
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electricity-driven sustainable synthesis of 2-aminobenzonitriles through C–C bond cleavage of isatins: post-functionalization via one-pot integration with enzyme catalysis
Kirti
Singh
a,
Shashi
Pandey
 b and
Vikas
Tyagi
*a
b and
Vikas
Tyagi
*a
aDepartment of Chemistry and Biochemistry, Thapar Institute of Engineering and Technology, Patiala-147004, Punjab, India
bDepartment of Chemistry, University of Lucknow, Lucknow-226007, Uttar Pradesh, India. E-mail: vikas.tyagi@thapar.edu
First published on 13th February 2026
Abstract
Herein, we report an electricity-mediated sustainable synthesis of 2-aminobenzonitriles, which serve as essential building blocks for numerous pharmaceuticals, by using isatins and hydroxylamine as starting materials. Furthermore, electricity served as an efficient and environmentally friendly mediator, promoting the C–C cleavage of isatin derivatives to afford the desired products in good to excellent yields. The scope and practicality of the method were validated through the screening of isatins bearing different substituents. Moreover, this electrochemical protocol was successfully integrated with an α-amylase-catalyzed aza-Michael addition in a one-pot system, providing pharmaceutically relevant β-aminocarbonyl compounds via C–N bond formation. Additionally, mechanistic insights obtained from control experiments and cyclic voltammetry studies suggest that 2-(2-aminophenyl)-2-oxoacetate serves as a key intermediate during the electrochemical step.
Introduction
The carbon–carbon bond serves as the fundamental backbone of organic molecules, and its cleavage or functionalization offers an effective way for structural modification.1 In recent years, considerable attention has been directed toward developing novel and efficient methods for achieving selective C–C bond cleavage. Besides, isatin is recognized as a crucial and privileged scaffold in synthetic chemistry, as it incorporates both a keto group and a lactam moiety within its structure.2 The coexistence of these functional groups, along with the easy availability of isatin, makes it a highly versatile starting material for generating clinically important molecules.3 Consequently, a wide range of chemical transformations are being carried out, targeting either the keto or the lactam functionality of isatin to broaden its synthetic and medicinal applications. However, the selective cleavage of C–C bonds in isatin remains relatively underexplored compared to other chemical transformations.4 Nevertheless, the molecular scaffolds generated through the selective cleavage of the C–C bond of isatin have significant importance, as they constitute the core structures of various pharmacologically active compounds, including several non-steroidal anti-inflammatory drugs (NSAIDs).5 Numerous methods have been previously reported for cleaving the carbon–carbon bond in isatin; however, most of these approaches are limited by drawbacks such as the use of costly transition metal catalysts, stoichiometric chemical oxidants, and harsh reaction conditions.6On the other hand, electrochemistry in organic synthesis has experienced remarkable growth and garnered widespread attention over the past decade, primarily due to its environmentally benign nature.7 Unlike conventional methods that rely on stoichiometric oxidants or reductants, electrochemical processes generate reactive intermediates directly via anodic oxidation or cathodic reduction, thereby minimizing hazardous reagents and improving the overall atom economy.8 Consequently, electrochemical methods have been successfully developed for the selective cleavage of carbon–carbon bonds in isatin.9 In this context, Wang et al. reported the electricity-driven cleavage of the C–C bond in isatin for the synthesis of anthranilic acid derivatives (Scheme 1a).10 Along with this, Liu et al. reported an efficient and convenient method for the ring opening of isatin using organic amines in an acidic buffer solution for the synthesis of methyl 2-ureidobenzoates (Scheme 1b).11 Moreover, the integration of electrosynthesis with enzyme catalysis has emerged rapidly, reflecting a shift towards sustainable and efficient chemical transformations.12 This merged system leverages the complementary strengths of both domains, where electrosynthesis provides a clean and green source of redox equivalents, and enzyme catalysis contributes exceptional selectivity and mild reaction conditions.13 With respect to this, Tyagi et al. reported the synthesis of chiral sulphur-containing organofluorine acids in good yields using fluorine-based unsaturated alkenes and thiophenols by combining electrosynthesis and biocatalysis (Scheme 1c).13b Additionally, Guan et al. reported the synthesis of 2,2-disubstituted 3-carbonyl indoles by integrating the non-natural catalytic activity of lipase with electrosynthesis, affording the products in good isolated yields as well as excellent enantio- and diastereoselectivities (Scheme 1d).13c
 | ||
| Scheme 1 Examples of oxidative ring opening of isatin and strategies to synthesize 2-aminobenzonitrile. | ||
Next, 2-aminobenzonitrile is an important moiety that has been used as a precursor for the synthesis of potent anti-inflammatory, antidiabetic, anti-HIV, antibacterial, and antiviral compounds, as shown in Fig. 1.14 Traditionally, 2-aminobenzonitriles have been synthesized via coupling or reduction of o-nitrobenzonitriles or related o-nitro derivatives.15–20 In this context, Sun et al. reported a rhodium-catalyzed cyanation of aromatic C–H bonds, followed by denitrosation of nitrosoarylamines, using the nitroso group as a directing moiety. This strategy enabled the synthesis of 2-(alkylamino) benzonitriles, with N-cyano-N-phenyl-p-methylbenzenesulfonamide serving as the cyano source (Scheme 1e).21 In addition to this, Zeng et al. demonstrated an aminocyanation method for the synthesis of bifunctional aminobenzonitriles by direct addition of aryl cyanamides to arynes, which embeds both amino and cyano groups simultaneously (Scheme 1f).22 Furthermore, Mo et al. reported a one-pot tert-butyl nitrite (TBN)-mediated nitrosation followed by Fe-catalyzed cleavage of the carbon–carbon bond for the synthesis of 2-aminobenzonitriles in excellent yields, using 2-aryl indoles as the starting material (Scheme 1g).23 While the aforementioned methods have been successfully established for the synthesis of 2-aminobenzonitriles, they still have certain limitations, such as many of these approaches rely on starting materials that are not easily accessible, which restricts their broader applicability.24 The frequent use of costly transition-metal catalysts further diminishes the practicality and economic feasibility of these protocols.25a,b Therefore, there is a need to devise alternative strategies for the synthesis of 2-aminobenzonitrile derivatives that not only employ readily available and inexpensive starting materials but also utilize more affordable catalytic systems, thereby offering a more sustainable approach.
 | ||
| Fig. 1 Important pharmaceuticals containing 2-aminobenzonitrile. | ||
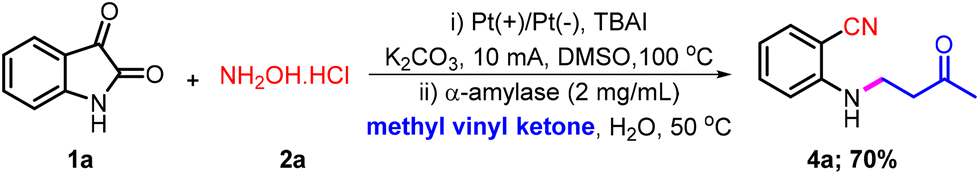
Inspired by the wide-ranging applications of 2-aminobenzonitriles and recognizing the limitations of conventional approaches, we propose an electricity-mediated, sustainable strategy for synthesizing 2-aminobenzonitrile derivatives from readily available isatins. Furthermore, by coupling the electrochemical step with enzyme catalysis, this approach enables an efficient two-step, one-pot synthesis of pharmaceutically significant β-amino carbonyl compounds (Scheme 1h).
Results and discussion
We began our study by establishing a model reaction in which isatin (1a) and hydroxylamine (2a) were electrolyzed using Pt/Pt electrodes and DMSO as the solvent at 120 °C. The reaction proceeded smoothly at a constant current of 10 mA in the presence of potassium iodide (KI) as the electrolyte and K2CO3 as the base, affording the desired product 3a in 69% yield (entry 1, Table 1). This might be due to the limited solubility of KI in organic solvents, whereas n-Bu4NI is highly soluble due to the lipophilic tetrabutylammonium cation, ensuring a uniform distribution of iodide ions throughout the reaction medium.25c To further improve the yield, several reaction parameters were optimized, as summarized in Table 1. First, various electrolytes were screened. The use of tetrabutylammonium iodide (n-Bu4NI) instead of KI significantly enhanced the yield to 82% (entry 2, Table 1). In contrast, when bromide salts such as KBr and n-Bu4NBr were used in place of KI, no product formation was observed (entries 3 and 4, Table 1), while lithium perchlorate as an electrolyte afforded the product only in a trace amount (entry 5, Table 1). Importantly, no product (3a) formation was observed in the absence of current, highlighting the essential role of electricity in the process (entry 6, Table 1). Next, the effect of the current was examined. Decreasing the current from 10 mA to 5 mA led to a noticeable drop in yield (entries 7 and 8, Table 1), whereas increasing the current to 15 mA resulted in no significant improvement in the yield of the product (entry 9, Table 1). Furthermore, solvent screening revealed that replacing DMSO with acetonitrile (ACN) or 1,2-dichloroethane (DCE) gave no product, while DMF and THF provided significantly reduced yields compared to DMSO (entries 10–13, Table 1). Next, the electrolyte concentration was also optimized. Lowering the loading of TBAI from 1.0 to 0.5 equiv. decreased the yield of 3a (entry 14, Table 1), whereas increasing it to 1.5 equiv. afforded 3a in 87% yield (entry 15, Table 1). Moreover, a further increase to 2.0 equiv. slightly reduced the yield of product 3a (entry 16, Table 1). Moreover, optimization of the base showed that replacing K2CO3 with Na2CO3 decreased reaction efficiency, while organic bases such as Et3N and DBU completely suppressed product formation (entries 17–19, Table 1). Notably, in the absence of a base, there was no product formation, confirming the requirement for a base in this protocol (entry 20, Table 1). Additionally, temperature studies revealed that lowering the reaction temperature to 80 °C decreased the yield to 55% (entry 21, Table 1), whereas operating at 100 °C yielded 3a in up to 87% yield (entry 22, Table 1). However, increasing the temperature further to 140 °C led to a reduced yield of 3a up to 79% (entry 23, Table 1). Finally, the stoichiometry of hydroxylamine was investigated. Reducing its amount to 0.5 equiv. or 1.0 equiv. decreased product (3a) formation (entries 24 and 25, Table 1), whereas 1.5 equiv. gave yields comparable to that for 2.0 equiv., establishing 1.5 equiv. as sufficient for optimal yield (entry 26, Table 1).|
|
||
|---|---|---|
| Entry | Divergence from standard conditions | Yieldb of 3a (%) |
| a Standard reaction conditions: isatin (1a) (20 mg, 1 equiv.), hydroxylamine (19 mg, 2 equiv.) (2a), KI (23 mg, 1 equiv.), K2CO3 (19 mg, 1 equiv.) in 1 mL of DMSO at a constant current of 10 mA in an undivided cell with Pt electrodes as the cathode and anode at 120 °C. b Yields were calculated using HPLC. | ||
| 1 | No changea | 69 |
| 2 | n-Bu4NI instead of KI | 82 |
| 3 | KBr instead of KI | Nr |
| 4 | n-Bu4NBr instead of KI | Nr |
| 5 | LiClO4 instead of KI | Trace |
| 6 | No current | Nr |
| 7 | 5 mA instead of 10 mA | 12 |
| 8 | 8 mA instead of 10 mA | 67 |
| 9 | 15 mA instead of 10 mA | 80 |
| 10 | ACN instead of DMSO | Trace |
| 11 | DCE instead of DMSO | Nr |
| 12 | DMF instead of DMSO | 19 |
| 13 | THF instead of DMSO | 23 |
| 14 | TBAI (0.5 equiv.) instead of TBAI (1 equiv.) | 49 |
| 15 | TBAI (1.5 equiv.) instead of TBAI (1 equiv.) | 87 |
| 16 | TBAI (2 equiv.) instead of TBAI (1 equiv.) | 79 |
| 17 | Na2CO3 instead of K2CO3 | 34 |
| 18 | Et3N instead of K2CO3 | Nr |
| 19 | DBU instead of K2CO3 | Nr |
| 20 | No base | Nr |
| 21 | 80 °C instead of 120 °C | 55 |
| 22 | 100 °C instead of 120 °C | 87 |
| 23 | 140 °C instead of 120 °C | 79 |
| 24 | 0.5 equiv. of hydroxylamine instead of 2 equiv. | 42 |
| 25 | 1 equiv. of hydroxylamine instead of 2 equiv. | 69 |
| 26 | 1.5 equiv. of hydroxylamine instead of 2 equiv. | 87 |
| 27 | 2.5 equiv. of hydroxylamine instead of 2 equiv. | 85 |

After optimizing the reaction conditions, the substrate scope of the electro-catalyzed reaction was thoroughly investigated, as shown in Scheme 2. First, we examined the influence of various electron-donating groups, such as –OCH3 and –CH3, at the C-5, C-6, and C-7 positions of isatin. Notably, the presence of an –OCH3 group at the C-6 position provided a good yield, i.e., 71% (3b, Scheme 2). In the case of a CH3 group at the C-5 position of isatin, the product (3c) was obtained in an isolated yield of 77%, whereas a CH3 group at the C-7 position led to a significantly lower yield of the corresponding product (3d). Next, we investigated the electron-withdrawing effect at the C-5 position, where an –NO2 group present at this position afforded the product in 58% yield (3e, Scheme 2). The influence of various halides, including –F, –Cl, and –Br, was examined at the C-4, C-5, and C-6 positions. It was observed that halide substitutions at the C-4 position resulted in lower yields (3g and 3i, Scheme 2). In contrast, isatins having F, Cl, and Br at the C-5 position produced products in good yields (3f, 3h, and 3j, Scheme 2). Additionally, halide substitutions at the C-6 position resulted in lower product efficiency (3k, Scheme 2). However, no product formation was observed when N-methyl-substituted isatin was employed in the reaction (3l, Scheme 2).
 | ||
| Scheme 2 Substrate scope of electricity-mediated synthesis of 2-aminobenzonitrile. Reaction conditions: isatin (1) (100 mg, 1 equiv.), hydroxylamine (2) (71 mg, 1.5 equiv.), TBAI (377 mg, 1.5 equiv.), K2CO3 (95 mg, 1 equiv.) in 5 mL of DMSO at a constant current of 10 mA in an undivided cell with Pt electrodes as the cathode and anode at 100 °C. | ||
In the second phase of our study, we conducted a reaction integrating electrochemical and α-amylase-catalyzed reaction conditions within a single vessel to synthesize a pharmaceutically relevant β-amino carbonyl compound (Scheme S1).26 Conversely, the one-pot procedure produced the aza-Michael product in a notably low yield of only 12%. To improve the yield of the integrated protocol, the reaction conditions were modified. In this context, after completion of the electrochemical reaction, as confirmed by TLC, α-amylase (2 mg mL−1), methyl vinyl ketone (1.5 equiv.), and water (1 mL) were added to the reaction mixture. The resulting mixture was stirred at 50 °C for 12 hours. Encouragingly, we obtained the aza-Michael product 4a in an improved yield, i.e., 45% (entry 1, Table 2). Furthermore, no reaction was observed when the enzyme concentration was reduced to 1 mg mL−1 (entry 2, Table 2). However, increasing the enzyme loading to 4 mg mL−1 and 6 mg mL−1 resulted in 56% and 52% isolated product yields, respectively (entries 3 and 4, Table 2). Additionally, the reaction medium was optimized for the enzymatic step by varying the amount of water. Reducing the volume of H2O from 1 mL to 0.5 mL decreased the yield of 4a to 39%. However, increasing the H2O volume to 2 mL improved the yield up to 61% (entry 6, Table 2). However, further increases beyond 2 mL did not significantly affect the yield of 4a (entry 7, Table 2). Furthermore, increasing the amount of methyl vinyl ketone up to 2.5 equivalents improved the isolated yield of the product (entries 8 and 9, Table 2). However, further increasing the amount of methyl vinyl ketone, i.e., 3.0 equivalents in comparison with isatin, did not significantly change the outcome of the reaction (entry 10, Table 2).
|
|
||
|---|---|---|
| Entry | Divergence from standard reaction conditions | Isolated yield of 4a (%) |
| Isatin (1a) (20 mg, 1 equiv.), hydroxylamine (14 mg, 1.5 equiv.) (2a), TBAI (75 mg, 1.5 equiv.), and K2CO3 (19 mg, 1 equiv.) in 1 mL of DMSO at a constant current of 10 mA in an undivided cell with Pt electrodes as the cathode and anode at 100 °C; after completion of the electrocatalytic step: α-amylase (2 mg mL−1), methyl vinyl ketone (18 μL, 1.5 equiv.), and 1 mL of water (v/v) at 50 °C, overnight, error = ±4% (reactions performed in triplicate). | ||
| 1 | No change | 45 |
| 2 | α-Amylase (1 mg mL−1) | No reaction |
| 3 | α-Amylase (4 mg mL−1) | 56 |
| 4 | α-Amylase (6 mg mL−1) | 52 |
| 5 | H2O (0.5 mL) | 39 |
| 6 | H2O (2 mL) | 61 |
| 7 | H2O (2.5 mL) | 60 |
| 8 | MVK (2 equiv.) | 68 |
| 9 | MVK (2.5 equiv.) | 76 |
| 10 | MVK (3 equiv.) | 74 |
Standard reaction conditions
Furthermore, the substrate scope of the integrated protocol was thoroughly investigated under the optimal electro-biocatalytic reaction conditions, as shown in Scheme 3. In this context, electron-donating groups like methoxy and methyl facilitated the synthesis of the aza-Michael products in high yields (4b–c, Scheme 3). Additionally, halide substituents such as F, Cl, and Br at the C-4 and C-5 positions were well tolerated, affording the desired products in isolated yields ranging from 55% to 75% (4d–h, Scheme 3). Additionally, Michael acceptors such as cyclohexanone and cyclopentanone were investigated, along with differently substituted isatins, and as a result, the corresponding products were obtained in low to moderate yields (4i–k, Scheme 3). However, only a trace amount of the product was observed when cyclopentanone was employed in combination with 5-NO2-substituted isatin (4l, Scheme 3). | ||
| Scheme 3 Substrate scope of the integrated electro-biocatalytic synthesis of β-amino carbonyl compounds. Reaction conditions: (i) isatin (1a) (100 mg, 1 equiv.), hydroxyl amine (71 mg, 1.5 equiv.) (2a), TBAI (367 mg, 1.5 equiv.), and K2CO3 (95 mg, 1 equiv.) in 5 mL of DMSO at a constant current of 10 mA in an undivided cell with Pt electrodes as the cathode and anode at 100 °C; (ii) after completion of the electrocatalytic step: α-amylase (4 mg mL−1), methyl vinyl ketone (150 μL, 2.5 equiv.), 10 mL water (v/v) at 50 °C, overnight. | ||
To investigate the rate and influence of electricity and the enzyme at different reaction stages leading to the formation of 3a and 4a, we conducted a systematic study by designing a series of controlled reactions under optimized conditions, as illustrated in Fig. 2. In the first experiment, substrates 1a and 2a were subjected to electrochemical conditions, resulting in the formation of 3a within 2 hours. Along with the formation of 3a, the consumption of isatin (1a) and the formation of isatin-3-oxime, an intermediate, were also monitored. The reaction showed complete consumption of isatin and formation of the corresponding isatin-3-oxime intermediate within 20 minutes. Subsequently, 3a was treated under enzymatic conditions, facilitating its conversion to 4a in 12 hours. Next, to validate the roles of electricity and the enzyme in the one-pot two-step protocol, we conducted different control experiments. First, an experiment was performed in the absence of electricity; however, the enzyme was still added to the reaction, and no formation of 3a was observed, confirming the necessity of electricity for the formation of 3a. Similarly, in the second experiment, electricity was employed without the addition of the enzyme, and no formation of 4a took place, thereby verifying the essential role of the enzyme in the aza-Michael addition step. Finally, in the third experiment, where electricity and enzyme were both absent, neither 3a nor 4a was formed. These control experiments suggested that electricity plays a role in the formation of product 3a, while the enzyme is necessary for the formation of 4avia an aza-Michael addition reaction.
 | ||
| Fig. 2 (a) Role of electricity and enzyme in the formation of 3a over time. (b) Role of electricity and enzyme in the formation of 4a over time; optimised reaction conditions: (i) isatin (1a) (20 mg, 1 equiv.), hydroxylamine (14 mg, 1.5 equiv.) (2a), TBAI (75 mg, 1.5 equiv.), K2CO3 (19 mg, 1 equiv.) in 1 mL of DMSO at a constant current of 10 mA in an undivided cell having Pt electrodes as the cathode and the anode at 100 °C; (ii) after completion of the electrocatalytic step: α-amylase (4 mg mL−1), methyl vinyl ketone (30 μL, 2.5 equiv.), and 2 mL of water (v/v) at 50 °C, overnight. | ||
Next, based on the information available in the literature, we propose a plausible reaction mechanism for the electricity-mediated synthesis of 2-aminobenzonitrile, as illustrated in Scheme 4.10,27 The process begins with the condensation of substrates 1a and 2a, resulting in the formation of isatin-3-oxime (I).28 At the cathode, water undergoes reduction, generating hydroxide ions. The electrogenerated hydroxide ion subsequently attacks intermediate I, producing intermediate II. Next, the base K2CO3 interacts with intermediate II, leading to electron redistribution and ring opening to yield intermediate III. Meanwhile, the iodide ion derived from tetrabutylammonium iodide (TBAI) is oxidised to molecular iodine (I2), which reacts with intermediate III, and provides intermediate IV.10 This intermediate is unstable and readily eliminates an iodine radical with the concurrent release of carbon dioxide, affording intermediate (V).27 The carbon-centred radical in (V) then loses an electron to generate intermediate (VI), which, upon dehydration, furnishes the desired product (3a).
 | ||
| Scheme 4 Proposed mechanism for the electro-synthesis of 2-aminobenzonitriles. | ||
Besides, to gain further insight into the proposed reaction mechanism, various control experiments were conducted. First, radical-scavenging experiments were conducted in which 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO) and butylated hydroxytoluene (BHT) were used as additives in the reaction. In the presence of TEMPO, product 3a was obtained in only 5% yield, whereas no reaction was observed with BHT, thereby indicating the involvement of radical species in the reaction mechanism (Scheme 5a). Furthermore, the electricity-driven reaction was initiated with the sodium salt of 2-(2-aminophenyl)-2-oxoacetate and hydroxylamine hydrochloride, yielding the product 3a in 65% yield (Scheme 5b). This experiment suggested the formation of intermediate III in the reaction, as determined by HRMS, and supports the proposed mechanistic pathway. Along with this, when the reaction of the sodium salt of 2-(2-aminophenyl)-2-oxoacetate was performed in the absence of electricity, there was no formation of product 3a, which depicts the necessity of electricity for this reaction (Scheme 5c). Furthermore, when N-methyl-substituted isatin was used in the electrochemical reaction, no formation of product 3a was observed (Scheme 5d). It was observed that no oxime formation occurred with N-methyl isatin, thereby suppressing the subsequent decarboxylation and electrochemical reduction steps. In addition to the above experiments, control reactions were performed to verify the enzyme's role in the formation of the aza-Michael products. In this context, the first experiment was conducted by adding starch in an equimolar amount, and in the second experiment, urea was used in a 4.0 equivalent ratio corresponding to the enzyme. As a result, product 4a was obtained in 29% and 7% isolated yields, respectively (Schemes 5e and f). Besides, when BSA was used as a catalyst in place of α-amylase, only a trace amount of 4a was observed (Scheme 5g). Subsequently, to verify the role of the active site in catalyzing the reaction, the enzyme was thermally denatured, and the product 4a was obtained only in a trace amount when denatured α-amylase was used as a catalyst (Scheme 5h).
 | ||
| Scheme 5 Control experiments. | ||
Furthermore, a cyclic voltammetry (CV) study was conducted to investigate the underlying mechanism involved in the electrosynthesis of 2-aminobenzonitrile (Fig. 3).27,29,30 The CV profiles of potassium (E)-2-(2-aminophenyl)-2-(hydroxyimino)acetate (blue line), which is an intermediate, tetrabutylammonium iodide (TBAI, red line), and a blank sample (LiClO4 in DMSO, black dotted line) were recorded using a platinum disc as the working electrode, a platinum wire as the counter electrode, and Ag/AgCl as the reference electrode. In the blank sample containing only the background electrolyte LiClO4 and DMSO, no oxidation peak was observed, confirming the electrochemical inactivity of the solvent under the applied conditions. The cyclic voltammogram of TBAI revealed two characteristic anodic peaks at 0.535 V and 0.752 V vs. Ag/AgCl. These features are assigned to the stepwise redox processes involving oxidation of iodide ions to I2 (I− → I2) followed by oxidation of triiodide species to iodine (I3− → I2).29 Notably, the CV of the mixture containing both TBAI and potassium (E)-2-(2-aminophenyl)-2-(hydroxyimino)acetate exhibited an excessive oxidation potential peak at 1.378 V. During the reaction mechanism, potassium (Z)-2-(2-aminophenyl)-2-(hydroxyimino)acetate salt is formed, which undergoes halogenation to generate a hypoiodite species due to in situ electrogenerated I2 and I3− in the presence of potassium salt.30
 | ||
| Fig. 3 Cyclic voltammetry using a Pt disc as the working electrode, Pt as the counter electrode & Ag/AgCl as the reference electrode; initial potential = zero volts, scan direction = 0 to +2 V (oxidative), room temperature. A cyclic voltammogram of potassium (E)-2-(2-aminophenyl)-2-(hydroxyimino)acetate (0.05 M) in DMSO with TBAI (0.01 M) and LiClO4 (0.1 M) (the background electrolyte); scan rate 0.05 V s−1, 0 to 2 V. | ||
Experimental
General information
All chemicals and solvents employed in this study were purchased from Sigma-Aldrich and used without further purification. α-Amylase from Aspergillus oryzae (≥150 U mg−1, CAS no.: 232-588-1, product code: A9857), utilized for the functionalization of 2-aminobenzonitriles, was also procured from Sigma-Aldrich and used as received.The electrochemical reactions were carried out using an OWON (P4305) instrument fitted with an undivided cell, a magnetic stirrer, and a set of platinum electrodes (2.5 cm length, 0.7 cm width and 0.05 mm thickness). Furthermore, the cyclic voltammetry study was performed using a DY2300 potentiostat instrument. The progress of the reaction was monitored using thin-layer chromatography (TLC, Silica 254G was coated on glass slides). The 1H and 13C NMR characterisation of compounds was carried out using a JEOL or a Bruker spectrometer, at 400 MHz and 500 MHz for 1H NMR and at 100 MHz and 125 MHz for 13C NMR. The spectra of samples were recorded in solvents CDCl3 and d6-DMSO using trimethyl silane as an internal standard (TMS). The chemical shift is denoted by δ (ppm) and the coupling constant is denoted by J (Hz). The chemical shift values of residual solvent peaks are as follows: for 1H NMR, in CDCl3 7.26 ppm and in d6-DMSO 2.50 ppm; for 13C NMR, in CDCl3 77.16 ppm and in d6-DMSO 39.51 ppm. The abbreviations used for peak descriptions in NMR are as follows: brs, broad singlet; s, singlet; d, doublet; t, triplet; dd, doublet of doublets. The HRMS data of the compounds were characterised using a QTOF mass spectrometer (XEVO G2 XS) in ESI(+ve) mode.
Characterisation data of synthesized compounds
Conclusion
In summary, we have developed an electricity-mediated, sustainable protocol for the efficient synthesis of 2-aminobenzonitriles from isatins and hydroxylamine as starting materials. This strategy overcomes the limitations of previously reported methods that often require harsh reaction conditions, metal catalysts, or costly substrates. A range of substituted 2-aminobenzonitriles were obtained in good yields directly from the corresponding substituted isatins. Furthermore, the electrochemically synthesized 2-aminobenzonitriles were successfully functionalized in a one-pot process by integrating electrochemical synthesis with α-amylase-catalyzed aza-Michael addition, affording clinically valuable β-amino carbonyl compounds. Furthermore, control experiments and cyclic voltammetry studies were conducted to gain mechanistic insight into the integrated protocol. Overall, this protocol highlights the potential of electrochemically driven C–C bond cleavage in isatin chemistry.Author contributions
Kirti Singh: writing – original draft, methodology, and investigation. Shashi Pandey: data characterisation and compilation of data. Vikas Tyagi: writing – original draft, project administration, investigation, and conceptualisation.Conflicts of interest
The authors declare no conflicts of interest.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5ob01983d.Acknowledgements
Financial support from the Department of Biotechnology, Ministry of Science and Technology, Government of India (BT/PR55062/BSA/33/294/2024), and the DST/INSPIRE Fellowship/2020/IF200048 is greatly acknowledged. The authors are also grateful to the Department of Science and Technology Grant (SR/FST/CS-II/2018/69) at the Department of Chemistry and Biochemistry, Thapar Institute of Engineering and Technology, Patiala, for providing the HRMS data.References
- (a) Y. F. Liang, M. Bilal, L. Y. Tang, T. Z. Wang, Y. Q. Guan, Z. Cheng, M. Zhu, J. Wei and N. Jiao, Carbon–carbon bond cleavage for late-stage functionalization, Chem. Rev., 2023, 123, 12313–12370 CrossRef CAS PubMed; (b) F. Chen, T. Wang and N. Jiao, Recent advances in transition-metal-catalyzed functionalization of unstrained carbon–carbon bonds, Chem. Rev., 2014, 114, 8613–8661 CrossRef CAS PubMed; (c) D. Ravelli, S. Protti and M. Fagnoni, Carbon–carbon bond forming reactions via photogenerated intermediates, Chem. Rev., 2016, 116, 9850–9913 CrossRef CAS PubMed; (d) P. Sivaguru, Z. Wang, G. Zanoni and X. Bi, Cleavage of carbon–carbon bonds by radical reactions, Chem. Soc. Rev., 2019, 48, 2615–2656 RSC; (e) I. Marek, A. Masarwa, P. O. Delaye and M. Leibeling, Selective carbon–carbon bond cleavage for the stereoselective synthesis of acyclic systems, Angew. Chem., Int. Ed., 2015, 54, 414–429 CrossRef CAS PubMed.
- (a) P. Nath, A. Mukherjee, S. Mukherjee, S. Banerjee, S. Das and S. Banerjee, Isatin: a scaffold with immense biodiversity, Mini-Rev. Med. Chem., 2021, 21, 1096–1112 CrossRef CAS PubMed; (b) A. Mondal, A brief review depending on the chemistry of isatin in single pot technique towards the construction of significant and valuable heterocyclic scaffolds, Lett. Org. Chem., 2024, 21, 929–957 CrossRef CAS; (c) P. Yadav, S. Berry and A. Bhalla, Chemical methods for the construction of spirocyclic β-lactams and their biological importance, Synthesis, 2025, 251–274 Search PubMed; (d) D. Karati, An insight into isatin and its hybrid scaffolds as anti-cancer agents: an explicative review, Discov. Chem., 2024, 1, 66 CrossRef; (e) I. M. Andrade, E. D. O. Lima Filho, C. F. Valadão, L. D. S. Forezi and F. de C. da Silva, Recent advances in isatin–thiazole hybrids: synthesis, structural design, and biological application, Chem. Biodiversity, 2025, e01989 CrossRef CAS PubMed.
- (a) R. Kakkar, Isatin and its derivatives: a survey of recent syntheses, reactions, and applications, MedChemComm, 2019, 10, 351–368 RSC; (b) R. E. Ferraz de Paiva, E. G. Vieira, D. Rodrigues da Silva, C. A. Wegermann and A. M. Costa Ferreira, Anticancer compounds based on isatin-derivatives: strategies to ameliorate selectivity and efficiency, Front. Mol. Biosci., 2021, 7, 627272 CrossRef PubMed; (c) R. S. Cheke, V. M. Patil, S. D. Firke, J. P. Ambhore, I. A. Ansari, H. M. Patel, S. D. Shinde, V. R. Pasupuleti, M. I. Hassan, M. Adnan and A. Kadri, Therapeutic outcomes of isatin and its derivatives against multiple diseases: recent developments in drug discovery, Pharmaceuticals, 2022, 15, 272 CrossRef CAS PubMed; (d) S. Bugalia, Y. Dhayal, H. Sachdeva, S. Kumari, K. Atal, U. Phageria, P. Saini and O. P. Gurjar, Review on isatin—a remarkable scaffold for designing potential therapeutic complexes and its macrocyclic complexes with transition metals, J. Inorg. Organomet. Polym. Mater., 2023, 33, 1782–1801 CrossRef CAS PubMed; (e) V. A. Shu, D. B. Eni and F. Ntie-Kang, A survey of isatin hybrids and their biological properties, Mol. Divers., 2025, 29, 1737–1760 CrossRef CAS PubMed.
- (a) C. Xu, F. C. Jia, Q. Cai, D. K. Li, Z. W. Zhou and A. X. Wu, Intramolecular decarboxylative coupling as the key step in copper-catalyzed domino reaction: facile access to 2-(1,3,4-oxadiazol-2-yl)aniline derivatives, Chem. Commun., 2015, 51, 6629–6632 RSC; (b) Y. Ma, Y. Zhu, D. Zhang, Y. Meng, T. Tang, K. Wang, J. Ma, J. Wang and P. Sun, Eco-friendly decarboxylative cyclization in water: practical access to the anti-malarial 4-quinolones, Green Chem., 2019, 21, 478–482 RSC; (c) F. C. Jia, Z. W. Zhou, C. Xu, Y. D. Wu and A. X. Wu, Divergent synthesis of quinazolin-4(3H)-ones and tryptanthrins enabled by a tert-butyl hydroperoxide/K3PO4-promoted oxidative cyclization of isatins at room temperature, Org. Lett., 2016, 18, 2942–2945 CrossRef CAS PubMed; (d) F. C. Jia, T. Z. Chen and X. Q. Hu, TFA/TBHP-promoted oxidative cyclisation for the construction of tetracyclic quinazolinones and rutaecarpine, Org. Chem. Front., 2020, 7, 1635–1639 RSC; (e) A. H. Kalbandhe, A. C. Kavale, P. B. Thorat and N. N. Karade, Oxidative cleavage of the C2–C3 bond in isatin using (diacetoxyiodo)benzene: a facile synthesis of carbamates of alkyl anthranilates, Synlett, 2016, 763–768 CAS.
- (a) N. U. A. Mohsin and M. Irfan, Selective cyclooxygenase-2 inhibitors: A review of recent chemical scaffolds with promising anti-inflammatory and COX-2 inhibitory activities, Med. Chem. Res., 2020, 29(5), 809–830 CrossRef CAS; (b) L. D. Brown, A. S. Girgis, S. Patel, N. Samir, M. F. Said, A. T. Baidya, R. Kumar, J. Moore, A. Khadanga, R. Sakhuja and S. S. Panda, Novel isatin conjugates endowed with analgesic and anti-inflammatory properties: design, synthesis and biological evaluation, Future Med. Chem., 2025, 17, 59–73 CrossRef CAS PubMed; (c) L. M. Zhou, R. Y. Qu and G. F. Yang, An overview of spirooxindole as a promising scaffold for novel drug discovery, Expert Opin. Drug Discovery, 2020, 15, 603–625 CrossRef CAS PubMed; (d) S. Chahal, P. Rani, Kiran, J. Sindhu, G. Joshi, A. Ganesan, S. Kalyaanamoorthy, Mayank, P. Kumar, R. Singh and A. Negi, Design and development of COX-II inhibitors: current scenario and future perspective, ACS Omega, 2023, 8, 17446–17498 CrossRef CAS PubMed; (e) R. A. Menezes and K. S. Bhat, Synthetic aspects, structural insights and pharmacological potential of pyrazole derivatives: an overview, Discover Appl. Sci., 2025, 7, 137 CrossRef CAS; (f) S. Mukherjee and M. Pal, Medicinal chemistry of quinolines as emerging anti-inflammatory agents: an overview, Curr. Med. Chem., 2013, 20, 4386–4410 CrossRef CAS PubMed.
- (a) P. V. Gandhi, S. R. Burande, M. S. Charde and R. D. Chakole, A review on isatin and its derivatives: synthesis, reactions and applications, J. Adv. Sci. Res., 2021, 12(04), 1–11 CAS; (b) A. Zafar, M. A. Iqbal, G. Iram, U. S. Shoukat, F. Jamil, M. Saleem, M. Yousif, Z. ul Abidin and M. Asad, Advances in organocatalyzed synthesis of organic compounds, RSC Adv., 2024, 14, 20365–20389 RSC; (c) C. Pradhan and B. Punji, Advancement in the C–H bond alkylation of (hetero)arenes catalyzed by the most abundant transition metal–iron, Org. Chem. Front., 2024, 11, 2397–2417 RSC.
- (a) Y. H. Budnikova, E. L. Dolengovski, M. V. Tarasov and T. V. Gryaznova, Electrochemistry in organics: a powerful tool for “green” synthesis, J. Solid State Electrochem., 2024, 28, 659–676 CrossRef CAS; (b) T. H. Meyer, I. Choi, C. Tian and L. Ackermann, Powering the future: how can electrochemistry make a difference in organic synthesis?, Chem, 2020, 6, 2484–2496 CrossRef CAS; (c) Q. Jing and K. D. Moeller, From molecules to molecular surfaces: exploiting the interplay between organic synthesis and electrochemistry, Acc. Chem. Res., 2019, 53, 135–143 CrossRef PubMed; (d) T. Ali, H. Wang, W. Iqbal, T. Bashir, R. Shah and Y. Hu, Electro-synthesis of organic compounds with heterogeneous catalysis, Adv. Sci., 2023, 10, 2205077 CrossRef CAS PubMed; (e) C. Ma, J. F. Guo, S. S. Xu and T. S. Mei, Recent advances in asymmetric organometallic electrochemical synthesis (AOES), Acc. Chem. Res., 2025, 58, 399–414 CrossRef CAS PubMed; (f) Z. Shi, C. Y. Wen, L. X. Yang, J. Li and X. Sun, Recent progress in electrochemical rearrangement reactions, Org. Chem. Front., 2025, 12, 2499–2524 RSC.
- (a) N. Li, R. Sitdikov, A. P. Kale, J. Steverlynck, B. Li and M. Rueping, A review of recent advances in electrochemical and photoelectrochemical late-stage functionalization classified by anodic oxidation, cathodic reduction, and paired electrolysis, Beilstein J. Org. Chem., 2024, 20, 2500–2566 Search PubMed; (b) J. Liu, L. Lu, D. Wood and S. Lin, New redox strategies in organic synthesis by means of electrochemistry and photochemistry, ACS Cent. Sci., 2020, 6, 1317–1340 CrossRef CAS PubMed; (c) L. F. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Electrocatalysis as an enabling technology for organic synthesis, Chem. Soc. Rev., 2021, 50, 7941–8002 Search PubMed; (d) X. Cheng, A. Lei, T. S. Mei, H. C. Xu, K. Xu and C. Zeng, Recent applications of homogeneous catalysis in electrochemical organic synthesis, CCS Chem., 2022, 4, 1120–1152 CrossRef CAS; (e) J. Q. Lu, Y. K. Wang, T. McCallum and N. Fu, Harnessing radical chemistry via electrochemical transition metal catalysis, iScience, 2020, 23, 101796 Search PubMed; (f) P. Li, Y. Wang, H. Zhao and Y. Qiu, Electroreductive cross-coupling reactions: carboxylation, deuteration and alkylation, Acc. Chem. Res., 2024, 58, 113–129 CrossRef PubMed; (g) C.-Y. Sun, T.-J. Lin, Y.-Y. Chen, H.-L. Li and C.-W. Chiang, Synergistic photoredox and electrochemical catalysis in organic synthesis and late-stage functionalizations, ChemCatChem, 2024, 16, e202301537 Search PubMed; (h) Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Single-atom catalysts: synthetic strategies and electrochemical applications, Joule, 2018, 2, 1242–1264 CrossRef CAS; (i) W. Guo, K. Zhang, Z. Liang, R. Zou and Q. Xu, Electrochemical nitrogen fixation and utilization: theories, advanced catalyst materials and system design, Chem. Soc. Rev., 2019, 48, 5658–5716 RSC; (j) M. Zhang, Q. Shi, X. Song, H. Wang and Z. Bian, Recent electrochemical methods in electrochemical degradation of halogenated organics: a review, Environ. Sci. Pollut. Res., 2019, 26, 10457–10486 CrossRef CAS PubMed; (k) B. P. Chaplin, The prospect of electrochemical technologies advancing worldwide water treatment, Acc. Chem. Res., 2019, 52, 596–604 CrossRef CAS PubMed.
- (a) S. H. Shi, Y. Liang and N. Jiao, Electrochemical oxidation induced selective C–C bond cleavage, Chem. Rev., 2020, 121, 485–505 CrossRef PubMed; (b) Y. Adeli, K. Huang, Y. Liang, Y. Jiang, J. Liu, S. Song, C.-C. Zeng and N. Jiao, Electrochemically oxidative C–C bond cleavage of alkylarenes for aniline synthesis, ACS Catal., 2019, 9, 2063–2067 CrossRef CAS; (c) S. Zhang, L. Li, J. Li, J. Shi, K. Xu, W. Gao, L. Zong, G. Li and M. Findlater, Electrochemical arylation of aldehydes, ketones, and alcohols: from cathodic reduction to convergent paired electrolysis, Angew. Chem., 2021, 133, 7351–7358 CrossRef.
- P. Qian, J. Liu, Y. Zhang and Z. Wang, Tunable electrosynthesis of anthranilic acid derivatives via a C–C bond cleavage of isatins, J. Org. Chem., 2021, 86, 16008–16015 CrossRef CAS PubMed.
- M. Zhao, J. Fu, Y. Sang, Z. Wang, W. Liu and C. Chen, Electrosynthesis of methyl 2-ureidobenzoates via a C2–C3 bond cleavage of isatins, Tetrahedron, 2023, 137, 133383 Search PubMed.
- (a) C. Ma, Y. Wang, K. Guo, R. Wu and Z. Zhu, Enzymatic electrosynthesis system based on multi-enzyme catalysis or coupled with microbial transformation, Catal. Sci. Technol., 2025, 15, 1390–1405 RSC; (b) L. Liu and D. Pant, Integrative electrochemical and biological catalysis for the mild and efficient utilization of renewable electricity and carbon resources, Sustainable Energy Fuels, 2024, 8, 460–480 Search PubMed; (c) H. Chen, O. Simoska, K. Lim, M. Grattieri, M. Yuan, F. Dong, Y. S. Lee, K. Beaver, S. Weliwatte, E. M. Gaffney and S. D. Minteer, Fundamentals, applications, and future directions of bioelectrocatalysis, Chem. Rev., 2020, 120, 12903–12993 CrossRef CAS PubMed; (d) R. Wu, F. Li, X. Cui, Z. Li, C. Ma, H. Jiang, L. Zhang, Y. H. P. J. Zhang, T. Zhao, Y. Zhang and Y. Li, Enzymatic electrosynthesis of glycine from CO2 and NH3, Angew. Chem., Int. Ed., 2023, 62, e202218387 CrossRef CAS PubMed.
- (a) K. Singh and V. Tyagi, Combining the nonnatural activity of lipase and electrocatalysis in one pot: sustainable and regioselective synthesis of C-3 alkylated oxindoles, ChemCatChem, 2025, 17, e202401182 CrossRef CAS; (b) P. Kaur and V. Tyagi, Merging electrosynthesis and biocatalysis to access sulfur-based chiral α-fluorinated carboxylic acids, J. Org. Chem., 2025, 90, 5378–5392 CrossRef CAS PubMed; (c) C. J. Long, H. Cao, B. K. Zhao, Y. F. Tan, Y. H. He, C. S. Huang and Z. Guan, Merging the non-natural catalytic activity of lipase and electrosynthesis: asymmetric oxidative cross-coupling of secondary amines with ketones, Angew. Chem., 2022, 134, e202203666 CrossRef; (d) I. Peñafiel, R. A. Dryfe, N. J. Turner and M. F. Greaney, Integrated electro-biocatalysis for amine alkylation with alcohols, ChemCatChem, 2021, 13, 864–867 CrossRef.
- (a) A. A. H. Abdel-Rahman, E. M. El-Ganzoury, I. F. Zeid, E. M. Zayed and W. A. El-Sayed, Quinazolines linked to sugar derivatives as nucleoside analogs: synthesis and biological aspects, Egypt. J. Chem., 2024, 67, 209–223 Search PubMed; (b) M. Benabdallah, O. Talhi, F. Nouali, N. Choukchou-Braham, K. Bachari and A. Silva, Advances in spirocyclic hybrids: chemistry and medicinal actions, Curr. Med. Chem., 2018, 25, 3748–3767 CrossRef CAS PubMed; (c) P. Yadav and A. Bhalla, Recent advances in green synthesis of functionalized quinolines of medicinal impact (2018–present), ChemistrySelect, 2022, 7, e202201721 Search PubMed; (d) W. Spillane and J. B. Malaubier, Sulfamic acid and its N- and O-substituted derivatives, Chem. Rev., 2014, 114, 2507–2586 CrossRef CAS PubMed; (e) V. T. Nguyen, D. P. Tran, T. T. Nguyen, K. D. Nguyen and H. V. Le, An efficient and green synthesis of 2-phenylquinazolin-4(3H)-ones via t-BuONa-mediated oxidative condensation of 2-aminobenzamides and benzyl alcohols under solvent- and transition metal-free conditions, Green Process. Synth., 2023, 12, 20228148 CrossRef CAS.
- (a) X. J. Yang, B. Chen, L. Q. Zheng, L. Z. Wu and C. H. Tung, Highly efficient and selective photocatalytic hydrogenation of functionalized nitrobenzenes, Green Chem., 2014, 16, 1082–1086 Search PubMed; (b) M. Nabi, K. Sharma, R. S. Wandre and A. B. Gade, One-pot oximation–Beckmann rearrangement under mild, aqueous micellar conditions, Green Chem., 2025, 27, 5332–5339 Search PubMed.
- H. Goksu, S. F. Ho, O. Metin, K. Korkmaz, A. Mendoza Garcia, M. S. Gultekin and S. Sun, Tandem dehydrogenation of ammonia borane and hydrogenation of nitro/nitrile compounds catalyzed by graphene-supported NiPd alloy nanoparticles, ACS Catal., 2014, 4, 1777–1782 Search PubMed.
- K. Kim and S. H. Hong, Photoinduced copper(I)-catalyzed cyanation of aromatic halides at room temperature, Adv. Synth. Catal., 2017, 359, 2345–2351 Search PubMed.
- B. Majhi and B. C. Ranu, Palladium-catalyzed norbornene-mediated tandem ortho-C–H amination/ipso-C–I cyanation of iodoarenes: regiospecific synthesis of 2-aminobenzonitrile, Org. Lett., 2016, 18, 4162–4165 CrossRef CAS PubMed.
- G. Dou and D. Shi, Efficient and convenient synthesis of indazol-3(2H)-ones and 2-aminobenzonitriles, J. Comb. Chem., 2009, 11, 1073–1077 CrossRef CAS PubMed.
- Z. Wei, J. Wang, S. Mao, D. Su, H. Jin, Y. Wang, F. Xu, H. Li and Y. Wang, In situ-generated Co0–Co3O4/N-doped carbon nanotubes hybrids as efficient and chemoselective catalysts for hydrogenation of nitroarenes, ACS Catal., 2015, 5, 4783–4789 CrossRef CAS.
- J. Dong, Z. Wu, Z. Liu, P. Liu and P. Sun, Rhodium(III)-catalyzed direct cyanation of aromatic C–H bond to form 2-(alkylamino)benzonitriles using N-nitroso as directing group, J. Org. Chem., 2015, 80, 12588–12593 Search PubMed.
- B. Rao and X. Zeng, Aminocyanation by the addition of N–CN bonds to arynes: chemoselective synthesis of 1,2-bifunctional aminobenzonitriles, Org. Lett., 2014, 16, 314–317 Search PubMed.
- W. L. Chen, S. Y. Wu, X. L. Mo, L. X. Wei, C. Liang and D. L. Mo, Synthesis of 2-aminobenzonitriles through nitrosation reaction and sequential iron(III)-catalyzed C–C bond cleavage of 2-arylindoles, Org. Lett., 2018, 20, 3527–3530 Search PubMed.
- (a) Q. Zhang and X. Jin, Recent advances in the fixation of CO2 to form N-containing heterocyclic compounds, Chem. – Eur. J., 2025, 31, e202500933 Search PubMed; (b) W. Huang, D. He, H. Jiang and W. Wu, Recent advances in the transformation of nitriles into diverse N-heterocycles, Chem. Soc. Rev., 2025, 54, 10724–10795 Search PubMed; (c) H. Li, H. Guo, Z. Fang, T. M. Aida and R. L. Smith, Cycloamination strategies for renewable N-heterocycles, Green Chem., 2020, 22, 582–611 RSC.
- (a) R. Tamatam, S. H. Kim and D. Shin, Transition-metal-catalyzed synthesis of quinazolines: a review, Front. Chem., 2023, 11, 1140562 Search PubMed; (b) G. Sivakumar, A. K. Suresh and E. Balaraman, Tandem multicomponent reactions for diverse heterocycles synthesis under 3d-transition metal catalysis, in Dehydrogenation Reactions with 3d Metals, Springer Nature Switzerland, Cham, 2023, pp. 129–171 Search PubMed; (c) N. Kumari, M. A. Hasan, B. D. Ward and L. Mishra, Reactivity of tetrabutylammonium iodide with a heteronuclear 6Cu(II)–4Na(I) complex: selective recognition of iodide ion, Ind. Eng. Chem. Res., 2013, 52, 15007–15014 CrossRef CAS.
- S. Dutt, V. Goel, N. Garg, D. Choudhury, D. Mallick and V. Tyagi, Biocatalytic Aza-Michael addition of aromatic amines to enone using α-amylase in water, Adv. Synth. Catal., 2020, 362, 858–866 Search PubMed.
- Q. Q. Wang, K. Xu, Y. Y. Jiang, Y. G. Liu, B. G. Sun and C. C. Zeng, Electrocatalytic Minisci acylation reaction of N-heteroarenes mediated by NH4I, Org. Lett., 2017, 19, 5517–5520 CrossRef CAS PubMed.
- (a) S. Sharma, Vaishali, A. Pandey, S. Rani, P. Sharma, K. Mishra, A. Kumar, H. Kumar and A. Dhyani, An untold story of ionic liquid for the isatin derivative synthesis, Curr. Org. Chem., 2025, 29, 1240–1255 Search PubMed; (b) G. Chen, H. J. Su, M. Zhang, F. Huo, J. Zhang, X. J. Hao and J. R. Zhao, New bactericide derived from isatin for treating oilfield reinjection water, Chem. Cent. J., 2012, 6, 90 CrossRef CAS PubMed.
- G. D. Allen, M. C. Buzzeo, C. Villagrán, C. Hardacre and R. G. Compton, A mechanistic study of the electro-oxidation of bromide in acetonitrile and the room-temperature ionic liquid 1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide at platinum electrodes, J. Electroanal. Chem., 2005, 575, 311–320 Search PubMed.
- I. Damljanović, D. Stevanović, M. Vukićević and R. D. Vukićević, Electrochemical bromochlorination of peracetylated glycals, Carbohydr. Res., 2011, 346, 2683–2687 Search PubMed.
| This journal is © The Royal Society of Chemistry 2026 |