
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new compound that extends the lifespan of worms, with favorable in vitro properties and a lack of toxicity in rodents
Heidi M.
Blank
a and
Michael
Polymenis
*ab
aDepartment of Biochemistry and Biophysics, Texas A&M University, 300 Olsen Blvd, College Station, TX 77843, USA. E-mail: michael.polymenis@ag.tamu.edu
bInstitute for Advancing Health Through Agriculture, 498 Olsen Blvd, College Station, TX 77840, USA
First published on 9th December 2025
Abstract
The development of geroprotectors, compounds that slow the biological aging process, is an important goal in modern medicine. These interventions hold the promise not only of extending lifespan, but more importantly, of extending healthspan—the period of life free from chronic disease. By targeting the fundamental mechanisms of aging, geroprotectors have the potential to simultaneously delay the onset and progression of multiple age-related conditions, thereby reducing the immense societal and economic burden of these illnesses. In this manuscript, we identified a new compound, MPOL_B_1, through a virtual screen targeting the proton-coupled folate transporter (PCFT). We describe the synthesis of MPOL_B_1 and two other lead compounds identified from the virtual screen. MPOL_B_1 had drug-like properties in vitro and a favorable safety profile in mice and rats. Most importantly, MPOL_B_1 increased the lifespan of the model organism C. elegans by up to 30%. Our findings suggest that MPOL_B_1 is a promising pro-longevity candidate that warrants further investigation, although its mechanism of action in animals requires additional studies.
1. Introduction
The enormous societal and economic impact of promoting human longevity motivates the search for compounds that extend the lifespan of model organisms from invertebrates, such as yeast, worms, and flies, to mammals, primarily mice. Results from those studies are cataloged in public databases.1,2 Research programs managed by the National Institute on Aging systematically assess the impact of drugs on the lifespan of mice3 and C. elegans worms.4 These efforts contribute to the field of geroscience by identifying promising compounds that may help extend a healthy human lifespan.Previous work in invertebrate model systems from several labs, including ours, implicated the conserved one-carbon (1C) metabolic pathways in longevity, and methionine restriction also extends longevity in many systems.5–17 Furthermore, lower methyl tetrahydrofolate (THF) levels are a common signature of pro-longevity pathways.18 We had reported that low-dose methotrexate, which limits the THF pools available for 1C reactions by inhibiting dihydrofolate reductase,19 extended the lifespan of yeast and worms.20 We further tested the generality of the idea that 1C pharmacologic interventions promote longevity. We inhibited ATIC (AICAR transformylase/inosine monophosphate cyclohydrolase) with an inhibitor previously shown to alleviate the metabolic syndrome in mice.21 The ATIC inhibitor increased the lifespan of worms.20 Following these observations, we found that restricting dietary folate in aged mice improved metabolic activity with no adverse effects.20
It is possible that as biosynthetic needs change in an individual's life – higher when young but lower when old, so does the role of folate-based metabolism in fulfilling those needs. Higher folate intake is beneficial in earlier stages of life when increased cell proliferation is necessary for proper development. Late in life, a lower folate intake may promote healthy aging. The contrasting effects of folate intake at different times in life can be explained by antagonistic pleiotropy, where traits beneficial early in life may have negative consequences later.22 This mirrors the well-established example of the target of rapamycin (TOR) pathway, which promotes growth and proliferation but accelerates aging.23 Similar to the TOR pathway, 1C metabolic pathways serve as a resource allocation platform, producing building blocks needed for biosynthesis. When dietary folate is restricted, metabolism shifts to a less anabolic state, which may promote longevity.20 Additionally, 1C metabolism impacts various hallmarks of aging, including genomic stability, epigenetic regulation, and mitochondrial function.24–27
Most animals, including humans, cannot synthesize folate on their own. Folate and its various forms, such as 5-methyltetrahydrofolate (the naturally occurring form that is absorbed through the intestinal mucosa), do not readily cross biological membranes. Instead, cells depend on receptors that bind folate to facilitate its entry. Dietary folate is primarily absorbed in the duodenum through the proton-coupled folate transporter (PCFT/SLC46A1).28,29 By pharmacologically adjusting folate uptake, one could harness the potential benefits of reduced 1C metabolism late in life without compromising its essential role in early development. Targeting the PCFT transporter may therefore represent a novel pro-longevity approach. Here, we present our results with a new compound identified from a virtual screen to bind PCFT. We describe its synthesis, its favorable, drug-like properties, and its ability to increase the lifespan of worms. This compound's effect was 2- to 3-fold greater than the lifespan extension by methotrexate we previously published,20 making it a promising pro-longevity candidate for further testing in mammals.
2. Results
2.1 Virtual screening for compounds that may bind PCFT
Since the high-resolution structure of PCFT/SLC46A1 has been determined (PDB: 7BC7) without and with an antifolate (pemetrexed),30 we performed a virtual screen through molecular docking to identify compounds that may bind PCFT, using the AutoDock 4 suite.31 The compound libraries included natural products and their derivatives (SuperNatural 3.0 (ref. 32)). Several preparatory steps were taken to ensure the PCFT target molecule and the ligands in the library were in the appropriate format for the docking algorithm to perform efficiently and accurately, as described in the Materials and methods.The candidate compounds identified by the computational docking algorithm were first ranked based on their ‘docking score’, a measure of the predicted binding energy between a ligand and PCFT – the more negative the docking score value, the more favorable the predicted binding affinity. The values of the 1000 compounds with the lowest docking scores ranged between −15.4 and −11.8. Each one of these candidates was then evaluated with the SwissADME platform,33 to gauge their potential drug-likeness. They were classified based on their molecular weight, complexity, alerts as lead, and predicted toxicity. We also further confined this set to unpatented compounds. A total of 19 compounds passed the above criteria (MW < 500, no drug-likeness violations, low complexity, predicted to be non-toxic), and they are described in the US20250002492A1 patent application filed by Texas A&M University. In the following sections of this manuscript we will describe the synthesis of three of these compounds (Table 1; PubChem compound identifiers (CIDs): 3723186, 74508575, 74577661) and experiments further focusing on one of them (CID: 3723186). We note that classic antifolates known to inhibit PCFT, such as pemetrexed, have a pyrrolo[2,3-d]pyrimidine core that mimics the pteridine ring system found in natural folates. Instead, the three compounds shown in Table 1 have different scaffolds. The compound with CID 3723186 has a rigid tricyclic core, giving it a significantly bulkier lipophilic profile than pemetrexed. The structural core of the other two compounds, an imidazopyridine ring system, is also distinct from the pteridine system of folic acid, albeit it is part of a broader class of heterocycles that could be developed as antifolates.
