
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and evaluation of carbagalactosyl 1,2-aziridines and -epoxides as glycosidase inhibitors
Yuqing Tian,
Hong-Ming Chen,
Lyann Sim,
Seyed A. Nasseri ,
Jolene P. Reid and
Stephen G. Withers*
,
Jolene P. Reid and
Stephen G. Withers*
Department of Chemistry, University of British Columbia, Vancouver, V6T 1Z1, Canada. E-mail: withers@chem.ubc.ca
First published on 15th December 2025
Abstract
The natural product cyclophellitol is a cyclitol epoxide that mimics a β-glucoside and functions as a mechanism-based inhibitor for retaining β-glucosidases by forming a stable covalent intermediate with the enzyme's catalytic nucleophile. The epoxide is located across the carbons equivalent to C1 and O5 of glucose, and multiple congeners have been synthesised, as well as the corresponding aziridines, and shown to inactivate their cognate glycosidases. In a quest for a potent covalent inactivator of an α-galactosaminidase that is used in the conversion of A type blood to O type through modification of the antigen, we synthesised the galacto-configured 1,2-α-aziridine in which the aziridine ring is located across the carbons equivalent to C1 and C2. While neither this nor the corresponding epoxide inactivated our enzyme or any of the α-galactosidases tested, the β-configured analogues were shown by mass spectrometry to function as covalent inactivators of several β-glycosidases and their inactivation behaviour characterised kinetically. Molecular modeling indicates that 1,2-α-aziridines adopt an unfavorable conformation in solution, preventing binding to the enzyme active site. The synthesis of these compounds and their activities as inhibitors and inactivators of glycosidases of different families will direct future attempts to develop inactivators beyond those of the more common cyclophellitol class.
Introduction
The antigens that define our ABO blood type on the surface of our red blood cells and tissues are oligosaccharides based upon the fucosyl-α-1,2-galactose (Fuc-α-1,2-Gal) terminal disaccharide of the H antigen found on O type cells. Extension of this structure with N-acetylgalactosamine attached α-1,3 to the galactose generates the A antigen, while equivalent extension with galactose yields the B-antigen. The ABO type of blood or organs must be carefully matched between donor and recipient to avoid serious immune responses due to anti-A or anti-B antibodies in non-matched recipient blood. The only exceptions are O type red blood cells or organs which can be donated to persons of essentially all types since antibodies against the H antigen are not present in A or B type persons. One consequence of this is that O-type blood and organs are always in short supply.One approach to improving the supply of O type blood and organs might be to enzymatically convert the A or B antigen to the H antigen using suitably active and specific glycosidases. Our search of human gut microbiome-derived metagenomic libraries for enzymes that will remove the terminal GalNAc of the A-antigen recently uncovered a pair of enzymes that work efficiently together. The first of these is a highly specific N-acetylgalactosamine deacetylase, which forms a terminal galactosamine that is then removed by a highly efficient α-galactosaminidase.1 Interestingly this latter enzyme, FpGalNase, also has a weak α-galactosidase activity. As part of our characterisation of this enzyme we solved its three-dimensional structure by X-ray crystallography and showed that the active site contains a cobalt that coordinates the substrate amine moiety.2 Most other enzymes of the CAZy GH36 family to which this enzyme belongs are α-galactosidases that have very similar active sites, but do not contain a metal.3 Enzymes from this family carry out hydrolysis with net retention of anomeric configuration through a β-glycosyl enzyme intermediate.4
In order to better characterise active site interactions in this enzyme, with a view to engineering its activity, we were interested in developing inhibitors that form a stable covalent intermediate that could be structurally characterised. A strategy used widely for trapping intermediates on glycosidases has involved 2-fluorosugars containing a reactive leaving group.5 The fluorine inductively destabilises the oxocarbenium ion transition state, slowing both formation and hydrolysis of this intermediate while the reactive leaving group ensures relatively rapid intermediate formation. However, that strategy is not applicable here due to the need for the 2-amine substituent and also because the strategy does not work well for α-glycosidases.6 While an equivalent approach using a 5-fluoroglycosyl fluoride might seem attractive,7,8 the presence of a free amine at the 2-position would likely render it unstable or at least very synthetically challenging.
