
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReductive deoxygenation of tertiary alcohols: a cobalt-catalyzed hydrosilylation approach
Swetarani
Meher
,
Ratiraman
Swain
,
Braja Kishore
Marndi
and
Bidraha
Bagh
 *
*
School of Chemical Sciences, National Institute of Science Education and Research (NISER), An OCC of Homi Bhabha National Institute, Bhubaneswar, Jatni, Khurda, Odisha 752050, India. E-mail: bidraha@niser.ac.in
First published on 8th December 2025
Abstract
Dehydrogenation of tertiary alcohols is both highly challenging and of notable importance in synthetic chemistry. We report a readily available Co2(CO)8 catalyzed hydrosilylation protocol for the reductive deoxygenation of tertiary alcohols involving a radical mechanism. Successful deoxygenation of various tertiary, secondary and primary alcohols demonstrated the broad applicability, including syntheses of fine chemicals. A plausible mechanistic path was proposed based on several control experiments.
Introduction
Deoxygenation of alcohols to alkanes is a key transformation for producing more stable, energy-dense, and versatile hydrocarbons, which are used as fuels, materials, and chemical intermediates.1 It is an important industrial process, with applications ranging from biofuel production to synthetic chemistry and pharmaceutical applications. However, reductive deoxygenation of alcohols presents significant challenges due to the high bond dissociation energy of the C–OH bond and the poor leaving ability of the hydroxyl group.2 Alcohol deoxygenation methods can be single-step and two-step strategies. The Barton–McCombie reaction, a classic method, involves a two-step process: first, conversion of the hydroxyl group into a better leaving group, followed by reduction with a stoichiometric amount of highly toxic Bu3SnH.3 Although modifications have been made, this two-step procedure still limits broader applicability. The search for sustainable and environmentally benign deoxygenation of alcohols has primarily focused on base metal catalysts4 and silanes as hydrogen sources.5 In 1988, the Chatgilialoglu group used (Me3Si)3SiH as a free radical hydride donor, replacing Bu3SnH with hydrosilane, a non-toxic hydrogen transfer reagent.6a In general, hydrosilanes offer advantages such as low toxicity, cost efficiency, and tunability through substituents on silicon.6 In 1997, the Lopez group reported the first deoxygenation using silanes in a catalytic variant of the Barton–McCombie reaction, although it required organotin compounds and alcohol prefunctionalization.7 This led to further exploration of silanes as hydride sources in catalytic systems with sustainable base metals. Titanium has been the most extensively investigated, with numerous titanocene-mediated catalytic systems utilized for the reductive deoxygenation of alcohols.8 In this context of using sustainable base metals, only a few research groups have reported progress. In 2014, Dang et al. employed a Cu-catalyst with Me3SiOSiMe3 as a hydrogen donor.9 However, this process proceeded via a two-step pathway involving a triflate intermediate. In 2017, Bauer et al. used a Mn–PNP pincer complex for this dehydrogenation, followed by Wolff–Kishner reduction utilizing hydrazine.10 However, this approach has limitations, as hydrazine is a toxic and hazardous reagent and it requires prolonged reaction times. In 2021, the Newman group reported an excellent work on the deoxygenation of various C–O containing functional groups using NiBr2·diglyme and Me2HSiOSiHMe2.11 However, this method required high catalyst loading and a stoichiometric amount of a base. Recently, the Gunanathan group used a Co–NNN pincer complex for the reductive deoxygenation of alcohols using Et2SiH2.12 However, this method requires a strong base and was mostly effective for reducing primary alcohols.Base metal catalysts with improved efficiency need to be developed for the hydrosilylative deoxygenation of alcohols. Cobalt is an important base metal, which is much cheaper and less toxic than its heavier analogues Rh and Ir (widely used as a catalyst). Along with other base metals, Co-catalysts have been extensively used in the hydrosilylation of various substrates such as alkynes, alkenes, allenes, amides, nitriles, aldehydes, ketones and esters (Scheme 1).13 Notably, carbonyl and carboxyl groups were reduced to the corresponding alcohols; further reduction/deoxygenation was not achieved. In the context of Co-catalyzed hydrosilylations, Co2(CO)8 is particularly important (Scheme 1). The application of Co2(CO)8 in the hydrosilylation of diverse organic substrates such as alkenes, alkynes, nitriles and α,β-unsaturated esters is well established.14 Importantly, Co2(CO)8 was used for the hydrosilylative deoxygenation of amides to amines.14e We previously reported the efficient use of Co2(CO)8 for the deoxygenation of nitroarenes to aromatic amines using PhSiH3 as a reducing agent.15 However, hydrosilylative deoxygenation of alcohols to alkanes using Co2(CO)8 is not documented. With regard to the deoxygenation of alcohols, reduction of tertiary alcohols to alkanes is both highly challenging and of notable importance. Herein, we report Co2(CO)8 catalyzed hydrosilylative deoxygenation of tertiary alcohols to the corresponding alkanes (Scheme 1).
