
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploration of post-print modification of 3D photo-printed materials for microfabrication by means of RAFT polymerization
Frank Marco
den Hoed
ab,
Tjon F.
Chen
a,
Virgilio
Mattoli
 b,
Luca
Ceseracciu
c and
Patrizio
Raffa
*a
b,
Luca
Ceseracciu
c and
Patrizio
Raffa
*a
aSmart and Sustainable Polymeric Products, Engineering and Technology institute Groningen (ENTEG), University of Groningen, Nijenborgh 4, Groningen, 4747 AG, The Netherlands. E-mail: p.raffa@rug.nl; Tel: +31503634465
bCenter for Materials Interfaces, Istituto Italiano di Tecnologia, Via R. Piaggio 34, 56025, Pontedera, Italy
cMaterials Characterization Facility, Istituto Italiano di Tecnologia, Via Morego, 30, 16163, Genova, Italy
First published on 27th January 2026
Abstract
A rather underappreciated aspect in the field of lithographic microfabrication is the post-print modification of 3D printed structures, which could be relevant to introduce or modify properties of both the bulk and the surface of materials. This study focusses on a strategy revolving around controlled polymerization, specifically reversible addition–fragmentation chain-transfer (RAFT) polymerization, to create a “living” resin material. We explored three different strategies which all focus on optimizing a different virtue of introducing a RAFT agent in microfabrication by two-photon lithography (TPP). The first method introduces a trithiocarbonate in a densely cross-linked material and changes the hydrophobicity of the surface in the chain extension process. To activate the chain extension, a zinc-based catalyst in the chain extension solution was irradiated by green light. The second approach instead altered the properties of the bulk of the structure. A different RAFT agent, a xanthate, was applied to increase the reaction rate and the cross-linking density was reduced. The property changes as a result of the chain extension were monitored by the change in the FT-IR spectrum and a lowering of the Young's Modulus of TPP printed microstructures. Finally, a tetra-functional self-polymerizing xanthate was synthesized, which allows for initiator-free polymerization and subsequent chain extension. These results show the successful modification of both bulk and surface properties of microstructures using RAFT control. Since all of the chain extensions are all light induced, this study opens avenues for localized property modification with focused light.
Introduction
Over the last half a century microfabrication has evolved from a niche to a fundamental part of technological progress. Micro-scale functional structures can be found in consumer, automotive and medical devices. New fabrication technologies have been driving this surge and the emergence of additive manufacturing, in particular lithographic techniques like 2-photon lithography, have provided product developers with unprecedented design freedom.1 Alongside the advances in fabrication approaches came new functionalities which can turn a micro-structure in a microdevice. Sensing, actuating, control and intelligence are features that are inevitably constricted by the materials applied to build the microstructure.2 Materials sensitive to (untethered) triggering by heat, light, electronic, magnetics, or other changes in their environment have been reported since the introduction of 3D printing on microscale.3–7 Furthermore, smart manufacturing approaches such as subtractive manufacturing and even the formation of biohybrids have been explored.8,9 Despite the rapid expansion of available TPP printable materials, a simple and versatile technique to post-functionalize printed structures is an aspect of the field that has received less attention.Additive manufacturing lithographic techniques with negative photoresists are generally employed because they provide geometric freedom, are easy to handle and are chemically resistant after the lithographically induced solidification reaction is completed and the covalent bonds are settled. In many applications this resistance is a virtue, as it prevents the structures from wear and undesired interactions with the environment. However, for post-cure modifications specific treatments are necessary,10 of which only few have been applied on microstructures. On TPP samples, the most widely used treatment is oxygen plasma, which results in clean and charged surfaces and can be used as a precursory step for chemical attachment. For instance, our group applied oxygen plasma to apply a super-hydrophobic layer on a TPP bioinspired gecko structure.11 Other examples of surface modification of TPP microstrucutres involve FIB treatment,12 masked gold-spattering,13 or UV irradiation. The principle of post-modification can be applied not only to modify the surface properties of a material, but also to change properties in the bulk of a structure. The most notable example for this is subtractive manufacturing, in which part of the fabricated material is removed in a later stage for structural reasons.9 However, it is debatable whether this is post-modification of the material, since this usually does not entail changes in the intrinsic properties of the polymer used. Another approach is the use of Diels–Alder reaction which instead can reversibly break covalent bonds. It has been applied in TPP, but the main application is found in self-healing materials rather than changing mechanical or surficial properties.14
Except for the masked modification of surfaces (which is only 2-dimensional), none of the approaches in both surface and bulk modification can be anchored to a specific area. This is a significant shortcoming that requires a different approach. Localized control over the degree of modification could generate a special gradient in properties. In addition, it offers a way to locally softening the material, thus allowing an ad hoc modulation of mechanical properties, useful for example to create joints and hinges in preformed microstructures.15 Furthermore, the nature of many fabrication processes curtail the variety of applicable materials. To overcome the previously described chemical resistivity of printed resist, we propose the use of reversible deactivation radical polymerization (RDRP).
RDRP refers to polymerization reaction where no termination happens. A so-called ‘dormant’ polymer chain can therefore be extended at a later stage, even in a different environment, allowing for the incorporation of a different monomer. ATRP (atom-transfer radical polymerization),16 NMP (nitroxide-mediated radical polymerization)17 and RAFT (reversible addition–fragmentation chain-transfer polymerization)18 were all discovered around three decades ago and have undergone significant leaps in understanding of the process and widened the choice of suitable materials. Fig. 1 presents a visual description of RAFT process. For as long as new radicals are generated in the reaction mixture, the reaction can continue. Consequently, the reaction can continue in a new environment enabling the possibility for precise design of block copolymers. New radicals are either generated by a separate radical initiator, catalyst or direct activation of the chain transfer agent. The latter is called photoiniferter process which is prompted by scission of the C–S bond (or S–S in the case of dithioesters).19 Depending on the specific type of transfer agent heat, light or a combination can bring about the cleavage allowing for monomer addition. It is a rather elegant method because a PI is no longer a requirement for chain transfer initiation resulting in a more constant radical concentration over time. Nevertheless, within the boundaries of the DLW process it may be difficult to realize high reaction speeds.
 | ||
| Fig. 1 Schematic of general RAFT reaction process (top). First an intiator introduces radicals to activate a polymerization reaction. These active chains are absorbed by the RAFT agent, which forms a reversible equilibrium shuttling the free radicals between the growing polymer chains and the agent. Since this process can be stopped and reactivated, it is possible to make a chain with one monomer R1 and continue in a later stage with another monomer R2. A schematic of a 2-step RAFT synthesis with methacrylates to form a block-copolymer is shown in the bottom scheme. The controlled process allows for precise design of chain length and low polydispersity. | ||
To allow for post-print modification of 3D printed micro- and nanostructures, a logical step is to introduce RDRP in TPP. However, traditional controlled polymerization is performed in an inert environment because oxygen would inhibit the growing chain permanently. Furthermore, a popular initiation agent is AIBN (azobisisobutyronitrile) which is activated by constant heating of the reaction mixture, curbing the potential of localized modifications. Recent development of photo-activated initiation and oxygen tolerant systems can fortunately provide the crucial tools for implementation of an RDRP process into microscale 3D printing.20–24 These have mainly been developed for RAFT, which is based on the reversible breaking of a covalent bond in di- or trithio-species.25 Recently, the group of Boyer have shown 3D structures on a macro-scale and showed the possibility to post-functionalize them changing the material surface to become more hydrophobic.26–28 The first instance of reactivation polymerization on the microscale was explored by Wu et al. who introduced a RAFT chain to a resist used in digital light processing.29 They incidentally exploited one of the benefits of introducing controlled polymerization by using NIPAM as a chain extension monomer. This thermosensitive material is not straightforward to implement in lithographic microfabrication without post-print modification. All of the mentioned examples crucially apply green light to activate the chain extension reaction. Finally, Jia et al. first employed controlled polymerization on a TPP fabricated structure with impressive ‘growth’ of a microstructure.30 They, on the other hand, used an NMP reaction and needed heat to activate the chain extension instead of photoactivation. The downside of this approach is that local chain extension is unfeasible as it requires prolonged warming. Furthermore, the reaction only proceeds with styrene as a monomer, limiting the property modification possibilities of the chain extension. It is desirable to widen the scope of monomers applicable for chain extension, because it allows for better tuning of properties in post-print modifications.
This research project focusses on finding approaches for surface and bulk post-print modification in TPP photoresists using RAFT. By employing different monomers, the aim is to tune the surface properties controllably from hydrophilic to hydrophobic. Furthermore, the resist composition was varied to tune the cross-linking density of the printed structures. With a low cross-linking density of the original structure, it is possible for new monomers to enter the bulk of the material and react with the reactivated RAFT agents.31 Therefore, this research seeks to modify the stiffness, or Young's Modulus, of a microstructure by controlled polymerization.
The current state-of-the-art TPP research has mostly neglected RDRP polymerization. This research offers instead the introduction of new monomers and shows the possibility to distinguish between chain extension at the surface or at the bulk. This could open up new ideas for TPP materials and expands the potential for microstructure applications.
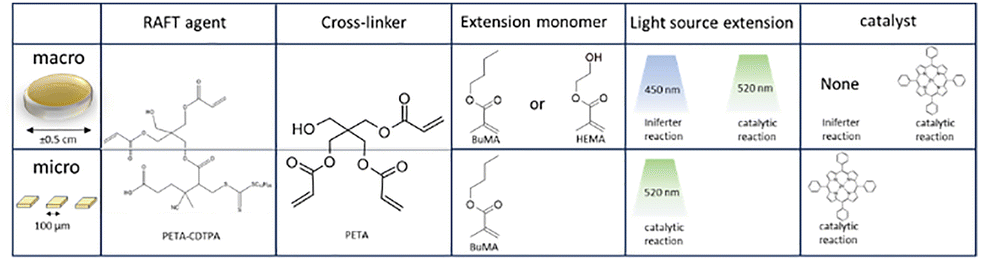
In this work, we explored potential advantages discussed in the introduction of post-modification using RAFT polymerization by looking at three different approaches, schematically shown in Fig. 2. For each approach a photopolymerizable mixture (commonly named resist) is prepared that contains materials suitable for RAFT controlled polymerization. These resists are designed for 2-photon lithography, but can also be polymerized by UV irradiation at a larger scale. All of the resists are therefore first scrutinized on the macroscale before scaling the process down aiming for similar effects. The macroscale samples are shaped like discs of 0.1 cm thick and a diameter of approximately 0.5 cm, while the microstructures prepared by TPP are shaped like pillars of varying size.
 | ||
| Fig. 2 Overview of the three approaches applied in this study. They differ in the RAFT agent that was applied, whether a photoinitiator was employed, the density of the cross-linking as a result of the chosen monomer and in the aim: bulk or surface modification. The first approach focusses on surface modification, the second on bulk modification and the final on the application of a photoiniferter proces removing the need for a photoinitiator. | ||
The first approach in Fig. 2 entails surface modification which is investigated by a resist containing functionalized (dodecylsulfanylthiocarbonyl)sulfanyl pentanoic acid (CDTPA), a standard RAFT agent for acrylates.32 The functionalization consists of an insertion of a single pentaerythritol triacrylate (PETA) molecule. This PETA-CDTPA is mixed with phenylbis(2,4,6-trimethylbenzoyl)phosphine oxide (TPO) as a photoinitiator and more Pentaerythritol triacrylate as a cross-linker. The many acrylate functional groups of PETA should result in a dense cross-linking, minimizing the penetration into the bulk as well as prevention of swelling. Therefore, this mixture is most suitable for surface modification, which we aimed to exploit in a chain extension reaction using either butyl methacrylate (BuMA) or hydroxy methacrylate (HEMA). Attachment of the hydrophobic BuMA should result in an increase of the contact angle of water while HEMA, containing a hydroxy group, the opposite can be expected.
In the second approach we sought instead to modify the bulk of the structure for which it is important that materials can easily penetrate the polymer matrix. The cross-linking density should thus be decreased and we therefore replaced PETA for PEGDMA, a dimer with an ethylene oxide spacer between the functional groups. Further, the RAFT agent EPEX ((S)-2-(Ethyl propionate)-(O-ethyl xanthate)) replaced CDTPA requiring more TPO photoinitiator in exchange for a potentially higher chain extension rate. In the chain extension step we investigated three different monomers with only one acrylate. Introducing mono-functional monomers in the bulk should further decrease the cross-linking density and introduce potentially softer materials both contributing to a lower toughness of the material.