An alternative approach is to use suitably substituted cyclitol epoxides or aziridines of the cyclophellitol family. Cyclophellitol is an epoxide-containing natural product that functions as a time-dependent covalent mechanism-based inhibitor of β-glucosidases9 (Fig. 1A). Many epimers of this molecule have been synthesised and shown, in most cases, to function as covalent inhibitors of their cognate glycosidases, including α-glycosidases. The equivalent aziridines (not yet known as natural products) have also been synthesised and shown to be similarly, indeed often more, potent as covalent inhibitors. However inhibitors of this general class will not form covalent bonds with glycosidases employing a neighbouring group assistance mechanism, such as those of GH20.10 Considerable work has been performed on these classes of molecules, primarily by the Overkleeft group in conjunction with the group of Davies. References to this work are found in the recent review.11
 | ||
| Fig. 1 Examples of (A) 1,5-cyclophellitol and (B) 1,2-aza-α-cyclophellitol acting as mechanism-based inhibitors of, respectively, retaining β- and α-glycosidases. | ||
The epoxide or aziridine in these “cyclophellitols” is located across carbons equivalent to C1 and O5 in the sugar being mimicked. We questioned whether, for our enzyme, a particularly effective inactivator might be an “α”-aziridine that bridged the carbons equivalent to C1 and C2 of galactosamine. In that case the nucleophilic carboxylate of the enzyme should attack C1 in the normal manner, and the amine moiety would end up coordinated by those residues ordinarily ligating the normal substrate amine (Fig. 1B). Compounds of this class in the gluco configuration have been synthesised previously (Shing,12 Overkleeft13) and generally found to be poorer inhibitors. Disaccharide versions containing α- and β-configured epoxides and aziridines were also synthesised as part of a mechanistic investigation of a GH99 endo-mannosidase, providing valuable support for an epoxide intermediate in the enzymatic reaction.14,15 However, no such studies had been performed with hexosaminidases which, as noted above, would seem to be particularly suitable target enzymes for this aziridine class of reagent. In this study we synthesise a full set of galacto configured “α” and “β”-1,2-epoxides and aziridines and explore their potential as inactivators of an α-galactosaminidase and a series of α- and β-galactosidases.
Results and discussion
Synthetic procedures
The most logical route for synthesis of the 1,2-epoxide and aziridine analogues of galacto-configured “cyclophellitol” involves preparation of the alkene (7) shown below as a key intermediate. Various routes to this compound class are published. The Shing synthesis accessed the alkene in 19% yield via a ten-step route from quinic acid.8 Overkleeft, on the other hand, used an eight-step route involving Claisen rearrangement of a protected glycal.9 A related Claisen rearrangement route was used for the disaccharide inhibitor by the Sollogoub group.14 We chose to access the “galacto” version through an asymmetric Diels Alder approach developed by Nishikawa.16 In this way (Scheme 1), the key intermediate (7) was synthesized in seven steps in 13% overall yield.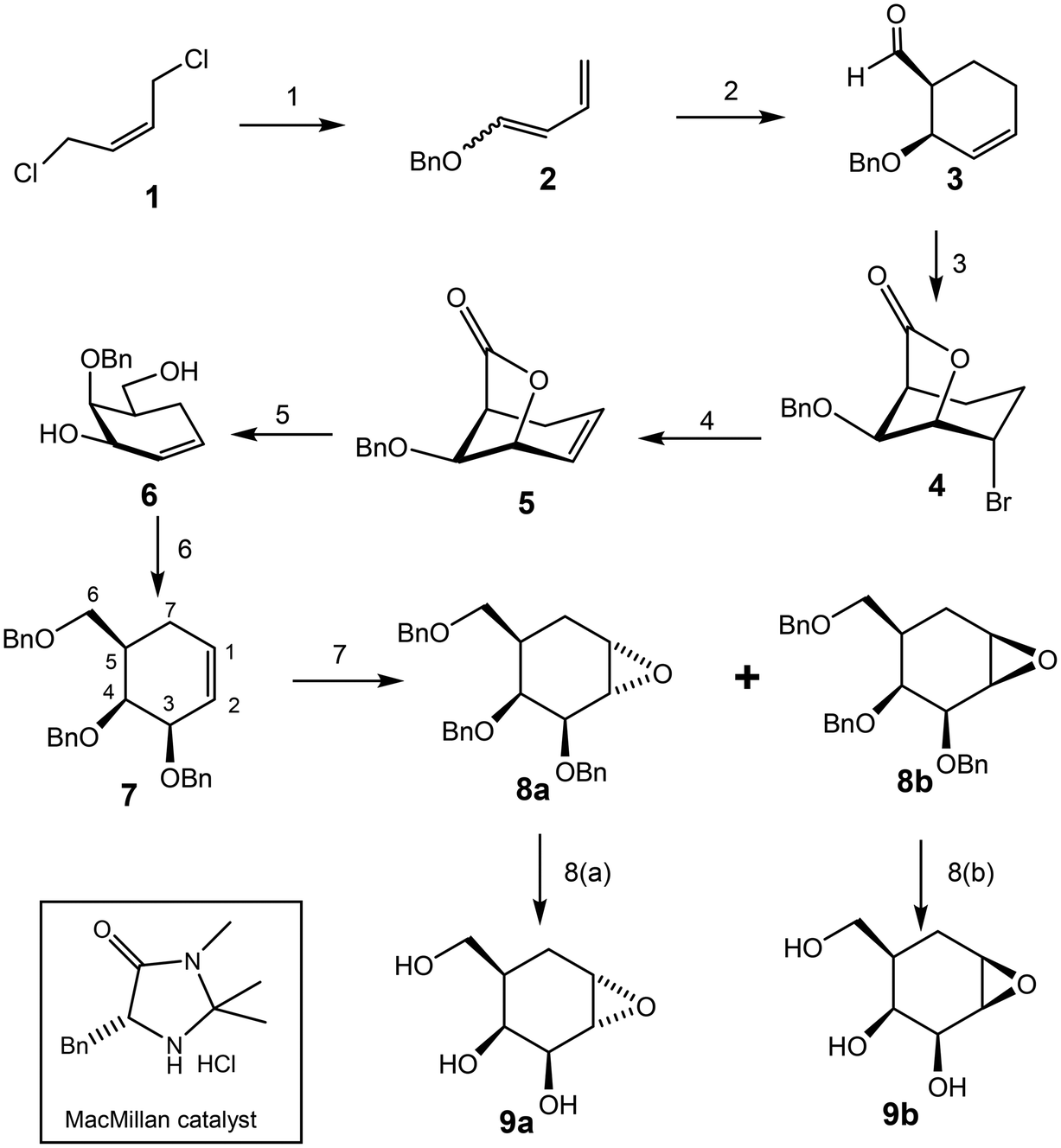 | ||
| Scheme 1 Synthesis of epoxides (9a & 9b) Reagents and conditions: (1) DMSO, 4,4′-thiobis(2-tert-butyl-5-methylphenol), NaH, BnOH, rt-50 °C, 82%. (2) MeOH, H2O, Acrolein, MacMillan catalyst, −10 °C, 22 h, 38%. (3) DMF, Oxone, rt; DCM, NaHCO3, NBS, rt, 63%. (4) THF, DBU, 50 °C, 86%. (5) Et2O, LiAlH4, −78 to −20 °C, 92%. (6) DMF, NaH, BnBr, 0 °C–rt, 92%. (7). DCM, m-CPBA, 0 °C-rt, 8a: 44%; 8b: 50%. (8(a)) MeOH/EtOAc = 5/1, Pd/C (10%), H2, rt, 24 h, 58%. (8(b)). MeOH/H2O/dioxane = 1/1/1, Pd(OH)2/C (20% wet), H2, rt, 2 h, 52%. | ||
Epoxidation of the double bond in 7 was performed with m-CPBA in DCM, affording “α”-epoxide 8a in 44% and “β” epoxide 8b in 50% yield, respectively.13 At this stage, it was very difficult to determine the configurations of the two anomers by NMR since the chemical shifts, coupling constants and NOEs are very similar for the two compounds thus we continued the synthesis with each compound separately expecting to assign configurations later. Deprotection of the sample comprising compound 8a was achieved by treatment with Pd/C, affording 9a in 58% yield, along with by-products of the over-reduced triol and the methyl ether formed upon attack on the epoxide by methanol. By contrast, only the over-reduced by-products were detected when compound 8b was treated under the same conditions. In order to access product 9b the reduction was performed using Pearlman's catalyst Pd(OH)2/C in a mixed solvent system (MeOH/H2O/dioxane), leading to the product with comparable yields to that of the other isomer.17
Azidolysis of the benzyl-protected α-epoxide 8a (Scheme 2) was achieved by stirring the compound with sodium azide for 48 h at 100 °C, affording 10a as the major product in 55% yield along with 10a′ as the minor product in 9% yield. Treatment of β-epoxide 8b with sodium azide under similar conditions (stirring for 22 h at 100 °C), afforded 10b in 92% yield.17,18 Attack of azide on the α-epoxide 8a necessarily occurred from the top face, which is very hindered. Indeed hindrance was such that ring opening occurred in a trans di-equatorial fashion via attack at C1 rather than di-axial through attack at C-2. By contrast attack of azide on the “β”-epoxide ring of 8b occurred at C-1 from the less hindered bottom face resulting in the trans-diaxial compound as the sole significant product. Notably this reaction was complete in a shorter period of time, consistent with the lesser hindrance. These reaction outcomes support the assignments of 8a and 8b.
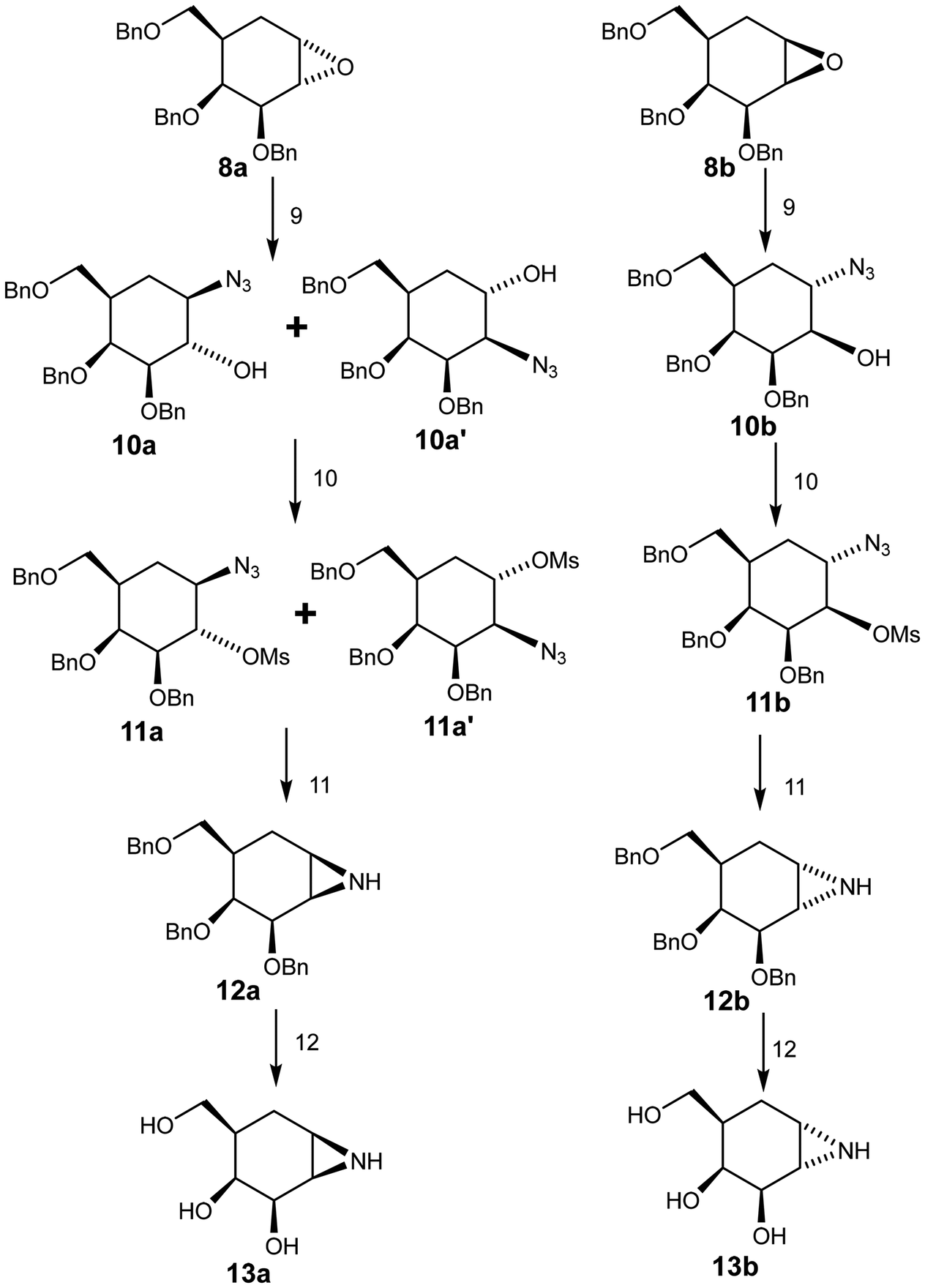 | ||
| Scheme 2 Synthesis of aziridines (13a & 13b) Reagents and conditions: 9. DMF, LiClO4, NaN3, 100 °C, 48 h, 10a: 55%, 10a′: 9%; 10b, 22 h, 92%. 10. THF (dry), MsCl, TEA, rt, 11a: 100%; 11a′: 94%; 11b: 100%. 11. THF (dry), LiAlH4, 0 °C–rt, 12a: 50%; 12b: 84%. 12. NH3, Na, THF (dry), −78 °C; 13a: 84%; 13b: 92%. | ||
Reaction of compounds 10a, 10a′ and 10b with methanesulfonyl chloride afforded the desired products 11a, 11a′ and 11b in good yields.19 The NMR spectra of these compounds provided further support for the assignments of 8a and 8b since the coupling constant J2,3 for 11a was 9.9 Hz (trans) while that for 11b was 2.8 Hz (cis). Selective reduction of the azide in compounds 11a and 11a′ with LiAlH4 followed by ring closure through in situ intramolecular displacement of the amino mesylates gave the same product 12a, the “β” aziridine. Similarly, 11b was converted to the “α” aziridine 12b. Removal of the benzyl protecting group was accomplished using a Birch reduction (Na, NH3) yielding the target aziridines 13a and 13b (Scheme 2), both of which proved to be unstable under neutral conditions. For stable storage it proved necessary to dissolve them in a basic (NaOH or NH4OH) aqueous solution and then lyophilize them.
Inactivation of galactosidases
The epoxides and aziridines synthesised (9a, 9b, 13a, 13b) were tested as inactivators and as reversible inhibitors for a range of α- and β-galactosidases from different families, with the results shown in Table 1 and Fig. S15–S19. Testing for inactivation was carried out by incubating the relevant reagent with each enzyme individually, removing aliquots at time intervals, and assaying for residual enzyme activity using the substrates indicated. To our considerable disappointment, none of the “α”-1,2-epoxides or aziridines functioned as time-dependent inactivators. While it would be tempting to ascribe this to the need for less favourable 1,2-di-equatorial ring opening in these cases, it is important to note that the equivalent “α”-1,5a-epoxides and aziridines, which would also need to open in a di-equatorial fashion, are indeed inactivators of α-glycosidases.11 Other factors must therefore be considered.| Enzyme | Family | Substrate | Epoxide | Aziridine | ||
|---|---|---|---|---|---|---|
| α | β | α | β | |||
| Data for all assays can be found in Fig. 2 and Fig. S15–19. Compounds denoted as “Reversible” inhibitors do not cause any time dependent inactivation, but inhibit the enzyme in a concentration dependent manner, with inhibition constants projected to be higher than 5 mM. | ||||||
| F. plautii α-galactosaminidase (FpGalNase)2 | GH36 | GalN-α-MU | No | No | No | Reversible |
| T. maritima α-galactosidase (TmGalA)4 | GH36 | Gal-α-pNP | No | No | No | Reversible |
| Coffee bean α-galactosidase20 | GH27 | Gal-α-pNP | No | No | No | No |
| Agrobacter sp. β-glucosidase/galactosidase (Abg)21 | GH1 | Glc-β-pNP | No | Covalent | No | Covalent |
| E. coli (LacZ) β-galactosidase22 | GH2 | Gal-β-pNP | No | Covalent | No | Reversible |
Inspired by a recent report by the Overkleeft group on gluco-configured aziridines and epoxides, which they found to adopt an unreactive conformation,13 we consider it most likely that our galacto-configured counterparts also adopt such a conformation. To probe this possibility, we calculated the most stable conformations of the “α”- and “β”-1,2-galacto-configured aziridines at the M062X/def2TZVP level of theory (Fig. S20). The lowest-energy DFT minima, which we refer to as the most stable conformations, correspond to a 4H5 conformation for the “α” galacto-configured aziridine both in its neutral and protonated forms while the “β” galacto-configured aziridine appears to adopt a conformation between 4E and 4H5. These are quite distinct from the 4H3 conformation adopted by the substrate at the glycosylation transition state, and also seen for the 1,5a-aziridines and epoxides.13 Although Ofman et al.13 used metadynamics simulations to compute full free-energy landscapes for the gluco-configured analogues, their lowest-energy regions show the same conformational preferences for the gluco-configured aziridines and epoxides. Their analysis further indicates that these 1,2 configured structures are more rigid and therefore less able to undergo the conformational changes necessary for effective reaction.
By contrast the Agrobacterium sp. β-glucosidase/galactosidase (Abg) was inactivated by both the “β”-1,2-galacto-configured aziridine and epoxide, while the Escherichia coli (LacZ) β-galactosidase was inactivated only by the “β”-1,2-galacto-configured epoxide (Fig. 2).
 | ||