 | ||
| Scheme 1 Base metal catalyzed hydrosilylation and hydrosilylative deoxygenation. | ||
Results and discussion
Unlike primary and secondary alcohols, tertiary alcohols lack a hydrogen atom on the carbon bearing the hydroxyl group, making conventional β-hydride elimination pathways inaccessible. Moreover, tertiary alcohols are generally more sterically hindered, which can impede substrate–catalyst interactions. This structural limitation necessitates alternative reaction mechanisms, such as radical or hydride transfer processes. Thermodynamically, the transformation is often uphill, as alkanes are more reduced than alcohols, necessitating the use of potent reducing agents such as hydrosilanes under metal catalysis. Motivated by the successful of use of Co2(CO)8 in various hydrosilylations, particularly in the challenging hydrosilylative deoxygenation of nitroarenes to anilines,15 we selected Co2(CO)8 as a potential catalyst for the hydrosilylative deoxygenation of tertiary alcohols. In the presence of heat or light, Co2(CO)8 generates a reactive Co(CO)4 radical, which might be well suited for the present purpose. Guided by this rationale, Co2(CO)8 was employed as a readily available catalyst for the hydrosilylative deoxygenation of 1,1-diphenylethanol (A1) as the standard substrate and PhSiH3 as the reducing agent (Table 1). Deoxygenation of tertiary alcohol A1 was tested by using two equivalents of PhSiH3 and 5 mol% of Co2(CO)8 at 120 °C in THF as the solvent and the reaction was run for 24 h (entry 1). We observed complete conversion of A1 to the desired deoxygenated product 2,2-diphenylethane (B1). When the catalyst loading was reduced to 3 mol%, complete deoxygenation of A1 to B1 (with an excellent isolated yield of 93%) was also achieved (entry 2). Further reduction of the catalyst loading to 2 mol% did not lead to complete deoxygenation of A1 and a significant amount of unreacted A1 was noted (entry 3). When the reaction time was reduced to 18 h, almost complete conversion of A1 to B1 was observed under 3 mol% catalyst loading (entry 4). However, a very small amount of A1 was left unreacted. Similarly, a small amount of unreacted A1 was also noted when the reaction was carried out at 100 °C for 24 h under 3 mol% catalyst loading (entry 6). Finally, we tested various silanes such as Ph2SiH2, Et3SiH, PHMS and TMDS; however, hydrosilylative deoxygenation of A1 did not reach completion (entries 7–10). We also tested different solvents without any improvement in the outcome (entry 11). Therefore, the conditions shown in entry 2 (in bold) were identified as the optimal reaction conditions for the hydrosilylative deoxygenation of A1 and a gram-scale reaction was also performed.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Co2(CO)8 (mol%) | Silane (2 eq.) | Temp. (°C) | Time (h) | Solv. | Yieldb (%) |
| a Reactions conducted with 0.5 mmol of A1, 1.0 mmol of silane and 2–5 mol% of Co2(CO)8 in 2 mL of solvent. b Yields were determined by GC using p-xylene as the internal standard. c Isolated yields. | ||||||
| 1 | 5/4 | PhSiH3 | 120 | 24 | THF | >99 |
| 2 | 3 | PhSiH 3 | 120 | 24 | THF | >99 (93 ) |
| 3 | 2 | PhSiH3 | 120 | 24 | THF | 84 |
| 4 | 3 | PhSiH3 | 120 | 18 | THF | 98 |
| 5 | 3 | PhSiH3 | 120 | 12 | THF | 90 |
| 6 | 3 | PhSiH3 | 100 | 24 | THF | 87 |
| 7 | 3 | Ph2SiH2 | 120 | 24 | THF | 78 |
| 8 | 3 | Et3SiH | 120 | 24 | THF | 80 |
| 9 | 3 | PHMS | 120 | 24 | THF | 36 |
| 10 | 3 | TMDS | 120 | 24 | THF | 42 |
| 11 | 3 | PhSiH3 | 120 | 24 | Benzene | 89 |
| 12 | 3 | PhSiH3 | 120 | 24 | Toluene | 75 |
| 13 | 3 | PhSiH3 | 120 | 24 | MeCN | 83 |
| 14 | 3 | PhSiH3 | 120 | 24 | Anisole | 90 |
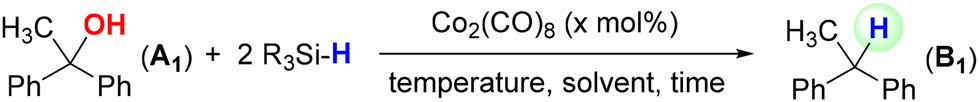
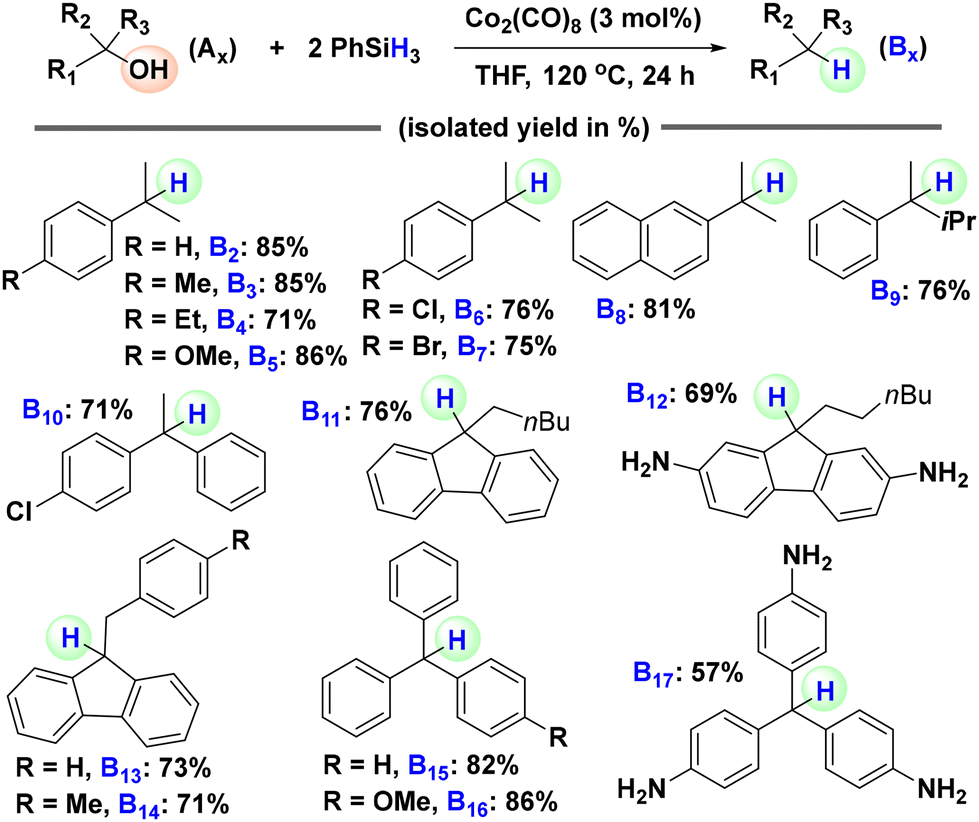