The third approach aimed to amplify the bulk swelling by using the reversible RAFT linkages as cross-linkers. Furthermore, this approach would not require any photoinitiator as the RAFT agent was used as a photoiniferter creating the radicals by itself.
Approach 1: surface modification
 | ||
| Fig. 3 Schematic representation of the RAFT process and its initiation methods. Radicals can either be introduced by photoinifersion (green) as the light energy breaks up the RAFT agent, or by a separate photoinitiator absorbing the light and thereby creating radicals. When there is an excess of photoinitiator, the RAFT agent is effectively a radical absorber and regular free radical polymerization occurs. | ||
Therefore, we performed a preparatory step, before adding the photoinitiator, comprising a reaction by a photoiniferter reaction. The C–S bond in virgin CDTPA is relatively weak and becomes slightly more stable after the insertion of a single monomer.34 This enables the possibility to connect all of the CDTPA to at least one monomer without extensive cross-linking which would make the resist very viscous. Fig. 4A schematically shows the reaction process and details can be found in the experimental part.
 | ||
| Fig. 4 (A) Scheme of precursor reaction introducing the acrylate group of a PETA monomer into the RAFT agent (B) conversion graph of CDTPA over time which is based on the integration of the Xanthate carboxyl group in the 1H NMR spectrum. (C) Comparison of the NMR spectrum at the start of the reaction (top) to the end (bottom). The proton peaks from the methyl group ‘c’ move from 1.9 to a split peak around 1.4 once an acrylate group has been introduced. | ||
NMR spectroscopy (Fig. 4C) allowed monitoring of the conversion (Fig. 4B) as the methyl group of the RAFT agent moves from 1.9 ppm to split peaks around 1.4 ppm; the splitting of the peak is probably due to the proton movement becoming restricted.34 The final conversion was over 90% while the change in signal from the acrylates did not display measurable change and the peaks did not broaden, which suggests that indeed only one (or very few) monomers reacted with each RAFT agent. In the SI, Fig. S1 shows the log scale version of the conversion, suggesting first order kinetics, but the acrylate integration was very large compared to the internal reference making the integration not sufficiently consistent to measure the relatively small change in acrylate concentration. For this reason, we repeated the reaction with a lower RAFT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PETA ratio (1:5.3 instead of 1:12.5) enabling better monitoring of the kinetics (Fig. S2). It confirms that the reaction proceeds via first order kinetics and also shows a reduction in reaction speed when the unreacted CDTPA starts to become scarcer. As the secondary monomer introduction should be slower, it is an indication of the success of single monomer introduction. However, the relatively high conversion of acrylates suggest that several acrylates have reacted with a single RAFT agent. After this synthesis the modified RAFT agent was used for the surface modification reactions described in the following paragraphs. The product from the precursor reaction (ratio 1:12.5) was mixed with the photoinitiator to finalize the resist as displayed in Fig. 2.
:PETA ratio (1:5.3 instead of 1:12.5) enabling better monitoring of the kinetics (Fig. S2). It confirms that the reaction proceeds via first order kinetics and also shows a reduction in reaction speed when the unreacted CDTPA starts to become scarcer. As the secondary monomer introduction should be slower, it is an indication of the success of single monomer introduction. However, the relatively high conversion of acrylates suggest that several acrylates have reacted with a single RAFT agent. After this synthesis the modified RAFT agent was used for the surface modification reactions described in the following paragraphs. The product from the precursor reaction (ratio 1:12.5) was mixed with the photoinitiator to finalize the resist as displayed in Fig. 2.
|
 | ||
| Fig. 5 (a) Contact angle change over time using a chain extension with BuMA. Blank samples are disc structures with only polymerized PETA, while the RAFT samples contain CDTPA. A further differentiation between the samples is the presence of a catalyst in the chain extension mixture. (b) Contact angle measurements over time of the same reaction with HEMA as a monomer for the chain extension. (c) FT-IR measurements before and after chain extension procedure with HEMA and BuMA. No significant change was found. (d) Contact angle change with HEMA as a monomer in regular (norm) conditions (40 °C, 500 mg mL−1 monomer), cooled reaction temperature of 17 °C and at a lowered concentration of 50 mg mL−1 monomer. (e) example of a cured disc with a water droplet to measure the contact angle and an image of disc and example of contact angle measurement. | ||
In the second step of the process the disc-samples were subjected to a chain extension process which aims to add a monomer with different surface properties than the original resist sample. The focus in our study lied on the contact angle of water on the disc which indicates the hydrophobicity of the sample surface. Butyl methacrylate (BuMA) and hydroxy ethylmethyacrylate (HEMA) are simple and effective monomers to tune the surface to become either more hydrophobic or hydrophilic, respectively. The methacrylate groups make them suitable for a RAFT polymerization with CDTPA.
For the chain extension reaction, the disc prepared in the first step was immersed in a solution of the second monomer (i.e. BuMA or HEMA), pre-dissolved in a solvent and irradiated with blue light (450 nm). This should trigger a photoiniferter activation of the CDTPA in the polymerized resist and activate the chain extension even in absence of a catalyst. In contrast with the photoiniferter reaction, we also performed the reaction in presence of the zinc-based photoactivated catalyst ZnTPP to see if an external source of radical is still necessary. Adding the catalyst should make the reaction not only faster, but also less sensitive to oxygen because the ZnTPP can consume it out of the solution. A potential disadvantage of this procedure is the possible unwanted polymerization of the second monomer in solution, rather than on the disc surface.
The effect of the chain extension reaction over time on the hydrophobicity of the samples was followed by contact angle measurements (Fig. 5). All samples are compared to a blank where the chain extension procedure was performed on a sample without a RAFT agent incorporated. This comparison is essential, as even in absence of RAFT agent-terminated chains, acrylates could still react via free radical mechanism, due to removal of inhibitors and the rise in temperature caused by the lamp. In these reference experiments we did not observe significant changes in the contact angles of the blank, as expected (Fig. 5a and b). Similarly, the reaction without catalyst did show an increase in contact angle indicating that the photoiniferter reaction is not fast enough or is inhibited by oxygen. Only when the ZnTPP catalyst is present in the mixture, the hydrophobicity increases indicating attachment of BuMA to the surface of the disc sample (BuMA is more hydrophobic than the original surface because the PETA contains alcohol groups) The constant slope of the contact angle over time (Fig. 5a, green line) furthermore suggests that the reaction is continuous, thus that the RAFT agent does not suffer oxygen inhibition.
Despite the enhanced surface hydrophobicity in the BuMA chain extension reaction, the corresponding FT-IR measurement in Fig. 5c does not show significant change in chemical composition compared to the starting spectrum. A possible explanation is that the measurement was performed with attenuated total reflectance (ATR) which is the most widely used tool for FT-IR sampling. Instead of measuring the absorbance through the entire sample ATR, uses a diamond with a high refractive index and brings the sample into close contact. The IR light is irradiated at the interface under an angle making it reflect and creating an evanescent wave which stretches into the sample. Depending on the sample material, some of the light is absorbed while the rest is reflected. This means that ATR FT-IR measures the spectrum at a small depth, typically between 0.5 and 5 µm from the internal reflection element.35 If the chain extension occurs only at the surface and not in the subsurface bulk, the reaction might not have a significant impact on the FT-IR spectrum. This explanation fits with the dense cross-linking of PETA which prevents penetration of dissolved materials into the bulk and fits the purpose of designing a surface chain extension resist. It is therefore plausible that the chain extension reaction only takes place at the surface. Thus, within the reaction timeframe and light power, this produces a very thin (<0.5 µm) surface layer.
In contrast with BuMA, hydroxy ethyl methacrylate (HEMA) has a strong affinity for water because of its higher polarity and the presence of hydroxyl side group. If attached, this should render the surface of the RAFT samples more hydrophilic. Fig. 5b does show a downward trend in contact angle both for iniferter and catalyst aided reactions decreasing of approximately 9° (final value 49° ± 4). However, the variation between the measurements is quite significant resulting in an overlap of the error bars with the blank control samples. Specifically the blank with catalyst chain extension shows a similar decrease in contact angle.
Furthermore, we found that the fluid had become thicker in this experiment indicating an increase in viscosity and probably some reaction happening in the solution. To reduce this effect, the reaction was repeated in cooled conditions (17 °C compared to 40 °C) and at a lower concentration (50 mg mL−1 compared to 500 mg mL−1). Both these measures individually were able to counter viscosity increase. Moreover, the lowering in concentration even improved the efficacy of the reaction slightly as it reached a contact angle of 46° ± 3 and 41° ± 1 respectively (Fig. 5d), which is approximately the expected value for a polyHEMA surface.36 The higher efficacy might be explained by the relative increase of DMSO concentration, which could change the RAFT reaction kinetics.37 Similar to the BuMA chain extension, the reactions performed with HEMA do not show a significant change in the FT-IR spectrum after the chain extension (Fig. 5c).
The clearest effect of the chain extension with the CDTPA resist discs was achieved with BuMA as a monomer for the chain extension and ZnTPP in the mixture to catalyse the reaction. To replicate this effect on the microscale, an array of cubes was printed with a height of approximately 5 µm (Fig. 6d). The exact height of each cube could be established with profilometry. Now, the conditions for chain extension could be duplicated from the experiment with discs by emersion of the samples in a mixture of monomer (BuMA), catalyst and solvent. We covered part of the microcube array in order to exclude the possibility of a heat driven side-reaction. The results from the profilometer are shown in Fig. 6b and c (average change in Fig. 6e). The image shows that some of the cubes have an increased height as monomer has reacted with the microstructure material, but none of the cubes has a change in width. Further, the covered cubes show no difference in size change over the chain extension time. This indicates that the reaction is driven from the surface and side-reactions in the solution play no role in the growth of the structure. Light is thus the distinct activator of the process.
 | ||
| Fig. 6 (a) Schematic description of the process to fabricate microstructures with integrated RAFT agents and subsequent chain extension at the surface. (b) Profilometric profile of printed squares containing PETA-CDTPA before chain extension in grey. Catalysed chain extension with BuMA increased the height of the structures displayed in yellow, but only in the area not covered by the mask. (c) Blank control experiment of chain extension on printed structures without a RAFT agent (grey) show significantly less growth (yellow). (d) Microscope image from samples in the profilometer. (e) Average size of RAFT and blank structures (grey) and average height increase (yellow). | ||
It is however noteworthy that the blank sample not containing any RAFT agent shows an increased height according to the profilometer. This is unexpected after the results from Fig. 5 not showing any change in contact angle and FT-IR spectrum. An explanation could be the difference in polymerization process of the macroscale sample compared the microstructures. While the acrylate conversion of the PETA molecule after several seconds of UV-exposure is close to 100% for the disc's, it has been shown that this can be as low as 20% of the acrylates for TPP fabricated samples.38–40 Consequently, reacting chains in the mixture can easily connect to free acrylates on the microstructure surface. A second consequence is that the matrix is less densely cross-linked, allowing for some penetration of the monomer which increases this effect. On the other hand, the RAFT reaction is significantly more extensive and the subsequent growth much more pronounced, with some of the cubes even doubling in size. The RAFT agent distinctively cannot get saturated and thus continue to include BuMA monomers and thereby growing the structure to become much taller than the blank. This shows the effectiveness of this approach with the application of the focused laser.
Approach 2: bulk modification
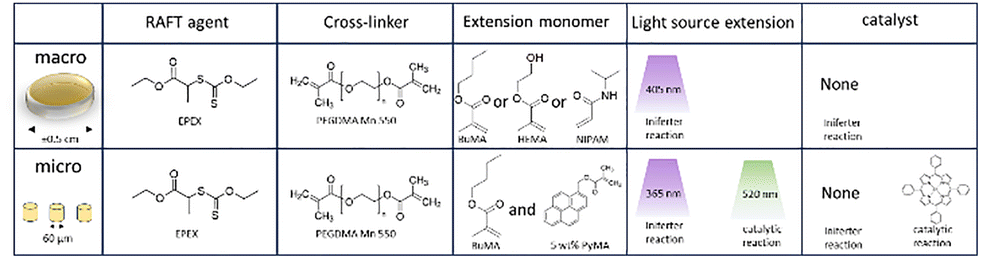
We have shown that with the prior CDTPA-containing resist it was possible to modify surface properties of the material in a post-printing setting. The bulk of the original structure however, does not undergo modification in these conditions. Yet, it could be desirable to induce change of properties in the inner volume of a microstructure either locally or in the formation as a whole. For instance, local alteration by introduction of monomers or moderation of cross-linking density are useful tools to create structural gradients. The previously discussed resist seems unsuitable for this aim as the reaction only proceeded at the surface. The trifunctional PETA monomer the resulting polymetric matrix has a relatively densely connected network which hinders penetration of the monomer and catalyst in the bulk. Consequently, for chain extension in the bulk a different composition of the resist is required starting with a cross-linker that allows for larger pores in the polymeric network. For this reason, we chose PEGDMA as a resist monomer, which is a molecule that has two acrylates connected by a short chain of ethylene glycol. It is a popular material to fabricate microstructure hydrogels and is known to swell in a variety of different solvents. Fig. 8 shows an overview of the macro and micro scale samples and how they were chain extensions.As an alternative to the trithiol agent CDTPA, we introduced a EPEX (Fig. 8), which is a xanthate type RAFT agent. Xanthates generally have a large absorption in high wavelength UV, in contrast with the green light and deep-UV absorption of trithiols. In addition, the C–S bond is comparatively weaker increasing the effectiveness of photolysis. Furthermore, xanthates are more stable, leading to less addition of acrylic radicals and thus faster polymerization. The intermediate xanthate radical inversely is less stable, driving fast fragmentation and therefore more propagating species.20 These improvements in reaction speed are likely to lead to an increased chain length distribution (PDI), but this is not an issue for the property modification applications. It follows from the higher polymerization speed combined with the large UV-absorption that a photoiniferter type of reaction becomes more prominent. Depending on the application, the need for catalytic support can be redundant. However, for the initial polymerization we always included TPO as a photoinitiator because the light exposure time is extremely short (Table 2).
|
 | ||
| Fig. 7 (a) FT-IR spectrum comparison of cured PEGDMA disk (black), after chain extension with HEMA on blank (pastel orange) and on EPEX containing sample (dark orange) (b) comparison of chain extension with BuMA on blank (pastel purple) and EPEX containing sample (dark purple) (c) comparison of chain extension with NIPAM on blank (pastel blue) and EPEX containing sample (dark blue) (d) contact angle of blank and EPEX containing sample before chain extension (green), after chain extension with HEMA (orange) and with BuMA (purple). | ||
The contact angle of the sample is consistent with the FT-IR results since the introduction of BuMA leads to a larger contact angle indicating increased hydrophobicity. The blank sample undergoes a slightly smaller but similar change. Even in the absence of a RAFT CTA it seems that some of the monomer still incorporates, presumably because there are still unreacted double bonds after curing.41–43 The hydrophilic monomer HEMA had a less significant effect on the contact angle, but the RAFT sample still shows the biggest change. Compared to the previous approach with the CDTPA RAFT agent, not only the contact angle, but also the FT-IR spectrum changed over the course of the chain extension. This indicates that the reaction occurs not just at the absolute surface but also in the polymer bulk. It would be expected that introduction of monomers in the entire sample would lead to a change in size. It should be noted however, that we did not measure a clear change in this regard, meaning that the reaction is probably still happening close to the surface. However, when translating this to the micro-scale the surface to volume ratio sharply increases. Profound infiltration of monomers in the entire structure is therefore decisively less complicated for the TPP printed structures.
 | ||
| Fig. 8 (a) Set-up of the nanocompression with a standard shape and size of the sample (60 µm diameter and height) (b) comparison of Young's Modulus of several samples printed with 2-photon polymerization. The toughness at the start is coloured in yellow, after chain extension with UV in purple and after ZnTPP catalysed extension in green. The blank samples have white stripes. A mixture of BuMA with 5 wt% 1-pyrene methylmethacrylate and DMSO was used for chain extension. | ||
The monomer mixture for chain extension contained beside BuMA also 5 wt% of pyrene methacrylate, a fluorescent molecule, introduced to better visualize the process. Fig. 9 shows microscope images from the printed micro-pillars after chain extension. After turning off the microscope light source, the cylinders are irradiated with a UV-lamp which induces fluorescence of the pyrene. The UV-chain extension samples remain dark, while the catalyst aided reaction lights up in blue as a consequence of the fluorescence. This is consistent with the changes in Young's Modulus, and suggests that the procedure with ZnTPP is more successful.
 | ||
| Fig. 9 Microscope images of printed pillars before chain extension (top row) and after chain extension (Bottom row). The white size indicator bar represents 100 µm. UV light was used to intiate the photoiniferter chain extension and on the blank, while green light was irradiated on the ZnTPP containing mixture. The experiments were performed with BuMA with 5 wt% 1-pyrene methylacrylate in DMSO. Only after the catalysed reaction, the sample shows fluorescence indicating attachment of the pyrene moiety. | ||
Approach 3: photoiniferter fabrication

|
 | ||
Fig. 10 (a) Schematic reaction overview, details are provided in the experimental part. (b) chain extension with BuMA induces a change of the FT-IR spectrum over time, here each graph is normalized to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peak at 1750 cm−1. (c) chain extension with NIPAM induces a very clear change in the FT-IR spectrum and continues to change strongly over time. The graphs are normalized to the CO peak at 1750 cm−1. O peak at 1750 cm−1. (c) chain extension with NIPAM induces a very clear change in the FT-IR spectrum and continues to change strongly over time. The graphs are normalized to the CO peak at 1750 cm−1. | ||
After preparing macro-scale disc shaped samples, the tetrafunctional RAFT could be subjected again to UV while immersed in a monomer. For both BuMA and NIPAM the reaction proceeds continuously over a couple of hours according to the FT-IR (Fig. 10c). Especially for NIPAM the gradual increase of integrated monomer is impressive as the graphs are normalized to the carbonyl group at 1750 cm−1. It shows that the chain extension effect with this approach is rather powerful. Despite the FT-IR suggesting that the bulk of the material is “filled” with the chain extended monomer, there is no significant growth of the disc and the well-known LCST behaviour of NIPAM in water could not be observed. It should be noted that this particular thermosensitive effect generally requires NIPAM chains of at least 30 units for a coil-to-globule transition, so in our system probably shorter chains are formed.44 The lack of LCST and size change indicates that the chain extension is mainly happening at the surface and the material simply does not allow for fast penetration and reaction of monomers in the bulk.