| Fig. 2 Time-dependent inhibition of “β”-1,2-galacto-configured epoxide and aziridine. Residual enzyme activities were measured after incubation with varying concentrations of the compounds for different time periods. During the incubation, protein concentrations were approximately 1.9 nM for Abg (with 0.1 mg mL−1 BSA) and 0.5 nM for LacZ β-galactosidase (with 0.1 mg mL−1 BSA). For activity assays, 200 μM Glc-β-pNP was used as the substrate for Abg in HEPES buffer (pH 7.4), and 100 μM Gal-β-pNP was used as the substrate for LacZ in HEPES buffer (pH 7.4) supplemented with 1 mM Mg2+. | ||
Consistent with the covalent nature of the reactions, inactivation occurred in a time-dependent manner, allowing determination of rate constants for inactivation (kinact) and dissociation constants (Ki) for binding according to the simple kinetic model below.

The formation of a covalent linkage was confirmed for Abg by intact mass electrospray ionisation mass spectrometry as shown in Fig. 3.
 | ||
| Fig. 3 Intact protein mass spectrometry of inactivated Abg. Observed increases in mass (Da) relative to apo enzyme are labeled on figures. Expected mass increases = 159 (aziridine), 160 (epoxide). | ||
Conclusions
In summary, although the 1,2-configured cyclophellitol analogues are highly attractive candidates as mechanism-based inactivators of retaining glycosidases, no inactivation of α-galactosidases by the α-configured derivatives was seen. More unexpectedly, the α-configured aziridine analogue also failed to inhibit the α-galactosaminidase. Computational studies suggest that this is due to an unfavourable conformation imposed by the three membered rings, that makes binding in the enzyme active site energetically unfavorable. However, time-dependent, covalent inhibition of β-galactosidases by the β-configured analogues is shown by mass spectrometric analysis and kinetic studies. Very similar findings were reported recently for the equivalent gluco-configured versions, highlighting the importance of ring conformation to ligand binding.11,13Experimental
Synthesis and characterization of compounds
Inhibition kinetics determination
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 30 min, 4 °C), the supernatant was loaded onto a nickel affinity column. Bound proteins were eluted using a gradient of 10–100% elution buffer (50 mM HEPES, 150 mM NaCl, 400 mM imidazole; pH 7.4), and fractions were analyzed by SDS-PAGE. Imidazole was removed and the buffer exchanged to the appropriate assay buffer using Amicon Ultra centrifugal filters (MWCO 30 kDa or 10 kDa, Millipore). α-galactosidase from green coffee beans was purchased from Sigma-Aldrich.
000 rpm, 30 min, 4 °C), the supernatant was loaded onto a nickel affinity column. Bound proteins were eluted using a gradient of 10–100% elution buffer (50 mM HEPES, 150 mM NaCl, 400 mM imidazole; pH 7.4), and fractions were analyzed by SDS-PAGE. Imidazole was removed and the buffer exchanged to the appropriate assay buffer using Amicon Ultra centrifugal filters (MWCO 30 kDa or 10 kDa, Millipore). α-galactosidase from green coffee beans was purchased from Sigma-Aldrich.For each inhibitor, sets of 85 μL PCR tubes were prepared containing the inhibitor at 2× the final assay concentration in the appropriate assay buffer. In parallel, another set of PCR tubes was prepared with the enzyme solution at 2× the final assay concentration in the same buffer. All tubes were pre-warmed at 25 °C for 10 minutes. Prior to initiating the inhibition reaction, substrate mixtures were prepared in a half-area 96-well plate, black plate for methylumbelliferyl (MU) substrate or clear plate for para-nitrophenyl (pNP) substrate. Each well contained 80 μL of substrate at 1.25× the final assay concentration in the appropriate buffer. The plate was pre-warmed at 25 °C for 5 minutes.
To initiate the reaction (t = 0), 85 μL of enzyme solution was added to 85 μL of inhibitor solution, mixed thoroughly (yielding a 170 μL mixture), and immediately, 20 μL of this mixture was transferred into 80 μL of substrate solution in the plate. The release of pNP or MU was monitored in real time for 5 minutes using a BioTek Synergy H1 plate reader. The initial reaction rate (V) was calculated from the linear portion of the curve. At subsequent incubation time points, additional 20 μL aliquots were taken from the enzyme-inhibitor mixture and added to fresh wells containing 80 μL of pre-warmed 1.25× substrate solution. All reaction rates were normalized to the relative activity of the t = 0 control sample to which no additional inhibitor compound was added. The inhibitor concentrations reported in the result figures refer to the concentrations during the 170 μL incubation phase in PCR tubes. When 20 μL of the enzyme-inhibitor mixture was added into 80 μL of substrate solution, the inhibitor concentration was 5 times diluted. All assays were performed in duplicate.
Conflicts of interest
There are no conflicts to declare.Data availability
Supplementary information (SI): cartesian coordinates of the lowest-energy structures and full NMR spectra. See DOI: https://doi.org/10.1039/d5ob01802a.Acknowledgements
S. G. W. thanks the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Canadian Glycomics Network, GlycoNet, for financial support. Financial support to J. P. R. was provided by the University of British Columbia, the Natural Sciences and Engineering Research Council of Canada (NSERC) and the CFI John R. Evans Leaders Fund. Computational resources were provided from the Digital Research Alliance of Canada and the Advanced Research Computing (ARC) center at the University of British Columbia. We thank Dr Shinya Fushinobu for providing access to his Cremer-Pople parameters calculator (https://enzyme13.bt.a.u-tokyo.ac.jp/CP/).References
- P. Rahfeld, L. Sim, H. Moon, I. Constantinescu, C. Morgan-Lang, S. J. Hallam, J. N. Kizhakkedathu and S. G. Withers, Nat. Microbiol., 2019, 4, 1475–1485 Search PubMed.
- Y. Tian, L. J. Worrall, L. Sim, F. Liu, S. A. Nasseri, P. Rahfeld, W. Mu, J. N. Kizhakkedathu, N. C. J. Strynadka and S. G. Withers, ACS Catal., 2024, 14, 13497–13508 Search PubMed.
- F. Fredslund, M. A. Hachem, R. J. Larsen, P. G. Sorensen, P. M. Coutinho, L. Lo Leggio and B. Svensson, J. Mol. Biol., 2011, 412, 466–480 Search PubMed.
- D. A. Comfort, K. S. Bobrov, D. R. Ivanen, K. A. Shabalin, J. M. Harris, A. A. Kulminskaya, H. Brumer and R. M. Kelly, Biochemistry, 2007, 46, 3319–3330 Search PubMed.
- I. P. Street, J. B. Kempton and S. G. Withers, Biochemistry, 1992, 31, 9970–9978 Search PubMed.
- R. M. Mosi and S. G. Withers, in Methods Enzymol, ed. D. L. Purich, Academic Press, 2002, vol. 354, pp. 64–84 Search PubMed.
- J. D. McCarter and S. G. Withers, J. Am. Chem. Soc., 1996, 118, 241–242 Search PubMed.
- D. J. Vocadlo, C. Mayer, S. M. He and S. G. Withers, Biochemistry, 2000, 39, 117–126 Search PubMed.
- S. G. Withers and K. Umezawa, Biochem. Biophys. Res. Commun., 1991, 177, 532–537 Search PubMed.
- D. J. Vocadlo and G. J. Davies, Curr. Opin. Chem. Biol., 2008, 12, 539–555 Search PubMed.
- M. Artola, J. Aerts, G. A. van der Marel, C. Rovira, J. D. C. Codee, G. J. Davies and H. S. Overkleeft, J. Am. Chem. Soc., 2024, 146, 24729–24741 Search PubMed.
- V. W.-F. Tai, P. H. Fung, Y. S. Wong and T. K. M. Shing, Tetrahedron: Asymmetry, 1994, 5, 1353–1362 Search PubMed.
- T. P. Ofman, J. J. A. Heming, A. Nin-Hill, F. Küllmer, E. Moran, M. Bennett, R. Steneker, A.-M. Klein, G. Ruijgrok, K. Kok, Z. W. B. Armstrong, J. M. F. G. Aerts, G. A. van der Marel, C. Rovira, G. J. Davies, M. Artola, J. D. C. Codée and H. S. Overkleeft, Chem. – Eur. J., 2024, 30, e202400723 Search PubMed.
- D. Lu, S. Zhu, L. F. Sobala, G. Bernardo-Seisdedos, O. Milet, Y. M. Zhang, J. Jiménez-Barbero, G. J. Davies and M. Sollogoub, Org. Lett., 2018, 20, 7488–7492 Search PubMed.
- L. F. Sobala, G. Speciale, S. Zhu, L. Raich, N. Sannikova, A. J. Thompson, Z. Hakki, D. Lu, S. S. K. Abadi, A. R. Lewis, V. Rojas-Cervellera, G. Bernardo-Seisdedos, Y. M. Zhang, O. Millet, J. Jiménez-Barbero, A. J. Bennet, M. Sollogoub, C. Rovira, G. J. Davies and S. J. Williams, ACS Cent. Sci., 2020, 6, 760–770 Search PubMed.
- N. Ushida, N. Nagai, M. Adachi and T. Nishikawa, Synlett, 2019, 30, 977–981 Search PubMed.
- S. P. Schröder, R. Petracca, H. Minnee, M. Artola, J. M. F. G. Aerts, J. D. C. Codée, G. A. van der Marel and H. S. Overkleeft, Eur. J. Org. Chem., 2016, 4787–4794 Search PubMed.
- D. Lu, S. Zhu, L. F. Sobala, G. Bernardo-Seisdedos, O. Millet, Y. Zhang, J. Jiménez-Barbero, G. J. Davies and M. Sollogoub, Org. Lett., 2018, 20, 7488–7492 Search PubMed.
- P. Serrano, A. Llebaria and A. Delgado, J. Org. Chem., 2005, 70, 7829–7840 Search PubMed.
- A. Zhu, Z.-K. Wang and J. Goldstein, Biochim. Biophys. Acta, 1995, 1247, 260–264 Search PubMed.
- J. B. Kempton and S. G. Withers, Biochemistry, 1992, 31, 9961–9969 Search PubMed.
- D. H. Juers, T. D. Heightman, A. Vasella, J. D. McCarter, L. Mackenzie, S. G. Withers and B. W. Matthews, Biochemistry, 2001, 40, 14781–14794 Search PubMed.
- F. W. Studier, Protein Expression Purif., 2005, 41, 207–234 Search PubMed.
| This journal is © The Royal Society of Chemistry 2026 |