| Compound CID | SMILES | Docking score | MW | Complexity | Synthetic accessibility | 2D structure |
|---|---|---|---|---|---|---|
| 3723186 | CC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)N1CCC(CC1)C(O)N2CCN3C(C2)C(O)Nc4ccc(cc4C3O)c5ccccc5 O)N1CCC(CC1)C(O)N2CCN3C(C2)C(O)Nc4ccc(cc4C3O)c5ccccc5 |
−12 | 460.5 | 818 | 3.92 |

|
| 74508575 | Cc1ccc(NC(O)N2Cc3nc[nH]c3CC2c4onc(n4)c5ccc6OCOc6c5)cc1 |
−11.9 | 444.4 | 706 | 4.21 |
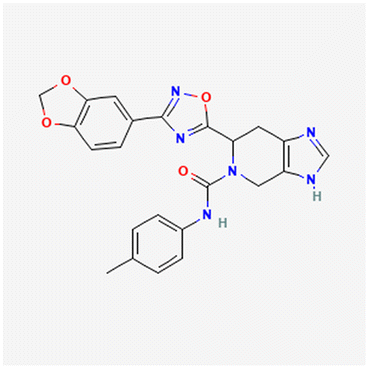
|
| 74577661 | Cc1cccc(c1)c2noc(n2)C3Cc4[nH]cnc4CN3C(O)Nc5cccc(F)c5 |
−11.8 | 418.4 | 641 | 4.11 |

|
2.2 Synthesis of candidate compounds
The three compounds that we synthesized were chosen because among the other 19 candidates they had low predicted values of ‘complexity’ and ‘synthetic accessibility’, based on the SwissADME platform.33 To simplify the naming, the compounds with CIDs 3723186, 74508575, 74577661 are referred as MPOL_B_1, MPOL_D_1, and MPOL_D_2, respectively, in this manuscript and in the US20250002492A1 patent application filed by Texas A&M University.The synthesis of MPOL_B_1 (13-(1-acetylpiperidine-4-carbonyl)-5-phenyl-1,9,13-triazatricyclo[9.4.0.03,8]pentadeca-3(8),4,6-triene-2,10-dione) is shown in Scheme 1. First, an amide coupling reaction between compound 1a and compound 2a, using HATU and triethylamine, produced compound 3a (53% yield). Then, a reduction of the nitro group of compound 3a using iron powder and ammonium chloride produced compound 4a, with a high yield of 93%. Compound 4a then underwent a Suzuki–Miyaura coupling reaction with phenylboronic acid, catalyzed by tetrakis(triphenylphosphine)palladium(0), yielding compound 5a in 67% yield. Next, a deprotection step using HCl in 1,4-dioxane quantitatively yielded compound 6a. The final product, MPOL_B_1, was obtained through a final HATU-mediated amide coupling between compound 6a and compound 7a. After purification by preparative HPLC, the yield was 10%.
 | ||
| Scheme 1 Reagents and conditions for the synthesis of MPOL_B_1. | ||
A convergent synthetic scheme was developed to prepare the two imidazo[4,5-c]pyridine derivatives, MPOL_D_1 and MPOL_D_2, from common intermediates. Synthesis of MPOL_D_1 (Scheme 2) began with a carbodiimide-mediated amide coupling between 1b and 2b using EDCI and HOBt to yield intermediate 3b in a 37% yield. Intermediate 4b was formed through a cyclization reaction in pyridine at 120 °C, at a 52% yield after purification by normal phase preparative chromatography. A subsequent HCl-mediated deprotection of 4b yielded intermediate 5b in 30% yield. The final product MPOL_D_1, (6S)-6-[3-(2H-1,3-benzodioxol-5-yl)-1,2,4-oxadiazol-5-yl]-N-(4-methylphenyl)3H,4H,5H,6H,7H-imidazo[4,5-c]pyridine-5-carboxamide, was made via a coupling reaction between 5b and 6b, followed by purification by reverse-phase preparative chromatography, providing the target compound as a white powder (23% yield).
 | ||
| Scheme 2 Reagents and conditions for the synthesis of MPOL_D_1. | ||
To make MPOL_D_2 (Scheme 3), (6S)-N-(3-fluorophenyl)-6-[3-(3-methylphenyl)1,2,4-oxadiazol-5-yl]-3H,4H,5H,6H,7H-imidazo[4,5c]pyridine-5-carboxamide, amide coupling using HATU and DIPEA produced compound 3c (58% yield). Cyclization formed intermediate 4c (52% yield), which, by HCl deprotection, led to intermediate 5c (51% yield). The final coupling and purification of MPOL_D_2 provided the target compound in 38% yield.
 | ||
| Scheme 3 Reagents and conditions for the synthesis of MPOL_D_2. | ||
All final compounds were characterized by 1H NMR spectroscopy, and the spectral data were consistent with their expected chemical structures (see Materials and methods, and Files S1–S4).
2.3 Testing the candidate compounds in folate transport
The three compounds were then evaluated for their ability to interfere with the transport of the sodium salt of [3,5,7,9-3H] folic acid in Caco-2 cell monolayers. These cells express the PCFT transporter and under the assay conditions (pH = 5.5), folate transport is mostly PCFT dependent.28,29 Folate uptake was quantified and normalized to total protein content for each well (see Materials and methods). The uptake rate was calculated in pmol mg−1 protein per min, and the inhibitory effect of each compound was expressed as a percentage of the uptake rate relative to a vehicle control (DMSO). The results from these assays are shown in Fig. 1. We conclude that MPOL_B_1 had the strongest inhibitory effect in folate uptake (42% inhibition at 50 μM) among the three new compounds we tested, albeit the effect was weaker than the known inhibitory effect of pemetrexed (62% inhibition at 50 μM). | ||
| Fig. 1 Folate uptake assay in Caco-2 cells. Uptake of folate in the presence of three different compounds (MPOL_B_1, MPOL_D_1, and MPOL_D_2) was measured as described in Materials and methods. Pemetrexed, a known positive control, and a vehicle control were also included for comparison. All compounds were tested at a concentration of 50 μM. The y-axis is the uptake rate in picomoles per milligram of protein per minute (pmol mg−1 protein per min). Each data point represents an individual measurement. | ||
2.4 Drug-like properties of MPOL_B_1 in vitro
The results of the folate transport assay led us to focus on the compound MPOL_B_1. We then experimentally evaluated its drug-like properties, which included several key variables to assess its suitability as a potential therapeutic agent. To understand its behavior within a biological system, we measured its protein binding in human and mouse serum to determine how much of the drug would be free and active versus bound to proteins. The Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D value was measured as a quantitative indicator of its lipophilicity, revealing how the compound distributes between lipid and aqueous environments. We also assessed its stability in human and mouse plasma and microsomes to understand how quickly it might be broken down in the bloodstream or metabolized by liver enzymes. To evaluate its intestinal absorption, we measured its permeability using a Caco-2 cell assay, which mimics the intestinal barrier. The results from this assay also included the basolateral-to-apical (B2A) permeability ratio (Papp), which indicated potential efflux, where the drug is actively pumped out of the cells.
D value was measured as a quantitative indicator of its lipophilicity, revealing how the compound distributes between lipid and aqueous environments. We also assessed its stability in human and mouse plasma and microsomes to understand how quickly it might be broken down in the bloodstream or metabolized by liver enzymes. To evaluate its intestinal absorption, we measured its permeability using a Caco-2 cell assay, which mimics the intestinal barrier. The results from this assay also included the basolateral-to-apical (B2A) permeability ratio (Papp), which indicated potential efflux, where the drug is actively pumped out of the cells.
We also evaluated the potential toxicity of MPOL_B_1. We performed a cytotoxicity assay in Hep2 cells to check if the compound is harmful to these cells, an indicator of general cellular safety. The Ames test was conducted to determine if the compound has the potential to cause genetic mutations. We also performed the hERG assay to evaluate its potential to inhibit potassium channels, as hERG channel inhibition can lead to serious cardiac arrhythmias. Lastly, we assessed its ability to inhibit human liver CYP isoforms, involved in drug metabolism, to identify potential drug–drug interactions.
The comprehensive results of these assays are summarized in Table 2, with the primary data presented in the SI figures, as referred to in the Materials and methods. Overall, the data indicate that MPOL_B_1 is safe in cell lines, is stable in biological fluids, exhibits moderate lipophilicity, shows limited potential for adverse interactions with other drugs, and has weak cell permeability with evidence of efflux. These properties suggest a favorable safety profile and potentially low systemic bioavailability, which may be beneficial for a drug designed to act primarily within the gut to target intestinal folate absorption.
| ASSAY | VALUE (mean ± SD) |
|---|---|
| Human plasma protein binding (2 μM) | 52.5 ± 8.0 (% unbound) after 6 h |
| Mouse plasma protein binding (2 μM) | 29.4 ± 5.9 (% unbound) after 6 h |
| LogD (octanol vs. PBS partitioning) (100 μM) |
1.26 ± 0.02 |
| Stability in human & mouse plasma (2 μM) | >90% after 4 h |
| Stability in human microsomes (2 μM) | >90% after 2 h |
| Stability in mouse microsomes (2 μM) | 83% after 2 h |
| Hep2 cells cytotoxicity (50 μM) | Not significant |
| Caco-2 permeability (A2B) (10 μM) | P app = 0.37 ± 0.04 (10−6 cm s−1) |
| Caco-2 permeability (B2A) (10 μM) | P app = 7.17 ± 0.31 (10−6 cm s−1) |
| hERG K+ channel inhibition (0.1–100 μM) | IC50 = 86.997 μM |
| Ames mutagenicity test (1.4 × 10−7 to 2.3 mM) | Not mutagenic |
| Human liver CYP %inhibition (50 μM). The isoforms tested are shown to the right, based on co-incubation with drugs known to be metabolized by each isoform | 1A2 = 5.2 ± 1.9, 2B6 = 0 ± 1.3, 2C8 = 4.9 ± 8.3, 2C9 = 11.4 ± 7.6, 2C19 = 19.4 ± 3.6, 2D6 = 16.9 ± 4.6, 3A4 = 22.5 ± 15.1 |
2.5 MPOL_B_1 pharmacokinetics in rodents
To initiate the preclinical evaluation of MPOL_B_1, we assessed its pharmacokinetic (PK) profile and potential toxicity in single-dose studies in mice and rats. This work could inform future dose selection for repeat-dose studies. Male CD1 mice were administered MPOL_B_1 via oral gavage at three doses (25, 50, and 100 mg kg−1). The concentration of MPOL_B_1 in serum samples was measured over 24 h by mass spectrometry (Fig. 2A). The PK profile revealed that systemic bioavailability became saturated at doses of 50 mg kg−1 and above, as indicated by a less-than-proportional increase in the area under the curve (AUC). For the highest dose tested (100 mg kg−1), the PK parameters were: AUClast = 2379 ± 1176 ng mL−1, Cmax = 416 ± 67 ng mL−1, Tmax = 1.7 ± 0.6 h, and T1/2 = 5.7 ± 4.1 h. | ||
| Fig. 2 Single-dose pharmacokinetics of MPOL_B_1 in mice and rats. A | Plasma concentration of MPOL_B_1 in mice. Following a single oral dose of MPOL_B_1 at 25, 50, or 100 mg kg−1, plasma concentrations were measured at various time points, as described in the Materials and methods. The y-axis shows the concentration of MPOL_B_1 in ng mL−1, while the x-axis is time in h. The red line represents the Loess regression fit of the data. B | Plasma concentration of MPOL_B_1 in rats. The plasma concentration-time profile of MPOL_B_1 was determined in rats after a single oral dose of 100 or 200 mg kg−1, as described in the Materials and methods. The plots were drawn as in A. | ||
Consistent with the limited systemic exposure, fecal samples collected 24 hours after administration showed high concentrations of the drug (5–10 mg g−1), suggesting that the majority of the orally administered drug was not absorbed and remained within the gastrointestinal tract. This may be a beneficial characteristic for an extracellular drug designed to target intestinal folate absorption.
In male Sprague–Dawley rats, systemic bioavailability was approximately 10 times lower than in mice (Fig. 2B). We tested two doses (100 and 200 mg kg−1), which yielded similar PK profiles and confirmed the low systemic absorption observed in mice. For these doses, the combined PK parameters were: AUClast = 222 ± 250 ng mL−1, Cmax = 52 ± 19 ng mL−1, Tmax = 1 ± 0.9 h, and T1/2 = 5.8 ± 2.8 h.
