
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of enantiomerically enriched β-N-amino (S)-α-alanine analogs via sequential reactions on a chiral Ni(II) complex
Emma A.
Khachatryan
a,
Lala A.
Stepanyan
b,
Hasmik R.
Gevorgyan
bc,
Armen S.
Sargsyan
b,
Anahit M.
Hovhannisyan
a,
Anna F.
Mkrtchyan
 *ab,
Andrei V.
Malkov
*d and
Ashot S.
Saghyan
*ab
*ab,
Andrei V.
Malkov
*d and
Ashot S.
Saghyan
*ab
aInstitute of Pharmacy, Yerevan State University, 1 Alex Manoogian Str., 0025 Yerevan, Armenia. E-mail: anna_mkrtchyan@ysu.am; Fax: +374 60 710 410; Tel: +37460 710 427
bScientific and Production Center “Armbiotechnology” of NAS RA, 14 Gyurjyan Str., 0056 Yerevan, Armenia
cScientific and Technological Center of Organic and Pharmaceutical Chemistry of NAS RA, 26 Azatutyan Ave., 0014 Yerevan, Armenia
dDepartment of Chemistry, Loughborough University, University Road, Loughborough, LE11 3TU, UK
First published on 9th December 2025
Abstract
A concise synthetic route to enantiomerically enriched β-N-substituted (S)-α-diaminopropanoic acids is reported. An acetylene group was introduced into the alanine side chain by nucleophilic Michael addition of substituted propargylamines to the dehydroalanine moiety in a chiral Ni(II) complex, which afforded β-N-propargylamino derivatives in high ee (≈90%). Subsequent Glaser, Sonogashira, and [3 + 2]-cycloaddition reactions delivered structurally diverse products with ee up to 99%. The isolated auxiliary (S)-BPB can be recovered and reused without any loss of enantiointegrity. Biological screening of the products identified (S)-3-(N-(1-phenylethyl)-N-(prop-2-ynyl)amino)-2-aminopropanoic acid as a moderate bacterial collagenase inhibitor (IC50 = 0.69 mM), demonstrating the potential of this platform for enzyme inhibitor development.
Introduction
α,β-Diamino acids represent a rare but highly significant class of non-proteinogenic amino acids that have attracted growing attention due to their unique structural and functional properties.1,2 For example, L-tambroline, an α,β-diamino acid incorporated into the non-ribosomal peptide tambromycin, plays a key structural role in the compound's antiproliferative properties.3 Among α,β-diamino acids, the most studied examples are L-quisqualic acid, L-lupinic acid, and L-willardine (Fig. 1). L-Quisqualic acid is a potent AMPA and mGlu receptor agonist used to investigate excitatory neurotransmission and excitotoxicity. L-Lupinic acid, a non-proteinogenic amino acid from Lupinus species, serves as a probe of nitrogen metabolism and polyamine biosynthesis. L-Willardine, a pyrimidine-containing AMPA agonist, is applied in studies of glutamatergic signaling and receptor structure–activity relationships.2 | ||
| Fig. 1 Selected bioactive natural α,β-diamino acids. | ||
The presence of α,β-diamino acid fragments in bioactive natural products such as streptothricins, viomycin, and capreomycin highlights their value as pharmacophores in antimicrobial and anticancer drug discovery.
Notably, incorporating these motifs into drug structures can prolong pharmacological action, supporting molecular design strategies that improve metabolic stability and biological activity.4,5 Likewise, the introduction of unnatural α-amino acids into peptide frameworks enables fine control over pharmacokinetic and pharmacodynamic properties—enhancing membrane permeability, enzymatic stability, and receptor selectivity—while improving antibacterial potency and reducing toxicity. Together, these features address key limitations of canonical peptides and offer promising avenues for combating antibiotic-resistant pathogens.6–8 Among unnatural α,β-diamino acids, derivatives featuring functionally substituted aliphatic, aromatic, or heterocyclic groups in the side chain attracted particular interest. Specifically, β-N-substituted alanine analogs are key structural components of several clinically relevant antibiotics, including Tuberactinomycins, Bleomycins, Edeines, and Capreomycins.9
Propargylamine motifs are also well-established in pharmaceutical chemistry.10–12 Notably, compounds such as pargyline, rasagiline, selegiline, and clorgiline containing an N-propargyl group, are potent inhibitors of monoamine oxidases A and B and have found direct clinical application in the treatment of neurodegenerative disorders, including Parkinson's disease13 and Alzheimer's disease.14–16 Derivatives of propargylamines are widely employed as synthetic building blocks.17–19 The broad utility of propargylamines stems from their unique structure, which combines a nucleophilic β-amino group with a reactive alkyne moiety, thereby enabling diverse chemical transformations. Their incorporation into peptides facilitates the formation of cyclic structures via click chemistry, allowing access to novel amino acids and peptidomimetics. C-Terminal propargyl amides also act as irreversible cysteine protease inhibitors through Michael-type addition, highlighting their pharmacological potential and suitability for further functionalization via alkyne coupling.20–22
The synthetic accessibility of α,β-diamino acids has historically posed a formidable challenge, primarily due to the inherent reactivity of vicinal amino groups and the difficulty of achieving stereocontrolled functionalization. Several strategies have been developed for the stereoselective synthesis of α,β-diamino acids. Early approaches, which date back to the mid-20th century, were largely limited to multistep sequences with poor stereoselectivity and low yields, confining their use to specialized applications.1,2
More recently, an efficient enzymatic Mannich-type protocol for accessing α,β-diamino acids has been reported, broadening the available biocatalytic repertoire.23,24 Among chemical methods, one of the most widely applied approaches is the asymmetric Mannich reaction, in which glycine Schiff bases or glycine equivalents undergo chiral-catalyst-controlled addition to imines, affording α,β-diamino acid derivatives with high enantio- and diastereoselectivity.25,26
Complementary routes to α,β-diamino acids employ haloalkanoate-based methodologies, particularly the nucleophilic substitution in α,β-dibromo esters, which delivers diamino succinates, piperazines, and aziridines as synthetically versatile intermediates.27 Enantioselective approaches based on alkenoates have also been developed, including osmium-mediated deamination,28,29 and electrophilic diamination with dichloro-sulfonamides.30,31 Further, Michael additions to dehydroalanine complexes32,33 and electrophilic amination of enolates provide highly modular routes to structurally diverse, enantiopure α,β-diamino acid frameworks.
The asymmetric synthesis of β-N-substituted analogues of (S)-α-alanine via nucleophilic addition of nitrogen nucleophiles to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of the dehydroalanine moiety of the complexes was developed in our group,34–36 albeit with limited scope.
C bond of the dehydroalanine moiety of the complexes was developed in our group,34–36 albeit with limited scope.
Our interest in alkyne-substituted amino acids and in exploring the synthetic potential of alkyne functionalities was highlighted using O-propargyl derivatives of β-hydroxy-α-amino acids such as serine and threonine.37,38 These derivatives were obtained with the aid of square-planar Ni(II) complexes of Schiff bases derived from dehydroamino acids (dehydroalanine and dehydroaminobutyric acid) and the chiral auxiliary (S)-2-N-(N-benzylprolyl)aminobenzophenone (BPB).
In the present study, a similar strategy was employed to introduce a propargylamine moiety into the amino acid side chain and to enable subsequent elaboration of the alkyne functionality through various cross-coupling reactions.
Results and discussion
The square-planar Ni(II) complex of the Schiff base derived from dehydroalanine and the chiral auxiliary (S)-BPB (1) was used as a template. This complex was synthesized according to a previously established procedure.34 The nucleophilic addition of amines 2a–e to the CC bond in 1 was carried out under basic catalysis conditions (Scheme 1).
 | ||
| Scheme 1 The addition of the propargylamines 2a–e to the CC bond of 1. | ||
The progress of the reaction was monitored by TLC by the disappearance of initial complex 1. The process is thermodynamically controlled, providing a high diastereomeric excess of the main (S,S)-diastereomer of the addition product. The structure and absolute configuration of the main diastereomeric complexes were rigorously established by physicochemical methods after chromatographic purification of the product.34–38 The (S)-absolute configuration of the α-carbon atom of the amino acid moiety was assigned using the positive sign of [α]D in analogy with previously reported, similar Ni(II) complexes of other amino acids.34–38
Additional verification was obtained by the circular dichroism (CD) spectra: positive Cotton effects of the main diastereomers 3a–e in the 500–580 nm region indicated (S)-absolute configuration of the α-carbon atom of the amino acid moiety (see experimental part). For comparison, the CD spectra of (S,S)- and (S,R)-diastereomers of the similar alanine complexes with the absolute configuration established by X-ray crystallography are also given. In addition to the points mentioned above, we also obtained the X-ray structure of the 3b complex, which further confirmed our previous description (Fig. 2).
 | ||
| Fig. 2 X-Ray structure of complex 3b. | ||
The ratio of diastereomers of the addition products was determined from the 1H NMR spectra by integrating the methylene protons signals of the N-benzylproline group. This was also verified by chiral HPLC analysis of the corresponding amino acids (4a–e), obtained by acid hydrolysis of a mixture of diastereomeric complexes followed by ion-exchange demineralization of the hydrolysate before chromatographic purification. (S,S)-Diastereomers of 3a–e were formed with high diastereomeric excess (86–92%) (Scheme 1). To confirm the stereocontrolling ability of (S)-BPB, the corresponding complexes were prepared using (R)-BPB, leading to the isolation of amino acids with the expected (R)-absolute configuration ((R)-4b and (R)-4d). These results demonstrate that the ligand exerts identical stereocontrol in the opposite enantiomeric series.
The pure target amino acids were isolated using a standard protocol: the diastereomeric mixture of complexes 3a–e was subjected to acid hydrolysis, followed by ion-exchange demineralization and crystallization from aqueous ethanol. The initial chiral auxiliary (S)-BPB was regenerated in a nearly quantitative yield (>96%) without loss of enantiomeric purity and was reused without any loss of chiral integrity.
Using this method, amino acids 4a–e were obtained in pure crystalline form, with high enantiomeric purities (ee 95–99%), as confirmed by chiral HPLC analysis, and in 30–52% chemical yield. The reported yields refer to the isolated recrystallized products; they could likely be improved through further optimization of the recrystallization procedure. At an industrial scale, additional material can be recovered from the mother liquor, thereby increasing the overall yield. These results indicate that the method is readily applicable for preparing other enantiomerically enriched amino acids bearing a propargylamino moiety.
The terminal alkyne groups in the side chain of the synthesized complexes can serve as valuable handles for further synthetic modification. In this study, complexes 3b and 3d were subjected to Sonogashira and Glaser cross-coupling reactions, a complex 3b was additionally employed in a [3 + 2] cycloaddition (Scheme 2).
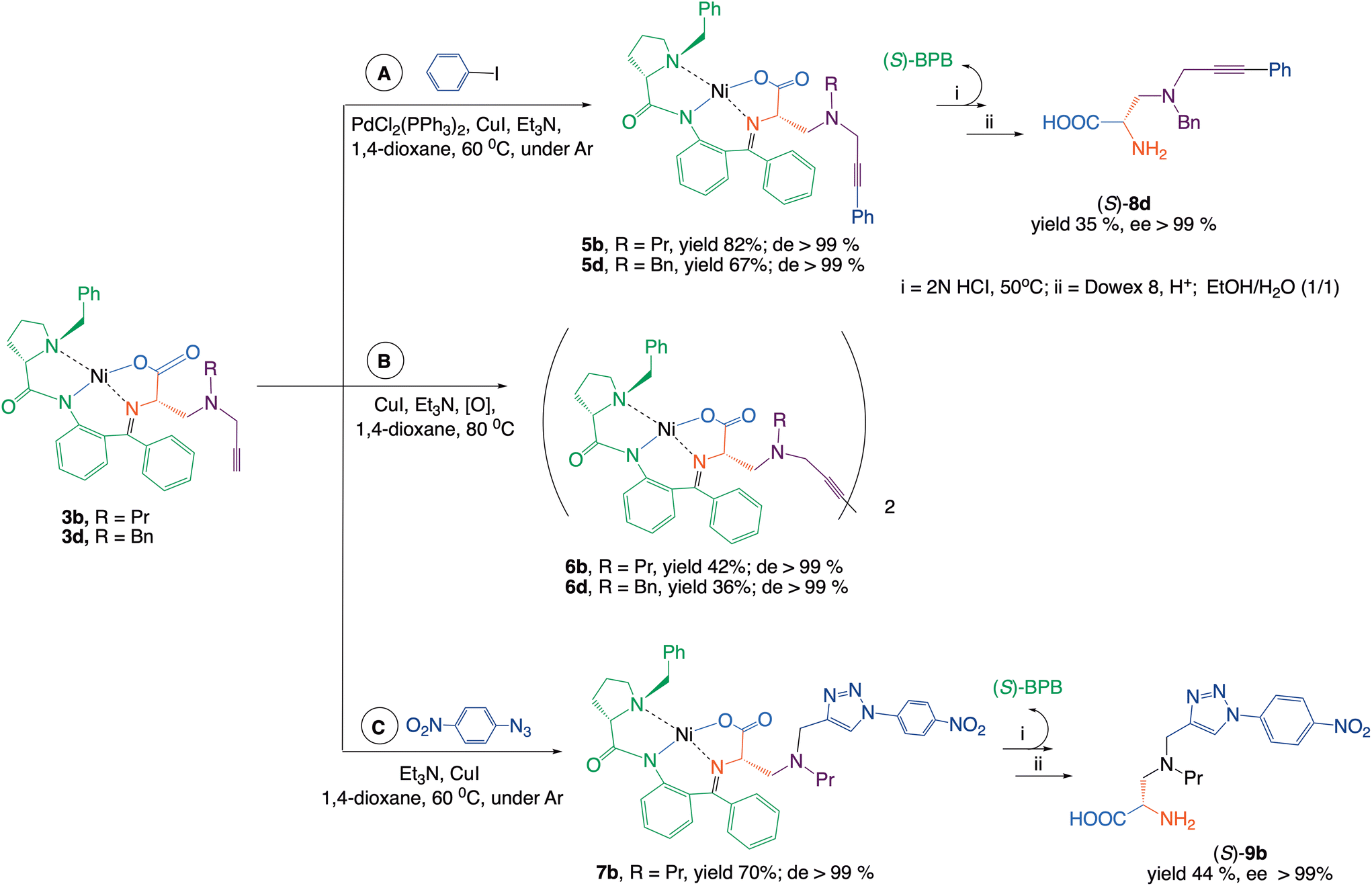 | ||
| Scheme 2 Cross-coupling and [3 + 2]-cycloaddition reactions of complexes 3b and 3d. | ||
Arylation of the terminal acetylene group in complexes 3b and 3d by Sonogashira coupling (Scheme 2, route A) gave rise to complexes 5b and 5d with good yield. During the reaction, a side product was formed, which was identified as a dimerization product. This observation prompted us to optimize a Glaser homocoupling reaction (route B), enabling the efficient synthesis of dimeric complex 6b and 6d in good yield. In a [3 + 2]-cycloaddition, 4-nitrophenylazide was reacted with the terminal alkyne group of complex 3b to furnish complex 7b in a good yield, which was converted to the respective amino acid 9b featuring a 1,2,3-triazole moiety in the side chain (route C).
In the cross-coupling and [3 + 2]-cycloaddition reactions, the initial 3b complex was used as pure (S,S)-diastereoisomer (Scheme 2). It is worth noting that the chiral α-carbon of these amino acid fragments does not directly participate in the formation of new carbon–carbon bond reactions.
However, weak basis conditions of the reaction may cause partial epimerization of this stereogenic center. Therefore, the chiroptical data of the intermediate complexes and the isolated final amino acids were checked. The NMR analysis of 5b–7b revealed that in the cross-coupling and [3 + 2]-cycloaddition reactions, the stereochemical integrity remained intact. This was additionally confirmed by the circular dichroism (CD) method (see the (SI)). The target amino acids were isolated from the diastereomeric mixture of complexes 5b–7b using standard procedures.3 The structure and enantiomeric purity of crystallized amino acids 8d and 9b were confirmed by analytical techniques.
Biological activity and docking
Molecular docking analysis revealed that all examined (S)-configured α-amino acid derivatives (4a–e) are capable of interacting with bacterial collagenase, as reflected by their calculated Gibbs free energy (ΔG) values (Table 1). Among the tested compounds, acid 4e, featuring a 1-phenylethyl substituent on the nitrogen atom, exhibited the strongest binding affinity with a ΔG of −6.048 kcal mol−1.| Obtained α-amino acids | ΔG |
|---|---|
| (S)-3-(N-Methyl-N-(prop-2-ynyl)amino)-2-aminopropanoic acid (4a) | −4.331 |
| (S)-3-(N-(Prop-2-ynyl)-N-propylamino)-2-aminopropanoic acid-4-ynoic acid (4b) | −3.989 |
| (S)-3-(N-Butyl-N-(prop-2-ynyl)amino)-2-aminopropanoic acid (4c) | −4.324 |
| (S)-3-(N-Benzyl-N-(prop-2-ynyl)amino)-2-aminopropanoic acid (4d) | −4.611 |
| (S)-3-(N-((S)-1-Phenylethyl)-N-(prop-2-ynyl)amino)-2-aminopropanoic acid (4e) | −6.048 |
| (S)-2-Amino-3-(benzyl(3-phenylprop-2-yn-1-yl)amino)propanoic acid (8d) | −6.821 |
| (S)-2-Amino-3-(((1-(4-nitrophenyl)-1H-1,2,3-triazol-4-yl)methyl)(propyl)amino)propanoic acid (9b) | −6.469 |
The docking results indicate that binding affinity correlates with the nature of the substituent on nitrogen. Compounds with bulkier or aromatic groups, as in 4d and 4e, exhibited stronger binding, likely due to enhanced van der Waals forces or π–π interactions within the enzyme's active site. In contrast, compound 4b, featuring a linear propyl chain, showed the weakest interaction (ΔG = −3.989 kcal mol−1), suggesting that smaller or less conjugated side chains may not engage in sufficient stabilizing interactions. For compound 4e, detailed interaction analysis (Fig. 3) showed that (S)-3-(N-(1-phenylethyl)-N-(prop-2-ynyl)amino)-2-aminopropanoic acid (4e) interacts with bacterial collagenase (ΔG = −6.048 kcal mol−1) through two hydrogen bonds and 2 π–π interactions. The carboxyl group forms a hydrogen bond with the free amino group of Gln215 (2.121 Å) while the amino group forms an additional hydrogen bond with the carboxyl group of Ala252 (2.145 Å). Aromatic π–π interactions occur with Tyr201 (4.541 Å) and Tyr198 (6.854 Å). Notably, this binding site lies within the activator domain, which is responsible for full enzymatic activity on collagen.39
 | ||
| Fig. 3 Molecular docking analysis of the interaction between collagenase and (S,S)-3-(N-(1-phenylethyl)-N-(prop-2-ynyl)amino)-2-aminopropanoic acid (4e). | ||
Determination of collagenase activity of compound 4e
Molecular docking analysis identified (S)-3-(N-(1-phenylethyl)-N-(prop-2-ynyl)amino)-2-aminopropanoic acid (4e) as a candidate for experimental evaluation of its effect on collagenase activity. Enzymatic assays were performed with 4e at concentrations ranging from 0.2 to 2 mM. The compound effectively inhibited collagenase activity, consistent with the predicted strong binding affinity, with an IC50 of 0.69 ± 0.014 mM, indicating moderate inhibitory potency. These findings suggest that compound 4e may serve as a promising lead compound for the development of collagenase inhibitors.Experimental
Materials
All starting reagents were obtained from commercial sources and used without further purification. The initial complexes 1 and substituted propargyl amines were prepared following literature protocols.40 TLC analyses were performed on glass plates coated with silica gel 60 F254. Column chromatography was performed on silica (60 × 120 mesh) using glass columns. Melting points (mp) were determined using an Electrothermal apparatus. 1H and 13C NMR spectra were recorded on a Varian Mercury-300 spectrometer (300 MHz and 75 MHz, respectively) using TMS as an internal standard (0.0 ppm). Elemental analyses were performed using a EURO EA 3000 instrument.The enantiomeric purity of the amino acids was determined by HPLC using a Waters Alliance 2695 HPLC System equipped with chiral phase column Nautilus-E, 5 μm, 4.6 mm × 250 mm (BioChemMak ST); and a mixture of 20% MeOH and 80% 88 mM aqueous solution of KH2PO4 was used as an eluent. The optical rotation was measured on a PerkinElmer Elmer-341 polarimeter. LCMS analysis was performed on Shimadzu LCMS 2020 with Prominence-I LC-2030C 3D. The CD analyses were carried out using Chirascan™ V100. The unit cells parameters of crystals were measured on an Enraf-Nonius automated diffractometer CAD-4 at room temperature using the diffraction angles of 24 reflections. Crystallographic data have been deposited at the CCDC under 2486969.
General procedure for nucleophilic Michael addition to complex 1
1.5 g (2.85 mmol) of complex 1, 1.97 g (14.25 mmol) of K2CO3, and 8.55 mmol of nucleophile were dissolved in 20 mL of acetonitrile. The reaction mixture was stirred at 50–60 °C. The progress of the reaction was monitored by TLC on silica, using a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of ethyl acetate and chloroform. Upon completion of the reaction, as confirmed by TLC monitoring (disappearance of the starting complex and appearance of the product), the reaction mixture was filtered to remove potassium carbonate and the filtrate was concentrated under reduced pressure to remove acetonitrile. The resulting residue was extracted with chloroform–water (3 × 30 mL). The extraction was continued until the aqueous phase reached a neutral pH. The combined organic layers were dried over anhydrous magnesium sulfate, filtered, and evaporated to dryness under reduced pressure. The crude products were purified by column chromatography on silica gel, eluting with a 2:1 mixture of acetone and chloroform, to afford complexes 3a,b and 3d,e. Complex 3c was recrystallized from acetone.
:1 mixture of ethyl acetate and chloroform. Upon completion of the reaction, as confirmed by TLC monitoring (disappearance of the starting complex and appearance of the product), the reaction mixture was filtered to remove potassium carbonate and the filtrate was concentrated under reduced pressure to remove acetonitrile. The resulting residue was extracted with chloroform–water (3 × 30 mL). The extraction was continued until the aqueous phase reached a neutral pH. The combined organic layers were dried over anhydrous magnesium sulfate, filtered, and evaporated to dryness under reduced pressure. The crude products were purified by column chromatography on silica gel, eluting with a 2:1 mixture of acetone and chloroform, to afford complexes 3a,b and 3d,e. Complex 3c was recrystallized from acetone.
General procedure for Sonogashira reaction
Iodobenzene (2.5 mL, 12.2 mmol) and 5 mL of triethylamine were placed in a pressure tube under an argon atmosphere, followed by the addition of PdCl2(PPh3)2 (0.1 g, 0.15 mmol) and copper iodide (0.058 g, 0.3 mmol) under argon. Then, 1,4-dioxane (5 mL) and complexes 3b (1.85 g, 3.05 mmol) or 3d (2.0 g, 3.05 mmol) were added to the reaction vessel. The reaction mixture was stirred at 60 °C. and was monitored by thin-layer chromatography (TLC) on silica gel using ethyl acetate/chloroform (3:1, v/v) as the eluent. Upon completion of the reaction, as indicated by TLC monitoring (disappearance of the starting complex and appearance of the product), the mixture was cooled to room temperature and diluted with ethyl acetate (30 mL). The organic layer was separated and washed successively with water (3 × 20 mL) and brine (20 mL). The combined organic extracts were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure to afford the crude product. A small portion of the products was purified by column chromatography for the characterization of complexes 5b and 5d.
General procedure for Glaser reaction
Complex 3b (1.85 g, 3.05 mmol) or 3d (2.00 g, 3.05 mmol) and copper(I) iodide (0.58 g, 3.05 mmol) were added to a mixture of 1,4-dioxane (5 mL) and triethylamine (5 mL). The reaction mixture was stirred at 80 °C, and the progress of the reaction was monitored by thin-layer chromatography (TLC) on silica gel using ethyl acetate/chloroform (3:1, v/v) as the eluent. Upon completion of the reaction, as indicated by TLC monitoring (disappearance of the starting complex and appearance of the product), the mixture was cooled to room temperature and diluted with ethyl acetate (30 mL). The organic layer was separated and washed successively with water (3 × 20 mL) and brine (20 mL). The combined organic extracts were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure to afford the crude product. A small portion of the crude residue was purified by column chromatography on silica gel (ethyl acetate/chloroform, 3:1, v/v) to yield the analytically pure compounds 6b and 6d for characterization.
General procedure for [2 + 3]-cycloaddition reaction
1-Azido-4-nitrobenzene (1.08 g, 6.5 mmol) was dissolved in 5 mL of 1,4-dioxane in a round-bottom flask under an argon atmosphere at room temperature. Copper iodide (0.062 g, 0.3 mmol) was added to the solution, and the mixture was stirred for 5 minutes. Then triethylamine (1.14 mL, 8.2 mmol) was added, and stirring was continued for 30 minutes. After that, complex 3b (2.0 g, 3.3 mmol) was added, and the reaction mixture was stirred at 60 °C and was monitored by TLC gel using ethyl acetate/chloroform (3:1, v/v) as the eluent. Upon completion of the reaction, as indicated by TLC monitoring (disappearance of the starting complex and appearance of the product), the mixture was cooled to room temperature and diluted with ethyl acetate (30 mL). The organic layer was separated and washed successively with water (3 × 20 mL) and brine (20 mL). The combined organic extracts were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure to afford the crude product. The filtrate was concentrated to dryness under reduced pressure, and the residue was recrystallized from acetone to yield a pure product. A small portion (0.1 g) was used for the characterization of complex 7b.
General procedure for isolation of amino acids
Diastereomeric mixtures of complexes 3a–e, 5b, 5d, 6b, 6d, and 7b (before chromatography) were suspended in CH3OH and then were added slowly to a vigorously stirred 4 M aqueous HCI at 50–60 °C.After decomposition of the complexes (ca. 20 min), the target amino acids were demineralized using cation-exchange resin Dowex 50-8 (H+) and then recrystallized from an aqueous ethanol. The structure, chemical, and optical purities of the target amino acids 4a–e, 8d, and 9b were established by physicochemical methods – NMR, HPLC, CD, [α]D.
Molecular docking
The 3D structures of the compounds were built using ChemBioOffice 2010 (ChemBio3D Ultra 12.0). Minimization of the ligand free energy was performed using the MM2 force field and the truncated Newton–Raphson method. The crystallographic structure of collagenase G (PDB ID: 2Y50) was used in the analysis. Water molecules were removed, and polar hydrogens were added according to the software producer's suggested protocol for macromolecule preparation.Docking of ligands to the enzymes was performed using AutoGrid 4 and AutoDock Vina 1.2.3 software.41 Ligands were ranked based on an energy-dependent score function, and protein–ligand interactions were modelled on a grid to speed up calculations.
Interaction modes were identified; bond types and lengths were also determined.
Determination of collagenase activity
Collagenase activity was determined by a method based on the measurement of free amino groups that are released during substrate hydrolysis.42 The reaction mixture consisted of 0.05 M HEPES buffer (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) with pH 7.2, 10 mg mL−1 gelatin and 0.025 mg mL−1 collagenase; the reaction was carried out at 37 °C. To determine the concentration of amino groups, the ortho-phthalaldehyde (OPA) reagent was used, consisting of 0.2 M borate buffer with pH 9.7, 0.1667 mg mL−1 OPA and 1.18 mM mercaptoethanol. Aliquots of 50 μL were taken every 30 minutes, and the reaction was stopped by adding 10 μL of 30% trichloroacetic acid. Then 1.5 mL of OPA reagent and 1.5 mL of water were added to the resulting sample, and the A340 value was recorded after 5 minute incubation at 27 °C.Conclusions
A highly selective asymmetric synthesis of a series of β-N-propargylamino substituted (S)-α-aminopropanoic acids has been developed via nucleophilic Michael addition of substituted propargyl amines to the CC bond of dehydroalanine in a square-planar NiII complex of its Schiff base with chiral auxiliary (S)-BPB. This approach afforded enantiomerically enriched (S)-β-(N-propargylamino)alanines featuring diverse alkyl side chains, all obtained with high diastereomeric excess (de ∼90%). The terminal alkyne group of some intermediates was further functionalized through Sonogashira cross-coupling with iodobenzene, Glaser dimerization, and [3 + 2]-cycloaddition with 4-nitrophenylazide. The latter reaction yielded (S)-β-(N-aminosubstituted)-α-alanine derivatives containing a 1,2,3-triazole ring linked to a p-nitrophenyl moiety. The final amino acids were obtained with high enantiomeric purity (ee > 98%).
Importantly, molecular docking and in vitro enzymatic assays revealed that one of the synthesized compounds, (S)-3-(N-(1-phenylethyl)-N-(prop-2-ynyl)amino)-2-aminopropanoic acid (4e), acts as a moderate inhibitor of bacterial collagenase, with an IC50 value of 0.69 ± 0.014 mM. These findings highlight the potential of β-N-aminosubstituted (S)-α-alanines as versatile scaffolds for the development of biologically active molecules, particularly enzyme inhibitors.
Author contributions
Emma A. Khachatryan (EAK) data curation, investigation, methodology, formal analysis; Lala A. Stepanyan (LAS) data curation, validation, visualization; Hasmik R. Gevorgyan (HRG) data curation, validation, writing – review & editing; Armen S. Sargsyan (ASSa) investigation, methodology, validation; Anahit M. Hovhannisyan (AMH) data curation, formal analysis, visualization; Anna F. Mkrtchyan (AFM) conceptualization, formal analysis, writing – original draft, writing – review & editing, resources, supervision, project administration; Andrei V. Malkov (AVM) formal analysis, methodology, visualization, writing – original draft, writing – review & editing; Ashot S. Saghyan (ASS) funding acquisition, methodology, project administration, supervision, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Data availability
Additional data related to the molecular docking studies are available from the corresponding author upon reasonable request. All experimental data supporting the findings of this study—including synthetic procedures, characterization data (NMR, HPLC, X-ray crystallography), and biological assay results—are provided in the Supplementary Information. The Supplementary Information also contains detailed experimental procedures, instrumentation details, NMR and MS spectra, HPLC analyses, X-ray crystallographic data, and molecular docking results. See DOI: https://doi.org/10.1039/d5ob01593f.CCDC 2486969 (3b) contains the supplementary crystallographic data for this paper.43
Acknowledgements
Support was provided by joint research projects SCS 21AG-1D013 and 23AA-1D021.References
- Y. Wu, T. Gu, J. Zhao, B. Li and L. Wang, Eur. J. Org. Chem., 2025, e202401299, DOI:10.1002/ejoc.202401299.
- A. Viso, F. R. Pradilla, M. Tortosa, A. García and A. Flores, Chem. Rev., 2011, 111(2), PR1–PR42, DOI:10.1021/cr100127y.
- S. Liu, J. Gao, Y. Zou, Y. Hai and J. Amer, Chem. Soc., 2024, 146(29), 20263–20269, DOI:10.1021/jacs.4c05581.
- A. S. Saghyan and P. Langer, Stoichiometric Asymmetric Synthesis of Non-proteinogenic Amino Acids, Wiley-VCH Verlag GmbH & Co. KGaA, 2016 Search PubMed.
- K. K. Sharma, K. Sharma, K. Rao, A. Sharma, G. K. Rathod, S. Aaghaz, N. Sehra, R. Parmar, B. VanVeller and R. Jain, J. Med. Chem., 2024, 67, 19932–19965 CrossRef CAS PubMed.
- J. L. Hickey, D. Sindhikara, S. L. Zultanski and D. M. Schultz, ACS Med. Chem. Lett., 2023, 14(5), 557–565 CrossRef CAS PubMed.
- M. Blaskovich, J. Med. Chem., 2016, 59(24), 10807–10836, DOI:10.1021/acs.jmedchem.6b00319.
- Y. Murakami, S. Ishida, Y. Demizu and K. Terayama, Digital Discovery, 2023, 2(5), 1347–1353, 10.1039/D3DD00090G.
- T. Takita, Y. Muraoka, T. Yoshioka, A. Fujii, K. Maeda and H. Umezawa, J. Antibiot., 1972, 25(12), 755–758, DOI:10.7164/antibiotics.25.755.
- J. W. Langston, I. Irwin, E. B. Langston and L. S. Forno, Science, 1984, 225(4669), 1480–1482, DOI:10.1126/science.6332378.
- J. J. Chen and D. M. Swope, J. Clin. Pharmacol., 2005, 45(8), 878–894, DOI:10.1177/0091270005277935.
- J. Birks and L. Flicker, Cochrane Database Syst. Rev., 2003 DOI:10.1002/14651858.CD000442.
- M. Baranyi, P. F. Porceddu, F. Gölöncsér, S. Kulcsár, L. Otrokocsi, Á. Kittel, A. Pinna, L. Frau, P. B. Huleatt, M. L. Khoo, C. P. Dunkel, P. Mátyus, M. Morelli and B. Sperlágh, Mol. Neurodegener., 2016, 11(1), 6, DOI:10.1186/s13024-015-0067-y.
- I. Bolea, A. Gella and M. Unzeta, J. Neural Transm., 2012, 120(6), 893–902, DOI:10.1007/s00702-012-0948-y.
- F. T. Zindo, J. Joubert and S. F. Malan, Future Med. Chem., 2015, 7(5), 609–629, DOI:10.4155/fmc.15.12.
- F. Mao, J. Li, H. Wei, L. Huang and X. Li, J. Enzyme Inhib. Med. Chem., 2015, 30(6), 995–1001, DOI:10.3109/14756366.2014.1003212.
- S. Arshadi, E. Vessally, L. Edjlali, R. Hosseinzadeh-Khanmiri and E. Ghorbani-Kalhor, Beilstein J. Org. Chem., 2017, 13, 625–638, DOI:10.3762/bjoc.13.61.
- N. Shachat and J. J. Bagnell, J. Org. Chem., 1963, 28(4), 991–995, DOI:10.1021/jo01039a028.
- R. D. Dillard and N. R. Easton, J. Org. Chem., 1964, 29(8), 2464–2467, DOI:10.1021/jo01031a518.
- H. Vilaça, P. Ferreira and N. M. Micaelo, Tetrahedron, 2014, 70(35), 5420–5427, DOI:10.1016/j.tet.2014.06.121.
- M. Wünsch, D. Schröder and T. Fröhr, Beilstein J. Org. Chem., 2017, 13(1), 2428–2441, DOI:10.3762/bjoc.13.240.
- A. Christoph and J. Rademann, Angew. Chem., Int. Ed., 2013, 52(32), 8210–8212, DOI:10.1002/anie.201303544.
- A. Viso, R. Fernández, A. García and A. Flores, Chem. Rev., 2005, 105(8), 3167–3196, DOI:10.1021/cr0406561.
- Y. Ouyang, S. Wang, D. Sorigue and T. K. Hyster, J. Am. Chem. Soc., 2025, 147(29), 25184–25190, DOI:10.1021/jacs.5c04097.
- R. G. Arrayás and J. C. Carretero, Chem. Soc. Rev., 2009, 38(7), 1940–1948, 10.1039/B820303B.
- G. Song, M. Jin, Z. Li and P. Ouyang, Org. Biomol. Chem., 2011, 9(20), 7144–7150, 10.1039/C1OB05848G.
- J. Zhang and Z. Tao, Synthesis, 2025,(20), 2997–3004, DOI:10.1055/a-2649-4768.
- K. Muñiz, A. Iesato and M. Nieger, Chem. – Eur. J., 2003, 9(22), 5581–5596, DOI:10.1002/chem.200305011.
- A. O. Chong, K. Oshima and K. B. Sharpless, J. Am. Chem. Soc., 1977, 99(10), 3420–3426, DOI:10.1021/ja00452a039.
- G. Li, H. S. Kim and H. X. Wei, Tetrahedron Lett., 2000, 41(45), 8699–8703, DOI:10.1016/S0040-4039(00)01579-3.
- H. X. Wei, S. Siruta and G. Li, Tetrahedron Lett., 2002, 43(21), 3809–3812, DOI:10.1016/S0040-4039(02)00694-9.
- M. Es-Sayed, C. Gratkowski, N. Krass, A. Meyers and A. Meijere, Synlett, 1992, 962–964, DOI:10.1055/s-1992-21545.
- A. S. Sagiyan, A. E. Avetisyan and S. M. Djamgaryan, Russ. Chem. Bull., 1997, 46(3), 483–486, DOI:10.1007/BF02495400.
- Y. N. Belokon’, A. S. Sagyan, S. M. Djamgaryan, V. I. Bakhmutov and V. M. Belikov, Tetrahedron, 1988, 44(17), 5507–5514, DOI:10.1016/S0040-4020(01)86056-7.
- A. S. Saghyan, G. M. Mkrtchyan, A. S. Dadayan, S. G. Petrosyan, A. V. Geolchanyan, H. M. Simonyan, A. F. Mkrtchyan, S. Mkrtchyan, A. Gevorgyan, V. O. Iaroshenko and P. Langer, Tetrahedron: Asymmetry, 2013, 24(4), 229–232, DOI:10.1016/j.tetasy.2013.01.014.
- A. S. Saghyan, H. M. Simonyan, S. G. Petrosyan, A. V. Geolchanyan, G. N. Roviello, D. Musumeci and V. Roviello, Amino Acids, 2014, 46(10), 2325–2332, DOI:10.1007/s00726-014-1782-3.
- E. A. Khachatryan, L. Yu. Sahakyan, A. S. Tovmasyan, G. S. Melikyan, H. A. Panosyan, A. F. Mkrtchyan, N. Shibata, A. V. Malkov and A. S. Saghyan, RSC Adv., 2025, 15(14), 10558–10564, 10.1039/d5ra00910c.
- A. S. Poghosyan, E. A. Khachatryan, A. F. Mkrtchyan, V. Mirzoyan, A. M. Hovhannisyan, K. R. Ghazaryan, E. V. Minasyan, P. Langer and A. S. Saghyan, Amino Acids, 2024, 56(1), 67, DOI:10.1007/s00726-024-03430-5.
- U. Eckhard, E. Schönauer and H. Brandstetter, J. Biol. Chem., 2013, 288(28), 20184–20194, DOI:10.1074/jbc.M112.448548.
- E. O. Chukhajian, L. V. Ayrapetyan, H. S. Mkrtchyan and H. A. Panosyan, Russ. J. Org. Chem., 2020, 56(2), 353–355, DOI:10.1134/S1070428020010311.
- J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, J. Chem. Inf. Model., 2021, 61(8), 3891–3898, DOI:10.1021/acs.jcim.1c00203.
- A. S. Sargsyan, L. T. Karapetyan, A. V. Mkhitaryan, L. A. Stepanyan, T. H. Sargsyan, Yu. M. Danghyan, A. V. Sargsyan, G. G. Oganezova and N. A. Hovhannisyan, Biomed. Khim., 2024, 70(6), 413–420, DOI:10.18097/pbmc20247006413.
- CCDC 2486969: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2pgwtw.
| This journal is © The Royal Society of Chemistry 2026 |