without phase separation (Fig. 10c). These results confirm that PCaPE2(Br) and PCaPE2(I) possess excellent emulsification stability, consistent with successful quaternization. | ||
| Fig. 10 (a) Dispersion behavior of PE-PDE2, PCaPE2(Br) and PCaPE2(I) in water; (b) miscibility test of n-butanol with water; (c) emulsification performance testing of PE-PDE2, PCaPE2(Br) and PCaPE2(I) in n-butanol–water system. | ||
Determination of the MIC of PCaPE(Br) and PCaPE(I)
For the bacteriostatic effect of PCaPE2(Br) against E. coli, the control group without PCaPE2(Br) showed luxuriant growth with abundant colonies (Fig. 11a). At a concentration of 0.3 mg mL−1, E. coli growth was significantly inhibited (Fig. 11b). When the concentration was further increased to 0.6 mg mL−1, bacterial growth was completely suppressed (Fig. 11c), indicating that the MIC of PCaPE2(Br) against E. coli is 0.6 mg mL−1. For S. aureus, the results were consistent: the blank control (without PCaPE2(Br)) exhibited luxuriant growth with abundant colonies (Fig. 11d). At 0.3 mg mL−1, S. aureus growth was significantly inhibited (Fig. 11e), and complete inhibition was observed at 0.6 mg mL−1 (Fig. 11f), demonstrating that the MIC of PCaPE2(Br) against S. aureus is also 0.6 mg mL−1. | ||
| Fig. 11 E. coli colonies after addition of different concentrations of PCaPE2(Br) for 24 h of incubation: (a) control group, (b) 0.3 mg mL−1, (c) 0.6 mg mL−1; S. aureus colonies after addition of different concentrations of PCaPE2(Br) for 24 h of incubation: (d) control group, (e) 0.3 mg mL−1, (f) 0.6 mg mL−1. | ||
For the bacteriostatic effect of PCaPE2(I) on E. coli, the control group without PCaPE2(I) demonstrated robust growth, with numerous colonies (Fig. 12a). When the PCaPE2(I) concentration reached 0.6 mg mL−1, a significant inhibition of E. coli growth was observed (Fig. 12b). Further increasing the concentration to 1.2 mg mL−1 resulted in complete suppression of colony growth in the medium (Fig. 12c), indicating that the MIC of PCaPE2(I) against E. coli is 1.2 mg mL−1. Similarly, in the inhibition experiments targeting S. aureus, the control group without PCaPE2(I) also exhibited luxuriant growth with abundant colonies (Fig. 12d). At a PCaPE2(I) concentration of 0.6 mg mL−1, S. aureus growth was significantly inhibited (Fig. 12e). When the concentration was elevated to 1.2 mg mL−1, colony growth in the medium was completely inhibited (Fig. 12f), demonstrating that the MIC of PCaPE2(I) against S. aureus is also 1.2 mg mL−1.
 | ||
| Fig. 12 E. coli colonies after addition of different concentrations of PCaPE2(I) for 24 h of incubation: (a) control group; (b) 0.6 mg mL−1; (c) 1.2 mg mL−1; S. aureus colonies after addition of different concentrations of PCaPE2(I) for 24 h of incubation: (d) control group; (e) 0.6 mg mL−1; (f) 1.2 mg mL−1. | ||
Taken together, the MIC of PCaPE2(Br) was determined to be 0.6 mg mL−1, while that of PCaPE2(I) was 1.2 mg mL−1. This indicates that PCaPE2(Br) exhibits superior antibacterial activity to PCaPE2(I), with a more potent inhibitory effect against E. coli and S. aureus.