The original intention of this tetrafunctional RAFT agent was to produce a photoinitiator-free resist. Despite our best attempts at exploring all parameters of the 780 2-photon lithography set-up like laser power and scan speed, attempts to print this material were unsuccessful. Therefore we can exclude that the concentration of RAFT agent was the limiting factor for photoiniferter reactions in TPP. Alternative solutions could be to drastically increase the laser power to a level beyond the commercial systems or use a different wavelength as did Wu et al. successfully,29 but this would force to shift away from commercially available TPP systems. Finally, a different RAFT agent could be selected with less rate-retardation, thus more propagation.
Conclusions
This study has explored the possibility to apply RAFT controlled polymerization as a strategy for post-print modification of polymeric materials. When we conceived the idea for this research, nothing similar had been performed on micro-scale 3D printed structures. However, some interest in this concept has emerged very recently in the shape of RAFT for digital light processing and NMP for 2-photon polymerization.29,30 Yet, we have taken another approach by focussing on property modification in both bulk and surface of structures, exploring several pathways. The method is transferable to other lithographic microfabrication technologies and has the potential to be applied for a wide range of monomers.The first approach presented successful surface modification on macro scale as well as TPP printed structures by altering the hydrophobicity of the surface. Contact angle and ATR-FTIR measurements suggest that the reaction was restricted to the surface, as it would logically result from the densely cross-linked material. A zinc-based catalyst was effective in initiating chain extension by green light.
The second method focussed on modifying the bulk properties of the printed structure, by reducing the cross-linking density and by using a xanthate based RAFT agent, instead of a trithiol compound, making the chain extension faster. Changes in the ATR FT-IR spectrum, lowering of the Young's Modulus and introduction of fluorescent monomers which all support successful bulk modification. There are however limitations to the current system. The experiments with NIPAM as a chain extension monomer do not display LCST behaviour and the change in Young's Modulus is rather small. It indicates a limited chain length of the added monomer particularly in the bulk and requires optimization in follow-up studies.
The third system explored in this study involved the synthesis of a tetra-functional self-polymerizing xanthate. The material could be applied for UV-triggered photoinifersion resulting in fast polymerization of a dense network, and avoids the use of external photocatalysts. Furthermore, using again UV-photoinifersion, chain extension with various monomers was possible. However, despite the maximized RAFT agent concentration, initiator-free polymerization with TPP was not achievable.
In conclusion, we have shown the effectiveness of RAFT agents as a means to post-print modify materials. We supported our conclusions with blank control samples, an aspect often overlooked in previous reports in this field. Interestingly, blank experiments also showed some modification, but convincingly less than the RAFT samples, suggesting that photoprinted materials can still contain a significant amount of reactive double bonds. This study can thus be a foundation for property modification of TPP printed microstructures. In addition, our restriction to light-induced chain extensions provides a future pathway to localized modification using focussed light.
Experimental
Materials
CDTPA (Boron Molecular, 95%), TPO (Sigma, 97%), PETA (Fischerscientific), potassium O-ethyldithiocarbonate (Sigma, >97%), ethyl 2-bromoproprionate (Sigma, 98%), triethylamine (Sigma, >99.5%), butyl methacrylate (Sigma, 99%), 1-pyrene methylmethacrylate (Sigma, 99%), hydroxy-ethylmethacrylate (TCI, >95%), 2-bromopropanoyl bromide (Sigma, 97%), pentaerythritol (Sigma, 98%), NIPAM (Sigma, >99%), potassium hydroxide (Sigma, >85%), carbon disulfate (Sigma, >99%), ethanol, DMSO, DCM, THF, DMF.PETA insertion in CDTPA RAFT agent
The reaction mixture was prepared by adding CDTPA, PETA (1:12.5 ratio; 300 mg and 3750 mg, respectively), and solvent (e.g., DMSO, THF, or DCM; 12 mL) to a 20 mL vial. Additionally, DMF was added as an internal reference (CDTPA:DMF - 1:3; 163 mg). The vial was equipped with a stirring egg and septum, followed by bubbling with argon gas for 30 minutes whilst stirring. Following this, the reaction mixture was placed in a cooling setup set at 17 °C (5 °C in case of DCM). Progress of the reaction was monitored through FT-IR and 1H-NMR spectroscopy. The photoiniferter reaction was initiated by blue light irradiation (450 nm wavelength, 13.8 mW m−2), which was continued until no signals of CDTPA is found in the NMR spectrum of the reaction mixture and was terminated by removing the light source. Upon reaction completion, the contents were washed with saturated saline solution and extracted with ethyl acetate. The ethyl acetate was evaporated, and the contents were further purified through silica column chromatography with ethyl acetate and petroleum ether as the eluent. Finally, this product was analysed with 1H-NMR spectroscopy (Fig. S1).
Synthesis of ((S)-2-(Ethyl propionate)-(O-ethyl xanthate)) (EPEX)
The schematic overview of the reaction is presented in Fig. S3. 10.14 g (63.3 mmol) of potassium O-ethyldithiocarbonate was added dropwise at 0 °C in inert atmosphere to a mixture of 10.2 g ethyl 2-bromoproprionate (56.3 mmol) previously dissolved in 100 mL of ethanol. The mixture was stirred overnight in the dark. In the morning 100 mL of MilliQ water were added and final solution was extracted using a mixture, ethyl acetate:pentane 1:2 (3 × 100 mL). The organic phase was dried under vacuum using a rotavap and the product was collected as transparent yellowish oil (around 10 g). Successful synthesis was confirmed with 1H-NMR 400 Hz spectroscopy (Fig. S4).
Synthesis of star-shaped RAFT-monomer
:ethyl acetate 8:2). The product was analysed using 1H-NMR and (Fig. S6) 13C-NMR spectroscopy 400 Hz (Fig. S7). All spectra were recorded at room temperature using an Agilent 400-m spectrometer MHz spectrometer. All 2D-NMR spectra were obtained using 4 scans and 256 increments.
Resist preparation
:12.5 ratio (CDTPA:PETA). Into this mixture the photoinitiator was introduced by adding dissolved TPO in a small amount of DCM followed by degassing in a vacuum chamber. For this process 4 wt% of TPO was used. Blank control samples had the same ratio of monomer and initiator but lacked the RAFT agent.
Initiator free polymerization. The tetra-functional RAFT agent was used without any photoinitiator nor additional monomer.
Disc-structure manufacturing
Generally the macro samples with any of the resists were prepared by applying approximately 150–300 mg of the mixture onto a glass mold. It was subsequently exposed to UV light (405 nm wavelength) for 30 seconds. The sample was left to be soaked/stored in ethanol overnight. After drying, the contact angle of water on the flat surface of the disc was measured as well as the FT-IR spectrum (Fourier transform infrared spectra were acquired on a Shimadzu-IRTracer 100 equipped with ATR (attenuated total reflectance) apparatus, a total of 32 scans at a resolution of 4 cm−1 in the range 4000–600 cm−1 were recorded).2-photon lithography
To print microstructures, a droplet of the same mixture was placed on a glass slide and placed in the printer, which was a Photonic Professional GT-2 from Nanoscribe. This device allows for localized polymerization of the resin using a femtosecond pulsed near-infrared laser. For samples with surface modification (approach 1) flat structures of approximately 5 µm in height were printed with a length and with of 100 µm. The bulk modification structures were cylindrical with a height and diameter of 60 µm. After the 2-photon lithographic printing process, non-reacted material was removed by immersion in propanol and dried afterwards.Chain extension
The chain extension of the disc shaped macrostructures was performed in an 8 mL reaction vial. Beside the structure, a mixture of 250 mg monomer, purified over aluminum oxide, and 0.5 mL DMSO was added to the vessel. In the catalysed instances of the reaction, ZnTPP was furthermore present in a 0.2 mg mL−1 concentration. The mixture was allowed to soak for 24 hours and was then flushed with argon. The vessel was then placed in the reaction box in which it was irradiated with blue light (without catalyst) or red light (with catalyst) with 13.8 mW m−2 of power. Intermittently the contact angle was monitored during which the disc was cleaned and the reaction mixture was refreshed. At the end of the chain extension, the FT-IR was taken using the ATR FT-IR.Chain extension on the micro-sized samples was performed by immersing the glass slide substrate in a reaction mixture with the same ratios. To speed up the reaction speed of the chain extension a 520 nm laser diode module with 35 mW of power on a spot with a ±150 µm diameter was used (Global Laser, RS127-1555, Acculase-PWM-520-35-S). The samples were irradiated for 6 minutes while immersed in the ZnTPP containing mixture. For the UV chain extension a 365 nm spot-lamp was used.
To measure the chain extension in the microstructures two strategies were applied. Firstly, the profile was examined before and after the reaction using a P-6 stylus profilometer (KLA Tencor, USA). The other was to measure the Young's Modulus using Nanocompression (Anton Paar nanoindenter equipped with a 100 µm-diameter tip, as schematically depicted in Fig. 10A). The latter requires relatively large structures of around 60 µm in height and diameter. Tests were performed with a loading and unloading rate of 20 mN min−1, to the maximum load of 10 mN, corresponding to strain of 3–6%, depending on the sample. Young's Modulus was calculated from the linear region of the loading curve.
Author contributions
The research was conceptualized by F. M. den Hoed and P. Raffa. The experiment work was executed by F. M. den Hoed and T. F. Chen, while the nanocompression experiment and analysis were performed by L. Ceseracciu. Writing of the original draft by F. M. den Hoed and reviewed by P. Raffa and V. Mattoli. Supervision from V. Mattoli and P. Raffa. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data that support the findings of this study are openly available in OSF at the following link: https://osf.io/jfrxp/overview?view_only=f3f1a0a20d63409fbb957ad8dd3e055e.Supplementary information, consisting of additional reactions scheme, kinetic studies performed with H-NMR, and additional NMR characterization, is available.45 See DOI: https://doi.org/10.1039/d5ma01403d.
Acknowledgements
F. D. H., P. R. and V. M. gratefully acknowledge support from the European Union Horizon 2020 research and innovation programme under the Future and Emerging Technologies Open Grant 5DNanoPrinting (ID: 899349).Notes and references
- F. M. den Hoed, M. Carlotti, S. Palagi, P. Raffa and V. Mattoli, Micromachines, 2024, 15, 275 CrossRef PubMed.
- J. Li and M. Pumera, Chem. Soc. Rev., 2021, 50, 2794 RSC.
- M. Carlotti and V. Mattoli, Small, 2019, 15, 1902687 CrossRef PubMed.
- S. Nocentini, D. Martella, C. Parmeggiani and D. S. Wiersma, Adv. Opt. Mater., 2019, 7, 1900156 CrossRef.
- M. Hippler, E. Blasco, J. Qu, M. Tanaka, C. Barner-Kowollik, M. Wegener and M. Bastmeyer, Nat. Commun., 2019, 10, 232 CrossRef PubMed.
- H. Zeng, D. Martella, P. Wasylczyk, G. Cerretti, J.-C. Gomez Lavocat, C.-H. Ho, C. Parmeggiani and D. S. Wiersma, Adv. Mater., 2014, 26, 2319 CrossRef CAS PubMed.
- Z. Liu, M. Li, X. Dong, Z. Ren, W. Hu and M. Sitti, Nat. Commun., 2022, 13, 2016 CrossRef CAS PubMed.
- N. Pellicciotta, O. S. Bagal, V. Carmona Sosa, G. Frangipane, G. Vizsnyiczai and R. Di Leonardo, Adv. Funct. Mater., 2023, 33, 2214801 CrossRef CAS.
- M. Carlotti, O. Tricinci and V. Mattoli, Adv. Mater. Technol., 2022, 7, 2101590 CrossRef CAS.
- S. K. Nemani, R. K. Annavarapu, B. Mohammadian, A. Raiyan, J. Heil, M. A. Haque, A. Abdelaal and H. Sojoudi, Adv. Mater. Interfaces, 2018, 5, 1801247 CrossRef.
- O. Tricinci, E. V. Eason, C. Filippeschi, A. Mondini, B. Mazzolai, N. M. Pugno, M. R. Cutkosky, F. Greco and V. Mattoli, Smart Mater. Struct., 2018, 27, 075009 CrossRef.
- P. Li, S. Chen, H. Dai, Z. Yang, Z. Chen, Y. Wang, Y. Chen, W. Peng, W. Shan and H. Duan, Nanoscale, 2021, 13, 1529 RSC.
- A. Ottomaniello, P. Vezio, O. Tricinci, F. M. den Hoed, P. Dean, A. Tredicucci and V. Mattoli, Nanophotonics, 2023, 12(8), 1557–1570 CrossRef PubMed.
- A. S. Quick, H. Rothfuss, A. Welle, B. Richter, J. Fischer, M. Wegener and C. Barner-Kowollik, Adv. Funct. Mater., 2014, 24, 3571 CrossRef CAS.
- P. Imrie, O. Diegel and J. Jin, Polymers, 2023, 276, 125944 CrossRef CAS.
- J. S. Wang and K. Matyjaszewski, Macromolecules, 1995, 28, 7572 Search PubMed.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661 CrossRef CAS PubMed.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559 CrossRef CAS.
- M. Hartlieb, Macromol. Rapid Commun., 2022, 43, 2100514 CrossRef CAS PubMed.
- B. Zhao, J. Li, Y. Xiu, X. Pan, Z. Zhang and J. Zhu, Macromolecules, 2022, 55, 1620 CrossRef CAS.
- A. Bagheri, C. W. A. Bainbridge, K. E. Engel, G. G. Qiao, J. Xu, C. Boyer and J. Jin, ACS Appl. Polym. Mater., 2020, 2, 782 CrossRef CAS.
- X. Shi, J. Zhang, N. Corrigan and C. Boyer, Mater. Chem. Front., 2021, 5, 2271 RSC.
- A. Bagheri, Macromolecules, 2023, 56, 1778 CrossRef CAS.
- J. Phommalysack-Lovan, Y. Chu, C. Boyer and J. Xu, Chem. Commun., 2018, 54, 6591 RSC.
- Q. Fu, K. Xie, T. G. McKenzie and G. G. Qiao, Polym. Chem., 2017, 8, 1519 RSC.
- A. Bagheri, K. E. Engel, C. W. A. Bainbridge, J. Xu, C. Boyer and J. Jin, Polym. Chem., 2020, 11, 641 RSC.
- K. Lee, N. Corrigan and C. Boyer, Angew. Chem., Int. Ed., 2021, 60, 8839 CrossRef CAS PubMed.
- Z. Zhang, N. Corrigan, A. Bagheri, J. Jin and C. Boyer, Angew. Chem., Int. Ed., 2019, 58, 17954 CrossRef CAS PubMed.
- X. Wu, B. Gross, B. Leuschel, K. Mougin, S. Dominici, S. Gree, M. Belqat, V. Tkachenko, B. Cabannes-Boué, A. Chemtob, J. Poly and A. Spangenberg, Adv. Funct. Mater., 2022, 32, 2109446 CrossRef CAS.
- Y. Jia, C. A. Spiegel, A. Welle, S. Heißler, E. Sedghamiz, M. Liu, W. Wenzel, M. Hackner, J. P. Spatz, M. Tsotsalas and E. Blasco, Adv. Funct. Mater., 2023, 33, 2207826 CrossRef CAS.
- C. W. A. Bainbridge, K. E. Engel and J. Jin, Polym. Chem., 2020, 11, 4084 RSC.
- G. Moad, RAFT Polymerization, John Wiley & Sons, Ltd, 2021, pp. 359–492 Search PubMed.
- J. Xu, C. Fu, S. Shanmugam, C. J. Hawker, G. Moad and C. Boyer, Angew. Chem., Int. Ed., 2017, 56, 8376 CrossRef CAS PubMed.
- C. Fu, Z. Huang, C. J. Hawker, G. Moad, J. Xu and C. Boyer, Polym. Chem., 2017, 8, 4637 RSC.
- H. Tiernan, B. Byrne and S. G. Kazarian, Spectrochim. Acta, Part A, 2020, 241, 118636 CrossRef CAS PubMed.
- M. Harata, M. Watanabe, S. Nagata, E. C. Ko, S. Ohba, T. Takato and A. Hikita, Regen. Ther., 2017, 7, 61 CrossRef PubMed.
- M. Benaglia, E. Rizzardo, A. Alberti and M. Guerra, Macromolecules, 2005, 38, 3129 CrossRef CAS.
- F. Burmeister, S. Steenhusen, R. Houbertz, U. D. Zeitner, S. Nolte and A. Tünnermann, J. Laser Appl., 2012, 24, 042014 CrossRef.
- J. B. Mueller, J. Fischer, F. Mayer, M. Kadic and M. Wegener, Adv. Mater., 2014, 26, 6566 CrossRef CAS PubMed.
- J. E. Johnson, Y. Chen and X. Xu, Opt. Express, 2022, 30, 26824 CrossRef CAS PubMed.
- K. Cicha, Z. Li, K. Stadlmann, A. Ovsianikov, R. Markut-Kohl, R. Liska and J. Stampfl, J. Appl. Phys., 2011, 110, 064911 Search PubMed.
- T. Baldacchini, M. Zimmerley, C.-H. Kuo, E. O. Potma and R. Zadoyan, J. Phys. Chem. B, 2009, 113, 12663–12668 Search PubMed.
- J. S. Oakdale, J. Ye, W. L. Smith and J. Biener, Opt. Express, 2016, 24, 27077–27086 CrossRef CAS PubMed.
- R. Singh, S. A. Deshmukh, G. Kamath, S. K. R. S. Sankaranarayanan and G. Balasubramanian, Comput. Mater. Sci., 2017, 126, 191 Search PubMed.
- V. K. Patel, A. K. Mishra, N. K. Vishwakarma, C. S. Biswas and B. Ray, Polym. Bull., 2010, 65, 97 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |