
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and evaluation of 14β-acyl substituted 17-cyclopropylmethyl-7,8-dihydromorphinone derivatives: mixed partial agonists at mu opioid and nociception/orphanin FQ peptide receptors
Mehrnoosh
Ostovar
a,
Keith
Olsen
b,
Gerta
Cami-Kobeci
a,
John R.
Traynor
bc,
Luka
Jeramaz
d,
Stewart B.
Kirton
e and
Stephen M.
Husbands
 *a
*a
aMedicinal Chemistry Section, Department of Life Sciences, University of Bath, Bath, BA2 7AY, UK. E-mail: s.m.husbands@bath.ac.uk
bDepartment of Pharmacology, and Edward F Domino Research Center, University of Michigan, Ann Arbor, MI 48109, USA
cDepartment of Medicinal Chemistry, University of Michigan, Ann Arbor, MI 48109, USA
dSchool of Health, Medicine and Life Sciences, University of Hertfordshire, Hatfield, Herts AL10 9AB, UK
eSchool of Applied and Health Science, London South Bank University, London, SE1 0AA, UK
First published on 12th February 2026
Abstract
Opioids remain the standard of care for management of severe pain, but adverse effects limit their use, particularly for the treatment of chronic pain. Compounds that have dual partial agonist activity at mu opioid (MOP) and nociceptin opioid peptide (NOP) receptors have been shown, in non-human primates, to display excellent analgesic activity with greatly reduced adverse effect profile. In this study we looked to increase the range of MOP/NOP dual acting compounds and, in particular, provide ligands with a greater diversity of MOP:NOP profiles. Reduction of the C6 carbonyl in the naltrexone scaffold to methylene resulted in a balanced MOP:NOP receptor profile in this series, in particular increasing potency at the NOP receptor. Ultimately, this will allow us to determine the optimal profiles for a range of therapeutic indications including pain and drug use disorders.
Introduction
Mu opioid (MOP) receptor agonists remain a mainstay of treatment for pain, particularly associated with post-operative care, cancer and orthopaedic pain. While undoubtedly effective when used appropriately, current standard of care opioids cause a range of significant side effects that put limitations on their use. In particular, they have abuse-liability, cause respiratory depression and constipation, and suffer from the development of tolerance (so that greater doses are required over time to remain efficacious). The current opioid epidemic is partly due to chronic pain sufferers transitioning from appropriate use of prescription opioids to their misuse and abuse and illustrates the need to develop new, strong analgesics with a greatly improved side effect profile.With these issues in mind, there has recently been interest in the development of opioids with mixed, partial agonist activity at the MOP and nociceptin opioid peptide (NOP) receptors. These compounds display strong analgesic activity in primate models of analgesia.1–4 Importantly, the lead compounds show significantly reduced abuse liability and physical dependence compared to standard of care opioids and do not cause respiratory depression, pruritus or constipation.
Initially, the close buprenorphine (1) analogue BU08028 (2) (Chart 1) was developed, displaying the desired increased NOP activity relative to buprenorphine, but equivalent pharmacology to buprenorphine at MOP, kappa (KOP) and delta (DOP) receptors.1,5 Key findings were potent, long-lived analgesia in primates and the lack of side effects normally associated with MOP receptor mediated analgesia. Thus, in primates BU08028 had no effects on respiration, did not induce itching and had a greatly improved abuse liability profile (very limited self-administration) and no effect on respiration. To confirm that this promising profile was not molecule specific, compounds from different chemical series, but with equivalent pharmacology were sought. This led to BU10038 (3a) which helped confirm that these startling effects were a result of the MOP/NOP partial agonist activity. As with 2, 3a showed excellent analgesic properties of long duration and greatly reduced levels of self-administration compared to oxycodone (predicting low/no abuse liability). It also showed no tolerance after repeated dosing and had no effects on physiological signs such as respiration even at doses 10–30 times higher than for analgesia.3 At the same time Zaveri and colleagues developed AT-121 (4), again having dual partial agonist activity and a structure distinct from that of 2 and 3a. 4 again profiled as a strong analgesic with improved side-effect profile.2
 | ||
| Chart 1 Several mu opioid and mixed mu/nociceptin opioid ligands. | ||
While this appears to be a promising strategy for the development of new analgesics, it is not clear if the ideal balance between MOP and NOP activities has been achieved to ensure maximal analgesic activity whilst minimising the undesirable activities. While 2 and 3a have somewhat higher affinity for MOP compared to NOP receptors, the opposite is true for 4. To this end, we now report on a series of close analogues of 3a, where an increase in relative NOP activity was sought.
Design
In the series of phenacetyl esters (5b) of naltrexone (5a), closely related to 3a, substitution of the side chain aryl ring had a marginal effect on affinity and efficacy for NOP but did modulate efficacy at MOP and KOP receptors.6 As the aim of the current study was to increase NOP affinity but leave MOP and KOP affinity and efficacy unchanged (and certainly not raised), rather than exploring ring-substitution of 3a, we looked to alternative ring systems (specifically, heterocyclic rings) as one approach and, more interestingly, the importance of the C6 keto group. It is well known that the carbonyl group of 5a can be subtly modified to a methylene (6: nalmefene), a hydroxy group (7: 6β-naltrexol) or removed (8: 6-deoxynaltrexone) without significantly affecting affinity for MOP receptors. As 6 has partial agonist activity at KOP receptors7 and 7 has reduced ability to enter the CNS,8 our focus was on removal of the carbonyl as 8 retains high affinity and very low/no efficacy at MOP and KOP receptors and should retain CNS access.9Results
Synthetic procedures
6-Deoxynaltrexone (8) was prepared using standard Wolff–Kishner conditions of hydrazine and KOH, as described previously.9 Introduction of the side chain was as previously reported for the phenacetyl analogues (Scheme 1).6 The 3-hydroxy group of 5a and 8 was protected with tert-butyldimethylsilyl chloride using a standard method10 followed by esterification of the 14-hydroxy group. The tendency of the C6-carbonyl to exist in its enol form meant that cleaner esterification was achieved with the appropriate anhydrides rather than with acyl chlorides. The anhydrides were synthesized from the corresponding phenylacetic acid and triphosgene.11 Thereafter the 3-hydroxy group was regenerated using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of methanol and HCl (6N) to give the target esters (3 and 11). (Scheme 1).
:1 mixture of methanol and HCl (6N) to give the target esters (3 and 11). (Scheme 1).