The growth curves of E. coli and S. aureus under the action of RCP and PCaPE
Fig. 13 shows the antibacterial activity of PCaPE2(Br), PCaPE2(I), and their counterparts RCP(Br) and RCP(I) agains E. coli and S. aureus over a 24-hour period. The blank groups of E. coli and S. aureus display a sharp rise in absorbance, indicating robust bacterial growth. PCaPE2(Br) and PCaPE2(I) exhibit almost flat absorbance curves throughout the 24-hour period, demonstrating potent and long-lasting bacteriostatic effects. This is attributed to their well-defined mesoporous structure, high hydrophilicity, and uniformly distributed quaternary amonium groups, which enable rapid and persistent interaction with bacterial surfaces. In contrast, the absorbance of bacterial suspensions containing RCP(Br) and RCP(I) first decreases and then increases. The randomly cross-linked structure of RCP leads to uneven distribution of quaternary ammonium groups and a lack ordered porosity, reducing the accessibility of antibacterial sites and ordered porosity, reducing the accessibility of antibacterial sites and hydrophilicity. Thus, RCP initially exerts some bacteriostatic effects but eventually fails to inhibit bacterial growth, with a subsequent rise in absorbance indicating microbial resurgence due to depleted or inefficient antibacterial activity. This marked contrast highlights that PCaPE, with its RAFT-derived block cross-linked architecture, outperforms RCP in both antibacterial potency and durability, owing to its optimized structure for bacterial interaction and sustained activity. | ||
| Fig. 13 The growth curves of E. coli and S. aureus in suspensions with RCP and PCaPE over a 24 h incubation period: (a) samples- E. coli; (b) samples- S. aureus. | ||
Inhibition rates of RCP and PCaPE against E. coli and S. aureus
In addition, the inhibition rates of RCP(Br), RCP(I), PCaPE2(Br) and PCaPE2(I) were calculated over different time intervals based on the A values of bacterial suspensions. As illustrated in Fig. 14, the inhibition rates of RCP(Br), RCP(I), PCaPE2(Br), and PCaPE2(I) differ notably, with PCaPE2(Br) and PCaPE2(I) demonstrating much higher and more stable performance. Their inhibition rates typically remain above 90% over the 24-hour period, with PCaPE2(Br) slightly outperforming PCaPE2(I) due to the smaller ionic radius of Br−, which enhances electrostatic interactions with bacterial surfaces. This superior performance is linked to their structured mesopores, strong hydrophilicity, and evenly distributed quaternary ammonium groups, which facilitate sustained interaction with bacterial surfaces. | ||
| Fig. 14 Inhibition rates of RCP(Br), RCP(I), PCaPE2(Br) and PCaPE2(I) against E. coli and S. aureus at 24 h. | ||
In contrast, RCP(Br) and RCP(I) exhibit lower initial inhibition rates, around 40–70%, which drop significantly over time, falling to 10–40% by the 24-hour mark as bacterial growth rebounds. Their randomly cross-linked structure causes an uneven distribution of active groups and poor porosity, reducing both the accessibility of antibacterial sites and the hydrophilicity of the material, leading to weaker and less durable efficacy. In summary, the structural control afforded by RAFT polymerization results in both higher initial antibacterial rates and long-term durability compared to the random cross-linked RCP system. Within each series, Br−-containing samples consistently outperform their I−-containing counterparts.
Comparative study on antibacterial durability of quaternized block cross-linked and random cross-linked polymers over multiple cycles
Fig. 15(a) shows the cyclic antibacterial performance of RCP(Br), RCP(I), PCaPE2(Br), and PCaPE2(I), over five consecutive cycles. The data reveal significant differences in antibacterial durability between the two polymer systems. PCaPE2(Br) and PCaPE2(I) exhibit exceptional cyclic stability. Both achieve 100% inhibition rate in the first cycle and maintain high efficacy even after five cycles. PCaPE2(Br) retains 97.0% of its initial activity while PCaPE2(I) remains at 96.0%. This minimal attenuation highlights the structural advantage of RAFT-derived block cross-linking. Well-defined mesopores facilitate consistent water retention and bacterial contact. Uniformly distributed quaternary ammonium groups ensure sustained antibacterial interactions. The slightly superior performance of PCaPE2(Br) over PCaPE2(I) across cycles further confirms that the smaller ionic radius of Br− reduces steric hindrance and enhances the stability of cation-bacterial membrane interactions. In contrast, RCP(Br) and RCP(I) show marked activity decay. Their initial antibacterial rates (82.3% and 78.6% respectively) are significantly lower than the PCaPE2 series. By the fifth cycle their retention rates drop to 67.1% and 61.5%. This rapid decline stems from their random cross-linked structure which causes uneven distribution of quaternary ammonium groups. Some active sites are encapsulated in dense polymer matrices reducing accessibility during repeated use. Additionally the lack of ordered porosity limits hydrophilicity and water retention weakening the reactivation of antibacterial sites after recovery steps. In a manner akin to the PCaPE2 series, RCP(Br) demonstrates superior antibacterial performance compared to RCP(I) within the same cycle. However, as the number of cycles increases, the overall disparity between RCP and PCaPE2 becomes more pronounced, highlighting the pivotal significance of a well-controlled polymer architecture in bolstering cyclic antibacterial stability. The low relative standard deviation (RSD < 1.2%) across all measurements confirms the reliability of the results reinforcing that the block cross-linked structure of PCaPE2 is key to achieving long-term reusable antibacterial performance. | ||
| Fig. 15 (a) Variation of inhibition rate retention for quaternized polymers over 5 antibacterial cycles, (b) photographs of the inhibition zones showing the growth inhibition of E. coli by PCaPE2(Br). | ||
To further verify the inhibitory effect of PCaPE2(Br) on E. coli growth and rule out potential interference from solvents and raw materials, the diameter of its inhibition zone against E. coli was determined. As shown in Fig. 15(b), PCaPE2(Br) formed a distinct inhibition zone with a diameter of approximately 27.5 mm, whereas no inhibition zones were observed for the other filter paper discs (i.e., control groups). These results confirm that only PCaPE2(Br) exhibited significant antibacterial activity, and the interference from 1,4-dioxane, PEMA, and PE-PDE was effectively excluded.
Structure–performance–mechanism relationship of antibacterial activity
The block cross-linked PCaPE series, represented by PCaPE2(Br) and PCaPE2(I), exhibits superior antibacterial properties, primarily attributed to the structural precision achieved via RAFT polymerization. This precision, absent in the randomly crosslinked RCP synthesized through conventional radical copolymerization, enables strict control over the molecular weight distribution of PCaPE precursors. Consequently, a narrow PDI and uniform crosslinking density are achieved during the quaternary ammonium salting process. This uniformity drives the formation of ordered porous aggregated structures, as confirmed by FESEM and BET analyses. PCaPE2(Br), in particular, features a high specific surface area and abundant mesoporous pores, generated through the amphiphilic self-assembly of hydrophobic PEMA fragments and hydrophilic quaternized DMAEA fragments.These structural advantages directly translate into enhanced antibacterial performance. The porous structure effectively captures bacteria via its high specific surface area, while enhanced hydrophilicity forms a hydration layer, ensuring close contact between quaternary ammonium groups and bacterial membranes.52,53 Positively charged quaternary ammonium groups bind to negatively charged membrane components (e.g., lipopolysaccharides (LPS) and peptidoglycan54–57), disrupting electrostatic balance.58,59 Hydrophobic PEMA fragments insert into the lipid bilayer, inducing intracellular leakage and bacterial death (Fig. 16). In contrast, the RCP lacks this structural mechanism due to its dense morphology and uneven functional group distribution, resulting in poor bacterial capture and membrane disruption.
 | ||
| Fig. 16 The inhibitory mechanisms of PCaPE against Gram-negative and Gram-positive bacteria. | ||
Conclusions
In conclusion, this study successfully employed RAFT polymerization to precisely synthesize quaternary ammonium salt-based block-crosslinked copolymers, PCaPE(Br) and PCaPE(I). Compared to randomly crosslinked polymers (RCP(Br) and RCP(I)), RAFT-regulated block-chain crosslinking in the PCaPE series enabled precise control over molecular weight distribution. This control facilitated the formation of ordered porous aggregates and ensured uniform functionalization. These structural features resulted in high and uniform exposure of antibacterial groups on the material surface, significantly enhancing contact efficiency between antibacterial moieties and bacterial cells and optimizing antibacterial performance. Consequently, the PCaPE exhibited approximately 20% higher antibacterial efficiency and 35% improved reusability stability compared to RCP. This clearly demonstrates the interdependence between structure, performance, and mechanism, highlighting the critical role of structural precision in achieving superior antibacterial efficacy.The results show that when the molar ratio of macromolecular chain transfer agent (PEMA) to crosslinking agent (EGDMA) is optimized to 1:6 (corresponding to PCaPE2(Br) and PCaPE2(I)), the material presents a porous and fluffy morphology with a high specific surface area. Both PCaPE2(Br) and PCaPE2(I) exhibit excellent emulsification stability and hydrophilicity. The synergistic effect of these two properties further enhances these copolymers’ antibacterial activity. Specifically, PCaPE2(Br) exerts significant inhibitory effects against E. coli and S. aureus at a concentration of 0.3 mg mL−1. It achieves complete bacterial inhibition when the concentration reaches 0.6 mg mL−1. In contrast, PCaPE2(I) demonstrates relatively weaker antibacterial activity. It only exhibits significant inhibition at a concentration of 0.6 mg mL−1 and requires a concentration of 1.2 mg mL−1 to achieve complete inhibition. This result confirms that PCaPE2(Br) has superior antibacterial performance. In conclusion, the quaternized structures of PCaPE2(Br) and PCaPE2(I) enhance antibacterial efficacy by increasing specific surface area, hydrophilicity, and emulsification stability. This structural advantage renders them highly promising candidates for novel antibacterial materials.
Author contributions
Chaoyue Shan, Haohao Zhang: Writing – original draft, writing – review & editing; Yan Gao: Conceptualization & methodology; Hui Wang: Supervision & validation; Li Han: Methodology; Yuanyuan Du, Wen He, & Xing Gao: Investigation; Mingyuan Pang: Methodology. Yen Leng Pak: Resources.Conflicts of interest
There are no conflicts to declare.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (82202240) and Taishan Scholar Project of Shandong Province (tsqn202211249).References
- L. Corse and A. Cartwright, Microbiology, 2024, 170, 001514 CrossRef CAS PubMed.
- A. L. Schilling, A. Rody and V. A.-O. Bossung, Geburtshilfe Frauenheilkd., 2023, 83, 192–200 CrossRef CAS.
- B. Chanda, M. Shamimuzzaman, A. Gilliard and K.-S. Ling, Virol. J., 2021, 18, 7 CrossRef CAS PubMed.
- Y. Wang, S. Wang, Y. Zhang, J. Mi and X. Ding, Polymers, 2020, 12, 2229 CrossRef CAS.
- C. Lian, J. Liu, W. Wei, X. Wu, T. Goto, H. Li, R. Tu and H. Dai, Bioact. Mater., 2024, 38, 181–194 CAS.
- M. Parcheta and M. Sobiesiak, Materials, 2023, 16, 4411 CrossRef CAS PubMed.
- X. Long, Y. He, A. Wang, X. Wang, A. Zhen, L. Wu, L. Yang, F. Luo, J. Li, Z. Li and H. Tan, Adv. Healthcare Mater., 2025, 14, 2500687 CrossRef CAS.
- R. Guo, Y. C. Kan, Y. Xu, L. Y. Han, W. H. Bu, L. X. Han, Y. Y. Qi and J. J. Chu, Front. Bioeng. Biotechnol., 2024, 12, 1414005 CrossRef PubMed.
- H. Zhao, X. Li, L. Zhang, Z. Hu, L. Zhong and J. Xue, Sci. Rep., 2021, 11, 12205 CrossRef CAS PubMed.
- I. Tzoumani, G. C. Lainioti, A. J. Aletras, G. Zainescu, S. Stefan, A. Meghea and J. K. Kallitsis, Materials, 2019, 12, 4067 CrossRef CAS PubMed.
- Z. Huang, J. Xie, T. Li, L. Xu, P. Liu and J. Peng, Polymers, 2024, 16, 2761 CrossRef CAS PubMed.
- S. Gottlieb, M. Fernández-Regúlez, M. Lorenzoni, L. Evangelio and F. Perez-Murano, Nanomaterials, 2020, 10, 974772 CrossRef PubMed.
- T. Yamazaki, T. Yamamoto and T. Hirose, Front. Mol. Biosci., 2022, 9, 974772 CrossRef CAS PubMed.
- I. M. Davletbaeva, O. O. Sazonov, I. M. Dzhabbarov, I. I. Zaripov, R. S. Davletbaev and A. V. Mikhailova, Polymers, 2022, 14, 2678 CrossRef CAS PubMed.
- M. M. Rahman, Membranes, 2023, 13, 164 CrossRef CAS PubMed.
- J. Luczak, M. Kroczewska, M. Baluk, J. Sowik, P. Mazierski and A. Zaleska-Medynska, Adv. Colloid Interface Sci., 2023, 314, 102864 CrossRef CAS PubMed.
- P. P. Kalelkar, M. Riddick and A. J. García, Nat. Rev. Mater., 2022, 7, 39–54 CrossRef CAS PubMed.
- M. A. Khattak, Z. Iqbal, F. Nasir, S. H. Neau, S. I. Khan, T. Hidayatullah, S. Pervez, M. Sakhi, S. R. Zainab, S. Gohar, F. Alasmari, A. Rahman, G. E. Maryam and A. Tahir, Pharmaceutics, 2023, 15, 2375 CrossRef.
- J.-A. Park, K. Y. Cho, C. H. Han, A. Nam, J.-H. Kim, S.-H. Lee and J.-W. Choi, Sci. Rep., 2019, 9, 383 CrossRef PubMed.
- K. Makri and S. Pispas, Polymers, 2024, 16, 1284 CrossRef CAS.
- F. Teixeira, A. M. Silva, S. Sut, S. Dall'Acqua, C. Delerue-Matos, B. Estevinho, P. C. Costa and F. Rodrigues, Antioxidant, 2023, 12, 1496 CrossRef CAS.
- W. Guo, P. Cao, F. Dai, Y. Li, R. Wang, P. Song and Y. He, Polym. Adv. Technol., 2022, 33, 2166–2175 CrossRef CAS.
- V. V. Kusumkar, M. Galamboš, E. Viglašová, M. Daňo and J. Šmelková, Materials, 2021, 14, 1083 CrossRef CAS PubMed.
- V. R. Kohout, C. L. Wardzala and J. R. Kramer, Adv. Drug Delivery Rev., 2022, 191, 114540 CrossRef CAS PubMed.
- M. S. Ganewatta, H. N. Lokupitiya and C. Tang, Polymers, 2019, 11, 1176 CrossRef PubMed.
- F.-P. Schmied, A. Bernhardt, C. Moers, C. Meier, T. Endres and S. Klein, Polymers, 2022, 14, 1281 CrossRef CAS PubMed.
- Z. Wang, F. Cui, Y. Sui and J. Yan, Beilstein J. Org. Chem., 2023, 19, 1580–1603 CrossRef CAS PubMed.
- L. Chen, Z. Wang, E. Fang, Z. Fan and S. Song, Chem. Sci., 2025, 16, 1250–1264 RSC.
- Y. Yang, Q. Hu, Q. Shao, Y. Peng, B. Yu, F. Luo, J. Chen, C. Xu, Z. Li, M. Tam, Z. Ju, R. Zhang, F. Xing, Z. Zha and H. T. Zhang, Adv. Sci., 2025, 12, e2410951 CrossRef PubMed.
- M. Zeng, M. Huo, Y. Feng and J. Yuan, Rapid Commun., 2018, 39, 1800291 CrossRef PubMed.
- Y. Liu, G. Jin, H. Miao and S. Xu, RSC Adv., 2024, 14, 15240–15248 RSC.
- C.-C. Li, Y.-L. Tu, S.-Y. Guo and J.-B. Shen, Chin. J. Polym. Sci., 2025, 43, 808–818 CrossRef CAS.
- R. Pitocchi, P. Cicatiello, L. Birolo, A. Piscitelli, E. Bovio, G. C. Varese and P. Giardina, Int. J. Mol. Sci., 2020, 21, 2913 CrossRef CAS PubMed.
- U. Griskeviciene, J. Dambrauskiene, M. Marksa, Z. Mazeliene, R. Vainoriene and L. Ivanauskas, Life, 2024, 14, 1191 CrossRef CAS PubMed.
- N. Słabisz, P. Leśnik, J. Janc, M. Fidut, M. Bartoszewicz, R. Dudek-Wicher and U. Nawrot, Front. Microbiol., 2024, 15, 1331628 CrossRef PubMed.
- Z. X. Yang, Y. H. Sun, J. G. He, H. Cao and G. Q. Jiang, Oncol. Lett., 2015, 10, 3443–3449 CrossRef CAS PubMed.
- T. Zheng, H. Chen, C. Wu, J. Wang, M. Cui, H. Ye, Y. Feng, Y. Li and Z. Dong, Pharmaceutics, 2023, 15, 1782 CrossRef CAS PubMed.
- L. Li, C. Zhang, L. Tian, Z. Wu, D. Wang and T. Jiao, Nanomaterials, 2023, 13, 583 CrossRef CAS PubMed.
- P. M. W. Chrószcz, R. I. M. Barszczewska, K. A. Kazek and P. I. Ślęzak, Materials, 2023, 16, 3855 CrossRef CAS PubMed; S. Liu, X. Wang, X. Yu, Q. Cheng, L. T. Johnson, S. Chatterjee, D. Zhang, S. M. Lee, Y. Sun, T. C. Lin, J. L. Liu and D. J. Siegwart, J. Am. Chem. Soc., 2021, 143, 21321–21330 CrossRef.
- J. Li, Y. Wang and X. Yu, Front. Chem., 2021, 9, 706311 CrossRef CAS PubMed.
- P. M. W. Chrószcz, R. I. M. Barszczewska, K. A. Kazek and P. I. Ślęzak, Materials, 2023, 16, 3855 CrossRef PubMed.
- K. Pathak, S. K. Misra, A. Sehgal, S. Singh, S. Bungau, A. Najda, R. Gruszecki and T. Behl, Polymers, 2021, 13, 2514 CrossRef CAS PubMed.
- G. Groslambert, V. Andrieux, M. Duquesnoy, R. Rullan, L. Khrouz, Q. S. Denis, S. N. Steinmann, B. T. Le, F. Chevallier, D. Frath and C. Bucher, Chem. Sci., 2025, 16, 9320–9325 RSC.
- X. Liu, Q. Cheng, Y. Zhu, S. Yu, Y. Hou, Z. Cui and S. Zhu, RSC Adv., 2021, 11, 34699–34709 RSC.
- G. M. Esteves, J. Esteves, M. Resende, L. Mendes and A. S. Azevedo, Antibiotics, 2022, 11, 235 CrossRef CAS PubMed.
- S. Otarbayeva and D. Berillo, Gels, 2024, 10, 753 CrossRef CAS PubMed.
- K. Michocka, K. Staszak, D. Gwiazdowska and D. Wieczorek, Molecules, 2019, 24, 4010 CrossRef CAS PubMed.
- A. Katerji, S. Trefi, Y. Bitar and A. Ibrahim, Heliyon, 2023, 9, e14555 CrossRef CAS.
- B. M. Cartwright, S. J. Fox, M. J. Underdown, W. A. Clark and J. A. Molnar, Antioxidants, 2023, 12, 1176 CrossRef CAS PubMed.
- Y. Yang, G. Ye, X. Qi, B. Zhou, L. Yu, G. Song and R. Du, Polymers, 2024, 16, 954 CrossRef CAS PubMed.
- R. Pangeni, S. Poudel, S. M. Herz, G. Berkbigler, A. S. Duerfeldt, M. I. Damaj and Q. Xu, Mol. Pharm., 2025, 22, 1641–1656 CrossRef CAS PubMed.
- X. Zhai, G. Wu, X. Tao, S. Yang, L. Lv, Y. Zhu, D. Dong and H. Xiang, RSC Adv., 2023, 13, 7798–7817 RSC.
- X. Mao, W. Liu, Z. Li, S. Mei and B. Zong, Polymers, 2024, 16, 1997 CrossRef CAS.
- H. Lin, Z. Gao, T. Shan, A. Asilebieke, R. Guo, Y. C. Kan, C. Li, Y. Xu and J. J. Chu, J. Orthop. Surg. Res., 2024, 19, 673 CrossRef PubMed.
- K. N. Shoudho, S. Uddin, M. M. H. Rumon and M. S. Shakil, ACS Omega, 2024, 9, 33303–33334 CrossRef CAS PubMed.
- H. Chopra, S. Bibi, Y. K. Mohanta, T. K. Mohanta, S. Kumar, I. Singh, M. S. Khan, P. R. Rauta, A. Alshammari, M. Alharbi and A. F. Alasmari, Gels, 2023, 9, 394 CrossRef CAS PubMed.
- A. Sukhdev, M. Challa, L. Narayani, A. S. Manjunatha, P. R. Deepthi, J. V. Angadi, P. M. Kumar and M. Pasha, Heliyon, 2020, 6, e03245 CrossRef CAS.
- A. Simamora, K. H. Timotius, H. Setiawan, F. A. Saputri, C. R. Putri, D. Aryani, R. A. Ningrum and A. Mun'im, Molecules, 2024, 29, 2093 CrossRef CAS PubMed.
- C.-H. Hung, K. H. Chan, W.-P. Kong, R.-L. Du, K. Ding, Z. Liang, Y. Wang and K.-Y. Wong, J. Med. Chem., 2025, 68, 8768–8785 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |