DOI:
10.1039/D5MH01550B
(Communication)
Mater. Horiz., 2026,
13, 480-490
Mechanically robust and high latent heat solid–solid phase change materials via a H-bonding collaborative strategy for energy storage and conversion
Received
12th August 2025
, Accepted 29th September 2025
First published on 30th September 2025
Abstract
Solid–solid phase change materials (SSPCMs) historically struggle to achieve an optimal balance between mechanical robustness and efficient thermal energy storage (TES) capacity. To overcome this limitation, we engineered a hierarchical hydrogen-bond array by integrating quadruple hydrogen-bonded UPy dimers (strong bonds) with urethane linkages (weak bonds). This synergistic network forms a crosslinked structure at room temperature, conferring exceptional mechanical strength (29.3 MPa) to the SSPCMs. Concurrently, the material achieves a high phase change component content (92 wt%) and delivers substantial latent heat (133.7 J g−1). This dual-functionality strategy yields comprehensive performance exceeding that of most previously reported SSPCMs. Furthermore, the dynamic hydrogen-bond network imparts multiple advanced functionalities, including excellent recyclability, shape memory, and self-healing capabilities. Critically, the hydrogen-bonding mechanism mitigates the aggregation of hydroxylated multi-walled carbon nanotubes (MWCNTs), ensuring uniform dispersion within the SSPCM matrix. This advancement facilitates practical implementation in photothermal conversion and low-pressure Joule heating applications. Our supramolecular design strategy thus establishes a new paradigm for sustainable energy storage materials that simultaneously possess high mechanical integrity and significant latent heat capacity.
New concepts
Solid–solid phase change materials (SSPCMs) hold potential for thermal energy storage (TES), yet face the mutually exclusive issue of mechanical robustness and high TES capacity. Covalently cross-linked SSPCMs exhibit strength but lack recyclability/self-healability, while supramolecular SSPCMs fail under high PCM loading. Herein, a hierarchical hydrogen-bond network (strong and weak hydrogen bonds) addresses this issue. Meanwhile, reversible hydrogen bonds enable recyclability, shape memory, and self-healing. Finally, the hydrogen-bond strategy eliminates the aggregation of hydroxylated MWCNTs at specific loadings, enhances photothermal conversion and low-voltage Joule heating, and bridges SSPCMs with practical devices. This provides a novel strategy for high-performance TES materials in sustainable energy and flexible systems.
|
1 Introduction
Organic phase change materials (PCMs) leverage latent heat absorption/release during solid–liquid transitions for TES, yet exhibit critical limitations for advanced applications. While organic PCMs have garnered significant research interest due to their inherent thermodynamic properties,1 their practical deployment in strategic domains, including deep space exploration, military systems, biomedical devices, electronics, and solar thermal conversion,2 is hindered by leakage and volumetric expansion during the phase transition.3 To overcome these constraints, polymeric solid–solid PCMs (SSPCMs) have emerged as a promising alternative, eliminating the need for encapsulation or structural supports while enhancing mechanical integrity.4 Conventional SSPCM synthesis typically involves chemical functionalization or supramolecular cross-linking of organic PCM precursors (e.g., fatty acids, fatty alcohols, or polyethylene glycol (PEG)) to construct polymer matrices that prevent leakage, retain thermal capacity, and improve mechanical performance. For instance, Shi et al.5 engineered cross-linked SSPCM using PEG as a soft segment and hexamethylene diisocyanate (HDI) as a cross-linker. By precisely controlling the PEG molecular weight and cross-linking density, their design optimized the synergistic interaction between PEG's phase-change characteristics and HDI's structural properties, yielding materials with exceptional flexibility capable of repeated bending, folding, and torsional deformation without failure.
While covalent cross-linked SSPCMs exhibit superior leakage resistance, structural stability, and mechanical integrities, their permanent network architecture precludes intrinsic self-healing capability.6 Consequently, supramolecular SSPCMs leveraging non-covalent interactions, including π–π stacking, hydrogen bonding, and metal coordination, have emerged as a promising alternative.7,8 Recent advances demonstrate diverse design strategies: Lin et al.9 engineered a Diels–Alder-based polyurethane network to encapsulate PEG, achieving multi-cycle stability and reprocessability; Chang et al.10 systematically investigated molecular weight effects by crosslinking isophorone diisocyanate (IPDI) with PEG variants; and Kong et al.11 established linear SSPCMs through π–π stacking-induced physical crosslinks, effectively suppressing PEG leakage while enhancing material toughness and achieving a maximum enthalpy of 123.4 J g−1. Nevertheless, supramolecular SSPCMs face critical limitations: recyclability often requires sophisticated molecular engineering, and weak non-covalent crosslinks typically compromise mechanical strength and structural stability.12 Although dynamic covalent bonds (e.g., disulfides, Diels–Alder adducts) partially mitigate these issues, they frequently reduce TES density or degrade thermal stability at high PCM loadings (>90 wt%). Hydrogen bonding networks present a viable pathway to reconcile these trade-offs through reversible yet robust interactions. However, current systems lack optimized energy dissipation mechanisms, resulting in either brittle fracture or diminished latent heat capacity.13
This study introduces a supramolecular design strategy that synergistically integrates quadruple hydrogen-bonded UPy dimers (UPy–UPy) with polyurethane-based urethane linkages to resolve the persistent trade-off between mechanical robustness and TES capacity in SSPCMs. Specifically, PEG served as the soft segment, IPDI as the hard segment precursor, and 5-(2-hydroxyethyl)-6-methyl-2-aminouracil (HMAU) as the UPy-functionalized crosslinker. At ambient temperature, these components self-assemble into a three-dimensional network through cooperative strong (UPy–UPy) and weak (urethane–urethane) hydrogen-bond interactions. This dual-network architecture simultaneously achieves exceptional mechanical strength (29.3 MPa) and high TES density (133.7 J g−1) at 92 wt% PEG loading, demonstrated by retained solid-state integrity after 30 min at 80 °C. The strategy leverages UPy–UPy bonds to maintain structural stability while maximizing PEG content for latent heat delivery, thereby surpassing the mechanical–thermal performance balance reported in >90% of prior SSPCM studies. Critically, the dynamic hydrogen-bond network enables homogeneous dispersion of hydroxylated MWCNTs, facilitating efficient photothermal conversion and low-pressure Joule heating. This multifunctional platform overcomes the traditional dichotomy between mechanical integrity and energy density, with demonstrated applicability in wearable thermal management systems, flexible electronics thermal regulation, and targeted hyperthermia therapies.
2. Results and discussion
2.1. Design and characterization of HPCMs
This study presents a supramolecular strategy for engineering high-performance SSPCMs through quadruple hydrogen-bonded UPy–UPy crosslinking. PEG functions as the soft segment, providing dynamic amorphous–crystalline phase transitions for TES, while IPDI and HMAU constitute the hard segment network (synthesis and 1H NMR characterization detailed in SI Fig. S1 and S2). The resulting hydrogen-bonded PCMs (HPCMs) (Fig. 1a and Fig. S3 for the detailed synthetic route) exhibit a dual-network architecture where: (i) PEG undergoes reversible crystalline–amorphous transitions during heating/cooling cycles to store/release latent heat, and (ii) the IPDI-HMAU hard segment forms a mechanically robust scaffold through cooperative strong (UPy–UPy) and weak (urethane–urethane) hydrogen bonds.14 This design leverages three critical features of the supramolecular network: (a) thermodynamic stability of quadruple hydrogen-bonded UPy dimers, (b) rapid bond reversibility enabling structural recovery, and (c) energy dissipation via weaker hydrogen bonds. As illustrated in Fig. 1b and c, when the temperature exceeds PEG's crystalline-to-amorphous transition point, energy is stored through melting; conversely, cooling below this threshold triggers recrystallization and heat release. Crucially, the dynamic crosslinking network prevents PEG leakage throughout the phase transitions. To systematically evaluate molecular weight effects on multifunctional performance, we synthesized a series of HPCMs with varying PEG molecular weights (Table S1), assessing their latent heat capacity, mechanical integrity, and thermal stability.
 |
| | Fig. 1 (a) The structural formula of HPCMs. (b) Schematic illustration of the working mechanism of the HPCMs on thermal energy storage/release and a stretching process. (c) Classification of strong hydrogen bonds and weak hydrogen bonds. | |
Fourier transform infrared (FT-IR) spectroscopy confirms the structural evolution during the synthesis of HPCMs. As shown in Fig. 2a, PEG exhibits characteristic absorptions at 1100 cm−1, corresponding to C–O–C symmetric stretching along with peaks at 3454 cm−1 for O–H stretching and 2876, 1461, 959, and 839 cm−1 for C–H vibrations. Critically, the HPCMs display complete disappearance of PEG's O–H stretching peak at 3447 cm−1 and IPDI's –NCO absorption at 2278 cm−1, confirming reaction completion.15 New peaks emerge at 1540 for N–H bending vibrations and at 1661, 1716, and 3336 cm−1 for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and N–H stretching vibrations in urethane and urea groups.16,17 The intensity of the 3336 cm−1 peak increases progressively from HPCM4K to HPCM20K, as shown in Fig. S5, indicating elevated urea content with higher PEG molecular weight. This suggests that increasing the molecular weight enhances hydrogen bonding within the system. These findings strongly support the successful synthesis of HPCMs. Further verification was obtained via1H NMR spectroscopy (Fig. S4). Peak deconvolution analysis of the 1650–1750 cm−1 spectral region quantified hydrogen bonding interactions in the synthesized materials. Hydrogen bonding modifies electron density and vibrational characteristics of carbonyl groups, producing spectral broadening that was resolved into hydrogen-bonded and free CO components through curve fitting. This analysis determined that HPCM10K contains 80.1% hydrogen-bonded carbonyl groups, as shown in Fig. 2b. Such high hydrogen-bonding density establishes a robust physical crosslinking network that uniformly distributes applied tensile stress throughout the material, preventing localized stress concentration and premature fracture. Consequently, these materials exhibit exceptional tensile strength. Furthermore, to further verify the hydrogen bond interactions, we collected the FTIR spectra of HPCM10K after heating from 30 to 170 °C. As shown in Fig. 2c, the two-dimensional perturbation–correlation moving-window (PCMW2D) spectroscopy reveals the temperature-dependent behavior of multiple hydrogen bonds. The absorbance intensities of the hydrogen bond-associated CO stretching vibration peaks—corresponding to urea groups around 1659 cm−1 and urethane groups around 1717 cm−1—gradually decrease with increasing temperature, indicating a gradual reduction in hydrogen bond strength as temperature rises. Specifically, the peak intensity of the CO stretching vibration of urethane groups decreases rapidly after approximately 119 °C. However, this characteristic (rapid intensity decrease) for the CO stretching vibration peak of urea groups only emerges after the temperature reaches as high as ∼142 °C. This observation suggests that the strong hydrogen bonds (UPy–UPy) exhibit higher thermal stability compared to the weak hydrogen bonds (urethane–urethane). When heated to 170 °C, the hydrogen bond-associated characteristic peaks still maintain a certain intensity, demonstrating significant stability of the hydrogen bonds in this system. Additionally, the absorbance intensity of the free N–H groups observed at 3323 cm−1 shows a positive correlation with temperature, i.e., it gradually increases as the temperature rises (Fig. 2d). This phenomenon arises because hydrogen bonds gradually dissociate with increasing temperature, thereby generating freer N–H groups. This result further provides evidence for the continuous dissociation of hydrogen bonds. Mechanical characterization confirmed these properties using standard tensile testing methodology. As shown in Fig. 2e, all HPCMs maintain elongation at break exceeding 700%. Tensile strength progressively increases from 14.6 to 29.3 MPa with rising PEG molecular weight, correlating directly with enhanced hydrogen bonding density. At strains below 30%, the stress–strain curves display linear elastic behavior where hard segments provide rigid structural support that prevents premature plastic deformation (Fig. S6a). As strain increases, the curves transition through a nonlinear regime characterized by gradually decreasing slope without abrupt yield point drop. This distinctive mechanical response stems from dynamic hydrogen bond dissociation that dissipates energy gradually, thereby avoiding stress concentration and catastrophic failure. The consistent high ductility aligns with the observed 700% minimum elongation. Yield strength similarly increases with molecular weight as shown in Fig. S6b. Notably, these materials retain 29.3 MPa tensile strength despite containing 92 wt% phase-change components, indicating exceptional mechanical performance for such high active material loading. Reproducible tensile properties across multiple specimens confirm uniform stress distribution and homogeneous hydrogen bond network formation throughout the material matrix as evidenced in Fig. S6c and d.
O and N–H stretching vibrations in urethane and urea groups.16,17 The intensity of the 3336 cm−1 peak increases progressively from HPCM4K to HPCM20K, as shown in Fig. S5, indicating elevated urea content with higher PEG molecular weight. This suggests that increasing the molecular weight enhances hydrogen bonding within the system. These findings strongly support the successful synthesis of HPCMs. Further verification was obtained via1H NMR spectroscopy (Fig. S4). Peak deconvolution analysis of the 1650–1750 cm−1 spectral region quantified hydrogen bonding interactions in the synthesized materials. Hydrogen bonding modifies electron density and vibrational characteristics of carbonyl groups, producing spectral broadening that was resolved into hydrogen-bonded and free CO components through curve fitting. This analysis determined that HPCM10K contains 80.1% hydrogen-bonded carbonyl groups, as shown in Fig. 2b. Such high hydrogen-bonding density establishes a robust physical crosslinking network that uniformly distributes applied tensile stress throughout the material, preventing localized stress concentration and premature fracture. Consequently, these materials exhibit exceptional tensile strength. Furthermore, to further verify the hydrogen bond interactions, we collected the FTIR spectra of HPCM10K after heating from 30 to 170 °C. As shown in Fig. 2c, the two-dimensional perturbation–correlation moving-window (PCMW2D) spectroscopy reveals the temperature-dependent behavior of multiple hydrogen bonds. The absorbance intensities of the hydrogen bond-associated CO stretching vibration peaks—corresponding to urea groups around 1659 cm−1 and urethane groups around 1717 cm−1—gradually decrease with increasing temperature, indicating a gradual reduction in hydrogen bond strength as temperature rises. Specifically, the peak intensity of the CO stretching vibration of urethane groups decreases rapidly after approximately 119 °C. However, this characteristic (rapid intensity decrease) for the CO stretching vibration peak of urea groups only emerges after the temperature reaches as high as ∼142 °C. This observation suggests that the strong hydrogen bonds (UPy–UPy) exhibit higher thermal stability compared to the weak hydrogen bonds (urethane–urethane). When heated to 170 °C, the hydrogen bond-associated characteristic peaks still maintain a certain intensity, demonstrating significant stability of the hydrogen bonds in this system. Additionally, the absorbance intensity of the free N–H groups observed at 3323 cm−1 shows a positive correlation with temperature, i.e., it gradually increases as the temperature rises (Fig. 2d). This phenomenon arises because hydrogen bonds gradually dissociate with increasing temperature, thereby generating freer N–H groups. This result further provides evidence for the continuous dissociation of hydrogen bonds. Mechanical characterization confirmed these properties using standard tensile testing methodology. As shown in Fig. 2e, all HPCMs maintain elongation at break exceeding 700%. Tensile strength progressively increases from 14.6 to 29.3 MPa with rising PEG molecular weight, correlating directly with enhanced hydrogen bonding density. At strains below 30%, the stress–strain curves display linear elastic behavior where hard segments provide rigid structural support that prevents premature plastic deformation (Fig. S6a). As strain increases, the curves transition through a nonlinear regime characterized by gradually decreasing slope without abrupt yield point drop. This distinctive mechanical response stems from dynamic hydrogen bond dissociation that dissipates energy gradually, thereby avoiding stress concentration and catastrophic failure. The consistent high ductility aligns with the observed 700% minimum elongation. Yield strength similarly increases with molecular weight as shown in Fig. S6b. Notably, these materials retain 29.3 MPa tensile strength despite containing 92 wt% phase-change components, indicating exceptional mechanical performance for such high active material loading. Reproducible tensile properties across multiple specimens confirm uniform stress distribution and homogeneous hydrogen bond network formation throughout the material matrix as evidenced in Fig. S6c and d.
 |
| | Fig. 2 (a) Fourier transforms infrared spectra of PEG and HPCM. (b) Hydrogen bond CO percentage of HPCM10K. (c) Synchronous PCMW2D spectrum of HPCM10K (1620–1740 cm−1). (d) Synchronous PCMW2D spectrum of HPCM10K (3200–3400 cm−1). (e) Stress–strain curve of HPCM at 25 °C. (f) HPCM10K stretches photos of different lengths. (g) HPCM10K can bend, twist, fold and crop photos of various shapes. | |
Fig. 2f demonstrates HPCM10K achieving 800% elongation which confirms its exceptional ductility. These mechanical properties arise from a three-stage energy dissipation mechanism beginning with crack propagation resistance through dissociation of weak urethane hydrogen bonds (ΔE ≈ 8 kcal mol−1), followed by energy absorption via UPy dimer dissociation (ΔE ≈ 25 kcal mol−1), and concluding with reinforcement through PEG chain alignment and crystallization (Fig. 1b). Furthermore, these flexible HPCMs can be easily twisted, folded, and cut into various shapes (Fig. 2g and Fig. S7), making them suitable for diverse application scenarios—such as those requiring complex geometries and flexible device designs. Of particular interest is the thermally activated shape memory function of HPCMs. As shown in Fig. S8, an HPCM10K strip was first heated to 80 °C (i), then plastically deformed into the number “6” under external force (ii) and subsequently cooled to room temperature to obtain a shape-fixed number “6” (iii). In a similar manner, it could also be plastically formed into the letter “m” (vii). Meanwhile, all these deformed shapes can be fully restored to their original state. This phenomenon is mainly attributed to the thermal response characteristics of PEG: when heated to 80 °C, the activity of the PEG chains inside HPCM10K is activated and their fluidity is regained, enabling the reshaping of the material. Afterward, when the material is cooled to room temperature, it can maintain the temporary deformed shape without external force. If the temperature is raised above 80 °C again, the PEG chains will regain fluidity, allowing the temporary shape to easily revert to its original state. This thermally activated shape memory function is expected to expand the applications of PCMs in wearable technologies and complex device configurations.
2.2. Phase transition and crystallization of HPCMs
The degree of crystallinity in PCMs significantly influences their enthalpy change capacity. X-ray diffraction (XRD) analysis revealed that both pure PEG and the synthesized HPCMs exhibit comparable diffraction peaks at 2θ = 19.5°, 23.7°, 26.6°, and 36.5°, corresponding to the (120), (112), (210), and (212) lattice planes, respectively (Fig. 3a). This indicates that the HPCMs retain a crystalline structure analogous to PEG, confirming that the crosslinking of hard segments does not disrupt the crystal structure of the PEG chains. To further investigate crystallinity, polarized optical microscopy (POM) was employed. At 25 °C, PEG10K, HPCM4K, and HPCM10K all displayed distinct spherical crystalline morphologies with cross-extinction patterns, corroborating the XRD findings and confirming the inheritance of PEG's crystalline structure (Fig. 3b). However, the presence of hard segments and hydrogen bonding suppresses PEG chain crystallization, resulting in smaller spherical crystals in the HPCMs. Notably, the spherical crystal size increases with higher PEG molecular weights. At 80 °C, the spherical structure of HPCM10K disappears, yet no liquid phase forms, demonstrating that even with 92% phase-change components, HPCMs maintain solid-state phase transitions (Fig. S9). Differential scanning calorimetry (DSC) was further used to analyze the enthalpy changes and phase transition temperatures. All samples exhibited similar endothermic and exothermic behaviors, confirming their latent heat storage capabilities (Fig. 3c, Fig. S10, and Table S3). The constraining effect of the hard segments and hydrogen bonds on the PEG chain mobility results in lower melting enthalpies for the HPCMs (HPCM4K: 96.08 J g−1, HPCM6K: 106.69 J g−1, HPCM10K: 127.79 J g−1, and HPCM20K: 133.70 J g−1) compared to their pure PEG counterparts (PEG4K: 190.21 J g−1, PEG6K: 188.42 J g−1, PEG10K: 185.67 J g−1, PEG20K: 177.08 J g−1) (Fig. 3d and Table S3). Despite the reduction, the HPCMs maintain remarkable latent heat performance. To quantify the effect of the hard segments, the phase transition latent heat efficiency (λ) was calculated. The λ values for HPCM4K, HPCM6K, HPCM10K, and HPCM20K were 54.90%, 61.55%, 74.81%, and 82.07%, respectively (Table S3). This trend of increasing efficiency with higher PEG molecular weight is attributed to a lower terminal hydroxyl group density, which reduces the number of hard segment binding sites and thus weakens their constraining effect on the PEG chains. The energy storage capacity of HPCM10K and HPCM20K was compared with other SSPCMs reported in the literature (Table S4).18–27 The results demonstrate that the prepared HPCMs exhibit superior enthalpy changes. Furthermore, when compared to the tensile strength and enthalpy changes of other SSPCMs and flexible PCMs, our materials demonstrate superior comprehensive performance, achieving an ultra-high stress of 29.3 MPa alongside a high enthalpy change of 133.7 J g−1 (Fig. 3e).28–46
 |
| | Fig. 3 (a) XRD patterns of PEG10K and HPCM. (b) POM images of PEG10K, HCPM4K and HPCM10K at 25 °C. (c) DSC curve of HPCM. (d) Enthalpy of crystallization and melting of HPCM. (e) HPCM20K was compared with other reported PCMs in terms of tensile stress and phase transition enthalpy. | |
2.3. Solid–solid characterization and leakage resistance of HPCMs
PCMs with crosslinked structures typically exhibit solid–solid phase transition behavior. To investigate this property in the HPCMs, rheological tests were conducted at elevated temperatures. As shown in Fig. 4a–d, the storage modulus (G′) remains greater than the loss modulus (G′′) across the entire tested frequency range (0.1 to 100 Hz) at a constant temperature of 120 °C. This indicates the presence of a stable, elastic-dominant network, strongly suggesting an intertwined or crosslinked structure within the material. Notably, even HPCM20K, which contains 92 wt% phase-change components, maintains a storage modulus that exceeds its loss modulus at 120 °C. This confirms the material's ability to retain excellent solid–solid phase transition properties despite its high PCM content. To further validate these findings, leakage tests were performed. HPCM samples of identical dimensions (10 × 10 × 5 mm) were placed on a hot plate at 30 °C and 80 °C for 30 minutes, during which no leakage was observed (Fig. 4e). In contrast, pure PEG10K leaked completely. When the temperature was increased to 100 °C and 120 °C for 30 minutes, the HPCMs maintained their structural integrity and solid-state properties, consistent with the rheological data. This stability is attributed to a synergistic effect of strong and weak hydrogen bonds, which form a dense dynamic crosslinking network that effectively prevents the leakage of the PEG chains. The leakage rate was quantitatively assessed by placing samples in Petri dishes on qualitative filter paper and incubating them at constant temperatures of 30 °C, 80 °C, 100 °C, and 120 °C for 30 minutes each. After cooling to room temperature, the samples were weighed. The leakage rate (γ) was calculated using eqn (1), defined as the ratio of the sample mass after heat treatment (mi) to the initial mass (m0). As shown in Fig. S11a, the leakage rate of the HPCMs increases gradually with temperature. This is primarily due to the partial disruption of the hydrogen bond network at high temperatures, coupled with enhanced PEG chain mobility, which collectively contributes to a gradual increase in leakage. Nevertheless, the leakage rates for all HPCM samples remain below 2.5%, demonstrating their excellent encapsulation capability. This performance is a result of the dense hydrogen-bonded network formed by the hard segments, which effectively confines the PEG chains and enables outstanding solid-state characteristics. Additionally, the HPCMs were immersed in N,N-dimethylacetamide (DMAc) at room temperature to assess solvent resistance. After five days, the pure PEG samples (PEG4K, PEG6K, PEG10K, and PEG20K) had dissolved completely, whereas the HPCMs exhibited only slight swelling after 15 days without dissolving (Fig. S11b). This result confirms the successful formation of a permanent crosslinked network in the HPCMs, providing a robust structural foundation for their solid–solid phase transition behavior.| | 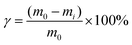 | (1) |
 |
| | Fig. 4 Rheological curves of (a) HPCM4K, (b) HPCM6K, (c) HPCM10K and (d) HPCM20K at 120 °C. (e) HPCM photographs of leakage tests performed at 30 min at 30, 80, 100 and 120 °C. | |
2.4. Thermal reliability and recyclability of HPCMs
Thermal stability is a critical parameter for evaluating the energy storage performance of PCMs. The thermal stability of the HPCMs was assessed using thermogravimetric analysis (TGA). As shown in Fig. S12, the initial decomposition temperatures (Td,onset) for HPCM4K, HPCM6K, HPCM10K, and HPCM20K were 284.2 °C, 290.6 °C, 290.6 °C, and 281.3 °C, respectively. The temperature at 50% mass loss (T50%) for these HPCMs was 386.8 °C, 385.6 °C, 386.4 °C, and 386.1 °C, respectively, indicating excellent thermal stability across a wide temperature range (up to 280 °C). To further evaluate the energy storage reliability and chemical stability, the HPCMs were characterized by DSC and FT-IR after 500 thermal cycles. The DSC curves of all HPCMs remained nearly identical before and after 500 cycles (Fig. 5a), with only minimal fluctuations in melting and solidification enthalpy (Fig. 5b). This demonstrates that the prepared HPCMs retain their energy storage capacity after extensive thermal cycling. The latent heat change rates for all HPCMs were below 2%, as detailed in Table S3, with HPCM20K exhibiting the lowest rate of 0.32%. This further confirms the reliable energy storage performance of the HPCMs. Furthermore, the FT-IR spectra of the HPCMs before and after 500 cycles show a perfect overlap between the cycled samples (dashed lines) and the original samples (solid lines) (Fig. 5c and Fig. S13), indicating excellent chemical stability after repeated thermal cycles.
 |
| | Fig. 5 (a) DSC curves of HPCMs before and after 500 thermal cycles (solid line is the curve before the thermal cycle of HPCMs; C-HPCMs is the curve after 100 thermal cycles (dashed line)). (b) Enthalpies of crystallization and melting of HPCMs and C-HPCMs. (c) Infrared spectra of HPCMs and C-HPCMs before and after 500 cycles of thermal cycling (solid line is the curve before thermal cycling of HPCMs; C-HPCMs is the curve after 500 cycles of thermal cycling (dashed line)). (d) Flowchart of the recovery of HPCM10K by the dissolution–crosslinking method. (e) Comparison of infrared spectra of HPCM10K before and after recovery. (f) Comparison of stress–strain curves before and after HPCM10K recovery. | |
Furthermore, we successfully demonstrated the efficient recovery of HPCMs through a process of degradation and re-crosslinking, facilitated by the reversible dissociation of hydrogen bonds. As shown in Fig. 5d, cut HPCM10K sheets were placed in a test tube containing DMAc and heated at 120 °C. Significant dissolution began within the first 5 minutes. After 10 minutes, the boundaries between the dissolving sheets blurred, and the mixture transitioned to a gel-like consistency. Following a total of 15 minutes of heating, the HPCM10K was completely dissolved, forming a homogeneous, pale-yellow solution. During this process, the hydrogen-bonded crosslinks dissociated into free amino and hydroxyl groups, inducing a gel-to-sol transition in the polymer network. Subsequently, heating at 80 °C to evaporate the solvent enabled the reconstruction of the crosslinked network structure, resulting in a reformed, intact HPCM film. The chemical integrity of HPCM10K throughout this degradation–recrosslinking cycle was investigated using FT-IR spectroscopy. As shown in Fig. 5e, the spectra of the recovered sample showed no significant differences from the initial material after the 120 °C treatment. Tensile tests further indicated that the recovered HPCM10K retained excellent mechanical properties, with a strain of 1784% and a tensile strength of 24.6 MPa (Fig. 5f). These values are close to those of the original sample (1931% strain and 26.8 MPa tensile strength), demonstrating that the material exhibits excellent mechanical stability after dissolution and recrosslinking. This process enables the rapid recovery of SSPCMs with the addition of a solvent. The recyclability of HPCMs was also confirmed via a conventional hot-pressing method. As shown in Fig. S14a and b, hot-pressing successfully restored the film state without inducing significant changes in the FT-IR spectra, indicating the stability of the chemical structure. Moreover, the stress–strain curves of the material subjected to 5 hot-pressing cycles showed no obvious decline in mechanical properties (Fig. S14c). Both stress and strain values remain above 90% of the original sample's values, confirming reliable recyclability. This robust recyclability effectively alleviates environmental burdens, minimizes resource waste, and helps prevent environmental pollution.
2.5. Photothermal conversion of composites
To evaluate the potential of HPCMs for solar energy applications, hydroxylated MWCNTs were incorporated into the HPCM10K matrix at loadings of 1 wt% and 3 wt% during synthesis (Fig. S15a). Morphological analysis revealed that pure HPCM10K possessed a smooth, flat cross-section (Fig. S15b), whereas the composite films exhibited uneven surfaces with protrusions. MWCNTs were sparsely distributed in the HPCM10K-1% composite (Fig. S15c) but formed a dense, continuous network in HPCM10K-3% (Fig. S15d); this morphology is conducive to enhanced photothermal conversion. The photothermal performance was evaluated using a solar simulator (xenon lamp, 100 mW cm−2, 1 sun) with sample temperatures monitored by an infrared camera (Fig. 6a). Under irradiation, the HPCM10K-1% film rapidly heated to ∼84 °C within 100 s, exhibiting a distinct phase transition plateau at ∼45 °C during heating (energy storage) and ∼39 °C during cooling (energy release; Fig. 6b). The HPCM10K-3% composite demonstrated a faster heating rate, reaching ∼94 °C, with more pronounced phase plateaus. In contrast, the blank HPCM10K sample reached only ∼37 °C with no observable plateaus. Infrared thermography confirmed the excellent shape stability of the HPCM-based composites during energy storage and release (Fig. 6c), while a pure PEG10K composite with 3% MWCNTs collapsed and leaked under identical conditions (Fig. 6d). The photothermal conversion efficiency, calculated using eqn (2),47 was 71.4% for HPCM10K-1% and 92.4% for HPCM10K-3% (Fig. 6e), a result consistent with the observed MWCNT distribution. Ultraviolet-visible-near infrared (UV-vis-NIR) spectroscopy corroborated these findings (Fig. S16a): HPCM10K showed a weak optical response across 200–2500 nm, while the addition of MWCNTs significantly enhanced light absorption, with HPCM10K-3% exhibiting the strongest absorption, a critical factor for efficient photothermal conversion. Furthermore, the higher MWCNT loading created continuous thermal conduction pathways via hydrogen bonding with the matrix, thereby accelerating heat transfer. This resulted in a higher thermal conductivity (Fig. S16b) and a shortened phase transition plateau.48 The HPCM10K-3% composite exhibited robust performance across irradiances from 25 to 150 mW cm−2, with surface temperatures reaching 40.2, 68.1, 99.3, and 120.1 °C, respectively (Fig. 6f). The phase transition time decreased while the efficiency increased from 90.7% to 93.1% (Fig. 6g), attributed to accelerated heat transfer at higher light intensities. Excellent cycling stability was confirmed over 4000 s, with minimal variation in the temperature-time curves and efficiency (Fig. 6h). The composite also demonstrated long-term reliability, maintaining a stable temperature of 99.3 °C for 5000 s under 100 mW cm−2 irradiation (Fig. 6i). Overall, these results indicate that this composite technology holds significant promise for advanced solar energy conversion applications.| | 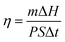 | (2) |
where η is the photothermal conversion efficiency, m and ΔH are the mass and melting enthalpy of the composite PCM, P and S are the power and optical surface area of the simulated sunlight, and Δt is the phase transition time, respectively.
 |
| | Fig. 6 (a) Schematic diagram of photothermal conversion. (b) Temperature vs. time curves (100 mW cm−2) for HPCM10K, HPCM10K-1% and HPCM10K-3%. (c) Infrared thermography of HPCM10K, HPCM10K-1% and HPCM10K-3%. (d) Physical and infrared thermography of PEG10K-3% (100 mW cm−2). (e) Photothermal conversion efficiency of HPCM10K, HPCM10K-1% and HPCM10K-3%. (f) Infrared thermography of HPCM10K-3% at different light intensities. (g) Saturation temperature and conversion efficiency of HPCM10K-3% at different light intensities. (h) HPCM10K-3% curves under multiple photothermal cycles. (i) Stability curve of HPCM10K-3% under 100 mW cm−2 illumination for up to 5000 s. | |
2.6. Electro-thermal conversion of the composite materials
In contrast to insulating PEG, the HPCM10K-X composites exhibit measurable electrical conductivity (Fig. 7a). Consequently, HPCM10K-3% not only demonstrates excellent photothermal conversion but also enables efficient Joule heating. The surface temperature evolution of HPCM10K-3% under an applied voltage ranging from 0.1 to 0.5 V is presented in Fig. 7b. The saturation temperature (Ts) increased significantly with applied voltage, consistent with Joule's law (Q = U2R−1t, where Q is Joule heat, U is voltage, t is time, and R is resistance). Specifically, the saturation temperatures of 26.1 °C, 31.2 °C, 33.8 °C, 39.7 °C, and 42.6 °C were achieved at 0.1, 0.2, 0.3, 0.4, and 0.5 V, respectively. These results indicate excellent electrothermal sensitivity and ideal low-voltage Joule heating characteristics. Furthermore, the temperature of HPCM10K-3% could be rapidly and stably controlled by modulating the external voltage between 0.1–0.5 V or vice versa, highlighting its potential as a responsive electric heater (Fig. 7c). The saturation temperatures at different voltages were visualized using gradient infrared thermography, which confirmed that the surface temperature increased immediately with driving voltage, demonstrating straightforward and precise Joule heating behavior (Fig. 7d). The infrared images also revealed a highly uniform temperature distribution across the composite surface, with minimal localized variation, a result attributed to the homogeneous dispersion of MWCNTs within the matrix. To assess long-term operational stability, cyclic heating/cooling tests were conducted under a 0.5 V driving voltage (Fig. 7e). The temperature profiles exhibited consistent shape and trend over multiple cycles, confirming outstanding repeatability and stability in heating performance. These results highlight the composite's advantages of low-voltage operation, rapid response, and cycling stability, thereby mitigating the instability risks associated with conventional high-voltage heating. This superior Joule heating functionality is promising for applications in medical thermotherapy and wearable devices. Finally, HPCM10K-3% was heated to 42 °C at 0.5 V and maintained this temperature steadily for 4000 s (Fig. 7f), demonstrating reliable long-term stable electrothermal performance.
 |
| | Fig. 7 (a) Conductivity of PEG, HPCM10K-1% and HPCM10K-3%. (b) Heating/cooling curves of HPCM10K-3% at different voltages. (c) The heating/cooling curves of HPCM10K-3% under different continuous voltages. (d) IR thermography of the saturation temperature of HPCM10K-3% at different voltages. (e) Cycling curves of HPCM10K-3% at repeated ramp-up/down temperatures. (f) Thermal stability curve of HPCM10K-3% at 0.5 V for up to 4000 s. | |
3. Conclusion
This study presents a synergistic strategy that integrates quadruple hydrogen bonds (UPy–UPy) with polyurethane hydrogen bonds to reconcile the longstanding trade-off between mechanical robustness and high latent heat in SSPCMs. The material was formulated with PEG as the soft segment, IPDI as the hard segment, and HMAU as a crosslinking agent. At room temperature, a cross-linked three-dimensional network forms through the cooperative action of UPy–UPy and urethane–urethane hydrogen bonds. This dual hydrogen-bonding system imparts exceptional mechanical strength (29.3 MPa) and ensures the retention of the solid-state characteristics, even with a PEG content of 92 wt% after exposure to 80 °C for 30 minutes. Meanwhile, the high PCM content yields a remarkable enthalpy of 133.7 J g−1. By strategically utilizing strong UPy–UPy hydrogen bonds for mechanical integrity and high-content PEG for latent heat, this design achieves a harmonious enhancement of both mechanical and TES properties, outperforming most previously reported materials. Furthermore, the hydrogen-bonding characteristics enabled the uniform dispersion of MWCNTs within the SSPCM matrix, facilitating successful applications in Joule heating and photothermal conversion. This approach not only resolves the critical balance between strength and latent heat but also expands the functional versatility of SSPCMs. The material demonstrates significant potential for future applications in hyperthermia, thermal management of flexible devices, and wearable devices.
4. Experimental section
Details of materials and experimental procedures are listed in the SI.
Author contributions
Zhiqiang Li: method; writing; write the original manuscript. Chunhua Ge: resources; access to funds; supervisor; review. Daming Feng: review. Xinyue Zhang: conceptualization. Lixue Zhou: data curation. Xiangdong Zhang: supervision; review.
Conflicts of interest
The authors declare no conflict of interest.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Supplementary information available: Supplementary Information includes detailed materials and methods (synthesis of HMAU, HPCMs, HPCM10K-X%, characterization protocols, statistical analysis), supplementary figures (synthesis routes, NMR/IR/DSC/TGA/SEM/mechanical property plots), supplementary tables (molar masses, crystalline structures, phase change properties, literature comparisons), and ref. 1 to support this work. See DOI: https://doi.org/10.1039/d5mh01550b.
Acknowledgements
This work was supported by the Special Funds for Basic Research Operating Expenses of Public Higher Education Institutions in Liaoning Province (LJ232410140033) and the Yingkou Municipal Enterprises Doctoral Shuangchuang Project (YKSCJH2023-008).
References
- Z. Chai, M. Fang and X. Min, Nano Energy, 2024, 124, 109437 CrossRef CAS.
- Y. Yao, X. Jiao, X. Xu, S. Xiong, Z. Song and Y. Liu, Nano Energy, 2024, 14, 2403117 CAS.
- A. Usman, F. Xiong, W. Aftab, M. Qin and R. Zou, Adv. Mater., 2022, 34, 2202457 CrossRef CAS PubMed.
- Z. Liu, X. Fu, L. Jiang, B. Wu, J. Wang and J. Lei, Sol. Energy Mater. Sol. Cells, 2016, 147, 177–184 CrossRef CAS.
- J. Shi, W. Aftab, Z. Liang, K. Yuan, M. Maqbool, H. Jiang, F. Xiong, M. Qin, S. Gao and R. Zou, J. Mater. Chem. A, 2020, 8, 20133–20140 RSC.
- X. Li, H. Wang, S. Yuan, S. Lin, S. Deng, Z. Du, X. Cheng and X. Du, Polymer, 2022, 250, 124885 CrossRef CAS.
- Z. Li, C. Ge, X. Li, L. Zhou, S. Liu and X. Zhang, Mater. Today Commun., 2024, 38, 108195 CrossRef CAS.
- Z. Li, C. Ge, D. Feng and X. Zhang, Colloids Surf. A, 2024, 703, 135304 CrossRef CAS.
- C. Lin, H. Ge, P. Ying, T. Wang, M. Huang, P. Zhang, T. Yang, J. Wu and V. Levchenko, ACS Appl. Polym. Mater., 2023, 5, 4190–4198 CrossRef CAS.
- J. Chang, H. Lu, Y. Xiao, S. Wu, Y. Xiao and Y. Wang, J. Appl. Polym. Sci., 2023, 141, e54950 CrossRef.
- T. Chong, N. Jingyi, Y. Yunyun, Z. Fanhao, H. Lei, L. Qiang, L. Jiahao, Z. Fuqi, K. Weibo and C. Xufu, Chem. Eng. J., 2021, 429, 132447 Search PubMed.
- J. van der Tol, G. Vantomme, A. Palmans and E. Meijer, Macromolecules, 2022, 55, 6820–6829 CrossRef CAS.
- J. Cui, W. Li, Y. Wang, H. Yu, X. Feng, Z. Lou, W. Shan and Y. Xiong, Adv. Funct. Mater., 2021, 32, 2108000 CrossRef.
- X. Yan, Z. Liu, Q. Zhang, J. Lopez, H. Wang, H. Wu, S. Niu, H. Yan, S. Wang and T. Lei, J. Am. Chem. Soc., 2018, 140, 5280–5289 CrossRef CAS.
- S. Chen, X. Bi, L. Sun, J. Gao, P. Huang, X. Fan, Z. You and Y. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 20591–20599 CrossRef CAS PubMed.
- Q. Jin, R. Du, H. Tang, Y. Zhao, W. Peng, Y. Li, J. Zhang, T. Zhu, X. Huang and D. Kong, Angew. Chem., Int. Ed., 2023, 62, e202305282 CrossRef CAS PubMed.
- Q. Yuan, T. Zhou, L. Li, J. Zhang, X. Liu, X. Ke and A. Zhang, RSC Adv., 2015, 5, 31153–31165 RSC.
- A. Yang, R. Huang, Z. Jiang, Y. Li, F. He, Q. Zhang, P. Zhou, Y. Wang, P. He and G. W. Yang, ACS Appl. Polym. Mater., 2023, 5, 1499–1508 CrossRef CAS.
- L. Li, J. Peng, L. Wang, J. Lai, C. Zhao, D. Xiang, H. Li, G. Yan, Z. Li and Y. Wu, J. Mater. Chem. A, 2024, 13, 3073–3083 RSC.
- M. Liu, Z. Huang, A. Bashir, Y. Zhang, H. Qian, X. Ouyang, Y. Hu, H. Chen and D. Chen, Chem. Eng. J., 2024, 500, 157322 CrossRef CAS.
- X. Du, L. Jin, S. Deng, M. Zhou, Z. Du, X. Cheng and H. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 42991–43001 CrossRef CAS.
- C. Lin, P. Ying, M. Huang, P. Zhang, T. Yang, G. Liu, J. Wu and V. Levchenko, Polym. Compos., 2021, 42, 6816–6826 CrossRef CAS.
- T. Wu, C. Wang, Y. Hu, X. Zeng and M. Song, Int. J. Heat Mass Transfer, 2023, 211, 124202 CrossRef.
- Z. Li, C. Ge, D. Feng, L. Zhou and X. Zhang, Appl. Surf. Sci., 2025, 690, 162632 CrossRef CAS.
- R. Huang, A. Yang, Z. Liu, F. Liu, T. Fang, F. He, Y. Li, Z. Jiang, H. Lan and K. Zhang, Colloids Surf., A, 2024, 698, 134544 CrossRef CAS.
- W. Kong, Y. Yang, A. Yuan, L. Jiang, X. Fu, Y. Wang, H. Xu, Z. Liu and J. Lei, Energy, 2021, 232, 121070 CrossRef CAS.
- Y. Zhao, T. Liu, Z. Wei, A. Yuan, Y. Chen, L. Jiang, J. Lei and X. Fu, Chem. Eng. J., 2023, 462, 142164 CrossRef CAS.
- B. Yang, X. Zhang, J. Ji, W. Hua and M. Jiang, J. Energy Chem., 2024, 97, 216–227 CrossRef CAS.
- H. Zhu, M. Gu, X. Dai, S. Feng, T. Yang, Y. Fan, J. Zhang, D. Fan, Y. Liu and Y. Lu, Chem. Eng. J., 2024, 494, 153235 CrossRef CAS.
- X. Geng, M. Qin, Z. Shen, F. Xiong, J. Di, C. Yang, Y. Wang, S. Gao and Q. Wang, Adv. Funct. Mater., 2024, 34, 2418848 Search PubMed.
- S. Gong, Y. Ding, X. Li, S. Liu, H. Wu, X. Lu and J. Qu, Composites, Part A, 2021, 149, 106505 CrossRef CAS.
- G. Zhu, W. Chen, X. Hu, W. Luo, Y. Ma, J. Wang, S. Tan, Y. Huang, J. Fan and X. Jiang, ACS Appl. Mater. Interfaces, 2024, 17, 2293–2303 CrossRef PubMed.
- B. Wu, Z. Liu, L. Jiang, C. Zhou and J. Lei, Adv. Polym. Technol., 2018, 37, 2997–3006 CrossRef CAS.
- F. Luo, Y. Xu, D. Wang, L. Zhan, Y. Feng, B. Lin, C. Zhai and H. Li, Mater. Horiz., 2025, 12, 4238–4247 RSC.
- L. Huang, Y. Yang, D. Yuan and X. Cai, J. Energy Storage, 2021, 36, 102343 CrossRef.
- A. Yang, R. Huang, Q. Zhang, Z. Jiang, Y. Li, F. He, P. Wang, G. He and W. Yang, ACS Sustainable Chem. Eng., 2024, 12, 7778–7790 CrossRef CAS.
- S. Song, H. Ai, L. Lv, Y. Guo, T. Han and L. Dong, Chem. Eng. J., 2023, 472, 144854 CrossRef CAS.
- X. Chen, X. Xu, J. Shen, N. Wen, J. Qian, Y. Su, X. Wang and F. Zhou, J. Energy Storage, 2024, 103, 114273 CrossRef.
- M. Liu, X. Li, R. Qian, X. Lu, Y. Liu and D. Zou, Adv. Funct. Mater., 2025, 35, 2415370 CrossRef CAS.
- S. Xue, G. Zhang, Y. Zhang, K. Wu and Q. Fu, Chem. Eng. J., 2024, 497, 154562 CrossRef CAS.
- J. Yu, L. Wang, D. Wang, L. Fang, H. Xie, W. Yu and Y. Li, J. Energy Storage, 2025, 123, 116786 CrossRef.
- J. Guo, S. Cai, X. Han, Y. Sun, C. Li, K. Zheng, Y. Xu, R. Li and C. Li, Chin. J. Polym. Sci., 2025, 43, 625–639 CrossRef CAS.
- L. Sun, Y. Huang, W. Chen, X. Jiang and X. Hu, J. Colloid Interface Sci., 2025, 695, 137831 CrossRef CAS PubMed.
- X. Ge, G. Tay, Y. Hou, Y. Zhao, P. Sugumaran, B. Thai, C. Ang, W. Zhai and Y. Yang, Carbon, 2023, 210, 118084 CrossRef CAS.
- S. Li, Y. Zhou, L. Wang, S. Wang, L. Bai, C. Feng, R. Bao, J. Yang, M. Yang and W. Yang, J. Mater. Chem. A, 2023, 11, 18832–18842 RSC.
- Y. Lin, Q. Kang, Y. Liu, Y. Zhu, P. Jiang, Y. Mai and X. Huang, Nano-Micro Lett., 2023, 15, 31 CrossRef CAS.
- K. Chen, Y. Li, Y. Feng, X. Li, Z. Li, S. Liu, C. Ge and X. Chen, J. Energy Chem., 2025, 107, 548–557 CrossRef CAS.
- Y. Wu, M. Chen, G. Hao, D. Qi, X. Zhang, Y. Li, Y. Huang and W. Yang, Adv. Mater., 2024, 36, 2311717 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Chunhua
Ge
*,
Daming
Feng
,
Chunhua
Ge
*,
Daming
Feng