With the optimized conditions for the hydrosilylative deoxygenation of 1,1-diphenylethanol in hand, we set out to explore the generality of this reduction protocol by applying it to the deoxygenation of various tertiary alcohols (Scheme 2). We started with 2-phenylpropan-2-ol and its derivatives with various electron-donating and electron-withdrawing functionalities attached to the phenyl ring. 2-Phenylpropan-2-ol was effectively reduced to the corresponding alkane, cumene (B2: 85%), with a good isolated yield. The derivatives having electron-donating groups such as methyl, ethyl and methoxy were also subjected to the optimized reaction conditions and efficient deoxygenations to the corresponding alkanes (B3: 85%, B4: 71%, and B5: 86%) were observed. o-Amino substituted 2-phenylpropan-2-ol was not compatible. Analogous compounds with weak electron-withdrawing groups such as chloro and bromo gave the corresponding alkanes (B6: 76% and B7: 75%) in decent yields. Then, we tested bulkier tertiary alcohols such as 2-(2-naphthyl)-2-propanol and 2-phenylpentan-2-ol and they were also effectively reduced to 2-isopropylnaphthalene (B8: 81%) and pentan-2-ylbenzene (B9: 76%), respectively. Similar to the hydrosilylation of the model substrate 1,1-diphenylethanol (A1), the hydrosilylation of 2-(4-chlorophenyl)-2-phenylethanol gave the desired product 2-(4-chlorophenyl)-2-phenylethane (B10: 71%) in a decent yield. Subsequently, we turned our attention to structurally more complex alcohols, focusing on cases where the deoxygenated products have potential practical applications. Four 9-alkyl-9-fluorenol with various functionalities were subjected to the optimized hydrosilylation conditions. All four were effectively deoxygenated to the corresponding 9-alkyl-9H-fluorenes (B11: 76%, B12: 69%, B13: 73%, and B14: 71%) with potential applications in pharmaceuticals, optoelectronics, semiconductors, and solar cells.16 Finally, we examined triphenylmethanol and two substituted derivatives. Under the optimized conditions, all of them were reduced to the corresponding triphenylmethane derivatives (B15: 82%, B16: 86%, and B17: 57%) with functional relevance. Leucopararosaniline (B18), in particular, finds diverse applications such as use as a radiochromic dye, an HCV helicase inhibitor, and a synthetic precursor for 3D COFs and polyisocyanate adhesives.17
 | ||
| Scheme 2 Hydrosilylative deoxygenation of tertiary alcohols. | ||
Successful hydrosilylative deoxygenation of tertiary alcohols motivated us to turn our attention to relatively easier primary and secondary alcohols. We initiated the reaction optimization for the reduction of primary alcohols using 4-methoxybenzyl alcohol (C1) as the model substrate (see the SI for details). The optimized reduction conditions for the deoxygenation of the primary alcohol C1 were as follows: 2 eq. of PhSiH3, 1 mol% of Co2(CO)8, THF, 40 °C, and 4 h. Similarly, the optimized conditions for the dehydrogenation of secondary alcohols were as follows: 2 eq. of PhSiH3, 1 mol% of Co2(CO)8, THF, 70 °C, and 12 h (see the SI for details). Next, we aimed to expand the substrate scope for both primary and secondary alcohols (Scheme 3). First, we targeted primary alcohols. Various substituted benzyl alcohols with electron-donating groups were deoxygenated to the corresponding toluene derivatives (D2–D12) in good yields. More complex alcohols such as 9-anthracenemethanol and 1-pyrenemethanol were also easily reduced to 9-methylanthracene (D13) and 1-methylpyrene (D14), respectively, commonly used as photoluminescent materials and OLEDs. A longer reaction time (24 h) and higher temperature (120 °C) were required for the reduction of benzyl alcohols with electron-withdrawing groups to the corresponding products (D15–D21). Similarly, 9-flurenylmethanol was reduced to 9-methyl-9H-fluorene, which is used in optoelectronics. Then, we tested a few secondary alcohols, starting with 1-phenylethanol and its substituted derivatives. They were reduced to the corresponding ethylbenzene derivatives (F1–F5) in decent to good yields. Diphenylmethanol (F6), fluorenol (F7) and 1-tetralol (F8) were also successfully deoxygenated. Finally, octopamine hydrochloride was deoxygenated to tyramine (F9), a naturally occurring trace monoamine that helps regulate blood pressure.
 | ||
| Scheme 3 Hydrosilylative deoxygenation of primary and secondary alcohols. | ||
The present hydrosilylative deoxygenation protocols can tolerate halide, phenolic, alkoxy and amine functional groups. We wanted to check if this hydrosilylation is selective in the presence of reducible functional groups. Therefore, several alcohols having reducible functionalities such as aldehyde, ketone, alkene, cyano and amide groups were subjected to cobalt catalyzed hydrosilylation in the presence of excess phenylsilane (Scheme 4). First, primary and secondary alcohols with aldehyde, ketone and amide groups were tested. We did not observe any chemoselectivity; the aldehyde, ketone and amide groups were also reduced along with the deoxygenation of the alcohol moieties and the completely reduced products (D6, D23, D23, F10 and F11) were isolated in decent yields. Primary and secondary alcohols with alkene and cyano groups gave a complex mixture of products that could not be separated. We also tested challenging aliphatic alcohols (Scheme 4). When subjected to cobalt catalyzed hydrosilylation, partial deoxygenation (40–70%) of the aliphatic alcohols was noted and the corresponding reduced products (D25, D26, D27 and F12) were isolated in poor yields.
 | ||
| Scheme 4 Hydrosilylative deoxygenation of aliphatic alcohols and alcohols with reducible groups. | ||
Finally, we investigated the plausible catalytic reaction path (Scheme 5). We carried out some control experiments to gain insight into this hydrosilylative dehydrogenation process (see the SI for details).
 | ||
| Scheme 5 Control experiments and the proposed mechanism for Co-catalyzed hydrosilylative deoxygenation of alcohol. | ||
First, deoxygenation of 4-chlorobenzyl alcohol under the standard optimized conditions was carried out in the absence of Co2(CO)8 (Scheme 5a). No conversion to the desired product D15 was observed, suggesting the active role of Co2(CO)8 as the catalyst. Next, the reduction of 4-chlorobenzyl alcohol under the optimized conditions was performed in the presence of excess mercury. Successful reduction to the desired product D15 was observed, indicative of a homogeneous pathway (Scheme 5b). Dehydrogenation of 4-chlorobenzyl alcohol under the optimized conditions was also performed in the presence of TEMPO as the radical scavenger (Scheme 5c). Extremely poor reduction (<5%) of the substrate was noted, hinting at a likely radical pathway for this process. The corresponding benzylic radical was trapped by TEMPO in this process and the trapped species was identified by mass analysis. To further verify the possibility of a radical pathway, we performed the catalytic hydrosilylative deoxygenation of 4-chlorobenzyl alcohol in the presence of 5,5-dimethyl-1-pyrroline N-oxide (DMPO), a well-known radical trapping agent. When a reaction proceeds through a radical route involving transient radical intermediates, DMPO can capture these species to generate a DMPO–radical adduct, which can be detected by EPR spectroscopy (Scheme 5d).18 This method has previously been employed to confirm radical mechanisms in various reactions.19 In our study, the cobalt-catalyzed hydrosilylative deoxygenation of 4-chlorobenzyl alcohol conducted with DMPO produced a distinct EPR signal corresponding to the DMPO–radical adduct, providing strong evidence for a radical pathway in this transformation. TM catalyzed hydrosilylations often generate metal-hydrides as reactive intermediates. The formation of (CO)4CoH from Co2(CO)8 in hydrosilylations is well known.18 To check this possibility, we performed a stoichiometric reaction of Co2(CO)8 with PhSiH3 in the absence of an alcohol substrate in THF-d8 (Scheme 5e). 1H NMR measurement of the reaction mixture showed the presence of cobalt-hydride resonance at −11.21 ppm. Next, we performed a deuterium labelling test with deuterated silane (Et3SiD, ca. 90% D) to confirm hydride transfer in this hydrosilylative deoxygenation (Scheme 5f). The deoxygenation of 9-anthracenemethanol to high deuterium enriched 9-methylanthracene provides direct evidence for hydride transfer in the present hydrosilylative deoxygenation. Based on the control experiments and previous reports,13–15 we proposed a plausible reaction pathway that begins with the formation of two Co(CO)4 radicals by homolytic bond cleavage of the Co–Co bond in Co2(CO)8 (Scheme 5g). Then Co(CO)4 radicals react with silane to form the cobalt–hydride and cobalt–silyl species X and Y, respectively. The cobalt-silyl intermediate Y reacts with alcohol to form silanol and the carbon-centered radical Z and regenerates a Co(CO)4 radical. The transfer of the hydrogen radical from the cobalt–hydride intermediate X to the carbon-centered radical Z yields the expected deoxygenated alkane product with the regeneration of the other Co(CO)4 radical.
Conclusions
In conclusion, we have developed an efficient Co-catalyzed hydrosilylation protocol for the reductive deoxygenation of alcohols to alkanes using readily available Co2(CO)8. This method is effective for tertiary alcohols, offering a rare yet highly selective transformation under additive-free conditions. This catalytic reaction also accommodates a broad range of primary and secondary alcohols, highlighting the versatility and generality of the protocol. The reaction proceeds under relatively mild conditions, exhibits good functional group tolerance, and delivers high yields of the corresponding alkanes. Several fine chemicals with applications in materials science have also been synthesized. Mechanistic studies, including radical trapping and spectroscopic evidence, suggest a plausible pathway involving cobalt-centered radicals and hydride species. Operational simplicity, scalability, and use of an Earth-abundant base metal catalyst make this strategy an attractive alternative to traditional deoxygenation approaches that often rely on prefunctionalized substrates or toxic reagents. This work not only provides a valuable tool for synthetic organic chemistry but also contributes to the growing field of sustainable base metal catalysis.Experimental
General experimental procedure for the deoxygenation of tertiary alcohols
In a dried pressure tube fitted with a magnetic stir bar, Co2(CO)8 (3–5 mol%), 1,1-diphenyl ethanol (0.099 g, 0.50 mmol), silane (1.00 mmol) and solvent (2 mL) were added successively under a nitrogen atmosphere. The reaction mixture was heated at the appropriate temperature (100/120 °C) in a preheated oil bath for 12 to 24 h. After cooling to r.t., p-xylene (62 µL, 0.50 mmol) was added to the resultant mixture. The mixture was then analyzed by GC to determine the conversion of 1,1-diphenyl ethanol to 1,1-diphenylethane. Thereafter, water (3 mL) was added to the mixture and the mixture was extracted with Et2O (3 × 5 mL). The organic layer was collected and dried over anhydrous Na2SO4. Thereafter, all volatiles were removed to give a crude product. When necessary, the crude product was purified by column chromatography using silica gel as the stationary phase and a mixture of hexane and ethyl acetate as the eluent.General experimental procedure for the deoxygenation of primary alcohols
In a dried pressure tube fitted with a magnetic stir bar, Co2(CO)8 (1/2/3 mol%), 4-methoxy benzylic alcohol (0.068 g, 0.50 mmol), silane (1.00 mmol) and solvent (2 mL) were added successively under a nitrogen atmosphere. The reaction mixture was heated at the appropriate temperature (40–120 °C) in a preheated oil bath for 4 to 24 h. After cooling to r.t., p-xylene (62 µL, 0.50 mmol) was added to the resultant mixture. The mixture was then analyzed by GC to determine the conversion of 4-methoxy benzylic alcohol to 4-methylanisole. Thereafter, water (3 mL) was added to the mixture and the mixture was extracted with Et2O (3 × 5 mL). The organic layer was collected and dried over anhydrous Na2SO4.Thereafter, all volatiles were removed to give a crude product. When necessary, the crude product was purified by column chromatography using silica gel as the stationary phase and a mixture of hexane and ethyl acetate as the eluent.General experimental procedure for the deoxygenation of secondary alcohols
In a dried pressure tube fitted with a magnetic stir bar, Co2(CO)8 (1/2/3 mol%), 4-(1-hydroxyethyl)-2-methoxyphenol (0.076 g, 0.50 mmol), phenylsilane (124 µL, 1.00 mmol) and THF (2 mL) were added successively under a nitrogen atmosphere. The reaction mixture was heated at the appropriate temperature (40–120 °C) in a preheated oil bath for 6 to 24 h. After cooling to r.t., p-xylene (62 µL, 0.50 mmol) was added to the resultant mixture. The mixture was then analyzed by GC to determine the conversion of 4-(1-hydroxyethyl)-2-methoxyphenol to 4-ethyl-2-methoxyphenol. Thereafter, water (3 mL) was added to the mixture and the mixture was extracted with Et2O (3 × 5 mL). The organic layer was collected and dried over anhydrous Na2SO4.Thereafter, all volatiles were removed to give a crude product. When necessary, the crude product was purified by column chromatography using silica gel as the stationary phase and a mixture of hexane and ethyl acetate as the eluent.General conditions for the substrate screening of primary, secondary, and tertiary alcohols
In a dried pressure tube fitted with a magnetic stir bar, Co2(CO)8 (1–5 mol%), alcohol substrates (0.50 mmol), phenylsilane (124 µL, 1.00 mmol) and THF (2 mL) were added successively under a nitrogen atmosphere. The reaction mixture was heated at 40–120 °C in a preheated oil bath for 4 to 24 h. After cooling to r.t., water (3 mL) was added to the mixture and the mixture was extracted with Et2O (3 × 5 mL). The organic layer was collected and dried over anhydrous Na2SO4. Thereafter, all volatiles were removed to give a crude product. The crude product was purified by column chromatography using silica gel as the stationary phase and a mixture of hexane and ethyl acetate as the eluent. When necessary, in the case of volatile alkane products, the 1H NMR yield was used to calculate the yield using 1,3,5-trimethoxybenzene as the standard (1/3 equiv.).Gram-scale synthesis of B1
In a dried pressure tube fitted with a magnetic stir bar, Co2(CO)8 (3 mol%), 1,1-diphenylethanol (1.98 g, 10 mmol), phenylsilane (2.48 mL, 20 mmol,) and THF (20 mL) were added successively under a nitrogen atmosphere. The reaction mixture was heated at 120 °C in a preheated oil bath for 24 h. After cooling to r.t., water (30 mL) was added to the reaction mixture and the mixture was extracted with Et2O (3 × 50 ml). The organic layer was collected and dried over anhydrous Na2SO4. Thereafter, all volatiles were removed and the crude product was purified by column chromatography using silica gel as the stationary phase and hexane as the eluent to give pure 1,1-biphenylethane (1.58 g, 87%).Reaction conditions for the deoxygenation of octapamine hydrochloride to tyramine
In a dried pressure tube fitted with a magnetic stir bar, Co2(CO)8 (10 mol%), the corresponding octapamine hydrochloride (0.50 mmol), phenylsilane (124 µL, 1.00 mmol) and THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOH (2 mL; 1:1) were added successively under a nitrogen atmosphere. The reaction mixture was heated at 90 °C in a preheated oil bath for 48 h. Thereafter, volatiles were removed and the crude product was purified by column chromatography using silica gel as the stationary phase and a mixture of DCM/MeOH/diethylamine = 20/1/1 as the eluent.
:EtOH (2 mL; 1:1) were added successively under a nitrogen atmosphere. The reaction mixture was heated at 90 °C in a preheated oil bath for 48 h. Thereafter, volatiles were removed and the crude product was purified by column chromatography using silica gel as the stationary phase and a mixture of DCM/MeOH/diethylamine = 20/1/1 as the eluent.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Data availability
The data underlying this study are available in the published article and its supplementary information (SI). Supplementary information: experimental procedures, characterization data and spectra, and mechanistic analysis. See DOI: https://doi.org/10.1039/d5ob01788b.Acknowledgements
We thank SERB New Delhi (CRG/2023/001341), DAE, and NISER (DPR: RIN4002) for financial support.References
- J. Q. Bond, A. A. Upadhye, H. Olcay, G. A. Tompsett, J. Jae, R. Xing, D. M. Alonso, D. Wang, T. Zhang and R. Kumar, Production of renewable jet fuel range alkanes and commodity chemicals from integrated catalytic processing of biomass, Energy Environ. Sci., 2014, 7(4), 1500–1523 Search PubMed.
- C. M. Hadad, P. R. Rablen and K. B. Wiberg, C–O and C–S Bonds: Stability, bond dissociation energies, and resonance stabilization, J. Org. Chem., 1998, 63, 8668 CrossRef CAS.
- (a) D. H. R. Barton and S. W. McCombie, A new method for the deoxygenation of secondary alcohols, J. Chem. Soc., Perkin Trans. 1, 1975, 1574 RSC; (b) C. Chatgilialoglu and C. Ferreri, Progress of the Barton–McCombie methodology: from tin hydrides to silanes, Res. Chem. Intermed., 1993, 19, 755 Search PubMed.
- (a) A. Cook, H. MacLean, P. St Onge and S. G. Newman, Nickel-Catalyzed Reductive Deoxygenation of Diverse C–O Bond-Bearing Functional Groups, ACS Catal., 2021, 11, 13337–13347 Search PubMed; (b) D. R. Chambers, A. Juneau, C. T. Ludwig, M. Frenette and D. B. C. Martin, C–O Bond Cleavage of Alcohols via Visible Light Activation of Cobalt Alkoxycarbonyls, Organometallics, 2019, 38, 4570–4577 CrossRef CAS.
- (a) A. Volkov, K. P. J. Gustafson, C. W. Tai, O. Verho, J. E. Bäckvall and H. Adolfsson, Mild Deoxygenation of Aromatic Ketones and Aldehydes over Pd/C Using Polymethylhydrosiloxane as the Reducing Agent, Angew. Chem., Int. Ed., 2015, 54, 5122–5126 CrossRef CAS PubMed; (b) H. Wang, L. Li, X. F. Bai, J. Y. Shang, K. F. Yang and L. W. Xu, Efficient Palladium-Catalyzed C-O Hydrogenolysis of Benzylic Alcohols and Aromatic Ketones with Polymethylhydrosiloxane, Adv. Synth. Catal., 2013, 355, 341–347 Search PubMed.
- (a) C. Chatgilialoglu, D. Griller and M. Lesage, Tris(trimethylsilyl)silane. A new reducing agent, J. Org. Chem., 1988, 53, 3641–3642 CrossRef CAS; (b) G. L. Larson and R. J. Liberatore, Organosilanes in Metal-Catalyzed, Enantioselective Reductions, Org. Process Res. Dev., 2021, 25, 1719–1787 CrossRef CAS.
- R. M. Lopez, D. S. Hays and G. C. Fu, Bu3SnH-catalyzed Barton-McCombie deoxygenation of alcohols, J. Am. Chem. Soc., 1997, 119, 6949–6950 CrossRef CAS.
- (a) H. R. Diéguez, A. López, V. Domingo, J. F. Arteaga, J. A. Dobado, M. M. Herrador, J. F. Quílez del Moral and A. F. Barrero, Weakening C–O Bonds: Ti(III), a New Reagent for Alcohol Deoxygenation and Carbonyl Coupling Olefination, J. Am. Chem. Soc., 2010, 132(1), 254–259 CrossRef PubMed; (b) Q. Lin, W. Tong, X.-Z. Shu and Y. Chen, Ti-Catalyzed Dehydroxylation of Tertiary Alcohols, Org. Lett., 2022, 24, 8459–8464 CrossRef CAS; (c) A. Caciuleanu, F. Voehringer and I. Fleischer, Titanium-catalysed deoxygenation of benzylic alcohols and lignin model compounds, Org. Chem. Front., 2023, 10, 2927–2935 RSC.
- H. Dang, N. Cox and G. Lalic, Copper-Catalyzed Reduction of Alkyl Triflates and Iodides: An Efficient Method for the Deoxygenation of Primary and Secondary Alcohols, Angew. Chem., Int. Ed., 2014, 53, 752–756 CrossRef CAS PubMed.
- J. O. Bauer, S. Chakraborty and D. Milstein, Manganese-Catalyzed Direct Deoxygenation of Primary Alcohols, ACS Catal., 2017, 7, 4462–4466 CrossRef CAS.
- A. Cook, H. MacLean, P. St Onge and S. G. Newman, Nickel-Catalyzed Reductive Deoxygenation of Diverse C–O Bond-Bearing Functional Groups, ACS Catal., 2021, 11, 13337–13347 CrossRef CAS.
- A. Kumar, S. Pattanaik, G. Joshi, M. K. Sahu, E. D. Jemmis and C. Gunanathan, Cobalt-Catalyzed Reductive Deoxygenation of Aldehydes, Ketones, Alcohols, and Ethers to Alkanes, ACS Catal., 2024, 14, 4249–4264 Search PubMed.
- (a) J.-W. Park, Cobalt-catalyzed alkyne hydrosilylation as a new frontier to selectively access silyl-hydrocarbons, Chem. Commun., 2022, 58, 491–504 RSC; (b) B. Cheng, P. Lu, H. Zhang, X. Cheng and Z. Lu, Highly Enantioselective Cobalt-Catalyzed Hydrosilylation of Alkenes, J. Am. Chem. Soc., 2017, 139, 9439–9442 CrossRef CAS PubMed; (c) Z. Yang, D. Peng, X. Du, Z. Huang and S. Ma, Identifying a cobalt catalyst for highly selective hydrosilylation of allenes, Org. Chem. Front., 2017, 4, 1829–1832 RSC; (d) H. Zhou, H. Sun, S. Zhang and X. Li, Synthesis and Reactivity of a Hydrido CNC Pincer Cobalt(III) Complex and Its Application in Hydrosilylation of Aldehydes and Ketones, Organometallics, 2015, 34, 1479–1486 CrossRef CAS; (e) A. Nurseiit, J. Janabel, K. A. Gudun, A. Kassymbek, M. Segizbayev, T. M. Seilkhanov and A. Y. Khalimon, Bench-Stable Cobalt Pre-Catalysts for Mild Hydrosilative Reduction of Tertiary Amides to Amines and Beyond, ChemCatChem, 2019, 11, 790 CrossRef CAS; (f) A. Sanagawa and H. Nagashima, Hydrosilane Reduction of Nitriles to Primary Amines by Cobalt–Isocyanide Catalysts, Org. Lett., 2019, 21, 287 CrossRef CAS.
- (a) K. Takeshita, Y. Seki, K. Kawamoto, S. Murai and N. Sonoda, The catalysed reaction of α,β-unsaturated esters with various hydrosilanes, J. Org. Chem., 1987, 52, 4864 CrossRef CAS; (b) T. Murai, T. Sakane and S. Kato, Cobalt carbonyl catalyzed reduction of aromatic nitriles with a hydrosilane leading to N,N-disilylamines, Tetrahedron Lett., 1985, 26, 5145 CrossRef CAS; (c) N. Chatani, T. Kodama, Y. Kajikawa, H. Murakami, F. Kakiuchi, S. Ikeda and S. Murai, Co2(CO)8-catalyzed hydrosilylation of oxygen-containing olefins: silylmetalation as a key step, Chem. Lett., 2000, 14 Search PubMed; (d) T. Konno, K.-i. Taku, S. Yamada, K. Moriyasu and T. Ishihara, Remarkable access to fluoroalkylated trisubstituted alkenes via highly stereoselective cobalt-catalyzed hydrosilylation reaction of fluoroalkylated alkynes, Org. Biomol. Chem., 2009, 7, 1167–1170 RSC; (e) T. Dombray, C. Helleu, C. Darcel and J. B. Sortais, Cobalt Carbonyl-Based Catalyst for Hydrosilylation of Carboxamides, Adv. Synth. Catal., 2013, 355, 3358–3362 CrossRef CAS; (f) A. Y. Khalimon, K. A. Gudun and D. Hayrapetyan, Base Metal Catalysts for Deoxygenative Reduction of Amides to Amines, Catalysts, 2019, 9, 490 CrossRef.
- S. Panda, A. Nanda, R. R. Behera and R. Ghosh, Cobalt catalyzed chemoselective reduction of nitroarenes: hydrosilylation under thermal and photochemical reaction conditions, Chem. Commun., 2023, 59, 4527–4530 RSC.
- (a) R. Saha, B. C. Hembram, S. Panda, R. Ghosh and B. Bagh, Iron-Catalyzed sp3 C–H Alkylation of Fluorene with Primary and Secondary Alcohols: A Borrowing Hydrogen Approach, J. Org. Chem., 2024, 89(22), 16223–16241 CrossRef CAS; (b) R. Saha, B. C. Hembram, A. Panda and B. Bagh, Iron-catalyzed peroxidation of fluorenes and 9-alkyl-9H-fluorenes and oxidative cleavage of the C=C bond in 9-alkylidene-fluorenes, Org. Biomol. Chem., 2025, 23, 4232–4239 RSC.
- (a) T. Ma, Y. Zhou, C. S. Diercks, J. Kwon, F. Gándara, H. Lyu, N. Hanikel, P. Pena-Sánchez, Y. Liu, N. J. Diercks, R. O. Ritchie, D. M. Proserpio, O. Terasaki and O. M. Yaghi, Catenated covalent organic frameworks constructed from polyhedral, Nat. Synth., 2023, 2, 286–295 CrossRef CAS; (b) Y. S. Soliman, S. M. Tadros, W. B. Beshir, M. M. Naoum, G. R. Saad and L. I. Ali, A Radichromic Dye of Tris-(4-Aminophenyl) Methane Incorporated into Polymeric Films for Radiation Dose Monitoring, Dyes Pigm., 2023, 217, 111387 CrossRef CAS.
- R. J. Klingler and J. W. Rathke, High-Pressure NMR Investigation of Hydrogen Atom Transfer and Related Dynamic Processes in Oxo Catalysis, J. Am. Chem. Soc., 1994, 116, 4772–4785 CrossRef CAS.
- (a) A. Manna, T. K. Dinda, S. Ghosh and P. Mal, CsPbBr3 in the Activation of the C−Br Bond of CBrX3 (X = Cl, Br) under Sunlight, Chem. Mater., 2023, 35, 628–637 CrossRef CAS; (b) R. Bhanja, S. K. Bera and P. Mal, Regioselective Synthesis of Phenanthridine-Fused Quinazolinones using 9-Mesityl-10-Methylacridinium Perchlorate Photocatalyst, Chem. Commun., 2023, 59, 4455–4458 RSC.
| This journal is © The Royal Society of Chemistry 2026 |