
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of α-chloro-β-ketoesters via ytterbium(III) triflate-mediated decarboxylative Claisen-type condensation
Yihao
Shi
 ,
Isao
Yoshikawa
,
Akihiro
Sakama
and
Kazuaki
Kudo
*
,
Isao
Yoshikawa
,
Akihiro
Sakama
and
Kazuaki
Kudo
*
Institute of Industrial Science, The University of Tokyo, 4-6-1 Komaba, Meguro-ku, Tokyo 1538505, Japan. E-mail: kkudo@iis.u-tokyo.ac.jp
First published on 11th February 2026
Abstract
A Yb(OTf)3-mediated decarboxylative Claisen-type condensation is established to access α-chloro-β-ketoesters. Operating under mild conditions, this method delivers products in 78–95% yields while circumventing the functional-group incompatibility and side reactions associated with conventional electrophilic chlorination of β-ketoesters. The synthetic utility of this method is further demonstrated by the synthesis of chlorinated 2-pyrones and asperphenylpyrone analogues, highlighting its potential for constructing structurally diverse and bioactive molecules.
Introduction
α-Chloro-β-ketoesters are widely used intermediates and building blocks in organic synthesis owing to their high reactivity. They serve as key precursors to bioactive chlorine-containing compounds, such as 3-chlorocoumarins,1 4-chloro-1,2-dithiole-3-thiones,2 and 5-chloropyrimidines.3 Thanks to the high reactivity of chlorine towards substitution, they have also been used for constructing aromatic heterocycles, including thiazoles,4 imidazoles,5 1,2,4-triazoles,6 furans,7 and imidazopyridines.8The synthesis of α-chloro-β-ketoesters is typically achieved through direct chlorination of β-ketoesters using electrophilic chlorinating reagents (Scheme 1a), such as SO2Cl2,9N-chlorosuccinimide (NCS),10 1,3-dichloro-5,5-dimethylhydantoin,11 trichloroisocyanuric acid,12 NH4Cl/O3,13 HCl/PhIO,14 and HCl/Cu(OTf)2.15 However, these methods often suffer from poor selectivity for α-monochlorination, affording mixtures of α-mono- and α,α-dichlorinated products, and exhibit limited functional-group compatibility under the required harsh acidic or oxidizing conditions. Consequently, there remains a clear need for the development of an efficient and versatile synthetic route to α-chloro-β-ketoesters.
 | ||
| Scheme 1 Strategies for the synthesis of α-chloro-β-ketoesters and decarboxylative Claisen-type condensation reaction. | ||
One feasible alternative strategy involves the Claisen condensation, employing chloroacetates or their equivalents. However, this approach has rarely been explored, not only due to the challenges posed by undesired self-condensation, but also due to complications induced by the α-chlorine under typical Claisen condensation conditions, such as dechlorination.16 In the 1980s, the Claisen condensation approach was first applied by Tanabe and Mukaiyama, to obtain a specific α-chloro-β-ketoester.17,18 Despite the low yield of 45%, they successfully prevented the loss of the base-sensitive α-chlorine by using Lewis acid TiCl2(OTf)2. In 2023, Presset and co-workers reported a Grignard reagent-mediated decarboxylative Claisen-type condensation method to access α-substituted β-ketoesters (Scheme 1b).19 By employing magnesium enolates of substituted malonic acid half oxyesters,20 they demonstrated the synthesis of a variety of α-substituted β-ketoesters, including one α-chloro-β-ketoester obtained in 91% yield. Nevertheless, the method also exhibits inherent limitations, particularly its poor functional-group compatibility and the tendency to induce halogen loss, arising from the harsh conditions provided by Grignard reagents.
Given these precedents, we envisioned establishing an efficient synthetic method for α-chloro-β-ketoesters through decarboxylative Claisen-type condensation under mild Lewis acidic conditions. In our previous work, the superior compatibility and reactivity of Lewis acid Mg(OTf)2 compared to Grignard reagents have been demonstrated for the construction of β-keto thioesters via decarboxylative Claisen-type condensation of malonic acid half thioesters (MAHTs).21 Iterative application of this method further enabled the construction of various naturally occurring polyketides, including 2-pyrone derivatives (Scheme 1c). On the other hand, a preliminary result reported by Presset and co-workers on the synthesis of α-substituted β-ketoesters showed that Mg(OTf)2 is ineffective in mediating the decarboxylative Claisen-type condensation of substituted malonic acid half oxyesters.19
In this study, we present a comprehensive investigation on Lewis acid-mediated decarboxylative Claisen-type condensation of chlorinated malonic acid half oxyester (Cl-MAHO) (Scheme 1d). This approach represents, to our knowledge, the first versatile alternative to electrophilic α-chlorination for the synthesis of α-chloro-β-ketoesters. Furthermore, we showcase the utility of this method and α-chloro-β-ketoesters by synthesizing chlorinated 2-pyrones and asperphenylpyrone analogues.
Results and discussion
Our study initially focused on the reaction between Cl-MAHO 1a and 3-phenylpropionic acid.22 As Claisen acceptors, combinations of carboxylic acids and coupling reagents have proven effective in our previous work and related decarboxylative Claisen-type condensation reactions.21,23–26 However, in the present case, none of the coupling reagents examined afforded the α-chloro-β-ketoester 3a in yields exceeding 35%, even when mediated by a Grignard reagent (Table S1). This poor reactivity is presumably attributable to the steric and inductive effects of the α-chlorine substituent, which reduce the nucleophilicity of 1a toward the Claisen acceptor.27,28We then altered the Claisen acceptor with 3-phenylpropionyl chloride (2a). The decarboxylative Claisen-type condensation between 1a and 2a, mediated by the Grignard reagent iPrMgBr, afforded 3a in 73% yield (Table 1, entry 1). Subsequently, studies regarding Lewis acids were performed. Instead of the Grignard reagent, a Lewis acid and an external base were employed in order to generate a metal–ester enolate complex from 1a, which was expected to further react with 2a to afford 3a. We screened several magnesium salts as Lewis acids for their effectiveness in the reaction. Compared to iPrMgBr, the combinations of Mg(OTf)2, MgCl2, and MgBr2 with N,N-diisopropylethylamine (DIPEA) showed reduced reactivity, giving 3a in 48%, 55%, and 62% yields, respectively (entries 2–4). Moreover, reactions employing other Lewis acids did not proceed (entries 5–9), and the acyl chloride 2a was recovered as the corresponding carboxylic acid after aqueous workup. The ineffectiveness of common metal salts prompted us to explore lanthanoid Lewis acids.29 Indeed, several lanthanoid triflates effectively mediated the reaction (entries 10–13), among which Yb(OTf)3 afforded the highest yield of 3a (81%, entry 12). This enhanced performance likely reflects the optimal balance of ionic radius, Lewis acidity, and oxophilicity of the ytterbium(III) ion in promoting this reaction.30,31
|
|
|||
|---|---|---|---|
| Entry | Lewis acid | Solvent | Yielda [%] |
| a Yields were determined by 1H NMR using 1,4-dimethoxybenzene as an internal standard. b n.r.: no reaction. c DMAP was used instead of DIPEA. d DBU was used instead of DIPEA. e Yb(OTf)3 (2 equiv.), 1a (2 equiv.), and DIPEA (2 equiv. + 1 equiv.) were used. f Isolated yield. | |||
| 1 | iPrMgBr | DME | 73 |
| 2 | Mg(OTf)2 | DME | 48 |
| 3 | MgCl2 | DME | 55 |
| 4 | MgBr2 | DME | 62 |
| 5 | LiCl | DME | 3 |
| 6 | ZnCl2 | DME | n.r.b |
| 7 | Cu(OTf)2 | DME | n.r. |
| 8 | Sc(OTf)3 | DME | Trace |
| 9 | CeCl3 | DME | Trace |
| 10 | La(OTf)3 | DME | 79 |
| 11 | Eu(OTf)3 | DME | 77 |
| 12 | Yb(OTf) 3 | DME | 81 |
| 13 | Lu(OTf)3 | DME | 62 |
| 14 | Yb(OTf)3 | DCM | 3 |
| 15 | Yb(OTf)3 | MeCN | 32 |
| 16 | Yb(OTf)3 | THF | 43 |
| 17 | Yb(OTf)3 | Dioxane | 42 |
| 18c | Yb(OTf)3 | DME | 72 |
| 19d | Yb(OTf)3 | DME | 79 |
| 20 | Yb(OTf) 3 | DME | 89 (86) |

Subsequently, we evaluated the effects of other reaction parameters. The investigation on a series of solvents revealed the essentialness of 1,2-dimethoxyethane (DME), as other common solvents diminished the yield significantly (entries 14–17). We speculate that DME functions not only as a solvent but also as a bidentate ligand that forms five-membered chelate rings with Yb(OTf)3.32,33 Such chelation may restrict excessive substrate coordination and suppress unproductive trans-coordination between reaction partners on the ytterbium center, thereby driving the reaction toward product formation. The use of other bases such as 4-(dimethylamino)pyridine (DMAP) (entry 18) and 1,8-diazabicyclo[5.4.0]-7-undecene (DBU) (entry 19) instead of DIPEA showed no significant effect on yield. Further investigation of reaction stoichiometry (Table S2) showed that using two equivalents of the metal–ester enolate complex (the combination of Cl-MAHO, Yb(OTf)3, and DIPEA) increased the yield to 89% (entry 20).
Based on literature precedents,27,34–36 a plausible mechanism for this reaction (Scheme 2) is proposed; it involves the initial formation of the Yb(III)–Cl-MAHO carboxylate A, followed by deprotonation to generate a nucleophilic species that reacts with acyl chloride 2 to afford intermediate B. Subsequent decarboxylation is envisaged to occur after the formation of the C(Cl)–C bond, and aqueous workup finally furnishes the α-chloro-β-ketoester 3. Notably, a total of 3 equivalents of DIPEA was employed (Table S2, entry 7), which may serve to deprotonate the carboxylic acid moiety of Cl-MAHO (1a → A) and to scavenge the HCl generated during the C(Cl)–C bond formation step (A → B).
 | ||
| Scheme 2 Mechanistic hypothesis for the formation of the products. | ||
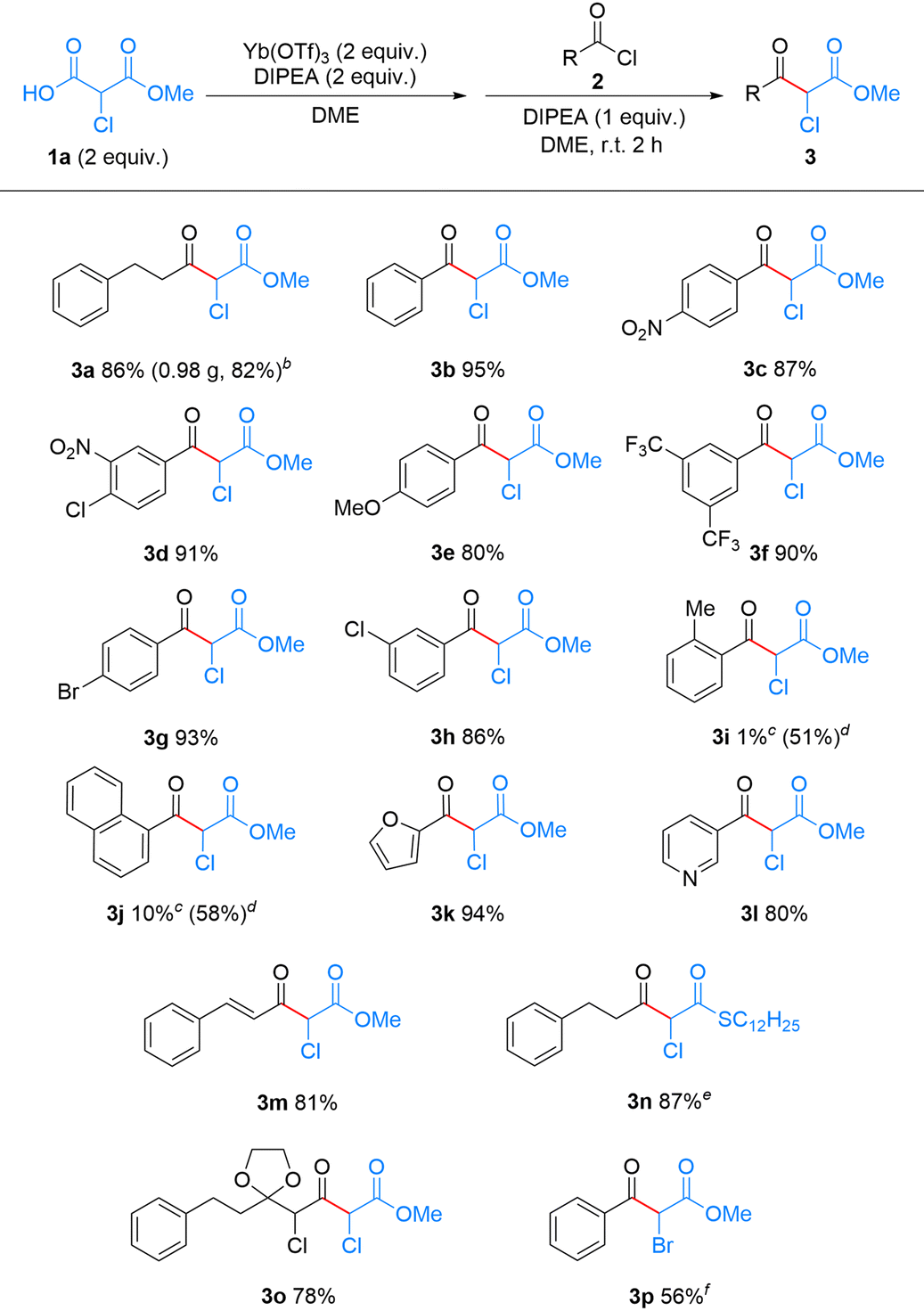
With the optimal conditions in hand, we then investigated the scope and generality of acyl chloride 2 in this reaction. As shown in Table 2, a scale-up experiment was performed with 5 mmol of 2a, which provided product 3a (0.98 g) in 82% yield. A series of benzoyl chlorides bearing various functional groups, such as nitro,19 chloro, bromo, trifluoromethyl, and methoxy groups,37 at the m-/p-positions smoothly afforded the corresponding α-chloro-β-ketoesters 3b–h in good yields, ranging from 80 to 95%. In contrast, sterically demanding acyl chlorides, such as o-methylbenzoyl chloride and 1-naphthoyl chloride, provided the desired products 3i and 3j only in 1% and 10% yields, respectively. We speculate that the key ytterbium complex intermediate is highly sterically demanding, such that acyl chlorides with substituents near the reaction center are likely to hinder the C–C bond formation with 1a. By comparison, this steric effect was much less pronounced in the magnesium-based Lewis acid-mediated reaction. Replacing Yb(OTf)3 with MgCl2 furnished 3i and 3j in 51% and 58% yields, respectively. This result is consistent with the lower coordination number and less sterically congested coordination environment typically associated with the magnesium ion.38,39
| a Unless otherwise specified, all reactions were carried out on a 0.1 mmol scale, and isolated yields are provided. b The reaction was performed on a 5 mmol scale of 2a. c 1H NMR yield (determined using 1,4-dimethoxybenzene as an internal standard). d MgCl2 was used instead of Yb(OTf)3. e S-Dodecyl-Cl-MAHT 1b was used instead of Cl-MAHO 1a. f Br-MAHO 1c and benzoyl bromide were used instead of Cl-MAHO 1a and benzoyl chloride. The reaction time was 24 h. |
|---|
|
We next applied this method to the synthesis of α-chloro-β-ketoesters that are considered to be challenging to access under previously reported acidic/oxidizing conditions. Heteroaryl-substituted products, such as 3k and 3l, were obtained in 94% and 80% yields, respectively. Cinnamoyl chloride, which bears a conjugated double bond, was also compatible, affording the unsaturated α-chloro-β-ketoester 3m in 81% yield. The oxidation-sensitive S-dodecyl-Cl-MAHT (1b) gave the α-chloro-β-ketothioester 3n in 87% yield.40 Moreover, this method is amenable to iterative application, allowing sequential extension of the α-chlorocarbonyl chain. For example, a second decarboxylative Claisen-type condensation using α-chloroacyl chloride 2o, prepared from 3a, afforded the α,γ-dichloro-β-ketoester 3o in 78% yield. The successful synthesis of these compounds demonstrates the broad compatibility and distinct advantage of our approach. Notably, no halogen loss was observed for the α-halogenated aliphatic substrate 2o, nor for aromatic substrates bearing halogen substituents (Br: 2g; Cl: 2d and 2h), in sharp contrast to Grignard reagent-mediated reactions.19
Encouraged by the success of this method in the synthesis of α-chloro-β-ketoesters, we also explored its feasibility with monomethyl bromomalonate (Br-MAHO, 1c) (Tables S3 and S4). By extending the reaction time to 24 h, α-bromo-β-ketoester 3p was synthesized in 56% yield from benzoyl bromide 2p.
Next, the synthetic versatility of our method was showcased by transforming the α-chloro-β-ketoester 3l into synthetically challenging biheteroaryl compounds.41,42 As shown in Scheme 3, the Pechmann condensation of 3l with m-cresol afforded the coumarin derivative 4 in 79% yield.43 In addition, the reaction of 3l with thiobenzamide furnished the thiazole derivative 5 in 76% yield.44 These transformations highlight a straightforward route to valuable, functionalized pharmacophore scaffolds.45–48
 | ||
| Scheme 3 Synthesis of coumarin and thiazole derivatives. | ||
Furthermore, we applied the present method to the construction of chlorinated 2-pyrones and their derivatives. Chlorinated 2-pyrones have recently attracted great interest for their potential bioactivities49,50 and synthetic utilities.51–53 Despite extensive research on 3-chloro-2-pyrones,54,55 both the synthesis and application of 5-chloro-2-pyrones remain unexplored, likely owing to the inherently higher reactivity of the C-3 position of 2-pyrones toward electrophilic chlorination. On the basis of our previously reported strategy for polyketide synthesis, we envisioned that 5-chloro-2-pyrones can be accessed by integrating the present method into the iterative chain-elongation sequence.25,26
The synthesis commenced from α-chloro-β-ketoester 3a (Scheme 4). To construct the triketide-derived 2-pyrone core, an additional iteration of the decarboxylative condensation was required. Treatment of 3a with (trimethylsilyl)diazomethane (TMSCHN2) smoothly afforded a mixture of E- and Z-isomers of methyl enol ether 6, which was then subjected to the ester hydrolysis.56–59 Recrystallization of the crude hydrolysis product provided the corresponding Z-configured carboxylic acid 7 in 55% yield. Subsequent decarboxylative condensation of 7 with S-dodecyl-MAHT furnished the triketide linear precursor 8. Acidic hydrolysis of the methyl enol ether followed by DBU-promoted Claisen-type lactonization generated the unstable intermediate 5-chloro-4-hydroxy-2-pyrone 9. Final methylation of the 4-hydroxy group of 9 yielded 5-chlorinated dihydro-5,6-dehydrokavain (DDK) 10. In total, starting from α-chloro-β-ketoester 3a, this five-step procedure succeeded in the synthesis of 5-chlorinated DDK 10 with an overall yield of 34%.
 | ||
| Scheme 4 Synthesis of 5-chloro-2-pyrone 10. | ||
Then, a series of derivatization reactions of 10 were investigated (Scheme 5). First, we took a closer look at the reaction conditions for the Suzuki–Miyaura coupling between 10 and (4-methoxyphenyl)boronic acid. It was found that the Buchwald phosphine ligand XPhos shows its efficiency in this reaction, which proceeded to provide 5-aryl DDK 11 in 89%.60 Next, we attempted a modification of the C-3 position of 10. In the presence of a catalytic amount of trifluoroacetic acid (TFA), treating 10 with N-bromosuccinimide (NBS) smoothly yielded 3-bromo-5-chlorinated DDK 12b in 91%. Likewise, 3,5-dichlorinated DDK 12a was obtained in 96% yield using NCS. Finally, sequential Suzuki–Miyaura couplings were performed on the different halogen-substituted DDK 12b, giving asperphenylpyrone analogue 14 in 64% yield.61
 | ||
| Scheme 5 Derivatization of 5-chloro-2-pyrone 10 and synthesis of its asperphenylpyrone analogue. | ||
Conclusions
In summary, we have developed an efficient method for synthesizing α-chloro-β-ketoesters through Yb(OTf)3-mediated decarboxylative Claisen-type condensation of Cl-MAHO. This strategy exhibits broad compatibility with a variety of functional groups and delivers yields of up to 95%. A comprehensive investigation of the reaction conditions revealed that both Yb(OTf)3 and DME are crucial for this transformation. The synthetic utilities of this strategy and the resulting α-chloro-β-ketoesters were demonstrated by the synthesis of coumarin and thiazole derivatives, 5-chloro-2-pyrone DDK and its derivatives. Overall, the present strategy circumvents the challenges of electrophilic chlorination, providing a complementary approach to existing methods and expanding the synthetic toolbox for the construction of chlorine-containing molecules.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5ob01966d.CCDC 2502623 contains the supplementary crystallographic data for this paper.62
Acknowledgements
This work was partially supported by a Grant-in-Aid for Scientific Research (C) from the Japan Society for the Promotion of Science (20K05487 for K. K.). We thank Dr Kengo Akagawa for his advice and discussion. We appreciate Mr Kohsaku Okuyama for his experimental assistance. MS spectral data were obtained at the Komaba Analysis Core, Institute of Industrial Science, the University of Tokyo. X-ray single crystallographic analysis was supported by the “Advanced Research Infrastructure for Materials and Nanotechnology in Japan (ARIM)” of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Grant Number JPMXP1223UT0002.References
- G. Li, D. Wang, M. Sun, G. Li, J. Hu, Y. Zhang, Y. Yuan, H. Ji, N. Chen and G. Liu, J. Med. Chem., 2010, 53, 1741 CrossRef CAS PubMed.
- D. A. Brown, S. Betharia, J. H. Yen, Q. Tran, H. Mistry and K. Smith, Bioorg. Med. Chem. Lett., 2014, 24, 5829 Search PubMed.
- L. Li, C. Zhou, M. Liu, P. Zhang, N. Zhang, J. Li, T. Li, X. Liu, S. Cheng, Q. Li and A. Liu, J. Heterocycl. Chem., 2019, 56, 3206 CrossRef CAS.
- J. Ann, H. S. Kim, S. A. Thorat, H. Kim, H. J. Ha, K. Choi, Y. H. Kim, M. Kim, S. W. Hwang, L. V. Pearce, T. E. Esch, N. A. Turcios, P. M. Blumberg and J. Lee, J. Med. Chem., 2020, 63, 418 CrossRef CAS PubMed.
- J. Dong, S. Chen, R. Li, W. Cui, H. Jiang, Y. Ling, Z. Yang and W. Hu, Eur. J. Med. Chem., 2016, 108, 605 CrossRef CAS PubMed.
- F. Bi, D. Song, Y. Qin, X. Liu, Y. Teng, N. Zhang, P. Zhang, N. Zhang and S. Ma, Bioorg. Med. Chem., 2019, 27, 3179 CrossRef CAS PubMed.
- A. Martin-Kohler, J. Widmer, G. Bold, T. Meyer, U. Séquin and P. Traxler, Helv. Chim. Acta, 2004, 87, 956 CrossRef CAS.
- A. Vakalopoulos, F. Wunder, I. V. Hartung, G. Redlich, R. Jautelat, P. Buchgraber, J. Hassfeld, A. V. Gromov, N. Lindner, D. Bierer, J. Gries, W. Kroh, H. Paulsen, J. Mittendorf, D. Lang, E. Becker-Pelster, D. Brockschnieder, V. Geiss, V. Li, A. Straub, A. Knorr, T. Mondritzki, H. Trübel, M. Raschke, M. Schaefer, D. Thomas, P. Sandner, J. P. Stasch and M. Follmann, J. Med. Chem., 2023, 66, 7280 CrossRef CAS PubMed.
- T. L. Gilchrist, O. A. S. Romero and R. C. Wasson, J. Chem. Soc., Perkin Trans. 1, 1989, 353 RSC.
- Y. Mei, P. A. Bentley and J. Du, Tetrahedron Lett., 2008, 49, 3802 CrossRef CAS.
- Z. Chen, B. Zhou, H. Cai, W. Zhu and X. Zou, Green Chem., 2009, 11, 275 RSC.
- A. K. Mishra, H. Nagarajaiah and J. N. Moorthy, Eur. J. Org. Chem., 2015, 2733 CrossRef CAS.
- P. Swamy, M. A. Kumar, M. M. Reddy and N. Narender, Chem. Lett., 2012, 41, 432 CrossRef CAS.
- T. Kitamura, Y. Tazawa, M. H. Morshed and S. Kobayashi, Synthesis, 2012, 1159 CrossRef CAS.
- K. Tsuchida, T. Kochi and F. Kakiuchi, Asian J. Org. Chem., 2013, 2, 935 CrossRef CAS.
- M. Yamaguchi, K. Shibato, H. Nakashima and T. Minami, Tetrahedron, 1988, 44, 4767 CrossRef CAS.
- Y. Tanabe and T. Mukaiyama, Chem. Lett., 1986, 15, 1813 CrossRef.
- Y. Tanabe, Bull. Chem. Soc. Jpn., 1989, 62, 1917 CrossRef CAS.
- T. Xavier, P. Tran, A. Gautreau, E. Le Gall and M. Presset, Synthesis, 2023, 598 CAS.
- R. E. Ireland and J. A. Marshall, J. Am. Chem. Soc., 1959, 81, 2907 Search PubMed.
- Y. Takeuchi, K. Akagawa and K. Kudo, J. Org. Chem., 2021, 86, 17307 CrossRef CAS PubMed.
- T. Xavier, S. Condon, C. Pichon, E. Le Gall and M. Presset, Org. Lett., 2019, 21, 6135 CrossRef CAS PubMed.
- C. Doebelin, Y. He and T. M. Kamenecka, Tetrahedron Lett., 2016, 57, 5658 CrossRef CAS PubMed.
- T. Hanari, N. Shimada, Y. Kurosaki, N. Thrimurtulu, H. Nambu, M. Anada and S. Hashimoto, Chem. – Eur. J., 2015, 21, 11671 CrossRef CAS PubMed.
- K. Akagawa and K. Kudo, Chem. Commun., 2017, 53, 8645 RSC.
- Y. Takeuchi, S. Kawasaki, K. Akagawa and K. Kudo, RSC Adv., 2022, 12, 5275 RSC.
- S. P. Bew, G. R. Stephenson, J. Rouden, J. Godemert, H. Seylani and L. A. Martinez-Lozano, Chem. – Eur. J., 2017, 23, 4557 Search PubMed.
- D. W. Brooks, L. D.-L. Lu and S. Masamune, Angew. Chem., Int. Ed. Engl., 1979, 18, 72 CrossRef.
- F. Berrué, S. Antoniotti, O. P. Thomas and P. Amade, Eur. J. Org. Chem., 2007, 1743 Search PubMed.
- K. Mikami, M. Terada and H. Matsuzawa, Angew. Chem., Int. Ed., 2002, 41, 3554 CrossRef CAS PubMed.
- K. P. Kepp, Inorg. Chem., 2016, 55, 9461 CrossRef CAS PubMed.
- T. E. Shaw, T. J. Diethrich, C. L. Stern, B. L. Scott, T. Jurca, T. M. Gilbert and A. P. Sattelberger, Dalton Trans., 2022, 51, 7856 Search PubMed.
- C. Vinas, R. Benakki, F. Teixidor and J. Casabo, Inorg. Chem., 1995, 34, 3844 CrossRef CAS.
- K. C. Fortner and M. D. Shair, J. Am. Chem. Soc., 2007, 129, 1032 CrossRef CAS PubMed.
- N. Blaquiere, D. G. Shore, S. Rousseaux and K. Fagnou, J. Org. Chem., 2009, 74, 6190 CrossRef CAS PubMed.
- M. Pinaud, T. Xavier, L. Lin, E. Le Gall and M. Presset, Eur. J. Org. Chem., 2025, e202500457 CrossRef CAS.
- W. Yang, Y. Cao, H. Cheng, Q. Sun and M. Ma, Lett. Org. Chem., 2020, 17, 788 CrossRef CAS.
- C. E. Holloway and M. Melnik, J. Organomet. Chem., 1994, 465, 1 CrossRef CAS.
- G. L. Tripodi, T. C. Correra, C. F. F. Angolini, B. R. V. Ferreira, P. Maître, M. N. Eberlin and J. Roithová, Eur. J. Org. Chem., 2019, 3560 CrossRef CAS PubMed.
- R. Borrmann, D. Zetschok and H. Wennemers, Org. Lett., 2022, 24, 8683 Search PubMed.
- J. Chen, W. Liu, L. Zhou and Y. Zhao, Tetrahedron Lett., 2018, 59, 2526 CrossRef CAS.
- A. Reichelt, J. M. Bailis, M. D. Bartberger, G. Yao, H. Shu, M. R. Kaller, J. G. Allen, M. F. Weidner, K. S. Keegan and J. H. Dao, Eur. J. Med. Chem., 2014, 80, 364 CrossRef CAS PubMed.
- C. Dubiella, H. Cui and M. Groll, Angew. Chem., Int. Ed., 2016, 55, 13330 CrossRef CAS PubMed.
- F. Denonne, F. Atienzar, S. Célanire, B. Christophe, F. Delannois, C. Delaunoy, M. L. Delporte, V. Durieu, M. Gillard, B. Lallemand, Y. Lamberty, G. Lorent, A. Vanbellinghen, N. Van Houtvin, V. Verbois and L. Provins, ChemMedChem, 2010, 5, 206 CrossRef CAS PubMed.
- P. Mutai, G. Breuzard, A. Pagano, D. Allegro, V. Peyrot and K. Chibale, Bioorg. Med. Chem., 2017, 25, 1652 CrossRef CAS PubMed.
- T. Kawate, N. Iwase, M. Shimizu, S. A. Stanley, S. Wellington, E. Kazyanskaya and D. T. Hung, Bioorg. Med. Chem. Lett., 2013, 23, 6052 Search PubMed.
- S. T. Dhumal, A. R. Deshmukh, M. R. Bhosle, V. M. Khedkar, L. U. Nawale, D. Sarkar and R. A. Mane, Bioorg. Med. Chem. Lett., 2016, 26, 3646 Search PubMed.
- P. C. Sharma, K. K. Bansal, A. Sharma, D. Sharma and A. Deep, Eur. J. Med. Chem., 2020, 188, 112016 CrossRef CAS PubMed.
- Z. S. Bhat, M. A. Rather, M. Maqbool, H. U. Lah, S. K. Yousuf and Z. Ahmad, Biomed. Pharmacother., 2017, 91, 265 Search PubMed.
- L. M. Deck, M. L. Baca, S. L. Salas, L. A. Hunsaker and D. L. Vander Jagt, J. Med. Chem., 1999, 42, 4250 Search PubMed.
- F. Bellina, A. Carpita, L. Mannocci and R. Rossi, Eur. J. Org. Chem., 2004, 2610 CrossRef CAS.
- I. J. S. Fairlamb, C. T. O'Brien, Z. Lin and K. C. Lam, Org. Biomol. Chem., 2006, 4, 1213 Search PubMed.
- P. Shah, M. D. Santana, J. García, J. L. Serrano, M. Naik, S. Pednekar and A. R. Kapdi, Tetrahedron, 2013, 69, 1446 CrossRef CAS.
- A. M. Prendergast and G. P. McGlacken, Eur. J. Org. Chem., 2017, 4827 CrossRef CAS.
- M. T. Nolan, L. M. Pardo, A. M. Prendergast and G. P. McGlacken, J. Org. Chem., 2015, 80, 10904 Search PubMed.
- A. Clerici, N. Pastori and O. Porta, Tetrahedron, 2001, 57, 217 CrossRef CAS.
- H. O. House and G. H. Rasmusson, J. Org. Chem., 1963, 28, 27 Search PubMed.
- C. R. Harris, S. D. Kuduk, A. Balog, K. Savin, P. W. Glunz and S. J. Danishefsky, J. Am. Chem. Soc., 1999, 121, 7050 CrossRef CAS.
- T. Aoyama, S. Terasawa, K. Sudo and T. Shioiri, Chem. Pharm. Bull., 1984, 32, 3759 CrossRef CAS.
- R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461 Search PubMed.
- S. Niu, L. Yang, T. Chen, B. Hong, S. Pei, Z. Shao and G. Zhang, Mar. Drugs, 2020, 18, 561 CrossRef CAS PubMed.
- CCDC 2502623: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2q05sq.
| This journal is © The Royal Society of Chemistry 2026 |