To assess safety, all animals in both species were monitored for 14 days following the single dose. There were no signs of illness, and no significant body weight loss was observed in any of the treated animals. At the end of the observation period, a gross necropsy was performed, revealing no organ pathologies or other abnormalities for any mice or rats that received the highest doses of MPOL_B_1. Some abnormalities were noted for some animals that received lower doses, but it is unclear that those were related to the administered compound. These results from the single-dose studies in both mice and rats support a safe profile and low systemic bioavailability for MPOL_B_1.
2.6 MPOL_B_1 extends the lifespan of worms
The in vitro properties and pharmacokinetic profile of MPOL_B_1 in rodents suggested favorable pharmacological properties. To further test its potential, we assessed its effect on longevity using the C. elegans model. This genetically tractable organism provides a powerful platform for screening compounds that modulate aging.34 Remarkably, animals continuously exposed to MPOL_B_1 had a longer maximal and mean lifespan by ∼30% at 50 and 100 μM (Fig. 3). We also tested MPOL_D_1 in the same dose range (1–100 μM), but it did not exhibit similar effects (Table S4). We note that the lifespan extension by MPOL_B_1 was 2- to 3-fold greater than the extension by methotrexate, which we published using the same assay,20 probably due to MPOL_B_1's low toxicity. These results suggest that MPOL_B_1 may hold promise as a longevity intervention, meriting further development in mammals. | ||
| Fig. 3 Effect of MPOL_B_1 on the lifespan of C. elegans. The y-axis in the Kaplan–Meier survival curve is the survival probability. The x-axis indicates the time in days. Three different concentrations of MPOL_B_1 were tested: 0 μM (black line, control), 50 μM (blue line), and 100 μM (red line). The median lifespan (in days) increased from 21.8 d for untreated animals (n = 86) to 26.5 d (n = 120) and 28.2 d (n = 112) for animals exposed to 50 μM and 100 μM of MPOL_B_1, respectively. The maximal lifespan also increased from 27 d to 35 d for both treatment doses. The p-values for the log-rank test assessing lifespan extension relative to the control group are also shown. | ||
3. Conclusion
Previous research, including our own, supports the idea that interventions targeting 1C metabolism may enhance longevity. Our findings with the novel compound MPOL_B_1 are encouraging, showing its potential to extend the lifespan of C. elegans. Additionally, the compound's safety profile observed in rodents suggests it may be a viable candidate for further development. Although the compound has low systemic availability (Fig. 2), as expected from its low permeability and high efflux ratio (Fig. S4), these properties are compatible with an intestinal site of action, where PCFT is localized. They may also be beneficial due to lower systemic toxicity. While this work introduces a new candidate for longevity research, our data do not provide any evidence that MPOL_B_1 modulates folate uptake or folate levels in animals (including worms). Furthermore, recent work has implicated the C. elegans FOLR-1 folate receptor in behavioral pathways, independently of 1C metabolism. Instead, FOLR-1 regulates neuronal activity in serotonergic neurons by regulating cellular calcium levels – a role that appears to be conserved in humans.35 Hence, the mechanisms by which MPOL_B_1 may exert its prolongevity effects remain unknown and await further experimentation. Further development of compounds that interact with PCFT would require validation of in vivo target engagement, after continuous oral administration, initially in mice, to evaluate their ability to modulate serum folate levels. Other experiments may also query if any healthspan metrics are improved or whether there are benefits in specific disease models. For example, selective PCFT inhibition may offer a novel strategy to harness the known anti-proliferative effects of folate reduction in rheumatoid arthritis without the systemic toxicities associated with classic antifolates like methotrexate.4. Materials and methods
4.1 Computational docking
The virtual screen was performed by Creative Biomart Inc. (Shirley, NY), using the AutoDock 4 suite, with the following functions. First, water molecules were removed from the structure to simplify the system and make the computations fast and efficient. Second, hydrogen atoms were added to PCFT, since hydrogens not represented in the structure, especially polar ones attached to oxygen or nitrogen atoms, often participate in protein–ligand binding through hydrogen bonds. Third, the “Compute Gasteiger Charges” function was selected. This function calculates and assigns partial atomic charges to each atom in the molecule using the Gasteiger method, which were then used to calculate the electrostatic interactions between the macromolecule and the ligand. Fourth, the command “AD4 Type Assign” categorized atoms based on their chemical properties (e.g., carbon, oxygen, nitrogen, etc.) and bonding environment (e.g., aromatic carbon, aliphatic carbon), ensuring that the interaction energy calculations are accurate for a wide range of chemical groups. Lastly, to make the screening conditions more efficient, precise, and biologically relevant, they were focused on the known pemetrexed binding site of PCFT. In the AutoDock Vina module, the binding pocket was defined using the PyMol Getbox plugin to place a virtual box around the residues that form the binding site.The ligand library was prepared as follows: first, since the ligands in the library were stored in a 2D format (SMILES), they were computationally converted to a 3D conformation from the 2D representation. Second, we used energy minimization with the MMFF94S (Merck molecular force field 94, static) type of force field to calculate the potential energy of a molecule and find the most stable, lowest-energy conformation for the ligand. Third, the optimized 3D structure was converted into a PDBQT file format, which contains all the necessary information – 3D coordinates, atom types, partial charges, and rotatable bonds, suitable for the docking software to accurately calculate the interactions and explore the conformational space of the ligand during the simulation.
4.2 Chemical synthesis
All chemical synthesis was done by Novalix Inc. (Strasbourg, France). Reagents were purchased from commercial suppliers and used without further purification. Anhydrous solvents were used as received unless otherwise stated. Reactions were monitored by thin-layer chromatography (TLC) on silica gel plates and by liquid chromatography-mass spectrometry (LC–MS). Flash column chromatography was performed on an Interchim Puriflash 430 system. Reverse-phase preparative high-performance liquid chromatography (HPLC) was performed on a C18 column using a gradient of acetonitrile and water with 0.2% ammonium bicarbonate as the mobile phase, unless noted otherwise. The certificates of analysis for each compound, along with the spectroscopic data, are provided in Files S1–S3. The detailed synthetic steps are in File S4.4.3 Folate uptake assays in Caco-2 cells
The transport assays were done by Novalix Inc (Strasbourg, France), as detailed below.Following pre-incubation, the solution was aspirated, and 300 μL of the pre-warmed working incubation solution (containing [3H]-folic acid with or without inhibitor) was added to each well to initiate the uptake assay. The incubation was carried out for 6 m, after which the reaction was stopped by rapidly washing the cells four times with 300 μL of ice-cold pH 5.5 buffer. The buffer was then removed, and 300 μL of pure water was added to each well to lyse the cells via three freeze–thaw cycles using liquid nitrogen and a 37 °C incubator. The cell lysates were then centrifuged at 3300g for 30 s. A 50 μL aliquot of the supernatant was transferred to an Isoplate-96 Microplate, mixed with 200 μL of scintillation fluid, and the radioactivity was measured for 1 m with a MicroBeta2 scintillation counter.