 | ||
| Scheme 1 Synthesis of 14-O-acylated naltrexone and 6-deoxynaltrexone analogues. Reagents and conditions: i) TBDMSCl, imidazole, DCM, rt ii) (R′CH2CH2CO)2O, toluene, 125 °C iii) TBAF, THF, rt. | ||
Pharmacological evaluation
Each analogue was tested for binding affinity in DOP-, KOP-, MOP-, and NOP receptors heterologously expressed in Chinese hamster ovary (CHO) cells (Table 1), followed by functional analysis in the [35S]GTPγS assay in the same cell lines (Table 2). All compounds retained high affinity at MOP, DOP and KOP receptors. Most of the new compounds showed increased NOP affinity and reduced MOR efficacy relative to buprenorphine (Table 1). 11a, 3a (the latter previously reported4) and 11d showed the highest NOP affinity and potency (Tables 1 and 2). No significant degree of agonist activity (stimulation <15% compared to standards) was observed at KOP or DOP receptors.| R | hNOP | hMOP | hKOP | hDOP | |
|---|---|---|---|---|---|
| K i (SEM)/nM | K i (SEM)/nM | K i (SEM)/nM | K i (SEM)/nM | ||
| Naloxone | 10 (1.4) | 0.6 (0.2) | 46 (9.9) | ||
| Nociceptin | 0.030 (0.006) | ||||
| Buprenorphine 1 | 1300 (321) | 1.3 (0.17) | 0.87 (0.43) | 10 (2.6) | |
| Naltrexone 5a | >10000 |
1.2 (0.15) | 3.7 (1.1) | 53 (12) | |
| 3a | Phenyl | 177 (40) | 0.16 (0.08) | 0.79 (0.40) | 0.42 (0.04) |
| 3b | 2-Furanyl | 229 (58) | 1.0 (0.67) | 0.35 (0.12) | 4.5 (1.2) |
| 3c | 3-Furanyl | 352 (67) | 0.16 (0.09) | 0.20 (0.14) | 4.3 (1.7) |
| 3d | 3-Thiophenyl | 240 (37) | 0.17 (0.10) | 0.14 (0.05) | 1.5 (0.6) |
| 3e | t-Butyl | >1000 | 0.77 (0.15) | 0.17 (0.04) | 13.5 (6.8) |
| 11a | Phenyl | 16 (5) | 0.16 (0.11) | 0.91 (0.69) | 7.5 (0.18) |
| 11b | 2-Furanyl | 120 (29) | 0.0082 (0.0011) | 0.019 (0.007) | 0.0013 (0.004) |
| 11c | 3-Furanyl | 127 (43) | 0.0024 (0.0008) | 0.035 (0.008) | 0.0031 (0.001) |
| 11d | 3-Thiophenyl | 35 (10) | 0.0068 (0.0022) | 0.007 (0.002) | 0.0044(0.0024) |
| 11e | t-Butyl | 563 (110) | 0.0067 (0.0007) | 0.032 (0.021) | 0.034 (0.14) |
| R | hNOP | hMOP | hKOP | hDOP | |||
|---|---|---|---|---|---|---|---|
| EC50 (SEM)/nM | E MAX (SEM) | EC50 (SEM)/nM | E MAX (SEM) | E MAX (SEM) | E MAX (SEM) | ||
| a Taken from ref. 4. NS = no stimulation. | |||||||
| DAMGO | 33 (3.1) | 100 (5.0) | |||||
| Nociceptin | 0.52 (0.061) | 98 (3.7) | |||||
| Buprenorphinea | >1000 | 35 (3.8) | 0.79 (0.31) | 34 (6.0) | <15% stim | <15% stim | |
| Naltrexone | NS | 5.5 (3.8) | −8 (3) | <15% stim | <15% stim | ||
| 3a | Phenyl | 160 (51) | 47 (9.0) | 1.5 (0.67) | 28 (4.4) | <15% stim | <15% stim |
| 3b | 2-Furanyl | 270 (79) | 42 (7.9) | 0.20 (0.044) | 24 (3.8) | <15% stim | <15% stim |
| 3c | 3-Furanyl | 286 (25) | 34 (3.3) | 0.18 (0.054) | 18 (1.3) | <15% stim | <15% stim |
| 3d | 3-Thiophenyl | 174 (82) | 49 (5.0) | 0.041 (0.019) | 22 (3.8) | <15% stim | <15% stim |
| 3e | t-Butyl | 472 (116) | 24 (4.4) | 0.23 (0.14) | 23 (4.4) | <15% stim | <15% stim |
| 11a | Phenyl | 0.97 (0.22) | 47 (11) | 3.9 (1.3) | 23 (7.1) | <15% stim | <15% stim |
| 11b | 2-Furanyl | 50 (7.6) | 35 (6.9) | 1.50 (0.80) | 28 (2.2) | <15% stim | <15% stim |
| 11c | 3-Furanyl | 49 (12) | 31 (5.0) | 0.88 (0.012) | 23 (4.4) | <15% stim | <15% stim |
| 11d | 3-Thiophenyl | 2.6 (0.64) | 48 (3.3) | 0.064 (0.020) | 26 (0.29) | <15% stim | <15% stim |
| 11e | t-Butyl | 359 (135) | 30 (7.0) | 0.79 (0.24) | 26 (0.29) | <15% stim | <15% stim |
The majority of compounds had higher affinity for each of the three classical opioid receptors than naltrexone. This was observed in the 6-keto series (3) where there was an approximate 10-fold increase across the receptors but was most striking for the heterocyclic 6-deoxy series (11) where affinities for all 3 classical opioid receptors were in the pM range. At NOP, where naltrexone had no measurable affinity, all but the t-butyl substituted compounds 3e and 11e, had good affinity, again with the 6-deoxy series showing higher affinity than their 6-keto counterparts, that was substantially higher (5–10-fold) than buprenorphine's for the same receptor. The clearest SAR related to removal of the carbonyl group, which led, in almost every case, to a substantial increase in affinity over the 6-keto equivalent for each receptor. The exception to this finding was 11a, where the only rise in affinity was at NOP receptors, leading to a more balanced MOP/NOP affinity profile than with any of the other compounds.
In functional assays the compounds profiled as MOP receptor partial agonists with efficacies similar to, or lower than, buprenorphine. All the new compounds were also partial agonists at NOP receptors and substantially more potent than buprenorphine. As predicted by their binding affinities, the 6-deoxy series (11) were more potent at the NOP receptor than their 6-keto counterparts; however, this was not the case at MOP receptors, where broadly similar potencies were found across the two series. The 6-deoxy compounds 11a (phenyl substituted) and 11d (3-thiophenyl substituted) stood out for their high potency at NOP receptors and the comparative (to other compounds in the series) low potency of 11a at MOP receptors meant that it profiled as a more balanced mixed MOP/NOP receptor partial agonist (4 nM versus 1 nM), approximately 100-fold more potent at NOPr than its keto counterpart 3a. Indeed, 11a was more potent at NOP than MOP receptors. Importantly, all compounds retained very low efficacy at KOP and DOP, so that the series has the desired MOP/NOP partial agonist, KOP/DOP antagonist profile.
In silico studies
Calculated logP values were obtained from a number of widely available in silico tools i.e. Flare (v7.2.0, Cresset), virtual logP,20 Molinspiration,21 MOE (v2022.02),22 SwissADME23,28 and VCCLab24 (Table 3). Using a variety of validated tools reduces the potential for bias in any single algorithm and allows for more robust consideration of any trends observed. As expected, the reduction of the C6 from a carbonyl (series 3) to methylene (series 11) resulted in an increase in hydrophobicity. This is demonstrated for each of the algorithms included in the study, albeit the difference is less pronounced with MOE and VCCLab when compared to the others. Within series the furan derivatives on average were substantially less lipophilic than their thiophenyl, phenyl and t-butyl analogues, but the absolute difference was less pronounced for the virtual logP algorithm when compared to the others.
P values
| Ligand | Flare | Virtual logP |
Mol-inspiration | MOE | Swiss-ADME | VCCLab | Mean | STDEV | Relative rank |
|---|---|---|---|---|---|---|---|---|---|
| Where italic font indicates the lowest, and bold font the highest, logP values calculated for each compound. |
|||||||||
| 3a (phenyl) | 3.7 | 3.593 | 3.86 | 3.02 | 3.64 | 4 | 3.64 | 0.3 | 8 |
| 3b (2-furanyl) | 2.7 | 3.595 | 2.87 | 2.37 | 2.99 | 3.28 | 2.97 | 0.4 | 9 |
| 3c (3-furanyl) | 2.7 | 3.507 | 2.8 | 2.37 | 2.98 | 3.34 | 2.95 | 0.4 | 10 |
| 3d (3-thiophenyl) | 3.8 | 4.004 | 3.44 | 2.99 | 3.69 | 4.06 | 3.66 | 0.4 | 6 |
| 3e (t-butyl) | 3.9 | 4.55 | 4.28 | 4 | 4.07 | 4.17 | 4.16 | 0.2 | 4 |
| 11a (phenyl) | 4.5 | 4.484 | 4.99 | 3.22 | 4.38 | 4.48 | 4.34 | 0.6 | 3 |
| 11b (2-furanyl) | 3.5 | 4.529 | 4 | 2.56 | 3.73 | 3.76 | 3.68 | 0.7 | 5 |
| 11c (3-furanyl) | 3.5 | 4.437 | 3.93 | 2.56 | 3.72 | 3.75 | 3.65 | 0.6 | 7 |
| 11d (3-thiophenyl) | 4.6 | 4.904 | 4.58 | 3.18 | 4.37 | 4.61 | 4.37 | 0.6 | 2 |
| 11e (t-butyl) | 4.7 | 5.298 | 5.41 | 4.2 | 4.71 | 5.24 | 4.93 | 0.5 | 1 |
The variations in NOP affinity (Ki) and potency (EC50) broadly followed the changes in lipophilicity – this was particularly noticeable within the 6-deoxo series 11. Thus, in each case, the Ph and thiophenyl analogs have greater potency than their furanyl counterparts. The exceptions to this, in both series, were the t-Bu containing analogs 3e and 11e, which both display highest lipophilicity but lowest potency within their series.
Absolute correlations between affinity and lipophilicity are difficult to ascertain,29,30 but it is often useful to use comparisons of relative rankings (i.e. Spearman's rankings) to determine whether general trends in lipophilicity are correlated to biological activities. Although this is not a perfect comparison by any means, as it assumes uniform distribution between the endpoints of each range of the data series concerned, it can highlight trends and provide a testable hypothesis such as the more lipophilic a compound the higher the biological affinity.
To this end, the mean calculated logP for each compound was ranked from 1 (most lipophilic) to 10 (least lipophilic). Similarly, the experimentally observed biological activities for each of the compounds against each of the opioid receptors were ranked from 1 (most active) to 10 (least active). The relative rank of mean lipophilicity versus relative rank for activity against each isoform (NOP, MOP, KOP and DOP) was plotted and an r2 value calculated (Table 4).
P vs. activity against hXOP isoforms
| Ligand | Relative rank mean logP |
Relative rank hNOP affinity | Relative rank hMOP affinity | Relative rank hKOP affinity | Relative rank hDOP affinity |
|---|---|---|---|---|---|
| 3a (phenyl) | 8 | 3.7 | 3.593 | 3.86 | 3.02 |
| 3b (2-furanyl) | 9 | 2.7 | 3.595 | 2.87 | 2.37 |
| 3c (3-furanyl) | 10 | 2.7 | 3.507 | 2.8 | 2.37 |
| 3d (3-thiophenyl) | 6 | 3.8 | 4.004 | 3.44 | 2.99 |
| 3e (t-butyl) | 4 | 3.9 | 4.55 | 4.28 | 4 |
| 11a (phenyl) | 3 | 4.5 | 4.484 | 4.99 | 3.22 |
| 11b (2-furanyl) | 5 | 3.5 | 4.529 | 4 | 2.56 |
| 11c (3-furanyl) | 7 | 3.5 | 4.437 | 3.93 | 2.56 |
| 11d (3-thiophenyl) | 2 | 4.6 | 4.904 | 4.58 | 3.18 |
| 11e (t-butyl) | 1 | 4.7 | 5.298 | 5.41 | 4.2 |
| R 2 values | — | 0.023 | 0.2292 | 0.2178 | 0.0194 |
Pairwise comparisons of relative ranks showed that there was no correlation between relative lipophilicity and relative affinity rankings across the dataset as a whole with r2 values ranging from 0.023 to 0.23.
It was possible that this analysis could have been skewed by variations in individual logP calculation algorithms. To this end pairwise Spearman rank correlation between rankings for each algorithm were carried out to identify any models that were potentially skewing the data. This showed that there was no significant variation between relative rankings for any of the algorithms at 95% confidence, and hence a general observation that lipophilicity is a proxy measure for biological affinity does not hold true for this set of compounds.
To explore this further, experiments were conducted to determine if there was a stronger correlation between relative rankings for calculated aqueous solubility of the compounds and their relatively ranked binding affinities. The algorithms investigated were VCCLabs,24 the TPSA model,25 Molinspiration,21 SILICOSIT,26 SwissADME27 and the logS calculation in MOE.22 Pairwise Spearman ranking showed correlation between the relative rankings at approximately 95% confidence for ESOL and the hMOP binding affinities (α = 0.054) and ESOL and the hDOP binding affinities (α = 0.054) but no correlation between ESOL and the other two isoforms hKOP and hNOP or any of the other solubilities generated by the remaining algorithms and the binding affinities for any isoform.
This suggests that affinity and potency is more nuanced than solely being a function of a holistic physicochemical property, and as such docking studies were conducted to investigate interactions between the molecules and the isoforms at the molecular level.
A comparison between PDB: 5DHH and an alternative NOP structure, PDB: 5DHG, showed that both had the same resolution (3.00 Å), both contained co-crystallized antagonist ligands, and had no Ramachandran outliers. Both structures contained mutations, but amino acid sequences were identical, and neither contained mutations in the binding pocket. As such either structure could be used. Another comparison was made, comparing 5DHH to 8F7X which represented the active state of NOP co-crystallized with nociceptin. A root mean square deviation (RMSD) of all atoms was taken and it revealed a structural difference of 1.37 Å. The volumes of 5DHH and 8F7X binding pockets were 932.9 Å3 and 1150 Å3 respectively. Using the sequence alignment tool in MOE, their sequences were found to be identical from 5DHH's residue 117 (glycine) to residue 414 (glycine) encompassing both of their binding pockets. MOE's “SiteView” feature demonstrates a key difference in their binding pockets where 8F7X contained more amino acid residues in its pocket (76) than 5DHH (59), consistent with the increased volume of the former. Ultimately, docking studies were performed using the inactive state of the NOP receptor (PDB: 5DHH), a choice justified by the low efficacy, partial agonist character of the ligands. The protein was downloaded from the RCSB Protein Data Bank and a full preparation of the protein–ligand complex performed on import into Flare. Docking (within Flare) used the ‘very accurate but slow’ docking mode in a region defined by the crystal structure ligand (SB-612111) and expanded to include protein atoms within 6 Å. A docking constraint was added so that the preferred binding modes allowed a salt bridge between the protonated N17 of the ligands and Asp130 of the protein active site. For comparison, docking was also carried out within MOE, again employing a docking constraint to favour binding poses with the salt bridge. Docking was then performed using the triangle matcher placement method and rigid receptor refinement with the binding site defined by the co-crystallized ligand. Flare and MOE produced very consistent binding poses with the C14 side chain phenyl ring extended into a region bounded by Tyr1313.33, Meth1343.36, Val2836.55, and Ile2195.42 amongst other residues (Fig. 1a) (superscript figures indicate Ballesteros Weinstein numbering). This is the region accessed by the dichlorophenyl ring of SB-612111 in the crystal structure (Fig. 1b). C6 of the new series of ligands projects towards Val2796.51, Tyr 3097.43, and Leu3017.35 in this pose.
 | ||
| Fig. 1 Left – Docking pose illustrated for 11a and Right – the equivalent crystal structure orientation of SB-612111 from PDB: 5DHH. | ||
Compounds 3a and 11a adopt closely related binding orientations, with both ligands positioning their shared aromatic scaffold in an area around Thr305, Asp130, Tyr309 amongst other residues and maintaining the same key N17 to Asp 130 salt bridge. The only structural distinction between the 3a and 11a is a carbonyl versus methylene at C6, but this does not alter the overall pose, and each ligand. In Fig. 2, surface complementarity between the NOP receptor protein and ligands 3a and 11a is displayed. Series 11 (6-deoxy) has improved electrostatic complementarity with the protein compared to the 6-keto analogues (series 3) but differences are small and do not account for the substantial differences in potency.
 | ||
| Fig. 2 Surface coloured by complementarity between ligands electrostatic potential and the protein (green = good, red = bad). Left) Region around C6 of 11a is highly complementary to the NOP receptor protein and Right) equivalent pose for 3a indicates poor complementarity between C6 region and the protein. | ||
The interactions of the most potent ligand (11a), and the least potent ligand (3e) with the binding pocket were also compared. While both ligands were predicted to form a salt bridge between the protonated N17 and Asp1303.32, 11a was a better fit for the pocket with both the core scaffold and the side chain well-buried within the binding site, resulting in minimal solvent exposure. With 3e the side chain and part of the core structure extended outward into the solvent-exposed region resulting in more solvent exposure.
Since all the new ligands bound with good affinity to the MOP a comparison was made between β-FNA, the co-crystallised ligand in 4DKL and the new series 3 and 11. 11e has an 88.78% Tanimoto similarity to β-FNA which is the highest out of any ligand in series 11 and series 3. The RMSD of the poses was 1.79 Å. Both ligands had a salt bridge between the protonated N17 and Asp1473.32. As expected their core structures overlap with each other and they both project towards Val3006.55, Val2365.42, Lys2335.39 with just the side chains extending towards a different regions of the binding pocket (SI).
Discussion
C14 analogs and derivatives of naltrexone have provided some of the most interesting opioid series, with a wide range of pharmacological profiles achievable. 14-O-Alkyl ethers of naltrexone (12: Chart 2) tend to have predominant MOP receptor antagonist activity12,13 though the 14-O-3-phenylpropyl ether is a high efficacy and high potency MOP agonist.14 Cinnamoyl esters (13), which differ from the lead, 3a, only in having a double bond in the side chain, are predominantly opioid receptor antagonists.15 The amide equivalents of the cinnamoyl esters are the 14-N-cinnamoylamino derivatives (14). These are similarly potent antagonists but of longer duration, as exemplified by the MOP-receptor selective irreversible antagonist methocinnamox (M-CAM; 14a)16–18 currently in preclinical development with potential as a relapse prevention agent and for overdose reversal and/or prevention.19 Closely related to the current series are the 14β-phenylacetyl substituted analogues (5b: Chart 1).6 These compounds profiled as partial agonists across each of the four opioid receptors, with affinities and potencies at NOP receptors very similar to buprenorphine. The current series of esters having, as predicted, partial agonist activity at MOP receptors, fall in the middle of this range.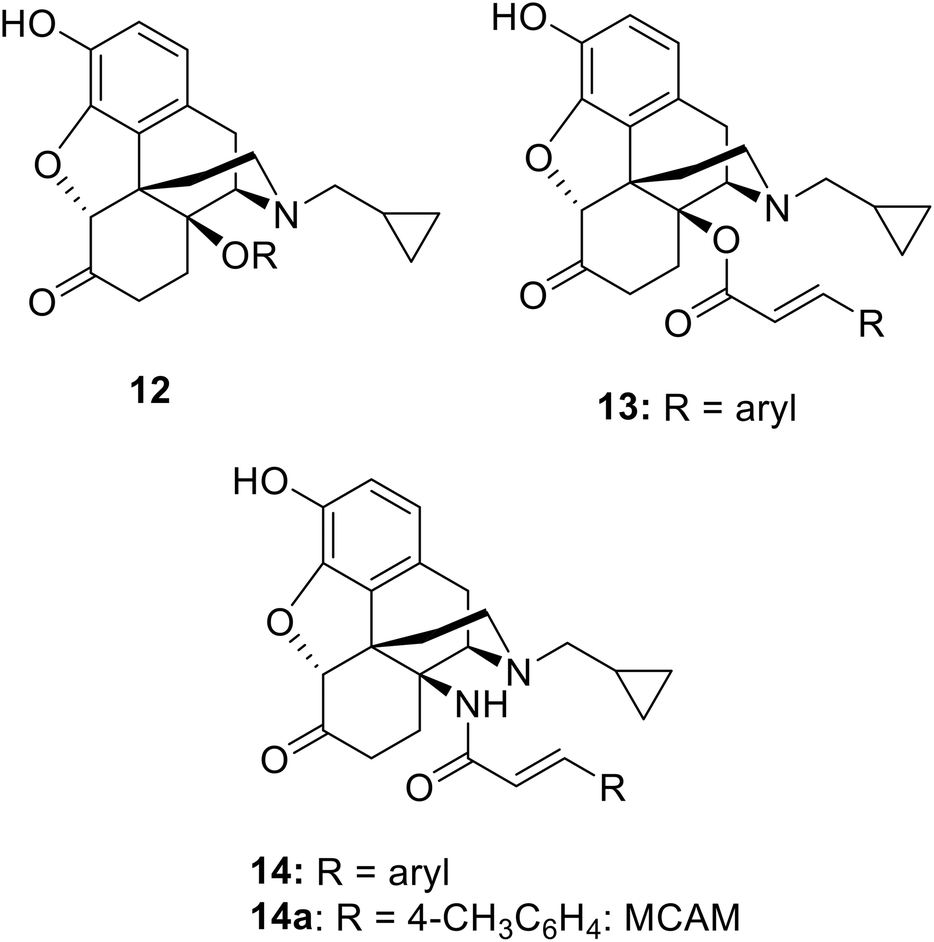 | ||
| Chart 2 Related C14-substituted analogues of naltrexone. | ||
A key priority in the current study was to further increase affinity and agonist activity at NOP receptors. The lead, 3a, has a highly desirable profile in non-human primates with evidence of agonist activity at both MOP and NOP receptors.3,4 Our in vitro findings differ slightly from the original report on 3a. Compared to Kiguchi et al.3 we obtained a 10-fold lower affinity and a 4-fold lower potency in the [35S]GTPγS assay at the NOP receptor.4 However, the levels of agonist efficacy are similar as are the binding affinities at the KOP and DOP receptors; both sets of data show no agonist activity at KOP or DOP receptors.
Introduction of heteroaryl rings in place of the phenyl ring of 3a resulted in increased selectivity towards the MOP receptor, as did replacement of the aryl ring with a t-butyl group. On reducing the C6 carbonyl to methylene, binding affinities for all receptors tended to increase, whereas in the [35S]GTPγS assay only potency at the NOP receptor was significantly altered (increased), leading to compounds with a more balanced MOP:NOP receptor profile and, in 11a, the first compound in this series identified as having higher potency at NOP- than MOP receptors. It will be of interest to see how this alters the in vivo profile of the compound. The fact that 11a has a lower affinity for the NOP receptor than the MOP receptor but a higher potency may relate to the different assay conditions for the two measures. Binding assays were performed in Tris buffer whereas the [35S]GTPγS assay was performed in a buffer containing 125 mM NaCl and 5 mM MgCl2, as well as GDP.
The increase in lipophilicity on going from the keto to the deoxo series is unlikely to explain the increase in potency of the latter series as the more lipophilic members of the keto series (3d and 3a) are substantially less potent than all of the deoxo series despite having greater lipophilicity than 11b and 11c. Increased lipophilicity does increase the difference in NOP affinity and potency between the reduced series 11 and the carbonyl containing series 3. Thus, 11a is 169 times more potent than 3a, there is a 68-fold difference for the thiophenes and a 5–6 fold difference for the furans.
The variation in potency and efficacy at MOP and NOP receptors through the series will allow future in vivo work to determine the optimal balance of MOP and NOP activity for strong analgesia with minimal side effects. For example, 3a, 11d and 11a have virtually identical efficacies to each other at MOP and NOP, but selectivity in potency varies from ∼100-fold for MOP (3a), ∼55-fold for MOP (11d) to 4-fold for NOP (11a).
Experimental
Synthesis and characterization
Reagents and solvents were purchased from Sigma-Aldrich or Alfa Aesar and used as received. 1H and 13C NMR spectra were obtained with a Brucker-400 MHz instrument (1H at 400 MHz, 13C at 100 MHz); residual solvent resonances were used as internal reference signals. ESIMS: microTOF (BRUKER). Column chromatography was performed using pre-packed columns on a Teledyne ISCO combiflash instrument. Ligands were tested as their hydrochloride salts, prepared by adding 5 equivalents of HCl (1 N solution in diethyl ether) to a solution of compound in anhydrous methanol. All reactions were carried out under an inert atmosphere of nitrogen unless otherwise indicated. All compounds were >95% pure.14β-(3-Phenylpropanoyl)-17-cyclopropylmethyl-7,8-dihydronoroxymorphinone (3a). Isolated as a white solid (97%). Rf 0.32 (at 30% EtOAc/hexane, 0.5% NH3); 1H NMR (CDCl3) δ 0.03–0.08 (2H, m), 0.42–0.46 (2H, m), 0.64–0.73 (1H, m), 1.43–1.50 (1H, m), 1.51 (1H, dt, J = 3.72 Hz & 14.44 Hz), 2.09–2.18 (3H, m), 2.20–2.54 (5H, m), 2.63–2.86 (3H, m), 2.98–3.02 (2H, m), 3.05 (1H, d, J = 18.2 Hz), 4.43 (1H, d, J = 5.52 Hz), 4.58 (1H, s), 6.55 (1H, d, J = 8.0 Hz), 6.68 (1H, d, J = 8.0 Hz), 7.14–7.31 (5H, m); 13C NMR, 400 MHz, (CDCl3) δ 3.84, 9.51, 23.06, 26.91, 30.10, 31.05, 35.44, 36.61, 43.87, 51.24, 55.57, 59.46, 82.55, 90.16, 117.89, 119.97, 125.29, 126.42, 128.05, 128.22, 128.25, 128.49, 128.59, 129.02, 138.60, 140.49, 143.30, 171.80, 208.51. HRMS, m/z for (C29H32NO5) [MH]+, calcd – 474.2280, found – 474.2245. Anal (C29H31NO5·HCl·H2O) C, H, N.
14β-(3-(Furan-2-yl)propanoyl)-17-cyclopropylmethyl-7,8-dihydronoroxymorphinone (3b). The product, free base, was obtained as a white foam (80%). 1H NMR (400 MHz, CDCl3) δ 7.29–7.26 (m, 1H), 6.70 (d, J = 8.0 Hz, 1H), 6.55 (d, J = 8.0 Hz, 1H), 6.25 (dd, J = 2.0, 3.2 Hz, 1H), 6.06–6.03 (m, 1H), 4.62 (s, 1H), 4.42 (d, J = 5.5 Hz, 1H), 3.08–3.0 (m, 3H), 2.86–2.74 (m, 3H), 2.66 (dd, J = 4.5, 11.8 Hz, 1H), 2.55–2.38 (m, 3H), 2.36–2.06 (m, 4H), 1.59 (td, J = 3.7, 14.5 Hz, 1H), 1.48 (dd, J = 2.5, 12.3 Hz, 1H), 0.78–0.66 (m, 1H), 0.52–0.4 (m, 2H), 0.1–(−)0.02 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 209.2, 171.3, 154.2, 143.4, 141.1, 138.9, 128.0, 124.8, 119.9, 118.3, 110.3, 105.3, 90.0, 82.7, 59.3, 55.5, 51.2, 43.8, 35.4, 33.7, 30.0, 26.9, 23.5, 20.0, 13.5, 9.4, 3.7. HRMS: calc. for C27H30NO6, 464.207313; found 464.2099. The corresponding HCl salt was obtained by using 1 M solution of HCl in Et2O. Anal (C27H29NO6·HCl·2H2O) C, H, N.
14β-(3-(Furan-3-yl)propanoyl)-17-cyclopropylmethyl-7,8-dihydronoroxymorphinone (3c). The product, free base, was obtained as a clear oil (78%). 1H NMR (400 MHz, CDCl3) δ 7.35 (t, J = 1.5 Hz, 1H), 7.30 (b s, 1H), 6.74 (d, J = 8.0 Hz, 1H), 6.58 (d, J = 8.0 Hz, 1H), 6.33 (s, 1H), 4.67 (s, 1H), 4.46 (d, J = 5.5 Hz, 1H), 3.08 (d, J = 18.3 Hz, 1H), 2.90–2.63 (m, 6H), 2.57–2.41 (m, 3H), 2.40–2.10 (m, 4H), 1.62 (td, J = 3.2, 13.8 Hz, 1H), 1.51 (dd, J = 2.5, 12.3 Hz, 1H), 0.98–0.83 (m, 1H), 0.78–0.66 (m, 1H), 0.55–0.40 (m, 2H), 0.14–0.00 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 209.0, 171.9, 143.5, 143.0, 139.1, 138.8, 128.1, 125.1, 123.6, 120.0, 118.2, 110.8, 90.1, 82.7, 59.4, 55.6, 51.3, 43.9, 35.7, 35.5, 30.1, 27.0, 23.9, 20.1, 17.3, 9.5, 3.7. HRMS: calc. for C27H30NO6, 464.207313; found 464.2103. The corresponding HCl salt was obtained by using 1 M solution of HCl in Et2O. Anal (C27H29NO6·HCl·0.5H2O) C, H, N.
14β-(3-(Thiophen-3-yl)propanoyl)-17-cyclopropylmethyl-7,8-dihydronoroxymorphinone (3d). The product, free base, was obtained as a clear oil (72%). 1H NMR (400 MHz, CDCl3) δ 7.29–7.23 (m, 1H), 7.04 (b s, 1H), 7.01 (dd, J = 1.5, 5.0 Hz, 1H), 6.73 (d, J = 8.0 Hz, 1H), 6.58 (d, J = 8.0 Hz, 1H), 4.63 (s, 1H), 4.46 (d, J = 5.5 Hz, 1H), 3.12–3.00 (m, 3H), 2.90–2.74 (m, 3H), 2.69 (dd, J = 4.77, 11.8 Hz, 1H), 2.58–2.23 (m, 5H), 2.23–2.10 (m, 2H), 1.60 (td, J = 3.7, 14.3 Hz, 1H), 1.50 (d, J = 10.0 Hz, 1H), 0.80–0.66 (m, 1H), 0.57–0.41 (m, 2H), 0.16–0.01 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 208.9, 171.2, 143.4, 140.9, 138.8, 127.8, 125.7, 124.9, 120.5, 119.9, 118.2, 90.0, 82.6, 59.4, 55.5, 51.2, 43.9, 36.0, 35.3, 30.0, 26.9, 25.6, 23.0, 20.9, 9.4, 3.7. HRMS: calc. for C27H30NO5S, 480.184469; found 480.1858. The corresponding HCl salt was obtained by using 1 M solution of HCl in Et2O. Anal (C27H29NO5S·HCl·2H2O) C, H, N.
14β-(4,4-Dimethylpentanoyl)-17-cyclopropylmethyl-7,8-dihydronoroxymorphinone (3e). The product, free base, was obtained as clear oil (30 mg, 37%). 1H NMR (500 MHz, CDCl3) δ 6.74 (d, J = 8.0 Hz, 1H), 6.60 (d, J = 8.0 Hz, 1H), 4.71 (s, 1H), 4.43 (d, J = 5.7 Hz, 1H), 3.08 (d, J = 18.6 Hz, 1H), 2.90–2.80 (m, 1H), 2.76–2.25 (m, 8H), 2.15 (td, J = 3.8, 12.0 Hz, 1H), 1.70–1.50 (m, 4H), 1.43–1.30 (m, 2H), 0.94 (s, 9H), 0.84–0.70 (m, 1H), 0.54–0.42 (m, 2H), 0.12–0.02 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 209.2, 173.5, 143.4, 138.8, 129.0, 125.3, 120.0, 118.2, 90.2, 82.1, 59.5, 55.8, 51.4, 43.8, 38.8, 35.8, 31.2, 29.1, 28.0, 23.9, 23.1, 9.50, 3.9, 3.7. HRMS: calc. for C27H36NO5, 454.259348; found 454.2614. The corresponding HCl salt was obtained by using 1 M solution of HCl in Et2O. Anal (C27H35NO5·HCl·0.5H2O) C, H, N.
14β-(3-Phenylpropanoyl)-17-cyclopropylmethyl-4,5-epoxy-3,14-dihydroxymorphinan (11a). Isolated as a white solid (92 mg, 94%). Rf 0.10, (at 30% EtOAc/hexane, 0.5% NH3), 1H NMR (400 MHz, CDCl3) δ 7.15–7.25 (m, 5H), 6.63–6.65 (d, J = 8.0 Hz, 2H), 6.49–6.51 (d, J = 8.0 Hz, 2H), 4.62 (t, J = 5.5 Hz, 1H), 4.28–4.29 (d, J = 4.0 Hz, 1H), 3.69–3.72 (t, J = 6.0 Hz, 2H), 2.97–3.01 (d, J = 20.0 Hz, 1H), 2.94–2.97 (m, 2H), 2.62–2.71 (m, 3H), 2.27–2.260 (m, 2H), 2.23–2.26 (m, 3H), 2.20–2.23 (m, 1H), 1.99–2.12 (m, 2H), 1.79–1.82 (t, J = 14.5 Hz, 1H), 1.26–1.32 (d, J = 8.0, 1H), 1.22–1.26 (t, J = 9.7 Hz, 1H), 0.56–0.60 (m, 1H), 0.39–0.41 (d, J = 8.0 Hz, 2H), −0.02–0.02 (d, J = 8.0 Hz, 2H); 13C NMR (100.6 MHz, CDCl3) δ 170.6, 147.0, 140.3, 133.4, 132.8, 131.1, 128.5, 128.4, 126.4, 122.0, 118.4, 90.2, 70.5, 62.6, 59.3, 56.7, 44.3, 35.6, 31.0, 30.7, 28.9, 23.1, 17.2, 9.5, 3.9, 3.8; HRMS: calc. for C29H33NO4, 450.2410; found 450.2378; anal (C29H33NO4·HCl·0.5H2O) C, H, N.
14β-(3-(Furan-2-yl)propanoyl)-17-cyclopropylmethyl-4,5-epoxy-3,14-dihydroxymorphinan (11b). The product, free base, was obtained as white foam (100%). 1H NMR (400 MHz, MeOD) δ 7.39 (s, 1H), 6.76 (d, J = 8.2 Hz, 1H), 6.71 (d, J = 8.2 Hz, 1H), 6.32 (dd, J = 1.76, 3.0 Hz, 1H), 6.14 (d, J = 3.3 Hz, 1H), 5.32 (d, J = 6.0 Hz, 1H), 4.77 (t, J = 7.3 Hz, 1H), 3.47–3.34 (m, 2H), 3.28–3.18 (m, 2H), 3.15–2.89 (m, 5H), 2.84 (td, J = 4.3, 13.0 Hz, 1H), 2.56 (td, J = 4.3, 13.0 Hz, 1H), 2.50–2.40 (m, 1H), 2.18–2.08 (m, 1H), 1.71 (dd, J = 3.5, 14.0 Hz, 1H), 1.44–1.25 (m, 4H), 1.10–0.98 (m, 1H), 0.87–0.77 (m, 1H), 0.76–0.68 (m, 1H), 0.56–0.46 (m, 2H); 13C NMR (100 MHz, MeOD) δ 174.4, 155.5, 144.3, 143.2, 142.7, 130.3, 122.1, 120.7, 119.8, 111.5, 106.8, 88.5, 84.8, 59.6, 59.1, 47.1, 35.1, 29.5, 29.0, 26.8, 25.3, 24.2, 17.4, 6.9, 6.4, 3.5. HRMS: calc. for C27H32NO5, 450.2280; found 450.2378. Anal (C27H31NO5·HCl·0.5H2O) C, H, N.
14β-(3-(Furan-3-yl)propanoyl)-17-cyclopropylmethyl-4,5-epoxy-3,14-dihydroxymorphinan (11c). The product, free base, was obtained as clear oil (93%). 1H NMR (400 MHz, MeOD) δ 7.41 (s, 1H), 7.37 (s, 1H), 6.75 (d, J = 8.0 Hz, 1H), 6.71 (d, J = 8.0 Hz, 1H), 6.40 (s, 1H), 5.32 (d, J = 5.5 Hz, 1H), 4.78–4.70 (m, 1H), 3.50–3.34 (m, 2H), 3.28–3.18 (m, 2H), 3.15–2.95 (m, 2H), 2.90–2.72 (m, 4H), 2.67–2.53 (m, 1H), 2.52–2.40 (m, 1H), 2.18–2.05 (m, 1H), 1.66 (d, J = 11.0 Hz, 1H), 1.44–1.22 (m, 4H), 1.13–1.00 (m, 1H), 0.87–0.76 (m, 1H), 0.75–0.65 (m, 1H), 0.64–0.55 (m, 1H), 0.55–0.45 (m, 1H); 13C NMR (100 MHz, MeOD) δ174.8, 144.3, 143.1, 140.7, 130.3, 125.1, 122.1, 120.7, 120.0, 112.1, 88.5, 84.5, 59.5, 58.9, 47.1, 37.4, 29.5, 28.9, 26.8, 25.4, 21.1, 17.4, 6.9, 6.5, 3.6. HRMS: calc. for C27H32NO5, 450.2280; found 450.2339. Anal (C27H31NO5·HCl·H2O) C, H, N.
14β-(3-(Thiophen-2-yl)propanoyl)-17-cyclopropylmethyl-4,5-epoxy-3,14-dihydroxymorphinan (11d). The product, free base, was obtained as clear oil (88%). 1H NMR (400 MHz, CDCl3) δ 7.30–7.24 (m, 1H), 7.08–6.98 (m, 2H), 6.72 (d, J = 8.0 Hz, 1H), 6.57 (d, J = 8.0 Hz, 1H), 4.71 (t, J = 6.8 Hz, 1H), 4.38 (d, J = 5.3 Hz, 1H), 3.11–2.99 (m, 3H), 2.84–2.62 (m, 3H), 2.52 (dd, J = 5.5, 18.3 Hz, 1H), 2.43 (d, J = 14.0 Hz, 1H), 2.38–2.22 (m, 3H), 2.14 (td, J = 3.7, 12.0 Hz, 1H), 2.10–2.00 (m, 1H), 1.47–1.20 (m, 5H), 0.8–0.67 (m, 1H), 0.55–0.4 (m, 2H), 0.14–0.0 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 171.7, 142.1, 141.1, 139.4, 130.8, 128.0, 125.9, 125.4, 120.4, 118.7, 116.8, 89.2, 84.0, 59.5, 56.0, 47.0, 44.6, 36.2, 30.2, 28.5, 26.2, 25.6, 23.1, 16.8, 9.5, 3.8, 3.7. C27H32NO4S, 466.2052; found 466.2163. The corresponding HCl salt was obtained by using 1 M solution of HCl in Et2O. Anal (C27H31NO4S·HCl·0.5H2O) C, H, N.
14β-(4,4-Dimethylpentanoyl)-17-cyclopropylmethyl-4,5-epoxy-3,14-dihydroxymorphinan (11e). The product, free base, was obtained as clear oil (94%). 1H NMR (400 MHz, CDCl3) δ 6.71 (d, J = 8.0 Hz, 1H), 6.55 (d, J = 8.0 Hz, 1H), 4.74 (t, J = 7.5 Hz, 1H), 4.31 (d, J = 5.3 Hz, 1H), 3.05 (d, J =18.3 Hz, 1H), 2.67 (dd, J = 4.5, 11.8 Hz, 1H), 2.55–2.40 (m, 2H), 2.39–2.24 (m, 5H), 2.17–2.06 (m, 2H), 1.60 (dd, J = 6.8, 8.0 Hz, 2H), 1.56–1.46 (m, 1H), 1.44–1.30 (m, 3H), 1.29–1.19 (m, 1H), 0.92 (s, 9H), 0.8–0.7 (m, 1H), 0.5–0.4 (m, 2H), 0.11–0.0 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 173.3, 142.1, 139.4, 130.9, 126.0, 118.7, 116.8, 89.2, 83.4, 59.6, 56.2, 47.1, 44.5, 38.8, 31.4, 30.2, 30.0, 29.1, 28.5, 26.3, 23.2, 17.0, 9.5, 3.9, 3.5. C27H38NO4, 440.2801; found 440.2899. The corresponding HCl salt was obtained by using 1 M solution of HCl in Et2O. Anal (C27H37NO4·HCl·1.5H2O).
Cell lines and cell culture
Human (h) DOP- and hNOP-receptor CHO cell lines were generous gifts from Larry Toll at the Torrey Pines Institute. The hMOP- and hKOP-CHO cell lines were generous gifts from John M. Streicher at the University of Arizona. All cells were cultured in 50:50 DMEM/F12 media with 10% heat-inactivated FBS and 1× penicillin/streptomycin supplement (all Gibco brand) in a 37 °C humidified incubator with 5% CO2 atmosphere. Propagation cultures were further maintained with 500 μg mL−1 G418. Cultures were propagated for no more than 30 passages. Cell pellets for experiments were prepared by growth in 15 cm2 plates, harvested with 5 mM EDTA in 50 mM Tris HCl pH 7.4. Cell pellets were resuspended in 50 mM Tris HCl pH 7.4 and homogenized with a tissue grinder for 15–30 seconds on ice. Crude membranes were spun down at 15000g for 30 min, washed once followed by recentrifugation. Membrane preparations were stored at −80 °C prior to use.
Competition radioligand binding
hDOP-, hKOP- or hMOP-receptor containing CHO membrane preparations were diluted to 10–20 μg in 50 mM Tris-HCl buffer (pH 7.4) and incubated with a fixed concentration of 3H-diprenorphine (0.2–0.4 nM) and varying concentrations of test compound. With hNOP receptor-CHO cells, 3H-nociceptin was used in an analogous manner. Reactions were performed in 400 μL volumes in 96 well plates and incubated at 37 °C for 3 hours. Reactions were terminated by rapid filtration through 24 well format GF/B filter mat (PerkinElmer) and washed with cold 25 mM Tris HCl pH 7.4 buffer, dried, and ECOLUME™ scintillation cocktail from MP Biomedicals (Santa Cruz, CA, USA) was added. Radioactivity retained on the filters was determined using a MicroBeta2 96-well format 6 detector scintillation counter (PerkinElmer). Ki values were calculated from the inhibitory concentration50 (IC50) of each competitor ligand using GraphPad Prism (v7.0).35S-GTPγS binding
Briefly, hDOP, hKOP, hMOP- or hNOP-receptor containing CHO membranes were combined with test compound, 50 pM [35S]GTPγS (Perkin Elmer), and 10–15 μg membrane homogenate in GTPγS assay buffer (50 mM Tris HCl pH 7.4, 125 mM NaCl, 5 mM MgCl2, 1 mM EDTA and 30 μM GDP). The 200 μL reaction was incubated at 30 °C for 50 minutes in 96 well plates, then collected by filtration and measured for reactivity as described for the binding experiments. Potency (EC50) and EMAX values were calculated using the three-variable log(agonist) vs. response curve in GraphPad Prism 7.0.Conclusions
Naltrexone analogues with C14-ester side chains have dramatically improved NOP partial agonist affinity/potency coupled with partial agonist activity at MOP receptors. Potency at NOP is further increased through removal of the C6-carbonyl. The mixed MOP:NOP partial agonist profile is retained across these series with the major differences between analogues being in the MOP:NOP selectivity. These series offer promising candidates for improved analgesics and potentially, addiction treatments and will allow for future studies to determine the optimal profile for these indications.Author contributions
MO, KO and GC-K performed the experiments and analyzed data. SMH, SBK, LJ performed the computational studies. SMH and JRT devised the study and with KO wrote the manuscript.Conflicts of interest
SMH is an inventor on the patent that describes these compounds.Data availability
Chemical characterisation data supporting this article have been uploaded as part of the supplementary information (SI).Supplementary information: 1H and 13C NMR spectra and image from docking of 11e to MOP. See DOI: https://doi.org/10.1039/d5md00685f
Acknowledgements
CHO-hDOPr were a generous gift from L. Toll; the hMOPr and hKOPr cells were a generous gift from J. Streicher. The authors gratefully acknowledge the Material and Chemical Characterisation Facility (MC2) at the University of Bath (https://doi.org/10.15125/mx6j-3r54) for technical support and assistance in this work. Funding provided by National Institute on Drug Abuse (NIDA) (Grants R01 DA07315 and R37 DA033397.Notes and references
- H. Ding, P. W. Czoty, N. Kiguchi, G. Cami-Kobeci, D. D. Sukhtankar, M. A. Nader, S. M. Husbands and M. C. Ko, A novel orvinol analog, BU08028, as a safe opioid analgesic without abuse liability in primates, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E5511–E5518 CAS.
- H. Ding, N. Kiguchi, D. Yasuda, P. R. Daga, W. E. Polgar, J. J. Lu, P. W. Czoty, S. Kishioka, N. T. Zaveri and M. C. Ko, A bifunctional nociceptin and mu opioid receptor agonist is analgesic without opioid side effects in nonhuman primates, Sci. Transl. Med., 2018, 10, eaar3483 Search PubMed.
- N. Kiguchi, H. Ding, G. Cami-Kobeci, D. D. Sukhtankar, P. W. Czoty, H. B. DeLoid, F.-C. Hsu, L. Toll, S. M. Husbands and M. C. Ko, BU10038 as a safe opioid analgesic with few side-effects after systemic and intrathecal administration in primates, Br. J. Anaesth., 2019, 122, e146–e156 Search PubMed.
- L. R. Gerak, D. R. Maguire, G. Cami-Kobeci, K. M. Olson, J. R. Traynor, S. M. Husbands, C. P. France, L. Acevedo, B. Belli and P. Flynn, OREX-1038: A potential new treatment for pain with low abuse liability and limited adverse effects, Behav. Pharmacol., 2022, 33, 377–394 CrossRef CAS PubMed.
- G. Cami-Kobeci, W. E. Polgar, T. V. Khroyan, L. Toll and S. M. Husbands, Structural determinants of opioid and NOP receptor activity in derivatives of buprenorphine, J. Med. Chem., 2011, 54, 6531–6537 CrossRef CAS PubMed.
- V. Kumar, W. Polgar, G. Cami-Kobeci, M. P. Thomas, T. Khroyan, L. Toll and S. M. Husbands, Synthesis, biological evaluation and SAR studies of 14b-phenylacetyl substituted 17-cyclopropylmethyl-7,8-dihydronoroxymorphinone derivatives: ligands with mixed NOP and opioid receptor profile, Front. Psychiatry, 2018, 9, 430 CrossRef PubMed.
- G. Bart, J. Schluger, L. Borg, A. Ho and J. M. Bidlack, Nalmefene Induced Elevation in Serum Prolactin in Normal Human Volunteers: Partial Kappa Opioid Agonist Activity?, Neuropsychopharmacology, 2005, 30, 2254–2262 CrossRef CAS PubMed.
- J. Yancey-Wrona, B. Dallaire, E. Bilsky, B. Bath, J. Burkart, L. Webster, D. Magiera, X. Yang, M. Phelps and W. Sadee, 6β-Naltrexol, a Peripherally Selective Opioid Antagonist that Inhibits Morphine-Induced Slowing of Gastrointestinal Transit: An Exploratory Study, Pain Med., 2011, 12, 1727–1737 CrossRef.
- M. P. Wentland, R. Lou, Q. Lu, Y. Bu, C. Denhardt, J. Jin, R. Ganorkar, M. A. VanAlstine, C. Guo, D. J. Cohen and J. M. Bidlack, Syntheses of novel high affinity ligands for opioid receptors, Bioorg. Med. Chem. Lett., 2009, 19, 2289–2294 CrossRef CAS PubMed.
- G. Besong, K. Jarowicki, P. J. Kocienski, E. Sliwinski and F. T. Boyle, Synthesis of (S)-(−)-N-acetylcolchinol using intramolecular biaryl oxidative coupling, Org. Biomol. Chem., 2006, 4, 2193–2207 RSC.
- M. C. Sheikh, S. Takagi, T. Yoshimura and H. Morita, Mechanistic studies of DCC/HOBt-mediated reaction of 3-phenylpropionic acid with benzyl alcohol and studies on the reactivities of ‘active ester’ and the related derivatives with nucleophiles, Tetrahedron, 2010, 66, 7272–7278 CrossRef CAS.
- H. Schmidhammer, L. Aeppli, L. Atwell, F. Fritsch, A. E. Jacobson, M. Nebuchla and G. Sperk, Synthesis and biological evaluation of 14-alkoxymorphinans. 1. Highly potent opioid agonists in the series of (−)-14-methoxy-N-methylmorphinan-6-ones, J. Med. Chem., 1984, 27, 1575–1579 CrossRef CAS PubMed.
- H. Schmidhammer, W. P. Burkard and L. Eggstein-Aeppli, Synthesis and biological evaluation of 14-alkoxymorphinans. 4. Opioid agonists and partial opioid agonists in a series of N-(cyclobutylmethyl)-14-methoxymorphinan-6-ones, Helv. Chim. Acta, 1989, 72, 1233–1240 CrossRef CAS.
- E. Greiner, M. Spetea, R. Krassnig, F. Schüllner, M. Aceto, L. S. Harris, J. R. Traynor, J. H. Woods, A. Coop and H. Schmidhammer, Synthesis and biological evaluation of 14-alkoxymorphinans. 18. N-substituted 14-phenylpropyloxymorphinan-6-ones with unanticipated agonist properties: extending the scope of common structure-activity relationships, J. Med. Chem., 2003, 46, 1758–1763 CrossRef CAS PubMed.
- H. Moynihan, A. R. Jales, B. M. Greedy, D. Rennison, J. H. Broadbear, L. Purington, J. R. Traynor, J. H. Woods, J. W. Lewis and S. M. Husbands, 14β-O-cinnamoylnaltrexone and related dihydrocodeinones are mu opioid receptor partial agonists with predominant antagonist activity, J. Med. Chem., 2009, 52, 1553–1557 CrossRef CAS PubMed.
- J. H. Broadbear, T. L. Sumpter, T. F. Burke, S. M. Husbands, J. W. Lewis, J. H. Woods and J. R. Traynor, Methocinnamox is a potent, long-lasting, and selective antagonist of morphine-mediated antinociception in the mouse: Comparison with clocinnamox, β-funaltrexamine, and β-chlornaltrexamine, J. Pharmacol. Exp. Ther., 2000, 294, 933–940 CrossRef CAS PubMed.
- D. R. Maguire, L. R. Gerak, J. H. Woods, S. Husbands, A. Disney and C. P. France, Long-lasting effects of methocinnamox on opioid self-administration in rhesus monkeys, J. Pharmacol. Exp. Ther., 2019, 368, 88–99 CrossRef CAS PubMed.
- L. R. Gerak, V. Minervini, E. Latham, S. Ghodrati, K. V. Lillis, J. Wooden, A. Disney, S. M. Husbands and C. P. France, Methocinnamox Produces Long-Lasting Antagonism of the Behavioral Effects of μ-Opioid Receptor Agonists but Not Prolonged Precipitated Withdrawal in Rats, J. Pharmacol. Exp. Ther., 2019, 371, 507–516 CrossRef CAS PubMed.
- C. P. France, G. P. Ahern, S. Averick, A. Disney, H. A. Enright, B. Esmaeli-Azad, A. Federico, L. R. Gerak, S. M. Husbands, B. Kolber, E. Y. Lau, V. Lao, D. R. Maguire, M. A. Malfatti, G. Martinez, B. P. Mayer, M. Pravetoni, N. Sahibzada, P. Skolnick, E. Y. Snyder, N. Tomycz, C. A. Valdez and J. Zapf, Countermeasures for Preventing and Treating Opioid Overdose, Clin. Pharmacol. Ther., 2021, 109, 578–590 CrossRef PubMed.
- P. Gaillard, P. A. Carrupt, B. Testa and A. Boudon, Molecular Lipophilicity Potential, a tool in 3D QSAR: Method and applications, J. Comput.-Aided Mol. Des., 1994, 8, 83–96 CrossRef CAS PubMed.
- Molinspiration Cheminformatics free web services, https://www.molinspiration.com/services/logp.html, Slovensky Grob, Slovakia, [Accessed online 15th April 2025] Search PubMed.
- Chemical Computing Group ULC, Molecular Operating Environment (MOE), 2022.02, Chemical Computing Group ULC, 910-1010 Sherbrooke St. W., Montreal, QC H3A 2R7, 2023 Search PubMed.
- A. Daina, O. Michielin and V. Zoete, iLOGP: a simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach, J. Chem. Inf. Model., 2014, 54, 3284–3301 CrossRef CAS.
- I. V. Tetko and V. Y. Tanchuk, Application of associative neural networks for prediction of lipophilicity in ALOGPS 2.1 program, J. Chem. Inf. Comput. Sci., 2002, 42, 1136–1145 CrossRef CAS PubMed.
- I. V. Tetko, J. Gasteiger, R. Todeschini, A. Mauri, D. Livingstone, P. Ertl, V. A. Palyulin, E. V. Radchenko, N. S. Zefirov, A. S. Makarenko, V. Y. Tanchuk and V. V. Prokopenko, Virtual computational chemistry laboratory - design and description, J. Comput.-Aided Mol. Des., 2005, 19, 453–463 CrossRef CAS PubMed.
- J. Ali, P. Camilleri, M. Brown, A. J. Hutt and S. B. Kirton, Revisiting the General Solubility Equation: In Silico Prediction of Aqueous Solubility Incorporating the Effect of Topographical Polar Surface Area, J. Chem. Inf. Model., 2012, 52, 420–428 CrossRef CAS PubMed.
- A. Lusci, G. Pollastri and P. Baldi, Deep architectures and deep learning in chemoinformatics: the prediction of aqueous solubility for drug-like molecules, J. Chem. Inf. Model., 2013, 53, 1563–1575 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 7, 42717 CrossRef.
- M. A. Parker, D. M. Kurrasch and D. E. Nichols, The role of lipophilicity in determining binding affinity and functional activity for 5-HT2A receptor ligands, Bioorg. Med. Chem., 2008, 16, 4661–4669 CrossRef CAS PubMed.
- A. Pirovano, M. A. Huijbregts, A. M. Ragas and A. J. Hendriks, Compound Lipophilicity as a Descriptor to Predict Binding Affinity (1/Km) in Mammals, Environ. Sci. Technol., 2012, 46, 5168–5174 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |