
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnusual C-2 selectivity of Grignard additions to N-Boc-protected isatin 3-imines
Alexandra Mary
Plakas
ab,
Kamogelo Rosinah
Butsi
ab,
Sahil
Lala
 bc,
Robyn Lynne
van Zyl
bc,
Manuel Antonio
Fernandes
a,
Songeziwe
Ntsimango
a,
Moira Leanne
Bode
a and
Amanda Louise
Rousseau
*ab
bc,
Robyn Lynne
van Zyl
bc,
Manuel Antonio
Fernandes
a,
Songeziwe
Ntsimango
a,
Moira Leanne
Bode
a and
Amanda Louise
Rousseau
*ab
aMolecular Sciences Institute, School of Chemistry, University of the Witwatersrand, Private Bag 3, PO WITS, 2050, South Africa. E-mail: Amanda.Rousseau@wits.ac.za
bWITS Research Institute for Malaria (WRIM), Faculty of Health Sciences, University of the Witwatersrand, 7 York Road, Parktown 2193, South Africa
cPharmacology Division, Department of Pharmacy and Pharmacology, Faculty of Health Sciences, University of the Witwatersrand, 7 York Road, Parktown 2193, South Africa
First published on 28th January 2026
Abstract
A series of N-tert-butoxycarbonyl-protected isatin 3-imines underwent an unexpected C-2 selective addition reaction with Grignard reagents, affording 3-imino-2-phenethylindolin-2-ols. The protecting group plays a role in this observed chemoselectivity, with N-benzyl and N-p-methoxybenzyl-protected isatin 3-imines undergoing C-3 addition under Grignard reaction conditions, affording 3-amino-3-phenethylindolin-2-ones. Both C-2 and C-3 addition products were assessed for antiplasmodial activity in vitro, with four compounds displaying activity in the sub-micromolar range against both drug-sensitive and drug-resistant Plasmodium falciparum strains.
Introduction
The oxindole scaffold is present in numerous natural and synthetic products that display a wide range of biological activities.1–5 In particular, 3,3-disubstituted 2-oxindoles have shown promise in the clinic, with selected examples including cipargamin, a potent antimalarial drug candidate currently in clinical trials,6 and nelivaptan, a vasopressin V1b receptor antagonist for the treatment of depression and anxiety7 (although suboptimal pharmacokinetic properties have halted further development) (Fig. 1). | ||
| Fig. 1 Oxindoles with potent biological activity, cipargamin and nelivaptan. | ||
The pharmacological activity of this class of compounds has attracted the interest of synthetic and medicinal chemists, and many synthetic methods have been described for the synthesis of compounds containing an oxindole core.8–13
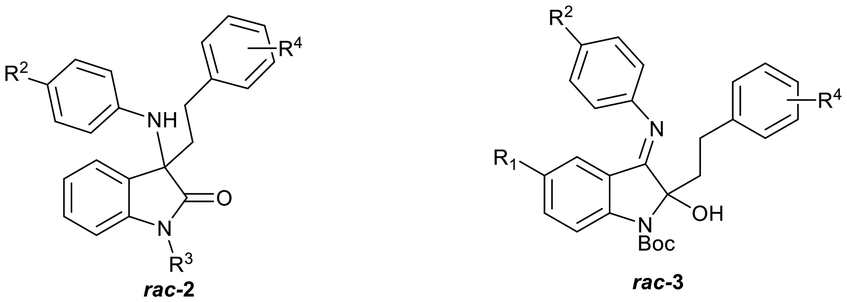
Previously, we reported the synthesis and antiplasmodial activity of a series of racemic spiroindolones 1 inspired by cipargamin, prepared by the Povarov reaction of ketimines with electron-rich alkenes (Fig. 2).14 We found that only spiroindolones bearing a methyl or a halogen substituent para to the ring nitrogen (i.e.1, R1 = Me or Hal) showed moderate antiplasmodial activity in vitro (1.31–4.20 µM), with all other compounds being inactive in this assay. Although these spiroindolones displayed only moderate activity in vitro, the interesting structure–activity relationships that were observed prompted us to consider the design of second-generation analogues. As the mode of action was not known, we considered strategies that would allow us to explore additional chemical space. One such strategy was to consider the virtual ring opening of the tetrahydroisoquinoline ring, affording oxindoles 2 with increased flexibility, while retaining the para-substituent R1. Herein, we report our synthetic approaches to a series of ring opened analogues 2, and the unexpected synthesis of indolin-2-ols 3.
 | ||
| Fig. 2 Spiroindolones 1 prepared previously,14 ring-opened analogues 2 and indolin-2-ols 3. | ||
Results and discussion
We envisaged preparing ring-opened analogues 2 as racemates initially, by reaction of ketimines 4 (derived from commercially available isatins 5), with suitably substituted Grignard reagents (Scheme 1). | ||
| Scheme 1 Planned approach for the synthesis of oxindoles 2. | ||
Our initial approach involved isatin N-Boc-protection, followed by reaction with para-substituted anilines to prepare ketimines 4. However, under these conditions, only the hemiaminal was isolated in low yields in each case. Although there is literature precedent for the use of hemiaminals in Grignard addition reactions,15 this also proved to be low-yielding in our hands. Furthermore, the N-Boc-protected isatin was found to be unstable at room temperature. By first reacting isatins 5 with para-substituted anilines to afford intermediate ketimines 6a–f, followed by N-Boc-protection, the protected ketimines 4a–f were isolated in reasonable yields (Scheme 2).16 In each case, the E-isomer was the major isomer formed, and in some cases only the E-isomer was isolated after purification.
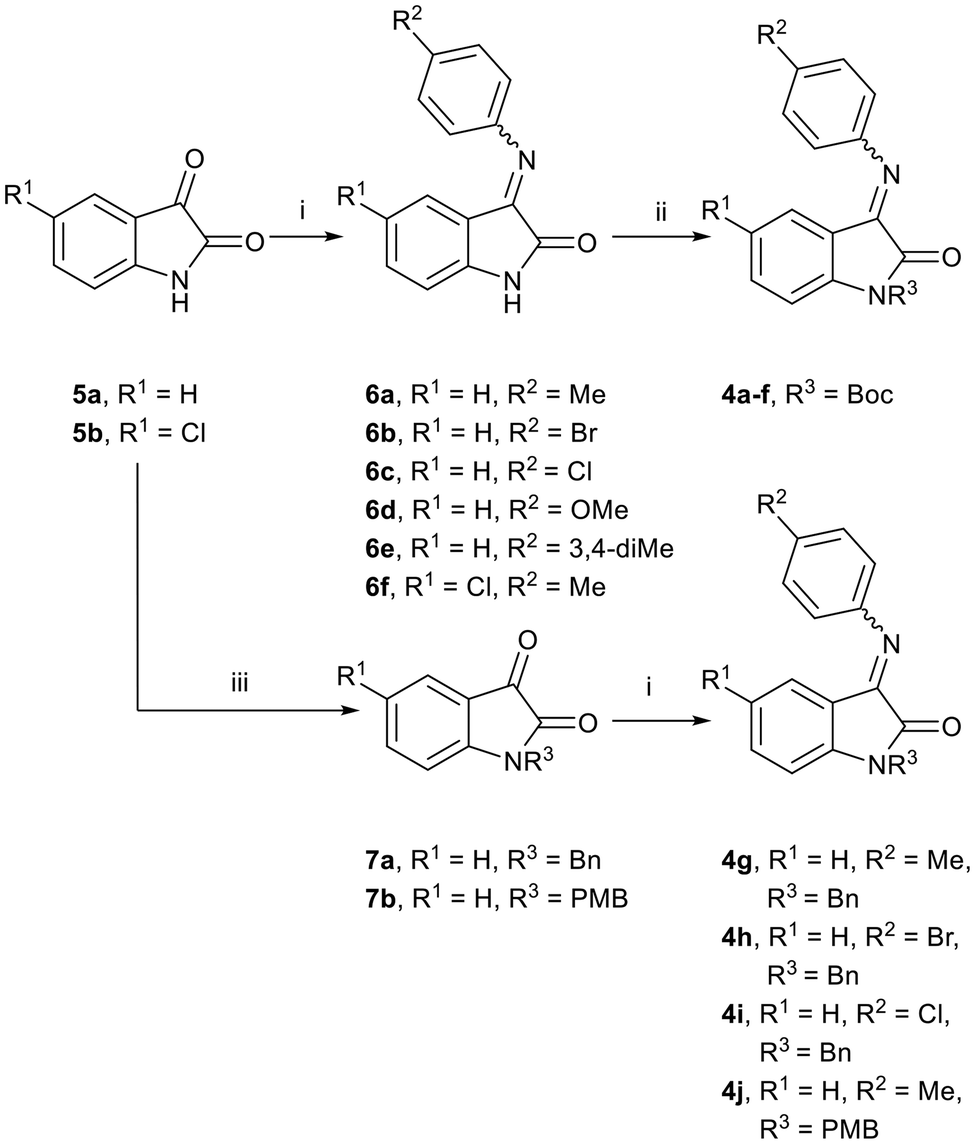 | ||
| Scheme 2 Reaction conditions: (i) substituted aniline, EtOH, AcOH, reflux, 5 h, 61–91%; (ii) Boc2O, THF, DMAP, rt, 3 h, 71–98%; (iii) (a) BnBr (for 7a) or 4-(OCH3)C6H4-CH2Br (for 7b), K2CO3, KI, DMF, reflux, 5 h, 55–74%. | ||
We then reacted the N-Boc-protected ketimines 4a–f with Grignard reagents derived from substituted phenethyl bromides, in an attempt to form oxindoles 2 (Scheme 3). This approach was based on the work of Lesma et al., where various Grignard reagents were successfully added to isatin-derived alkyl- and sulfinyl-imines, achieving yields ranging from 49–77%.15 However, to our surprise, Grignard addition to our ketimine substrates 4a–f occurred at the C-2 carbonyl of the indolin-2-one core, and not the expected C-3 imine, affording 3-imino-2-phenethylindolin-2-ols 3 instead (Scheme 3 and Table 1).
 | ||
| Scheme 3 Reaction conditions: (i) substituted phenethylmagnesium bromide, MgBr2, THF, −40 °C–rt, 18 h, 10–53%; (ii) substituted phenethylmagnesium bromide, MgBr2, DCM, −40 °C–rt, 18 h, 2–12%. | ||
|
|
|||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | Yield % |
| 2a | — | Me | Bn | 2-Cl | 5 |
| 2b | — | Br | Bn | 4-OMe | 6 |
| 2c | — | Cl | Bn | 4-OMe | 12 |
| 2d | — | Me | PMB | 2-OMe | 2 |
| 3a | H | Me | — | 4-OMe | 35 |
| 3b | H | Me | — | 4-Cl | 38 |
| 3c | H | Me | — | 3-Cl | 19 |
| 3d | H | Me | — | 2,4-diCl | 10 |
| 3e | H | Br | — | 4-OMe | 45 |
| 3f | H | Br | — | 4-Cl | 34 |
| 3g | H | Br | — | 3-Cl | 39 |
| 3h | H | Br | — | 2,4-diCl | 27 |
| 3i | H | Cl | — | 4-OMe | 50 |
| 3j | H | Cl | — | 4-Cl | 53 |
| 3k | H | Cl | — | 3-Cl | 48 |
| 3l | H | Cl | — | 2,4-diCl | 15 |
| 3m | H | OMe | — | 4-OMe | 32 |
| 3n | H | OMe | — | 4-Cl | 39 |
| 3o | H | OMe | — | 3-Cl | 40 |
| 3p | H | OMe | — | 2,4-diCl | 33 |
| 3q | H | 3,4-diMe | — | 4-Cl | 15 |
| 3r | 5-Cl | 3,4-diMe | — | 3-Cl | 18 |
| 3s | 5-Cl | Me | — | 4-OMe | 13 |
| 3t | 5-Cl | Me | — | 3-OMe | 12 |
| 3u | 5-Cl | Me | — | 2-OMe | 19 |
| 3v | 5-Cl | Me | — | 4-Cl | 37 |
| 3w | 5-Cl | Me | — | 3-Cl | 49 |
| 3x | 5-Cl | Me | — | 2-Cl | 42 |
| 3y | 5-Cl | Me | — | 2,4-diCl | 22 |
| 3z | 5-Cl | Me | — | 4-F | 48 |
| 3aa | 5-Cl | Me | — | 2-F | 23 |
This was confirmed by single-crystal X-ray diffraction on two analogues, one of which is shown in Fig. 3a. The reaction was repeated on ketimines substituted with both electron-donating and electron-withdrawing groups using a range of substituted phenethylmagnesium bromides, affording analogues 3a–3aa in varying yields of 10–53%.
 | ||
| Fig. 3 ORTEP diagrams of the X-ray crystal structures of (a) tert-butyl 3-((4-bromophenyl)imino)-2-hydroxy-2-(4-methoxyphenethyl)indoline-1-carboxylate 3e and (b) 1-benzyl-3-((4-chlorophenyl)amino)-3-(4-methoxyphenethyl)indolin-2-one 2c. Thermal ellipsoids are shown at the 50% probability level. | ||
The yield of isolated product was found to be low in most cases and attempts to improve the yield of the reaction by varying the solvent, temperature and number of equivalents of the Grignard reagent added, were not successful. In every case, both starting material and several side products were isolated, including the N-Boc deprotected starting material 6, as well as isatin 5. The product of Wurtz homocoupling of the phenethyl bromides used to prepare the Grignard reagent was also identified in some cases in low yields (3–7%), however, the low yield of desired products is not likely to be due to inefficient preparation of the phenethylmagnesium bromide derivatives as the Grignard reagent was prepared in excess. Interestingly, the β-hydride elimination product was not isolated from these reactions. However, we did isolate secondary amines in some instances, which could have formed by the reaction of either the β-hydride elimination product or unreacted phenethyl bromide with aniline formed in situ by imine hydrolysis.
To the best of our knowledge, this unexpected C-2 chemoselectivity in reactions of N-Boc isatin-derived imines has not been reported previously. However, similar reactivity has been observed in N-acyl isatins with nucleophiles, typically leading to ring opening by cleavage of the isatin C-2–N bond.17–20
By comparison, isatin and N-alkyl isatins have been shown to undergo nucleophilic addition at the C-3 carbonyl with hard amine and alcohol nucleophiles.18 We therefore speculated that the N-protecting group may have played a role in the unexpected chemoselectivity of the reaction. To further investigate this, we prepared N-benzyl- and N-p-methoxybenzyl-protected ketimines 4g–j (Scheme 2) and subjected these to Grignard reaction conditions. Interestingly, these reactions did not proceed at all in tetrahydrofuran, however, very low yields of product were isolated when the reaction was carried out in dichloromethane, affording analogues 2 (Scheme 3, four examples, 2a–d), with starting material recovered in each case. Further attempts to improve the yield of the reaction by the addition of LiCl to the Grignard reagent, or by the addition of CuI to the ketimine prior to the addition of the Grignard reagent, were not successful. Despite the very low yields obtained for these reactions, we were able to obtain a single crystal X-ray structure for one analogue, which confirmed that in these instances, Grignard addition had taken place at the C-3 imine carbon to afford rac-2c (Fig. 3b). When the Grignard reaction was tested on the unprotected imines 6, no reaction was observed.
Apart from the N-protecting group, scaffolds 2 and 3 have similar electronic functionalities; C2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/C3–N and C3N/C2–O, and therefore, only give rise to minor 13C NMR spectroscopic chemical shift differences. 3-Amino-3-phenethylindolin-2-ones 2 contain characteristic C2O and C3–N functionalities, which give rise to signals in their 13C NMR spectra at 177.4–177.8 ppm and 64.0–65.3 ppm, respectively. In contrast, 2-phenethyl-3-imino-indolin-2-ols 3 contain characteristic C3N and C2–O moieties, which give rise to signals appearing at 165.1–171.1 ppm and 91.6–95.6 ppm in the 13C NMR spectra, respectively.
O/C3–N and C3N/C2–O, and therefore, only give rise to minor 13C NMR spectroscopic chemical shift differences. 3-Amino-3-phenethylindolin-2-ones 2 contain characteristic C2O and C3–N functionalities, which give rise to signals in their 13C NMR spectra at 177.4–177.8 ppm and 64.0–65.3 ppm, respectively. In contrast, 2-phenethyl-3-imino-indolin-2-ols 3 contain characteristic C3N and C2–O moieties, which give rise to signals appearing at 165.1–171.1 ppm and 91.6–95.6 ppm in the 13C NMR spectra, respectively.
To explore this unusual chemoselectivity further, we tested the addition of allylmagnesium bromide to both N-benzyl-protected ketimine (4i) and N-Boc-protected ketimine (4c), however, only the benzyl-protected ketimine (4i) afforded the expected addition product upon reaction with allylmagnesium bromide (see SI). Reaction of 4c with allylmagnesium bromide gave the deprotected imine 6c as the major product. We did not confirm the mechanism by which this deprotection occurred, however, it is possible that chelation with the Lewis acid may have promoted hydrolysis upon work-up, or that Grignard addition may have occurred at the Boc-carbonyl carbon instead of the isatin C-2 carbonyl.
We then postulated that Grignard additions to N-Boc-protected benzylidene ketimines 10 would result in C-2 addition. To this end, we prepared benzylidene isatin 10via a Knoevenagel condensation of oxindole and p-tolualdehyde (Scheme 4). To our surprise, treatment of benzylidene ketimines 10 under the same Grignard reaction conditions shown in Scheme 3 were unsuccessful, with only starting material recovered in each case. This suggests that the C-3 electron-withdrawing group present in the N-Boc-protected ketimines 4a–f is also essential for activating the C-2 carbon towards nucleophilic attack.
 | ||
| Scheme 4 Reaction conditions: (i) p-tolualdehyde, piperidine, EtOH, under Ar, reflux, 4 h., 24 h, 80%; (ii) Boc2O, DMAP, THF, under Ar, rt, 24 h, 73%. | ||
We therefore deduce that in the case of N-alkyl protected substrates, the C-2 carbonyl participates in amide resonance, reducing the electrophilicity of the C-2 carbon while enhancing the electrophilicity of the C-3 carbon (Fig. 4a). In contrast, for N-acyl protected substrates, while the resonance form described in Fig. 4a is possible, there is an additional form in which the nitrogen engages in resonance with the acyl carbonyl (Fig. 4b). We speculate that this, together with the electron-withdrawing effect of the C-3 imine, increases the electrophilicity of the C-2 carbonyl carbon. In the presence of the Lewis acid MgBr2, which can coordinate to both the N-Boc carbonyl and the isatin C-2 carbonyl, this facilitates reaction at C-2 with phenethylmagnesium bromides, affording 3-imino-2-phenethylindolin-2-ols 3 after work-up (Fig. 4b).
 | ||
| Fig. 4 Comparison of the resonance behaviour in (a) N-alkyl and (b) N-acyl protected isatin imines and implications for chemoselectivity. | ||
Finally, we set about preparing the original targeted compounds 2 by reacting isatin-derived imine 6a with substituted phenylacetylene nucleophiles (Scheme 5) as demonstrated by Xie et al.21 The desired C-3 addition products, 3-amino-3-ethynylindolin-2-ones 11a–e, were afforded in yields of 40–63%.
 | ||
Scheme 5 Reaction conditions: (i) HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C-PhR,1 KOtBu, CuI, THF, r.t., 24 h, 40–63%; (ii) H2, Pd/C (5%), Na2CO3, EtOH, r.t., 200 mbar, 70–82%. C-PhR,1 KOtBu, CuI, THF, r.t., 24 h, 40–63%; (ii) H2, Pd/C (5%), Na2CO3, EtOH, r.t., 200 mbar, 70–82%. | ||
Initial attempts to reduce substrates 11a–e with traditional Pd/C catalysed hydrogenation conditions led to the removal of the aniline moiety. However, the desired 3-amino-3-phenethylindoline-2-ones 2e–i were afforded in yields of 70–82% by the addition of sodium carbonate. C-3 addition was confirmed by NMR spectroscopy, where the characteristic C2O and C3–N 13C NMR spectroscopic chemical shifts were observed at 179.0–179.2 ppm and 64.0–64.2 ppm, respectively.
We assessed a selection of the Boc-protected 2-phenethylindolin-2-ols 3 and 3-amino-3-phenethylindoline-2-ones 2 for antiplasmodial activity in a whole cell P. falciparum screen against a drug-sensitive parasite strain (NF54). Those compounds that were found to inhibit parasite growth by 60% or more at a concentration of 10 µM were then assessed further and IC50 values obtained. From these assays, we were able to determine that the 3-imino-2-phenethylindolin-2-ols 3 were more potent inhibitors of parasite growth than the 3-amino-3-phenethylindolin-2-ones 2, as only one of the 3-amino-3-phenethylindoline-2-ones 2 tested inhibited parasite growth by more than 50% at a concentration of 10 µM (see SI). By comparison, all but one of the 2-phenethylindolin-2-ols 3 inhibited parasite growth by more than 50% at a concentration of 10 µM, with four compounds displaying sub-micromolar activity against the NF54 strain in vitro (IC50 0.11–0.85 µM, Fig. 5). The remaining compounds were active in the low micromolar range (1.03–7.83 µM, see SI). A similar trend was observed against the chloroquine-resistant FCR3 strain, with four compounds displaying moderate activity (IC50 0.35–0.63 µM). Compounds were also tested for cytotoxicity against the human embryonic kidney epithelial cell line HEK293 in an MTT cell viability assay, with no significant cytotoxicity observed.
 | ||
| Fig. 5 Most potent compounds identified from the P. falciparum whole cell assay. | ||
Conclusions
Attempts to prepare a series of 3-amino-3-phenethylindolin-2-ones via Grignard addition to N-Boc-protected isatin-derived imines led to the discovery of an interesting chemoselectivity, yielding 3-imino-2-phenethylindolin-2-ols instead. While the Grignard addition reactions to N-Boc-protected isatin imines were C-2 selective, those to N-Bn- and N-PMB-protected isatin imines favoured C-3 addition, establishing the importance of the N-protecting group in this observed chemoselectivity. Furthermore, Grignard additions to benzylidene indolin-2-one substrates lacking the imine moiety did not proceed, confirming the imine as essential for C-2 selectivity. This Grignard-based approach offers a novel strategy for functionalising the C-2 carbonyl of 3-iminoindolin-2-ones, yielding the biologically relevant indolin-2-ol scaffold. The target 3-amino-3-phenethylindolin-2-ones were subsequently prepared via alkynylation and hydrogenation of isatin-derived imines in yields up to 52%. Compounds from each series were evaluated for antiplasmodial activity against the drug-sensitive NF54 strain of the parasite. Four compounds exhibited moderate activity in the sub-micromolar range against both drug-sensitive and drug-resistant strains of Plasmodium facliparum. In future studies, the synthesis of chiral oxindole products could be considered.Author contributions
Conceptualisation, ALR and AMP; synthesis of compounds KRB and AMP; crystallography, MAF; biological data, SL and RLvZ; writing, ALR and AMP; co-supervision of AMP and KRB, SN and MLB.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: experimental procedures, characterization data and copies of NMR spectra. See DOI: https://doi.org/10.1039/d5ob01909e.Ref. 22–39 appear in the SI.
CCDC 2513396 and 2513397 contain the supplementary crystallographic data for this paper.40a,b
Acknowledgements
We thank the NRF and the University of the Witwatersrand for generous funding to enable the purchase of a dual-wavelength hybrid diamond anode X-ray diffractometer (Bruker D8 Discovery Bio equipped with Mo and Cu X-ray sources) under NEP Grant No 129920. We acknowledge the WITS Pharmacology Division McGill Trust for research funding support.References
- F. Salim and R. Ahmad, Stud. Nat. Prod. Chem., 2015, 45, 485–525 CrossRef.
- B. Yu, D.-Q. Yu and H.-M. Liu, Eur. J. Med. Chem., 2015, 97, 673–698 CrossRef CAS PubMed.
- Y.-T. Yang, J.-F. Zhu, G. Liao, H.-J. Xu and B. Yu, Curr. Med. Chem., 2018, 25, 2233–2244 Search PubMed.
- S. S. Panda, R. A. Jones, P. Bachawala and P. P. Mohapatra, Mini-Rev. Med. Chem., 2017, 17, 1515–1536 Search PubMed.
- M. Kaur, M. Singh, N. Chadha and O. Silakari, Eur. J. Med. Chem., 2016, 123, 858–894 Search PubMed.
- M. Rottmann, C. McNamara, B. K. S. Yeung, M. C. S. Lee, B. Zou, B. Russell, P. Seitz, D. M. Plouffe, N. V. Dharia, J. Tan, S. B. Cohen, K. R. Spencer, G. E. González-Páez, S. B. Lakshminarayana, A. Goh, R. Suwanarusk, T. Jegla, E. K. Schmitt, H.-P. Beck, R. Brun, F. Nosten, L. Renia, V. Dartois, T. H. Keller, D. A. Fidock, E. A. Winzeler and T. T. Diagana, Science, 2010, 329, 1175–1180 CrossRef CAS PubMed.
- M. Schönberger, C. Leggett, S. W. Kim and J. M. Hooker, Bioorg. Med. Chem. Lett., 2010, 20, 3103–3106 CrossRef PubMed.
- G. M. Ziarani, R. Moradi and N. Lashgari, Tetrahedron, 2018, 74, 1323–1353 CrossRef.
- B. Yu, H. Xing, D.-Q. Yu and H.-M. Liu, Beilstein J. Org. Chem., 2016, 12, 1000–1039 Search PubMed.
- J.-S. Yu, F. Zhou, Y.-L. Liu and J. Zhou, Synlett, 2015, 2491–2504 CAS.
- G. M. Ziarani, P. Gholamzadeh, N. Lashgari and P. Hajiabbasi, ARKIVOC, 2013, 2013, 470–535 Search PubMed.
- M. Zhao, N.-K. Li, Y.-F. Zhang, F.-F. Pan and X.-W. Wang, Tetrahedron, 2016, 72, 1406–1414 Search PubMed.
- H. Pellissier, Synthesis, 2019, 1311–1318 CrossRef CAS.
- B. Mathebula, K. R. Butsi, R. L. van Zyl, N. C. Jansen van Vuuren, H. C. Hoppe, J. P. Michael, C. B. de Koning and A. L. Rousseau, Chem. Biol. Drug Des., 2019, 94, 1849–1858 CrossRef CAS PubMed.
- G. Lesma, N. Landoni, T. Pilati, A. Sacchetti and A. Silvani, J. Org. Chem., 2009, 74, 4537–4541 Search PubMed.
- Z. Al Marhoon, A. Abdel-Megeed, E. N. Sholkamy, M. Rafiq, H. Siddiqui and A. El-Faham, Asian J. Chem., 2014, 26, 7665–7672 CrossRef.
- J. F. M. da Silva, S. J. Garden and A. C. Pinto, J. Braz. Chem. Soc., 2001, 12, 273–324 CrossRef CAS.
- W. C. Cheah, K. Wood, D. S. Black and N. Kumar, Tetrahedron, 2011, 67, 7603–7610 Search PubMed.
- A. El-Faham, S. N. Khattab, H. A. Ghabbour, H.-K. Fun and M. R. H. Siddiqui, Chem. Cent. J., 2014, 8, 27 CrossRef PubMed.
- V. Suryanti, R. Zhang, V. Aldilla, M. Bhadbhade, N. Kumar and D. S. Black, Molecules, 2019, 24, 4343 Search PubMed.
- D. Xie, X. Xu, S. Long, X.-Y. Tang and L. Wang, J. Org. Chem., 2022, 87, 7852–7863 CrossRef CAS PubMed.
- I. Allous, S. Comesse, M. Sanselme and A. Daïch, Eur. J. Org. Chem., 2011, 5303–5310 CrossRef CAS.
- P. K. Prasad, R. G. Kalshetti, R. N. Reddi, S. P. Kamble and A. Sudalai, Org. Biomol. Chem., 2016, 14, 3027–3030 Search PubMed.
- X. Guo, P. Zhang, M. Chen, T. Li, C. Hou, X. Que, L. Xu, Z. Zhou, Q. Wang and Z. Wang, Bioorg. Chem., 2024, 153, 107757 CrossRef CAS PubMed.
- J. Tian, C. Li and H. Lv, J. Org. Chem., 2025, 90, 12023–12028 Search PubMed.
- S. K. Sridhar, M. Saravanan and A. Ramesh, Eur. J. Med. Chem., 2001, 36, 615–625 Search PubMed.
- F. Yu, P. Li, B. Wang and K. Han, J. Am. Chem. Soc., 2013, 135, 7674–7680 CrossRef CAS PubMed.
- K. Gopalaiah and A. Tiwari, Eur. J. Org. Chem., 2020, 7229–7237 CrossRef CAS.
- P. You, M. Liu, K. Zhang, F. Yang, Z. Tan and F. Chen, Org. Biomol. Chem., 2024, 22, 4466–4471 RSC.
- W. Trager and J. B. Jensen, Science, 1976, 193, 673–675 CrossRef CAS PubMed.
- C. Lambros and J. P. Vanderberg, J. Parasitol., 1979, 65, 418–420 CrossRef CAS PubMed.
- M. T. Makler and D. J. Hinrichs, Am. J. Trop. Med. Hyg., 1993, 48, 205–210 Search PubMed.
- R. L. van Zyl, A. M. Viljoen and A. K. Jäger, S. Afr. J. Bot., 2002, 68, 106–110 Search PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 Search PubMed.
- R. L. Van Zyl, S. T. Seatlholo and A. M. Viljoen, S. Afr. J. Bot., 2010, 76, 662–667 CrossRef CAS.
- Bruker, APEX4 version 2021.4-1 data collection software which includes SAINT version 8.40B, SADABS-2016/2, and XPREP version 2014/2, Bruker AXS Inc., Madison, Wisconsin, USA, 2021 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. A:Found. Adv., 2015, 71, 3–8 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. C:Struct. Chem., 2015, 71, 3–8 Search PubMed.
- (a) CCDC 2513396: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2qcd9t; (b) CCDC 2513397: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2qcdbv.
| This journal is © The Royal Society of Chemistry 2026 |