The uptake rate (pmol mg−1 protein per min) was calculated using the following equation: Uptake rate = Conc./(Protein × Time), where: Conc. is the concentration of drug in cell lysate (nM); Protein is the protein concentration of cell lysate (mg mL−1); Time is the incubation time (m). The percentage transport (relative to the vehicle control) was calculated using the following formula: Percentage Transported (%) = R(+inhibitor)/R(vehicle control) × 100, where: R(+inhibitor) stands for the uptake rate of substrate in the presence of test compound or positive control inhibitor, and R(vehicle control) stands for the uptake rate of substrate in the vehicle control. The percentage inhibition was then determined as: Percentage Inhibition (%) = 100 − Percentage Transported (%).
4.4 In vitro assays for MPOL_B_1
The assays were done by Charles River Laboratories (Wilmington, MA) according to industry standards as described below. All reagents used were purchased from commercial vendors. A summary of the results are in Table 2.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) D assay (octanol vs. PBS partition) assays.
To ensure equilibrium and that n-octanol and PBS were mutually saturated before the experiment, the two phases were vigorously mixed together and allowed to separate before use. The test compounds were prepared as stock solutions in DMSO. All compounds (MPOL_B_1, pemetrexed disodium heptahydrate, chlorpromazine – lipophilic control, warfarin – moderate control, atenolol – hydrophilic control) were tested at 100 μM. The mixture in each case was vigorously shaken for 60 m at 25 °C to allow each compound to reach equilibrium between the two phases. The samples were centrifuged to separate the organic layer (top) from the aqueous layer (bottom). The concentration of each compound in both phases was quantified using LC–MS. The ratio of each compound's concentration in the n-octanol phase to its concentration in the aqueous (PBS) phase was then log10-transformed to calculate the LogD value, which is shown in Fig. S2.
D assay (octanol vs. PBS partition) assays.
To ensure equilibrium and that n-octanol and PBS were mutually saturated before the experiment, the two phases were vigorously mixed together and allowed to separate before use. The test compounds were prepared as stock solutions in DMSO. All compounds (MPOL_B_1, pemetrexed disodium heptahydrate, chlorpromazine – lipophilic control, warfarin – moderate control, atenolol – hydrophilic control) were tested at 100 μM. The mixture in each case was vigorously shaken for 60 m at 25 °C to allow each compound to reach equilibrium between the two phases. The samples were centrifuged to separate the organic layer (top) from the aqueous layer (bottom). The concentration of each compound in both phases was quantified using LC–MS. The ratio of each compound's concentration in the n-octanol phase to its concentration in the aqueous (PBS) phase was then log10-transformed to calculate the LogD value, which is shown in Fig. S2.
Similarly, to evaluate each compound's metabolic stability and its susceptibility to cytochrome P450 (CYP) enzymes, the compounds (at 2 μM) were incubated with a suspension of liver microsomes from human or mouse (strain CD1) at 37 °C. The CYP cofactor nicotinamide adenine dinucleotide phosphate (NADPH) was added to initiate the enzymatic activity of the CYP enzymes. Aliquots were taken at the same time points as above, quenched, and analyzed by LC–MS, to yield the results shown in Fig. S3.
To measure absorption (apical-to-basolateral (A→B, A2B) permeability), each compound was added to the apical (donor) chamber. The basolateral (receiver) chamber was filled with fresh buffer, and the plate was incubated at 37 °C for 2 h. Samples were then collected from the basolateral chamber at specific time points to measure the flux of the compound. To measure efflux, or active pumping out of the cells (basolateral-to-apical (B→A, B2A) permeability), the procedure was the same except that each compound was added to the basolateral chamber, while the apical chamber was filled with fresh buffer.
After incubation, the concentration of the compound in both the donor and receiver chambers was quantified using LC–MS. The apparent permeability coefficient (Papp, in cm s−1) was calculated for each direction using the following equation: Papp = C0 × AΔQ/Δt, where ΔQ/Δt is the rate of compound transport across the monolayer, C0 is the initial concentration of the compound in the donor chamber. A is the surface area of the cell monolayer membrane.
In addition to MPOL_B_1 (tested at 10 μM), control drugs tested in both directions were warfarin, ranitidine, talinolol (each also tested at 10 μM), and a mixture of talinolol (10 μM) plus verapamil (25 μM). The results are shown in Fig. S4.
![[O with combining low line]](https://www.rsc.org/images/entities/char_004f_0332.gif)
![[E with combining low line]](https://www.rsc.org/images/entities/char_0045_0332.gif)
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
![[D with combining low line]](https://www.rsc.org/images/entities/char_0044_0332.gif)
![[g with combining low line]](https://www.rsc.org/images/entities/char_0067_0332.gif)
![[u with combining low line]](https://www.rsc.org/images/entities/char_0075_0332.gif)
![[i with combining low line]](https://www.rsc.org/images/entities/char_0069_0332.gif)
![[d with combining low line]](https://www.rsc.org/images/entities/char_0064_0332.gif)
![[e with combining low line]](https://www.rsc.org/images/entities/char_0065_0332.gif)
![[l with combining low line]](https://www.rsc.org/images/entities/char_006c_0332.gif)
![[n with combining low line]](https://www.rsc.org/images/entities/char_006e_0332.gif)
![[4 with combining low line]](https://www.rsc.org/images/entities/char_0034_0332.gif)
![[7 with combining low line]](https://www.rsc.org/images/entities/char_0037_0332.gif)
![[1 with combining low line]](https://www.rsc.org/images/entities/char_0031_0332.gif) ). The negative control was DMSO, used as the vehicle for MPOL_B_1. The positive controls were sodium azide (NaAz), 9-aminoacridine hemihydrate (9AC), 2-nitrofluorene (2NF), 4-nitroquinoline N-oxide (NQO), 2-aminoanthracene (2AA), benzo[a]pyrene (BaP). MPOL_B_1 was prepared as a stock solution in DMSO (50 mg mL−1), and all lower-level formulations were made directly from this stock or intermediate formulations. All formulations (and vehicle aliquot) were prepared on the day of use at ambient temperature. The bulk positive controls were also dissolved in DMSO. Toxic effects were considered positive for inducing reverse mutations if a compound induced a dose-dependent increase in revertant frequency that is at least 2-fold higher than its concurrent negative control in tester strains TA98, TA100, and WP2 uvrA, or at least 3-fold higher than its concurrent negative control in tester strains TA1535 and TA1537. If none of these criteria were met, the results were considered negative. The results without or with metabolic activation are in Tables S2 and S3, respectively.
). The negative control was DMSO, used as the vehicle for MPOL_B_1. The positive controls were sodium azide (NaAz), 9-aminoacridine hemihydrate (9AC), 2-nitrofluorene (2NF), 4-nitroquinoline N-oxide (NQO), 2-aminoanthracene (2AA), benzo[a]pyrene (BaP). MPOL_B_1 was prepared as a stock solution in DMSO (50 mg mL−1), and all lower-level formulations were made directly from this stock or intermediate formulations. All formulations (and vehicle aliquot) were prepared on the day of use at ambient temperature. The bulk positive controls were also dissolved in DMSO. Toxic effects were considered positive for inducing reverse mutations if a compound induced a dose-dependent increase in revertant frequency that is at least 2-fold higher than its concurrent negative control in tester strains TA98, TA100, and WP2 uvrA, or at least 3-fold higher than its concurrent negative control in tester strains TA1535 and TA1537. If none of these criteria were met, the results were considered negative. The results without or with metabolic activation are in Tables S2 and S3, respectively.
4.5 Single-dose pharmacokinetic (PK) assays in rodents
Animals from both species (n = 3 per group) were sourced from an approved vendor and allowed to acclimate to the facility for at least two days before the study commenced. Mice were housed individually in metabolic cages to facilitate fecal collection and were fasted for 4 h before dosing, with food returned 2 h post-dose. Rats were also housed in metabolic cages, but they were fasted overnight, and food was returned 4 h post-dose. Water was provided ad libitum for both species throughout the study. Body weights for all animals were recorded prior to dose administration and on days 7 and 14. Dose volumes (10 mL kg−1) were calculated based on each animal's body weight on day 1.
Fecal samples were collected over 24 h post-dose for all animals. The collection tubes, placed on ice packs, were changed at the 8 h time point. The fecal samples were transferred to pre-weighed conical tubes, and their weights were recorded. Samples were stored at −70 °C until they were transferred for analysis by LC–MS.
4.6 C. elegans lifespans
All the assays were performed at 20 °C using C. elegans strain N2 and a bacterial strain (OP50) commonly used as food for the worms, as described previously.20,34 The standard OP50 bacterial strain used in nematode experiments is a strain of Escherichia coli (E. coli B) that is a uracil auxotroph. As a result of the uracil auxotrophy, OP50 grows a thin, slow-growing lawn on the nematode growth medium (NGM) plates used for culturing C. elegans, facilitating the movement of the animals and their observation. We note that the bacteria used to feed the worms in these experiments were killed by ultraviolet radiation to exclude any impacts from bacterial metabolism. MPOL_B_1 and MPOL_D_1 were first dissolved in DMSO and then added to the media used to prepare the plates after autoclaving (the media were kept in a 50 °C water bath until the plates were poured). Mock-treated control plates contained only DMSO. The reported lifespans were compiled from several independent experiments and analyzed with the survival R language package. Survival data were constructed using the Surv() function. This function defined the outcome as a survival object by specifying the time until the event of interest (death, in this case). This survival object was then used as the response variable in subsequent survival models, such as the Kaplan–Meier estimation with the survfit() function. No experiments were excluded from the analysis. The data for both drugs and all doses tested are in Table S4.Abbreviations
| 1C | One-carbon metabolism |
| ACN | Acetonitrile |
| AICAR | 5-Aminoimidazole-4-carboxamide ribonucleotide |
| ATIC | AICAR transformylase/inosine monophosphate cyclohydrolase |
| ATP | Adenosine triphosphate |
| AUC | Area under the curve |
| BaP | Benzo[a]pyrene |
| BCA | Bicinchoninic acid |
| C. elegans | Caenorhabditis elegans |
| CID | PubChem compound identifier |
| C max | Maximum concentration |
| CYP | Cytochrome P450 |
| DCM | Dichloromethane |
| DMEM | Dulbecco's modified Eagle medium |
| DMF | N,N-Dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| EDCI | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| Et2O | Diethyl ether |
| Et3N | Triethylamine |
| FBS | Fetal bovine serum |
| HATU | N,N,N′,N′-Tetramethyl-O-(7-azabenzotriazol-1-yl)uronium hexafluorophosphate |
| HB-PS | HEPES-buffered physiological saline solution |
| HBSS | Hank's balanced salt solution |
| hERG | Human ether-à-go-go-related gene |
| HLM | Human liver microsome |
| HOBt | 1-Hydroxybenzotriazole |
| HPLC | High-performance liquid chromatography |
| IACUC | Institutional Animal Care and Use Committee |
| LC–MS | Liquid chromatography–mass spectrometry |
| MeOH | Methanol |
| MES | 2-(N-Morpholino)ethanesulfonic acid |
| MMFF94S | Merck molecular force field 94, static |
| MW | Molecular weight |
| Na2CO3 | Sodium carbonate |
| Na2SO4 | Anhydrous sodium sulfate |
| NaAz | Sodium azide |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NEAA | Non-essential amino acids |
| NH4Cl | Ammonium chloride |
| NMR | Nuclear magnetic resonance |
| NQO | 4-Nitroquinoline N-oxide |
| Papp | Apparent permeability coefficient |
| PBS | Phosphate-buffered saline |
| PCFT | Proton-coupled folate transporter |
| Pd(PPh3)4 | Tetrakis(triphenylphosphine)palladium(0) |
| PDB | Protein Data Bank |
| PK | Pharmacokinetics |
| PO | Per oral |
| P-type ATPase | P-type adenosine triphosphatase |
| RED | Rapid equilibrium dialysis |
| RT | Room temperature |
| SD | Sprague–Dawley |
| SMILES | Simplified molecular-input line-entry system |
| TEER | Transepithelial electrical resistance |
| TFA | Trifluoroacetic acid |
| THF | Tetrahydrofolate |
| TLC | Thin-layer chromatography |
| T max | Time to reach maximum concentration |
| TOR | Target of rapamycin |
| 2AA | 2-Aminoanthracene |
| 2NF | 2-Nitrofluorene |
| 9AC | 9-Aminoacridine hemihydrate |
Author contributions
H. M. B. and M. P.: worm lifespans; M. P.: writing – original draft; H. M. B. and M. P.: writing, review and editing; M. P.: study management, funding acquisition.Conflicts of interest
The compounds MPOL_B_1, MPOL_D_1, and MPOL_D_2 are described in the US20250002492A1 patent application filed by Texas A&M University, with M. P. listed as the lead inventor. M. P. is the founder and manager of Krateos Therapeutics LLC.Data availability
All relevant data are within the manuscript and its accompanying files.Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d5md00780a.
Acknowledgements
This research was funded by a grant (M2502317) from the USDA – Agricultural Research Service and the Institute for Advancing Health Through Agriculture to M. P. M. P. was also supported by a grant from the National Institutes of Health (R01GM123139).Notes and references
- D. Barardo, D. Thornton, H. Thoppil, M. Walsh, S. Sharifi, S. Ferreira, A. Anžič, M. Fernandes, P. Monteiro, T. Grum, R. Cordeiro, E. A. De-Souza, A. Budovsky, N. Araujo, J. Gruber, M. Petrascheck, V. E. Fraifeld, A. Zhavoronkov, A. Moskalev and J. P. de Magalhães, The DrugAge database of aging-related drugs, Aging Cell, 2017, 16, 594–597 CrossRef CAS PubMed.
- A. Moskalev, E. Chernyagina, J. P. de Magalhães, D. Barardo, H. Thoppil, M. Shaposhnikov, A. Budovsky, V. E. Fraifeld, A. Garazha, V. Tsvetkov, E. Bronovitsky, V. Bogomolov, A. Scerbacov, O. Kuryan, R. Gurinovich, L. C. Jellen, B. Kennedy, P. Mamoshina, E. Dobrovolskaya, A. Aliper, D. Kaminsky and A. Zhavoronkov, Geroprotectors.org: a new, structured and curated database of current therapeutic interventions in aging and age-related disease, Aging, 2015, 7, 616–628 CrossRef CAS PubMed.
- R. A. Miller, D. E. Harrison, C. M. Astle, R. A. Floyd, K. Flurkey, K. L. Hensley, M. A. Javors, C. Leeuwenburgh, J. F. Nelson, E. Ongini, N. L. Nadon, H. R. Warner and R. Strong, An Aging Interventions Testing Program: study design and interim report, Aging Cell, 2007, 6, 565–575 CrossRef CAS.
- M. Driscoll, C. A. Sedore, B. Onken, A. L. Coleman-Hulbert, E. Johnson, P. C. Phillips and G. Lithgow, NIA Caenorhabditis Intervention Testing Program: identification of robust and reproducible pharmacological interventions that promote longevity across experimentally accessible, genetically diverse populations, GeroScience, 2025, 47, 2791–2816 Search PubMed.
- N. Maitra, C. He, H. M. Blank, M. Tsuchiya, B. Schilling, M. Kaeberlein, R. Aramayo, B. K. Kennedy and M. Polymenis, Translational control of one-carbon metabolism underpins ribosomal protein phenotypes in cell division and longevity, eLife, 2020, 9, e53127 Search PubMed.
- C. Barcena, P. M. Quiros, S. Durand, P. Mayoral, F. Rodriguez, X. M. Caravia, G. Marino, C. Garabaya, M. T. Fernandez-Garcia, G. Kroemer, J. M. P. Freije and C. Lopez-Otin, Methionine Restriction Extends Lifespan in Progeroid Mice and Alters Lipid and Bile Acid Metabolism, Cell Rep., 2018, 24, 2392–2403 CrossRef CAS PubMed.
- F. Cabreiro, C. Au, K.-Y. Leung, N. Vergara-Irigaray, H. M. Cochemé, T. Noori, D. Weinkove, E. Schuster, N. D. E. Greene and D. Gems, Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism, Cell, 2013, 153, 228–239 CrossRef CAS PubMed.
- J. E. Johnson and F. B. Johnson, Methionine restriction activates the retrograde response and confers both stress tolerance and lifespan extension to yeast, mouse and human cells, PLoS One, 2014, 9, e97729 CrossRef PubMed.
- R. Kozieł, C. Ruckenstuhl, E. Albertini, M. Neuhaus, C. Netzberger, M. Bust, F. Madeo, R. J. Wiesner and P. Jansen-Dürr, Methionine restriction slows down senescence in human diploid fibroblasts, Aging Cell, 2014, 13, 1038–1048 CrossRef.
- B. C. Lee, A. Kaya and V. N. Gladyshev, Methionine restriction and life-span control, Ann. N. Y. Acad. Sci., 2016, 1363, 116–124 CrossRef CAS.
- B. C. Lee, A. Kaya, S. Ma, G. Kim, M. V. Gerashchenko, S. H. Yim, Z. Hu, L. G. Harshman and V. N. Gladyshev, Methionine restriction extends lifespan of Drosophila melanogaster under conditions of low amino-acid status, Nat. Commun., 2014, 5, 3592 CrossRef PubMed.
- R. A. Miller, G. Buehner, Y. Chang, J. M. Harper, R. Sigler and M. Smith-Wheelock, Methionine-deficient diet extends mouse lifespan, slows immune and lens aging, alters glucose, T4, IGF-I and insulin levels, and increases hepatocyte MIF levels and stress resistance, Aging Cell, 2005, 4, 119–125 Search PubMed.
- N. Orentreich, J. R. Matias, A. DeFelice and J. A. Zimmerman, Low methionine ingestion by rats extends life span, J. Nutr., 1993, 123, 269–274 Search PubMed.
- C. Ruckenstuhl, C. Netzberger, I. Entfellner, D. Carmona-Gutierrez, T. Kickenweiz, S. Stekovic, C. Gleixner, C. Schmid, L. Klug, A. G. Sorgo, T. Eisenberg, S. Buttner, G. Marino, R. Koziel, P. Jansen-Durr, K. U. Frohlich, G. Kroemer and F. Madeo, Lifespan extension by methionine restriction requires autophagy-dependent vacuolar acidification, PLoS Genet., 2014, 10, e1004347 CrossRef PubMed.
- L. Sun, A. A. Sadighi Akha, R. A. Miller and J. M. Harper, Life-span extension in mice by preweaning food restriction and by methionine restriction in middle age, J. Gerontol., Ser. A, 2009, 64, 711–722 CrossRef PubMed.
- K. Zou, S. Rouskin, K. Dervishi, M. A. McCormick, A. Sasikumar, C. Deng, Z. Chen, M. Kaeberlein, R. B. Brem, M. Polymenis, B. K. Kennedy, J. S. Weissman, J. Zheng, Q. Ouyang and H. Li, Life span extension by glucose restriction is abrogated by methionine supplementation: Cross-talk between glucose and methionine and implication of methionine as a key regulator of life span, Sci. Adv., 2020, 6, eaba1306 CrossRef PubMed.
- E. Enriquez-Hesles, D. L. Smith, N. Maqani, M. B. Wierman, M. D. Sutcliffe, R. D. Fine, A. Kalita, S. M. Santos, M. J. Muehlbauer, J. R. Bain, K. A. Janes, J. L. Hartman, M. D. Hirschey and J. S. Smith, A cell-nonautonomous mechanism of yeast chronological aging regulated by caloric restriction and one-carbon metabolism, J. Biol. Chem., 2021, 296, 100125 CrossRef PubMed.
- A. Annibal, R. G. Tharyan, M. F. Schonewolff, H. Tam, C. Latza, M. M. K. Auler, S. Grönke, L. Partridge and A. Antebi, Regulation of the one carbon folate cycle as a shared metabolic signature of longevity, Nat. Commun., 2021, 12, 3486 CrossRef PubMed.
- J. W. Williams, J. F. Morrison and R. G. Duggleby, Methotrexate, a high-affinity pseudosubstrate of dihydrofolate reductase, Biochemistry, 1979, 18, 2567–2573 CrossRef PubMed.
- H. M. Blank, S. E. Hammer, L. Boatright, C. Roberts, K. E. Heyden, A. Nagarajan, M. Tsuchiya, M. Brun, C. D. Johnson, P. J. Stover, R. Sitcheran, B. K. Kennedy, L. G. Adams, M. Kaeberlein, M. S. Field, D. W. Threadgill, H. L. Andrews-Polymenis and M. Polymenis, Late-life dietary folate restriction reduces biosynthesis without compromising healthspan in mice, Life Sci. Alliance, 2024, 7, e202402868 CrossRef PubMed.
- D. J. Asby, F. Cuda, M. Beyaert, F. D. Houghton, F. R. Cagampang and A. Tavassoli, AMPK Activation via Modulation of De Novo Purine Biosynthesis with an Inhibitor of ATIC Homodimerization, Chem. Biol., 2015, 22, 838–848 CrossRef PubMed.
- G. C. Williams, Pleiotropy, Natural-Selection, and the Evolution of Senescence, Evolution, 1957, 11, 398–411 CrossRef.
- M. V. Blagosklonny, Revisiting the antagonistic pleiotropy theory of aging: TOR-driven program and quasi-program, Cell Cycle, 2010, 9, 3151–3156 CrossRef PubMed.
- G. S. Ducker and J. D. Rabinowitz, One-Carbon Metabolism in Health and Disease, Cell Metab., 2017, 25, 27–42 CrossRef PubMed.
- M. Reina-Campos, M. T. Diaz-Meco and J. Moscat, The complexity of the serine glycine one-carbon pathway in cancer, J. Cell Biol., 2020, 219(1), e201907022 Search PubMed.
- A. Rosenzweig, J. Blenis and A. P. Gomes, Beyond the Warburg Effect: How Do Cancer Cells Regulate One-Carbon Metabolism?, Front. Cell Dev. Biol., 2018, 6, 90 CrossRef PubMed.
- C. Lopez-Otin, M. A. Blasco, L. Partridge, M. Serrano and G. Kroemer, The hallmarks of aging, Cell, 2013, 153, 1194–1217 CrossRef PubMed.
- A. Qiu, M. Jansen, A. Sakaris, S. H. Min, S. Chattopadhyay, E. Tsai, C. Sandoval, R. Zhao, M. H. Akabas and I. D. Goldman, Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption, Cell, 2006, 127, 917–928 CrossRef PubMed.
- M. Visentin, N. Diop-Bove, R. Zhao and I. D. Goldman, The intestinal absorption of folates, Annu. Rev. Physiol., 2014, 76, 251–274 Search PubMed.
- J. L. Parker, J. C. Deme, G. Kuteyi, Z. Wu, J. Huo, I. D. Goldman, R. J. Owens, P. C. Biggin, S. M. Lea and S. Newstead, Structural basis of antifolate recognition and transport by PCFT, Nature, 2021, 595, 130–134 Search PubMed.
- S. Forli, R. Huey, M. E. Pique, M. Sanner, D. S. Goodsell and A. J. Olson, Computational protein-ligand docking and virtual drug screening with the AutoDock suite, Nat. Protoc., 2016, 11, 905–919 Search PubMed.
- K. Gallo, E. Kemmler, A. Goede, F. Becker, M. Dunkel, R. Preissner and P. Banerjee, SuperNatural 3.0-a database of natural products and natural product-based derivatives, Nucleic Acids Res., 2023, 51, D654–D659 CrossRef PubMed.
- A. Daina, O. Michielin and V. Zoete, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 7, 42717 Search PubMed.
- G. L. Sutphin and M. Kaeberlein, Measuring Caenorhabditis elegans Life Span on Solid Media, J. Visualized Exp., 2009, e1152 Search PubMed.
- R. S. Peesapati, B. L. Austin-Byler, F. Z. Nawaz, J. B. Stevenson, S. A. Mais, R. N. Kaya, M. G. Hassan, N. Khanal, A. C. Wells, D. Ghiai, A. K. Garikapati, J. Selhub and E. T. Kipreos, A specific folate activates serotonergic neurons to control C. elegans behavior, Nat. Commun., 2024, 15, 8471 Search PubMed.
| This journal is © The Royal Society of Chemistry 2026 |