
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHarnessing oligonucleotide architecture for potent multivalent inhibition of human carbonic anhydrases
Eslam M.
Abbass
 ab,
Federico
Ladu
ac,
Justine
Mansot
a,
Mathieu
Noël
a,
Alessio
Nocentini
d,
Mario
Sechi
c,
Claudiu T.
Supuran
d,
Michael
Smietana
*a and
Jean-Yves
Winum
*a
ab,
Federico
Ladu
ac,
Justine
Mansot
a,
Mathieu
Noël
a,
Alessio
Nocentini
d,
Mario
Sechi
c,
Claudiu T.
Supuran
d,
Michael
Smietana
*a and
Jean-Yves
Winum
*a
aIBMM, University of Montpellier, CNRS, ENSCM, Montpellier, France. E-mail: michael.smietana@umontpellier.fr; jean-yves.winum@umontpellier.fr
bChemistry Department, Faculty of Science, Ain Shams University, Cairo, Egypt
cDepartment of Medicine, Surgery and Pharmacy, University of Sassari, 07100 Sassari, Italy
dNeurofarba Department, Section of Pharmaceutical and Nutraceutical Sciences, University of Florence, Via Ugo Schiff 6, Sesto Fiorentino, Florence, 50019, Italy
First published on 6th February 2026
Abstract
This study presents the design and synthesis of a novel class of carbonic anhydrase inhibitors (CAIs) using oligonucleotides as an original multivalent platform. Single-stranded oligonucleotides bearing one to four terminal alkyne moieties were prepared by solid-phase synthesis using the commercially available 2′-O-propargyl uridine phosphoramidite. In parallel, four different azide-functionalized CA inhibitor derivatives were generated from established coumarin and benzenesulfonamide pharmacophores. The resulting components were introduced into the oligonucleotides through Cu-catalyzed azide–alkyne cycloaddition (CuAAC), yielding a library of 17 oligonucleotide-based CA inhibitors with defined mono- to tetravalent architectures. This modular approach highlights the versatility of oligonucleotides as programmable platforms for the spatially controlled presentation of pharmacophores, opening new avenues for the development of potent and selective multivalent enzyme inhibitors. Among the resulting constructs, the divalent benzenesulfonamide conjugate 19 exhibited the most pronounced multivalency effect, showing a 3.5-fold increase in potency (rp = 3.5) against the tumor-associated isoform hCA IX, with a Ki of 69 nM compared to its monovalent analogue 12 (Ki = 245 nM).
Introduction
Carbonic anhydrases (CAs, EC 4.2.1.1) are a superfamily of ubiquitous zinc metalloenzymes that catalyze the reversible hydration of carbon dioxide into bicarbonate and protons, thereby playing central roles in acid–base homeostasis, respiration, electrolyte secretion, and biosynthetic pathways in both prokaryotic and eukaryotic organisms.1,2 In humans, 15 catalytically active isoforms have been identified, each with distinct tissue distribution, subcellular localization, and physiological roles.1–3 Several isoforms are druggable targets: for example, hCA II is implicated in glaucoma, epilepsy, and idiopathic intracranial hypertension, while the tumor-associated isoforms hCA IX and hCA XII are upregulated during hypoxia and contribute to the acidification of the tumor microenvironment, promoting invasion, metastasis, and therapeutic resistance.1–5Consequently, small-molecule CA inhibitors (CAIs) have advanced into clinical use for conditions such as glaucoma, altitude sickness, and epilepsy, with sulfonamide derivatives representing the most clinically successful class.4–6 The sulfonamide SLC-0111 is a pioneering compound designed to selectively inhibit carbonic anhydrase isoforms IX and XII, which are commonly overexpressed in hypoxic tumor environments.5–7 It is currently being tested in a phase 1b/2 clinical trial for metastatic pancreatic cancer that expresses CA IX, in combination with gemcitabine (ClinicalTrials.gov ID: NCT03450018). Additional phase 2 studies are planned, including trials evaluating its use alongside chemotherapy in patients with glioblastoma.
Other chemotypes, especially heterocycles such as coumarins and boron-containing inhibitors, have also been developed to expand chemical diversity and improve pharmacological profiles.4,8 Despite this progress, achieving high isoform selectivity remains an enduring challenge due to the strong conservation of the CA active-site architecture.1 Isoform-selective inhibition is particularly critical when targeting tumor-associated isoforms hCA IX and hCA XII to avoid systemic side effects linked to inhibition of ubiquitous isoforms such as hCA I and hCA II.5–7
To address these challenges, multivalent inhibition strategies have gained increasing attention. By presenting multiple ligands on a single scaffold, multivalency can enhance binding avidity, promote cooperative interactions with target surfaces, and provide improved isoform discrimination through geometric complementarity.9 These strategies have been successfully applied in enzyme inhibition, protein–ligand recognition, and drug delivery.10 In the field of carbonic anhydrases, engineered multivalent platforms have been designed to present multiple functional groups on a single scaffold, enabling enhanced binding affinity and selectivity toward specific CA isoforms.11 Such platforms often incorporate sulfonamide or coumarin-based ligands, arranged on cyclopeptide,12 dendrimers,13 porphyrin14 or silica or gold nanoparticle surfaces.15–17 By exploiting multivalency, researchers achieve cooperative interactions that significantly improve inhibition compared to monovalent counterparts.
Among available molecular scaffolds, synthetic oligonucleotides represent a particularly versatile and programmable platform. Their predictable secondary structures, sequence-defined spatial addressability, and facile chemical modification enable nanometer-scale positioning of functional groups.18 Solid-phase synthesis allows incorporation of modified nucleotides, such as alkyne-functionalized residues, which can be coupled with orthogonal conjugation strategies.19 In particular, the Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) enables efficient and modular attachment of small-molecule to oligonucleotides under mild conditions.19–22
Multivalent strategies have previously been explored to enhance carbonic anhydrase inhibition using a variety of molecular scaffolds, including cyclopeptides, dendrimers, porphyrins, and inorganic or hybrid nanoparticle-based platforms.10–17 In these systems, multivalency was shown to improve binding affinity and, in some cases, isoform selectivity through cooperative or avidity-driven effects.9–11 However, such scaffolds often rely on less defined architectures, limited control over ligand spacing, or reduced modularity.
In contrast, the oligonucleotide-based approach described herein offers a sequence-defined and programmable scaffold that enables precise control over valency and spatial arrangement of pharmacophores at the nanometer scale.18–22 This level of architectural control allows systematic evaluation of spacing-dependent multivalency effects and facilitates rapid diversification through post-synthetic modification.19–22 As such, oligonucleotides represent a complementary and highly adaptable platform for the design of multivalent carbonic anhydrase inhibitors, distinct from previously reported systems.
Here, we describe the rational design and synthesis of a library of oligonucleotide-based multivalent CA inhibitors assembled via click chemistry. Using 2′-O-propargyl uridine phosphoramidite building blocks, we prepared alkyne-modified oligonucleotides of differing architectures and conjugated them with azide-derivatized sulfonamide and coumarin inhibitors. This modular strategy afforded 17 distinct constructs, enabling systematic exploration of mono- to tetravalent assemblies. These results highlight the value of oligonucleotide-based platforms as programmable scaffolds for engineering next-generation multivalent inhibitors with the potential to achieve isoform-selective targeting of carbonic anhydrases.
Results and discussion
Chemistry
The synthetic strategy employed for the preparation of multivalent carbonic anhydrase (CA) inhibitors can be conceptually likened to a modular assembly, composed of three key components. The first involves the synthesis of oligonucleotide sequences incorporating one or more alkyne functionalities. The second consists of the design and synthesis of azide derivatives based on known potent CA inhibitors. The final step is the copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC), which enable the site-specific conjugation of the azide-functionalized inhibitors into the oligonucleotides 19.In the first stage of this strategy, five different 12-mer oligonucleotide sequences were synthesized using the standard phosphoramidite chemistry. The 12-mer sequences used in this study were selected to avoid stable secondary structures, providing flexible and solvent-exposed presentation of the attached inhibitors. While short sequences of this length may show coincidental matches in genome-wide BLAST analyses, these sequences were designed purely as proof-of-concept models, and any apparent complementarity does not imply biologically relevant interactions. Commercially available phosphoramidite building blocks dA, dG, dC, and 2′-O-propargyl uridine phosphoramidite were employed to introduce the CA inhibitors via post-synthetic click chemistry (Scheme 1). The 2′-O-propargyl modification was chosen to enable conjugation at the sugar moiety while avoiding direct modification of the nucleobase, which could more strongly perturb the local nucleobase environment. In single-stranded oligonucleotides, where canonical major and minor grooves are not defined, this attachment mode allows a flexible and solvent-exposed presentation of the inhibitor. The first sequence (5′-GCC AGC UGA CCG-3′) 1 was designed as a reference to assess the impact of multivalency in subsequent modified sequences. To investigate divalent presentation, two oligonucleotides were prepared, each featuring two alkyne-bearing uridine residues. In the first construct, the modified nucleotides were positioned three bases apart (5′-GCU AGC UGA CCG-3′) 2, while in the second they were separated by six bases (5′-GCU AGC TGA UCG-3′) 3. For higher-valency architectures, tri- (5′-GCU AGC UGA UCG-3′) 4 and tetravalent (5′-GCU AUC UGA UCG-3′) 5 sequences were also prepared. These single-stranded oligonucleotides were designed to avoid stable secondary structures and the attached inhibitors are expected to be largely solvent-exposed. All sequences have comparable overall lengths, ensuring that observed differences in inhibition arise from the number and distribution of modified uridines rather than from strand length. The spacing and progressive introduction of uridines, together with the relatively high A/U content, are intended to provide a flexible presentation of the inhibitors and to increase the statistical probability of productive interactions with the carbonic anhydrase active site and how increasing valency and spatial arrangement influence carbonic anhydrase inhibition. The structural integrity and purity of all synthesized oligonucleotides was confirmed by MALDI-TOF mass spectrometry and RP high-performance liquid chromatography (Table S1).
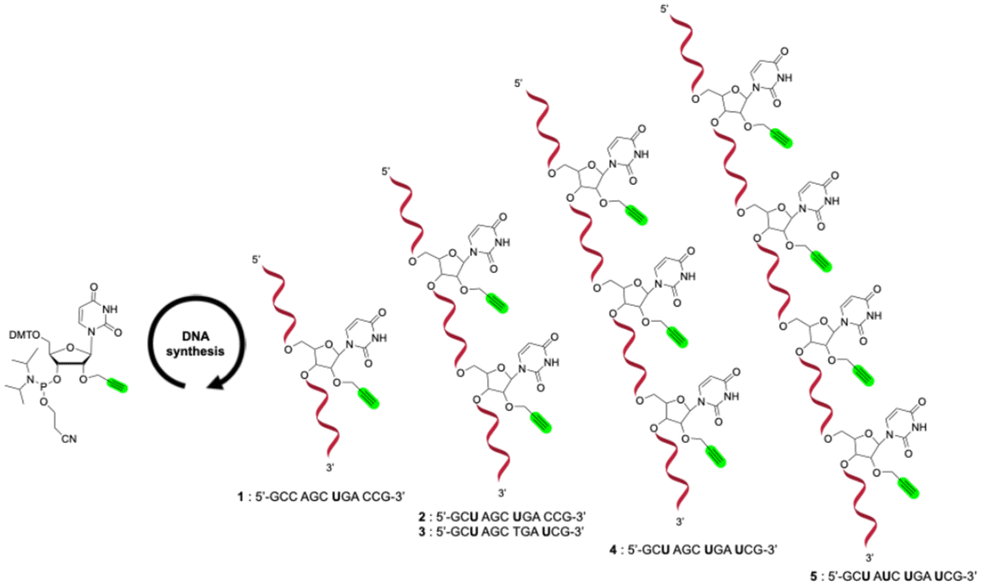 | ||
| Scheme 1 Synthesis of modified oligonucleotide sequences. | ||
In the next stage, azide functionalities were introduced onto well-established carbonic anhydrase inhibitor scaffolds selected for their documented binding affinities and inhibition profiles across various CA isoforms. Specifically, coumarin and benzenesulfonamide cores were chosen due to their well-characterized and distinct mechanisms of action. Coumarins act as suicide inhibitors: they undergo hydrolysis within the CA active site to generate 2-hydroxycinnamic acids, which subsequently occupy the enzyme's catalytic pocket, conferring isoform selectivity. In contrast, benzenesulfonamides are classical zinc-binding inhibitors that coordinate directly with the active-site zinc ion, resulting in strong and broad-spectrum inhibition. Based on these two structural classes, four azide-functionalized derivatives were synthesized as outlined in (Scheme 2).23 Compound 6 was derived from a coumarin core, whereas 7, 8 and 9 originated from benzenesulfonamide structures.
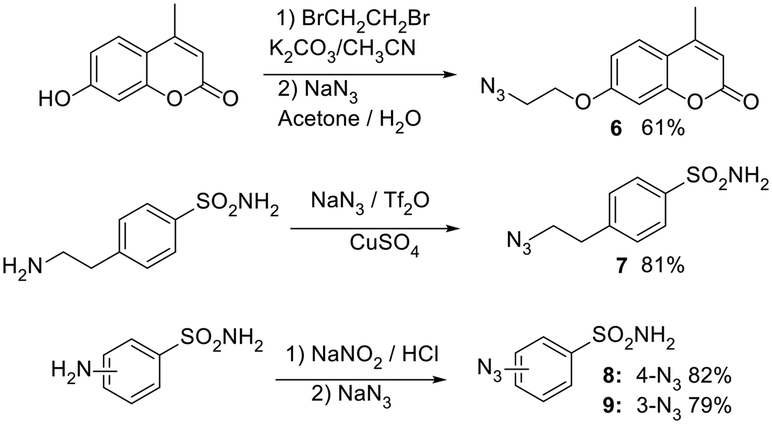 | ||
| Scheme 2 Synthesis of azide derivatives 6–9 based on CA inhibitors. | ||
Subsequently, a copper-mediated azide–alkyne cycloaddition (CuAAC) was used to conjugate the synthesized azide derivatives 6–9 to the alkyne-modified oligonucleotide platforms 1–5, generating a library of 17 mono- to tetravalent carbonic anhydrase inhibitor (CAI) constructs (Scheme 3). The post-synthetic functionalization was chosen because it is straightforward, efficient, and routinely mastered in our laboratory,24 whereas pre-synthesized phosphoramidites bearing the CA inhibitors would require more complex synthetic steps. All conjugations were performed in solution using an ascorbate-free Cu(0) nanoparticle-mediated protocol, in which the oligonucleotide scaffolds were combined with the appropriate azide partners in a water/methanol/TEAAc buffer pH 7 (v/v) in the presence of THPTA as a stabilizing tris(triazolylmethyl)amine ligand. Under these conditions, the Cu(0) surface gradually generates catalytically active Cu(I) at the solvent interface, enabling formation of the characteristic 1,4-disubstituted triazole linkage that couples each CAI unit to the nucleic acid framework under mild conditions. All constructs were purified by RP HPLC and structurally confirmed by MALDI-TOF mass spectrometry, with observed m/z values in excellent agreement with theoretical masses (Table S2).
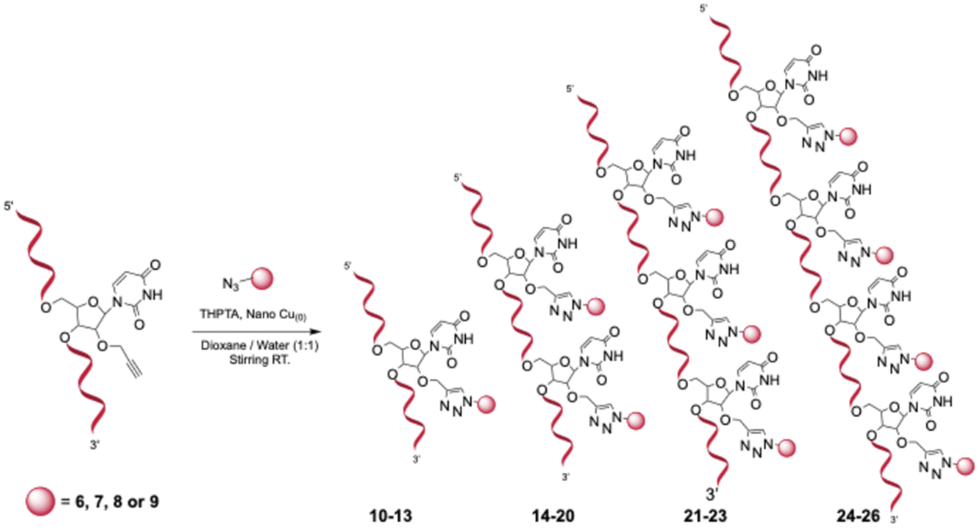 | ||
| Scheme 3 CuAAC click ligation between the modified sequences and the azides of CAs inhibitors. | ||
Inhibitory activity
The inhibitory activities of the synthesized oligonucleotide sequences were systematically evaluated in vitro using a stopped-flow carbon dioxide hydration assay, targeting several human carbonic anhydrase (hCA) isoforms. Their selectivity profiles were established by quantifying inhibitory effects against five specific hCA isoforms: the ubiquitous cytosolic forms hCA I and II, the membrane-associated isoform hCA IV, and the tumor-associated transmembrane isoforms hCA IX and XII. The potency of these novel compounds was assessed relative to acetazolamide AAZ, a classical sulfonamide-based carbonic anhydrase inhibitor (Table 1).| Cmpd | Clicked sequence | K i (nM) | ||||
|---|---|---|---|---|---|---|
| hCA I | hCA II | hCA IV | hCA IX | hCA XII | ||
| a Errors were in the range of ±10% of the reported values (data not shown). rp: relative potency. rp/n: relative potency per sulfonamide/coumarin inhibitor; Ux: uridine ligated with inhibitor x. | ||||||
| 12 | 5′-GCC AGC U8GA CCG-3′ | 1552 | 643.2 | 502.1 | 245.0 | 101.7 |
| 19 | 5′-GCU8 AGC TGA U8CG-3′ | 1913 | 434.4 | 423.9 | 69.2 | 85.6 |
| r p | 0.81 | 1.48 | 1.18 | 3.54 | 1.19 | |
| r p/n | 0.40 | 0.74 | 0.59 | 1.7 | 0.59 | |
| 16 | 5′-GCU8 AGC U8GA CCG-3′ | 1589 | 469.8 | 404.1 | 358.9 | 170.8 |
| r p | 0.98 | 1.36 | 1.24 | 0.68 | 0.60 | |
| r p/n | 0.49 | 0.68 | 0.62 | 0.34 | 0.30 | |
| 23 | 5′-GCU8 AGC U8GA U8CG-3′ | 2101 | 339.5 | 370.4 | 192.3 | 280.1 |
| r p | 0.74 | 1.89 | 1.36 | 1.28 | 0.36 | |
| r p/n | 0.24 | 0.36 | 0.45 | 0.43 | 0.12 | |
| 26 | 5′-GCU8 AU8C U8GA U8CG-3′ | 2719 | 286.5 | 618.7 | 218.6 | 70.9 |
| r p | 0.57 | 2.24 | 0.81 | 1.12 | 1.43 | |
| r p/n | 0.14 | 0.56 | 0.20 | 0.28 | 0.36 | |
| 13 | 5′-GCC AGC U9GA CCG-3′ | 3907 | 1373 | 1654 | 531.4 | 367.4 |
| 20 | 5′-GCU9 AGC TGA U9CG-3′ | 3117 | 1738 | 1116 | 388.7 | 463.6 |
| r p | 1.25 | 0.79 | 1.48 | 1.37 | 0.79 | |
| r p/n | 0.63 | 0.39 | 0.74 | 0.68 | 0.40 | |
| 10 | 5′-GCC AGC U6GA CCG-3′ | >10 μM | >10 μM | >10 μM | 682 | 3420 |
| 14 | 5′-GCU6 AGC U6GA CCG-3′ | >10 μM | >10 μM | >10 μM | 731 | 2230 |
| r p | — | — | — | 0.93 | 1.53 | |
| r p/n | 0.47 | 0.77 | ||||
| 17 | 5′-GCU6 AGC TGA U6CG-3′ | >100 μM | >100 μM | >100 μM | 554 | 1093 |
| r p | — | — | — | 1.23 | 3.12 | |
| r p/n | 0.62 | 1.56 | ||||
| 21 | 5′-GCU6 AGC U6GA U6CG-3′ | >10 μM | >10 μM | >10 μM | 1450 | 1880 |
| r p | — | — | — | 0.47 | 1.82 | |
| r p/n | 0.16 | 0.61 | ||||
| 24 | 5′-GCU6 AU6C U6GA U6CG-3′ | >10 μM | >10 μM | >10 μM | 2210 | 1300 |
| r p | — | — | — | 0.31 | 2.63 | |
| r p/n | 0.10 | 0.88 | ||||
| 11 | 5′-GCC AGC U7GA CCG-3′ | 762 | 386 | 894 | 99.7 | 129 |
| 15 | 5′-GCU7 AGC U7GA CCG-3′ | 554 | 243 | 1650 | 124 | 78.2 |
| r p | 1.38 | 1.56 | 0.54 | 0.80 | 1.65 | |
| r p/n | 0.69 | 0.80 | 0.27 | 0.40 | 0.83 | |
| 18 | 5′-GCU7 AGC TGA U7CG-3′ | 775 | 201 | 1283 | 78.0 | 55.2 |
| r p | 0.98 | 1.92 | 0.70 | 1.28 | 1.34 | |
| r p/n | 0.49 | 0.96 | 0.35 | 0.64 | 1.17 | |
| 22 | 5′-GCU7 AGC U7GA U7CG-3′ | 1560 | 498 | 1980 | 84.0 | 141 |
| r p | 0.49 | 0.78 | 0.45 | 1.19 | 0.91 | |
| r p/n | 0.16 | 0.26 | 0.15 | 0.40 | 0.46 | |
| 25 | 5′-GCU7 AU7C U7GA U7CG-3′ | 1740 | 310 | 1430 | 148 | 103 |
| r p | 0.44 | 1.25 | 0.63 | 0.67 | 1.25 | |
| r p/n | 0.11 | 0.31 | 0.16 | 0.17 | 0.31 | |
| AAZ | 250 | 12 | 74 | 25 | 5.7 | |
To quantitatively assess multivalency effects, we calculated the relative potency (rp) and the relative potency per pharmacophore (rp/n) for each multivalent construct. The rp value represents the fold improvement in inhibitory activity of a multivalent conjugate compared to its monovalent analogue bearing the same inhibitor chemotype. It is defined as: rp = Ki,mono/Ki,multi, where Ki,mono is the inhibition constant of the corresponding monovalent conjugate, and Ki,multi is the inhibition constant of the multivalent construct. Because absolute potency increases with the number of attached inhibitor units, we additionally report rp/n, which normalizes rp by the valency (n) of the conjugate.
This parameter reflects the per-pharmacophore efficiency and enables discrimination between true cooperative multivalency (rp/n > 1), additive effects (rp/n ≈ 1), and steric or conformational penalties (rp/n < 1). Reporting rp and rp/n together provides a rigorous framework for interpreting how scaffold architecture and inhibitor spacing contribute to enhanced or diminished binding interactions.
Previous studies demonstrated that 4-azidobenzenesulfonamide 8 is a more effective carbonic anhydrase inhibitor than 3-azidobenzenesulfonamide 9.21 As shown in Table 1, the monovalent conjugate 12 exhibited superior inhibitory activity against hCA I, II, IV, IX, and XII, with Ki values (nM) of 1552, 643.2, 502.1, 245.0, and 101.7, respectively. In contrast, conjugate 13 displayed Ki values of 3907, 1373, 1654, 531.4, and 367.4 nM.
Conjugate 19 showed enhanced activity compared to monovalent 12, particularly against hCA IX (Ki = 69.2 nM to 245 nM), corresponding to a multivalency factor (rp/n) of 1.7. Construct 20 exhibited slight improvements over monovalent 13 for hCA I, IV, and IX, but no significant multivalency effect (rp/n = 0.39–0.74), with Ki values of 3117, 1738, 1116, 388.7, and 463.6 nM.
These results confirm that conjugates based on 8 outperform those based on 9, likely due to reduced steric hindrance in the 4-substituted derivative, which facilitates CA binding. Consequently, additional multivalent inhibitors derived from compound 8 were synthesized.
Divalent construct 16 exhibited lower activity than construct 19 and showed no measurable multivalency effect. This reduced potency is likely due to steric constraints: in 16, the two benzenesulfonamide moieties are separated by three bases, whereas in 19 they are spaced six bases apart, reducing crowding and improving affinity for hCA II, IX, and XII.
The trivalent inhibitor 23 displayed moderate-to-low activity (Ki = 2101, 339.5, 370.4, 192.3, and 280.1 nM), while the tetravalent inhibitor 26 was most potent against hCA XII (Ki = 70.9 nM) and moderately active against hCA I, II, IV, and IX (Ki = 2719, 286.5, 618.7, and 218.6 nM). For inhibitors based on 7-(2-azidoethoxy)-4-methyl-2H-chromen-2-one 6, mono-, di-, tri-, and tetravalent sequences were synthesized. All showed negligible activity against hCA I, II, and IV, and only moderate-to-weak activity against hCA IX and XII. Steric hindrance strongly influenced potency: the less hindered divalent sequence 17 (six-base spacing) was most active (Ki = 554 nM, rp/n = 1.56), whereas the more hindered divalent sequence 14 (three-base spacing) showed reduced activity (Ki = 731 nM) with no multivalency effect. Activity further decreased in trivalent (21, Ki = 1450 nM) and tetravalent (24, Ki = 2210 nM) conjugates. Finally, sequences based on 4-(2-azidoethyl)benzene sulfonamide 7 exhibited moderate activity against hCA I, II, and IV, and good activity against hCA IX and XII. Steric effects were again evident: the least hindered divalent sequence 18 was most potent, with Ki values of 78 and 55 nM for hCA IX and XII, respectively, and a multivalency effect on hCA XII (rp/n = 1.17).
Compared with acetazolamide (AAZ), a potent yet non-selective inhibitor of multiple hCA isoforms, the oligonucleotide-based inhibitors exhibit lower overall potency but a more distinct inhibition profile, displaying enhanced selectivity towards hCA IX and hCA XII.
Conclusion
The systematic evaluation of oligonucleotide–benzenesulfonamide conjugates demonstrates that both substitution pattern and multivalency critically influence carbonic anhydrase inhibition. Conjugates based on ligation with 4-azidobenzenesulfonamide consistently outperform those derived from 3-substituted analogues, underscoring the role of reduced steric hindrance in enhancing target engagement. Multivalent architectures further amplify potency, particularly against hCA IX and XII, with optimal spacing between inhibitor moieties being essential to minimize steric interference and maximize cooperative binding. While higher valency constructs do not uniformly improve activity, strategic design, balancing valency, and spatial arrangement, enables significant gains in selectivity and affinity. These findings establish oligonucleotide scaffolds as versatile platforms for engineering potent, isoform-selective CA inhibitors and highlight structural parameters that should guide future development of nucleic acid-based multivalent therapeutics.Experimental
Materials and methods
The syntheses of the oligonucleotides were performed on solid support using an automated DNA synthesizer (Applied Biosystems 394). Crude oligonucleotides were analyzed by RP-HPLC (Macherey Nagel Nucleodur C18 ec, 3 μm, 100 Å, 4.6 × 75 mm, buffer A: 50 mM TEAAc pH 7 in 1% CH3CN, buffer B: 50 mM TEEAc in 80% CH3CN, 1 mL min−1 flow rate, detection at 260 nm) and purified by RP-HPLC (Macherey Nagel Nucleodur C18 ec, 7 μm, 100 Å, 8 × 125 mm, same buffer A and B, 4 ml min−1 flow rate, detection at 260 nm). Oligonucleotides fractions pure were desalted by co-evaporation with mix water/acetonitrile (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) and transferred to a 2 mL Eppendorf-vial and lyophilized from water. MALDI-TOF mass spectra were recorded on a AXIMA Assurance (Shimadzu) using 2,4,6-trihydroxyacetophenone as a saturated solution in a mixture of acetonitrile/0.1 M ammonium citrate solution (1:1, v/v) for the matrix. Analytical samples were mixed with the matrix in a 1:5 (v/v) ratio, crystallized on a 100-well stainless-steel plate and analyzed.
:1, v/v) and transferred to a 2 mL Eppendorf-vial and lyophilized from water. MALDI-TOF mass spectra were recorded on a AXIMA Assurance (Shimadzu) using 2,4,6-trihydroxyacetophenone as a saturated solution in a mixture of acetonitrile/0.1 M ammonium citrate solution (1:1, v/v) for the matrix. Analytical samples were mixed with the matrix in a 1:5 (v/v) ratio, crystallized on a 100-well stainless-steel plate and analyzed.
Synthesis of oligonucleotides
Synthesis of azide derivatives
First step: synthesis of 7-(2-bromoethoxy)-4-methyl-2H-chromen-2-one. In a solution of 7-hydroxy-4-methyl-2H-chromen-2-one (2 g, 11.35 mmol) and K2CO3 (9.4 g, 204.34 mmol) in acetonitrile, (100 mL), 1,2-dibromoethane (6.4 g, 51.08 mmol) was added dropwise, and the solution was stirred for 2 h at reflux. Subsequently, the mixture was filtered, and the solvent evaporated. The solid obtained was solubilized in DCM, and the organic phase was washed with brine and dried with anhydrous Na2SO4, filtered, and evaporated to get a white solid that was purified with flash chromatography (CH3Cl/ethyl acetate 9
:1), affording the product in 43% yield. 1H NMR and 13C NMR spectra matched those reported in the literature.24
Second step: synthesis of 7-(2-azidoethoxy)-4-methyl-2H-chromen-2-one. In a solution of 7-(2-bromoethoxy)-4-methyl-2H-chromen-2-one (0.5 g, 1.77 mmol) in acetone (20 mL), NaN3 (0.230 g, 3.53 mmol, 2 eq.) in water (5 mL) was added dropwise and the solution was stirred for 8 h at reflux. The mixture was reduced under pressure, solubilized in ethyl acetate, and washed with water and brine. The organic phase was dried over anhydrous Na2SO4, filtered, and evaporated to obtain a white solid that was purified with flash chromatography (CH3Cl/ethyl acetate 7
:3), affording the product in 61% yield. 1H NMR and 13C NMR spectra matched those reported in the literature.25
Solution B: 4-(2-aminoethyl)benzenesulfonamide (0.250 g, 1.25 mmol) was solubilized in water (4 mL) and methanol (8 mL) and CuSO4 (0.028 g, 0.175 mmol, 0.14 eq.) was added. Solution A was added dropwise to solution B at 0 °C, let warm to rt, and stirred overnight. The solvent was evaporated, and the solid solubilized in ethyl acetate, washed with water and brine, dried over anhydrous Na2SO4, and reduced to obtain a white solid that was purified with flash chromatography (CH2Cl2/MeOH 9.5:0.5), affording the product in 81% yield. 1H NMR and 13C NMR spectra matched those reported in the literature.23
:1). The isolated yields were 82% for compound 8 and 79% for compound 9 respectively. 1H NMR and 13C NMR spectra matched those reported in the literature.23
After reverse-phase HPLC purification, samples were analyzed by MALDI-TOF mass spectrometry. Residual copper is readily detected through the presence of copper adducts. When observed, samples were treated with QuadraPure™ beads in aqueous solution and re-analyzed by mass spectrometry until no copper adducts were detected, prior to lyophilization.
For each inhibitor, at least six independent stopped-flow traces were collected, focusing on the initial 5–10% of the reaction to calculate initial velocities. Uncatalyzed reaction rates were determined under identical conditions and subtracted from the observed total rates. Inhibitor stock solutions (0.1 mM) were prepared in deionized water and diluted with assay buffer to working concentrations as low as 0.01 nM. Prior to measurements, enzyme and inhibitor solutions were preincubated for 15 min at room temperature to allow formation of the enzyme–inhibitor complex.
Inhibition constants (Ki) were calculated using nonlinear regression analysis (GraphPad Prism 3) and the Cheng–Prusoff equation, with values reported as means from at least three independent determinations.27,28 Enzyme concentrations ranged between 6 and 14 nM. All hCA isoforms were recombinant proteins produced in-house as previously described.28
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included into the supplementary information (SI).Supplementary information: HPLC chromatograms and MALDI-TOF mass spectra of synthesized compounds 1–5 and 10–26. See DOI: https://doi.org/10.1039/d5md01167a.
Acknowledgements
This work was financed by a fellowship awarded under Call 10 STDF-IFE, funded through a partnership between the French Embassy in Egypt – French Institute of Egypt (IFE) and the Science and Technology Development Fund (STDF), granted to EMA. The Agence Nationale de la Recherche is gratefully acknowledged for funding (D-CYSIV, ANR-2015-CE29-0021-01). M. Sechi was supported by AIRC (IG 2021 – ID 26211).References
- K. D'Ambrosio, A. Di Fiore, V. Alterio, E. Langella, S. M. Monti, C. T. Supuran and G. de Simone, Chem. Rev., 2025, 125, 150 CrossRef PubMed.
- C. T. Supuran, Bioorg. Med. Chem. Lett., 2023, 93, 129411 CrossRef CAS PubMed.
- C. T. Supuran, ACS Med. Chem. Lett., 2025, 16, 1889–1895 Search PubMed.
- A. Nocentini, A. Angeli, F. Carta, J.-Y. Winum, R. Zalubovskis, S. Carradori, C. Capasso, W. A. Donald and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2021, 36, 561–580 CrossRef CAS PubMed.
- A. Angeli, F. Carta, A. Nocentini, J.-Y. Winum, R. Zalubovskis, A. Akdemir, V. Onnis, W. M. Eldehna, C. Capasso, G. de Simone, S. M. Monti, S. Carradori, W. A. Donald, S. Dedhar and C. T. Supuran, Metabolites, 2020, 10, 412 CrossRef CAS.
- S. Giovannuzzi and C. T. Supuran, Trends Pharmacol. Sci., 2025, 46, 836 Search PubMed.
- C. T. Supuran, Pharmacol. Rev., 2025, 77, 100004 Search PubMed.
- S. Giovannuzzi, A. Nikitjuka, B. R. Pereira Resende, M. Smietana, A. Nocentini, C. T. Supuran and J.-Y. Winum, Bioorg. Chem., 2024, 143, 106976 Search PubMed.
- M. Mammen, S. K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754 Search PubMed.
- N. Kanfar, E. Bartolami, R. Zelli, A. Marra, J.-Y. Winum, S. Ulrich and P. Dumy, Org. Biomol. Chem., 2015, 13, 9894 Search PubMed.
- F. Carta, P. Dumy, C. T. Supuran and J.-Y. Winum, Int. J. Mol. Sci., 2019, 20, 5352 CrossRef CAS PubMed.
- N. Kanfar, M. Tanc, P. Dumy, C. T. Supuran, S. Ulrich and J.-Y. Winum, Chem. – Eur. J., 2017, 23, 6788 CrossRef CAS PubMed.
- F. Carta, S. M. Osman, D. Vullo, A. Gullotto, J.-Y. Winum, Z. AlOthman, E. Masini and C. T. Supuran, J. Med. Chem., 2015, 58, 4039 CrossRef CAS PubMed.
- A. Merabti, M. Roger, C. Nguyen, A. Nocentini, P. Gerbier, S. Richeter, M. Gary-Bobo, C. T. Supuran, S. Clement and J.-Y. Winum, Eur. J. Org. Chem., 2022, 21, e202101538 CrossRef.
- M. Stiti, A. Cecchi, M. Rami, M. Abdaoui, V. Barragan-Montero, A. Scozzafava, Y. Guari, J.-Y. Winum and C. T. Supuran, J. Am. Chem. Soc., 2008, 130, 16130 Search PubMed.
- N. Touisni, N. Kanfar, S. Ulrich, P. Dumy, C. T. Supuran, A. Mehdi and J.-Y. Winum, Chem. – Eur. J., 2015, 21, 10306 Search PubMed.
- N. Kanfar, A. Mehdi, P. Dumy, S. Ulrich and J.-Y. Winum, Chem. – Eur. J., 2017, 23, 17867 Search PubMed.
- S. B. Yeldell and O. Seitz, Chem. Soc. Rev., 2020, 49, 6848–6865 RSC.
- N. Z. Fantoni, A. H. El-Sagheer and T. A. Brown, Chem. Rev., 2021, 121, 7122 Search PubMed.
- G. Pourceau, A. Meyer, J. J. Vasseur and F. Morvan, J. Org. Chem., 2009, 74, 1218 CrossRef CAS PubMed.
- A. Meyer, G. Pourceau, J. J. Vasseur and F. Morvan, J. Org. Chem., 2010, 75, 6689 CrossRef CAS.
- M. Debiais, J. J. Vasseur, S. Müller and M. Smietana, Synthesis, 2020, 52, 2962 CAS.
- C. Ismail, A. Nocentini, C. T. Supuran, J.-Y. Winum and R. Gharbi, Arch. Pharm., 2022, 355, e2100405 Search PubMed.
- J. Mansot, S. Aubert, N. Duchemin, J.-J. Vasseur, S. Arseniyadis and M. Smietana, Chem. Sci., 2019, 10, 2875 Search PubMed.
- Y. C. Duan, Y. C. Ma, E. Zhang, X. J. Shi, M. M. Wang, X. W. Ye and H. M. Liu, Eur. J. Med. Chem., 2013, 62, 11 CrossRef CAS PubMed.
- R. G. Khalifah, J. Biol. Chem., 1971, 246, 2561 CrossRef CAS PubMed.
- A. Nocentini, A. Bonardi, C. Bazzicalupi, V. Alterio, D. Esposito, S. M. Monti, M. Smietana, G. de Simone, C. T. Supuran, P. Gratteri and J.-Y. Winum, J. Med. Chem., 2023, 66, 8118 Search PubMed.
- S. Giovannuzzi, A. Nikitjuka, A. Angeli, M. Smietana, M. L. Massardi, M. Turati, R. Ronca, A. Bonardi, A. Nocentini, M. Ferraroni, C. T. Supuran and J.-Y. Winum, J. Med. Chem., 2024, 67, 18221 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |