DOI:
10.1039/D5MA01125F
(Paper)
Mater. Adv., 2026,
7, 3254-3267
NiCo LDH-derived defect-engineered Se-doped NiCoP mesoporous nanoflowers for enhanced oxygen evolution reaction
Received
30th September 2025
, Accepted 6th February 2026
First published on 23rd February 2026
Abstract
The rational design and development of doped and defect-engineered electrocatalysts are vital for enhancing the oxygen evolution reaction (OER) during water electrolysis for sustainable energy conversion. In this study, a NiCo LDH precursor was first synthesized via a solvothermal route and anchored on nickel foam (NF), followed by a one-pot phospho-selenization process to simultaneously introduce Se doping and P vacancy into a bimetallic phosphide nanoflower architecture (NCP–Se/NF). The optimized NCP–Se/NF catalyst requires the smallest overpotential of only 260 mV to achieve a current density of 10 mA cm−2. It exhibits the lowest Tafel slope of 28.17 mV dec−1 compared with bimetal phosphide (NCP/NF) and the parent LDH (NCL/NF). It exhibits long-term durability, maintaining 84.1% of its original activity even after 50 hours of operation under anodic conditions in an alkaline electrolyte. The synergistic effect of Se doping and P vacancy manipulation alters the electronic redistribution around the Ni/Co sites, which subsequently optimizes the d-band centre, providing a more exposed active site, minimizing the diffusion path length, and reducing the kinetic energy barrier for the OER. The P vacancy is evidenced by XPS and EPR analyses. Furthermore, the use of CTAB as a surfactant template in synthesis generates mesopores within the nanoflower structure, significantly increasing the specific surface area and facilitating the electrolyte penetration, which in turn promote rapid oxygen bubble release. This work highlights the use of combined heteroatom doping and vacancy-engineering strategies to provide an effective route for the design of high-performance electrocatalysts for efficient water oxidation.
1. Introduction
From the beginning of civilization and industrialization, fossil fuels have been the primary source for meeting the majority of the energy requirements. Undoubtedly, fossil fuels have brought exceptional economic growth and development across the globe; however, these benefits are accompanied by intolerable environmental pressure and problems that the blue planet and its occupants are suffering from. Moreover, fossil fuel deposits are running out, and someday in the future, they will be completely depleted.1,2 Therefore, hard work and infinite efforts have been made by trained brains to find a solution to this problem. Energy conversion from renewable sources is found to be an outstanding and encouraging solution for significantly reducing the dependency on traditional fossil fuels.3,4 The several energy systems governed by electrochemical reactions can be broadly classified into fuel cells, water splitting and metal–air batteries, which are among the most efficient and reliable energy conversion and storage technologies. Among them, the electrocatalytic oxygen evolution reaction (OER) along with the hydrogen evolution reaction (HER) through water splitting is the most promising and widely acceptable strategy globally. Enhancing the OER via electrocatalysis is crucial for overcoming the fundamental kinetic bottleneck that hinders the efficacy of water-splitting systems. By enhancing the OER electrocatalysis, the energy barrier can be reduced, resulting in lower anodic overpotential, which in turn reduces the operating voltage and energy consumption of the electrolyzer. It also enables the stable operation of the electrolyzer at high current densities by suppressing electrode degradation, allowing for high-rate operation without excessive heat generation and performance decay. The OER is a pH-dependent reaction, as in acidic and neutral environments, two H2O molecules are oxidized to O2 and H+ species, while in basic conditions, the OH− species are oxidized and converted to O2 and H2O molecules.5,6 The production of O2 molecules from the OER involves a transfer of four proton-coupled electrons. From a kinetic perspective, the OER is a multi-step reaction with the transfer of a single electron in each step. Thus, the accumulation of energy at each step makes the OER kinetically sluggish and requires high overpotential. The sluggish kinetics have limited the commercialization of these energy-conversion technologies.7,8 To minimize the loss of energy in these energy-conversion technologies, the exploration of highly active electrocatalysts is urgently required to facilitate the OER and reduce the overpotential.9 Generally, Ir/Ru-based oxides are known to be effective OER electrocatalysts. Still, their high cost, natural scarcity, and poor stability render them unsuitable for extensive applications.10,11 Enormous efforts have been devoted to seeking efficient, nonprecious and cost-effective electrocatalysts.
Transition metal phosphides (TMPs) are promising OER electrocatalysts due to their low cost, excellent corrosion resistance, and efficient electron conduction, which facilitates effective charge transport.12,13 TMPs exhibit an adaptable and tunable structure with a stoichiometric ratio of metals to phosphorus. During the OER process, the oxidation of phosphorus promotes the conversion of active species into oxides/hydroxides, thereby lowering the kinetic energy barrier and improving the catalytic efficiency.14,15 Compared to single-metal phosphides, the bimetallic phosphide benefits from the synergistic interaction between two metals, which promotes rapid electron transfer, improves conductivity, and increases the number of active sites for electrocatalytic reactions. Among various transition metal phosphides, NiCoP is regarded as one of the most effective metal phosphides for the OER, owing to the cooperative effect of Ni and Co atoms and their similar atomic radii, which facilitate their combination to form a ternary transition metal phosphide. This improved electronic structure is favourable for the adsorption/desorption of intermediates during the OER process.15,16 Despite their advantages, NiCoP remains unable to meet ideal electrocatalytic operational requirements due to the insufficient number of active sites. Therefore, diverse approaches, including heterostructure formation,17 nanostructured morphology,18 heteroatom doping,19 and vacancy engineering,14 as well as noble metal modification,20 have been employed to enhance the intrinsic activity of TMPs. Some investigations have found that heteroatom doping of TMPs is an effective strategy to enhance their electrochemical activities. Incorporating heteroatoms manipulates the electronic structure, thereby optimizing the adsorption energy of reaction intermediates, which is strongly correlated with the charge distribution at interfacial active sites.21 Regulating the charge density near the Fermi level at active metal sites is closely associated with their d-orbitals arrangement. The d-band centre of the active metal serves as a descriptor of the charge density, as well as the catalytic activity. Shifting it toward the Fermi level (Ef) can regulate the intermediate adsorption/desorption and modify their bonding/antibonding interactions on the catalyst surface.22 For instance, Le et al. prepared Ru-doped NiCoP efficient electrocatalysts for robust water electrolysis. Their strategy was to modulate the Ru electronic configuration on the NiCoP framework, consequently exposing maximized active sites. Ru atoms can modulate the d-band centre and enhance the adsorption of intermediate species on the catalyst surface.23 Very recently, Zhang and his co-worker modulated the electronic structure of NiCoP nanosheets by Ce doping towards efficient overall water splitting. Their study illustrates that Ce doping regulates the 3d orbital electrons of Ni/Co in TMPs by its occupied 4f and 5d orbitals and enhances the structural stability, improving electrocatalytic processes.24
Apart from this, selenium (Se) has been utilized as a dopant in electrocatalyst design because its 3d orbital energy level is close to the energy levels of the 3s and 3p orbitals that participate in bonding with metal atoms. Such strong bonding optimizes the electronic configuration of active-site atoms, lowering the reaction potential barrier for the electrocatalytic water oxidation reaction.25 On the other hand, introducing vacancy defects has proven to be a powerful approach for tuning interfacial chemical properties while increasing the availability of active sites, facilitating charge transfer by adjusting the electronic configuration of metal atoms, which in turn reduces the energy barriers of intermediates and reactants. In particular, the P vacancies in TMPs create unsaturated metal sites neighbouring the vacancy. These unsaturated metal atoms generate new electronic states near the Fermi level due to a change in the d orbital, which optimizes the OER kinetics. Moreover, both doping strategy and vacancy engineering have been employed as effective approaches to modulate the electronic environment and optimize the adsorption/desorption energies of reactive intermediates (*O, *OH, *OOH) in TMPs.14,19 For example, Yan et al. developed V-doped NiCoP nanosheets with P vacancies, which exhibited synergistic effects and enabled faster electron transfer for the OER.26 The Ce–NiCoP@C/NF electrocatalyst was fabricated by Su et al. for the overall water splitting process. Their group demonstrated that Ce doping induces lattice distortion in NiCoP, improves its structural stability, and facilitates strong bonding of the carbon layer on the NiCoP surface, which accelerates the electrolysis of water.27 Although the aforementioned strategies yield excellent electrocatalytic activity, their complex synthesis procedures continue to impede further progress in electrochemical applications.
In addition, both theoretical and experimental evidence have confirmed that the rational design of electrocatalysts with various morphologies plays a crucial role in enhancing their electrochemical performance.2,28 Exclusively, the mesoporous nanoflower architecture supported on a conductive substrate derived from layered double hydroxide (LDH) exhibits superior electrochemical activity. The LDH-derived material not only possesses the synergistic advantages of LDHs (such as their intrinsic layered structure and tunable metal cation composition) but also delivers more stable mechanical properties, which are advantageous for increasing the lifespan of electrodes. Also, the uniform dispersion of multivalent metals present in the laminar structure of LDHs can create abundant active sites in the derived compound. Furthermore, the formation of abundant mesopores on the surface of the LDH-derived catalyst can further enhance the OER performance by reducing the diffusion path length of electrons and the electrolyte, which enables the rapid liberation of O2 bubbles. Nevertheless, there is limited literature on LDH-derived bimetallic phosphides for electrocatalytic water splitting processes.29,30 However, studies exploring the application of the LDH-derived one-pot-assisted Se-doping induced P vacancy in mesoporous bimetallic phosphide nanoflower catalysts for the OER are rare.
In this work, we have fabricated a NiCo LDH-derived Se-doped NiCoP mesoporous nanoflower hybrid for the OER. First, Ni foam (NF)-anchored NiCo LDH is synthesized via the solvothermal technique using CTAB as a structure director. Then, by using the phospho-selenization technique, a Se-doped NiCoP mesoporous nanoflower hybrid (NCP–Se) was fabricated. Interestingly, the one-pot phospho-selenization reaction not only incorporates Se heteroatoms but also induces abundant lattice defects and P vacancies at the heterointerfaces of bimetallic phosphide (NCP). Furthermore, the CTAB micelle acts as a surfactant template, creating mesopores on the flakes of the NCP–Se nanoflower hybrid after pyrolysis at high temperatures. The optimum NCP–Se electrocatalyst exhibits enhanced OER performance in an alkaline electrolyte compared to NCP and the parent NCL. It exhibits the lowest overpotential values of 260, 320, and 346 mV to achieve 10, 50, and 100 mA cm−2 current densities, respectively, and demonstrates long-term durability. These outstanding results are attributed to the synergetic effects of Se doping and P vacancies, which can profoundly regulate the electronic structure of metal active sites, promote faster electron transfer, and enrich the density of reactive sites. Furthermore, the mesopores in NCP–Se not only noticeably improve conductivity but also efficiently restrict volume expansion throughout the electrochemical OER. The present research opens new avenues for the development of advanced OER electrocatalysts for water splitting.
2. Results and discussion
The NCP–Se/NF mesoporous nanoflower was synthesized through a two-step process, namely a solvothermal method followed by pyrolysis, as illustrated in Scheme 1. The complete fabrication process is explained in SI. Initially, the NCL supported on the NF substrate was prepared using the solvothermal method, following the proposed mechanism. In preparation for NCL/NF, CTAB acts as both a structure-directing agent and a surfactant template. Primarily, when CTAB is dissolved in an aqueous methanol solution, it forms various water-in-oil spherical micelles. Co, Ni and nitrate ions are condensed around the hydrophilic head groups of the CTAB micelles, which perform as a surfactant template. Under solvothermal conditions, hydroxyl ions are produced through a redox reaction. Then, hydroxyl ions interact with Ni2+ and Co2+ metal cations to generate hydroxide monomers, which rapidly nucleate to create basic particles.31 The metal hydroxide matrix forms a continuous network around micelles anchored to the chains of the NF surface to produce aggregation cores. By increasing the reaction time, these hydroxide nanoflakes aligned along the c-axis, and subsequently, a nanoflower structure of NCL formed on the NF surface. Next, one-pot phospho-selenisation was implemented to convert the NCL/NF into NCP–Se/NF. Initially, NaH2PO2 (P source, 20 times to NCL) decomposes into PH3 gas, which reacts with the NiCo precursor (NCL/NF) under high temperature. Se powder (4 times to NCL) converts into Se vapour and reacts with metal hydroxide to form NF-supported Se-doped bimetal phosphide (NCP–Se/NF). Additionally, for comparison purposes, two more catalysts were prepared (Fig. S1(a)) by varying the Se powder to parent NCL weight ratio to 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (NCP–Se3) and 5:1 (NCP–Se5). Moreover, a CTAB control material was synthesized, named as NCP–Se (WC)/NF. All the catalysts were evaluated for their OER activity, out of which NCP–Se/NF exhibited the best activity (Fig. S1(b), (c) and Fig. 5(a) and (b)). Hence, for further physicochemical characterization and other electrocatalytic activities, the NCL/NF, NCP/NF, and NCP–Se/NF catalysts are considered. Due to the large atomic size and higher electronegativity of Se in comparison to that of P, in the pyrolysis process, Se enters and disrupts the NCP lattice by partially substituting the P atom. Therefore, compared to the case for the pure NCP/NF, both P vacancies and Se doping are simultaneously introduced into the NCP–Se/NF catalyst. In addition, the surfactant templating agent, CTAB, creates highly ordered mesopore channels in the flakes of the NCP–Se/NF nanoflower material by utilizing its molecular micelle aggregates after pyrolysis at high temperatures.32
:1 (NCP–Se3) and 5:1 (NCP–Se5). Moreover, a CTAB control material was synthesized, named as NCP–Se (WC)/NF. All the catalysts were evaluated for their OER activity, out of which NCP–Se/NF exhibited the best activity (Fig. S1(b), (c) and Fig. 5(a) and (b)). Hence, for further physicochemical characterization and other electrocatalytic activities, the NCL/NF, NCP/NF, and NCP–Se/NF catalysts are considered. Due to the large atomic size and higher electronegativity of Se in comparison to that of P, in the pyrolysis process, Se enters and disrupts the NCP lattice by partially substituting the P atom. Therefore, compared to the case for the pure NCP/NF, both P vacancies and Se doping are simultaneously introduced into the NCP–Se/NF catalyst. In addition, the surfactant templating agent, CTAB, creates highly ordered mesopore channels in the flakes of the NCP–Se/NF nanoflower material by utilizing its molecular micelle aggregates after pyrolysis at high temperatures.32
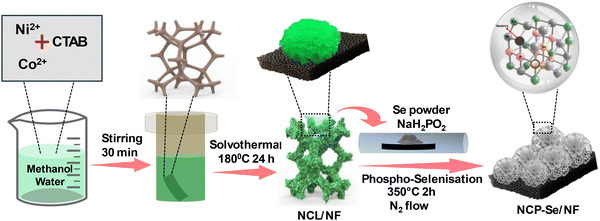 |
| | Scheme 1 Schematic of the synthesis of the NCP–Se/NF nanoflower. | |
After the completion of the substantial synthesis process, the working electrodes of NCL/NF and their derived materials over NF were first analyzed using the XRD technique to verify the successful formation of catalysts with a crystal structure and phase purity. As shown in Fig. 1(a), the NCL/NF spectrum primarily exhibits three characteristic signals at 2θ values of 44.5°, 51.8°, and 76.4°, indexed to the (111), (200), and (220) planes of the NF (JCPDS#04-0850), respectively. Moreover, the NCL/NF produces many more peaks centred at 10.9°, 22.2°, 33.4°, 34.3°, 38.3°, 59.8° and 61.2°, corresponding to the (003), (006), (101), (012), (015), (110) and (113) basal planes of NCL (JCPDS# 00-38-0715), respectively, with the rhombohedral phase.31 The interlayer spacing of (d003) is found to be 0.78 nm, in accordance with CO32− intercalation. This result demonstrates the successful growth of NCL on the NF substrate through the solvothermal process. The strong intensity of the substrate peaks suppressed the intensity of all desired materials’ peaks. After phosphidation, the diffraction peaks of NCP/NF are well matched with the NiCoP phase (JCPDS#00-71-2336) and positioned at 40.9°, 44.7°, 47.5°, 54.2°, 54.8°, 66.7°, 73.1°, and 75.15°, credited to the (111), (201), (210), (300), (002), (310), (311) and (400) crystal planes, respectively, as represented in Fig. 1(b).30 The absence of parent diffraction peaks indicates complete conversion of the NCL precursor to NCP on the NF surface. Meanwhile, some additional diffraction peaks were observed in the XRD spectrum of NCP–Se/NF (Fig. 1(b)) at 33.0°, 44.5°, 50.2°, and 59.7°, 61.2°, 69.3° and 70.7°, matching the (101), (102), (110), (103), (201), (202) and (004) crystal planes, respectively, in accordance with the (NiCo)Se2 phase (JCPDS #01-070-2851).33 A small intense diffraction at 49.0° for both the NCP/NF and NCP–Se/NF materials was due to the Ni5P4 phase.34 The XRD outcomes suggest the successful incorporation of Se in the lattice of bimetal phosphide in the NCP–Se/NF catalyst. Furthermore, to determine the functional group of the as-prepared catalyst, FTIR analysis was performed. Fig. 1(c) displays the FTIR spectra of the NCL, NCP and NCP–Se catalysts. In the case of NCL, a broad band at 3200–3600 cm−1 is allocated, corresponding to the O–H stretching of the metal hydroxide layer, whereas the 1625 cm−1 band was assigned to the H–O–H deformation mode. The asymmetric and symmetric stretching vibration bands of intercalated carbonate ions were observed at 1386–1450 cm−1 and 1300 cm−1, respectively. Similarly, the peak at 1050–1150 cm−1 was assigned to the C–O stretching. The meta-oxygen vibration band was observed between 500 and 800 cm−1. In particular, the Ni/Co–O and Ni–O–Co stretching bands were observed at 527 and 645 cm−1, respectively.35 After pyrolysis, the less intense oxygenated functional group vibrational bands were observed, as represented in Fig. 1(c). Specifically, the –OH stretching intensity significantly decreased, confirming the successful transformation of NCL to NCP and the NCP–Se-derived nanohybrid catalyst. The presence of the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (990–1227 cm−1) and P–O bonds (900 cm−1) in the material was due to the surface oxidation of bimetal phosphide.36 Ni/Co–P and Ni/Co–Se vibrational bands were detected at 544 and 711 cm−1 for both the NCP and NCP–Se-derived nanohybrid catalysts, respectively.37,38 These FTIR outcomes demonstrated the successful incorporation of Se anion into the NCP lattice and the formation of the NCP–Se catalyst. There was strong bonding between Ni/Co and heteroatoms (P and Se), which exposes more active sites for water electrolysis and enhances the intrinsic catalytic activity by reducing the energy barrier. The N2 adsorption/desorption technique was adopted to evaluate the surface area and porosity of the as-prepared catalyst. Fig. 1(d) and Fig. S2 show the N2 isotherms of the NCL with NCP–Se and NCP with NCP–Se(WC) materials, respectively. It is clearly observed that four materials display characteristics of a type-IV isotherm with H3 hysteresis loops in the relative pressure range of 0–1.0, validating their mesoporous nature.35 The BET surface areas of the NCL, NCP, and NCP–Se nanoflower catalysts were found to be 15.5, 22.7, and 26.7 m2 g−1, respectively. These outcomes reveal that, after phosphidation, the NCP possesses a higher surface area than the parent NCL due to the mesoporous structure. Moreover, the highest specific surface area was observed for the NCP–Se material compared to those for the NCP and NCL materials. In simultaneous phospho-selenisation treatment, the incorporation of Se induces P vacancies or defect sites, causing the lattice expansion of NCP and creating more mesopores, which lead to an increase in the surface area. In addition, the pore diameters were calculated using the Barrett–Joyner–Halenda (BJH) method, and the corresponding results are presented in the inserted image of Fig. 1(d) and Fig. S2. NCL, NCP and NCP–Se exhibited characteristic BJH pore sizes of 19.9, 20.2, and 23.7 nm and pore volumes of 0.010, 0.018, and 0.020 cm3, respectively. Additionally, BET analysis was performed on the CTAB control catalyst, i.e., NCP–Se (WC), and the results revealed a very small surface area of 11.4 m2 g−1 and a pore diameter of 5.7 nm (Fig. S2(b)). This result indicates that the presence of CTAB provides a nanoflower morphology with a large surface area, as well as a more ordered mesoporous structure, to the NCP–Se material. The CTAB micelles create a void in the flakes of the NCP–Se nanoflower material; consequently, the mesoporous channel with a large pore diameter and pore volume is developed after the removal of the CTAB template during pyrolysis, which is in good agreement with the TEM and FESEM micrographs.32 This high specific surface and distinct distribution of mesopores in the flakes of the NCP–Se nanoflower structure are beneficial for allowing effective mass/charge transfer and enhancing the communication between the electrolyte and electrode, thereby providing more exposed active sites for improving the OER.
O (990–1227 cm−1) and P–O bonds (900 cm−1) in the material was due to the surface oxidation of bimetal phosphide.36 Ni/Co–P and Ni/Co–Se vibrational bands were detected at 544 and 711 cm−1 for both the NCP and NCP–Se-derived nanohybrid catalysts, respectively.37,38 These FTIR outcomes demonstrated the successful incorporation of Se anion into the NCP lattice and the formation of the NCP–Se catalyst. There was strong bonding between Ni/Co and heteroatoms (P and Se), which exposes more active sites for water electrolysis and enhances the intrinsic catalytic activity by reducing the energy barrier. The N2 adsorption/desorption technique was adopted to evaluate the surface area and porosity of the as-prepared catalyst. Fig. 1(d) and Fig. S2 show the N2 isotherms of the NCL with NCP–Se and NCP with NCP–Se(WC) materials, respectively. It is clearly observed that four materials display characteristics of a type-IV isotherm with H3 hysteresis loops in the relative pressure range of 0–1.0, validating their mesoporous nature.35 The BET surface areas of the NCL, NCP, and NCP–Se nanoflower catalysts were found to be 15.5, 22.7, and 26.7 m2 g−1, respectively. These outcomes reveal that, after phosphidation, the NCP possesses a higher surface area than the parent NCL due to the mesoporous structure. Moreover, the highest specific surface area was observed for the NCP–Se material compared to those for the NCP and NCL materials. In simultaneous phospho-selenisation treatment, the incorporation of Se induces P vacancies or defect sites, causing the lattice expansion of NCP and creating more mesopores, which lead to an increase in the surface area. In addition, the pore diameters were calculated using the Barrett–Joyner–Halenda (BJH) method, and the corresponding results are presented in the inserted image of Fig. 1(d) and Fig. S2. NCL, NCP and NCP–Se exhibited characteristic BJH pore sizes of 19.9, 20.2, and 23.7 nm and pore volumes of 0.010, 0.018, and 0.020 cm3, respectively. Additionally, BET analysis was performed on the CTAB control catalyst, i.e., NCP–Se (WC), and the results revealed a very small surface area of 11.4 m2 g−1 and a pore diameter of 5.7 nm (Fig. S2(b)). This result indicates that the presence of CTAB provides a nanoflower morphology with a large surface area, as well as a more ordered mesoporous structure, to the NCP–Se material. The CTAB micelles create a void in the flakes of the NCP–Se nanoflower material; consequently, the mesoporous channel with a large pore diameter and pore volume is developed after the removal of the CTAB template during pyrolysis, which is in good agreement with the TEM and FESEM micrographs.32 This high specific surface and distinct distribution of mesopores in the flakes of the NCP–Se nanoflower structure are beneficial for allowing effective mass/charge transfer and enhancing the communication between the electrolyte and electrode, thereby providing more exposed active sites for improving the OER.
 |
| | Fig. 1 XRD patterns of the (a) NCL/NF, (b) NCP/NF, and NCP–Se/NF catalysts. (c) FT-IR spectra of the NCL, NCP, and NCP–Se catalysts. (d) N2 adsorption–desorption isotherms (pore-size distribution curve was inserted) of the NCL and the NCP–Se material. | |
The external morphology and internal nanostructure of the as-prepared catalyst were characterized by FESEM and TEM/HRTEM analyses. The micrographs of the bare NF and NCL/NF (Fig. S3(a) and (b)) suggest that NCL has been fruitfully developed on the NF substrate under the solvothermal condition. The different magnification FESEM images of NCL/NF in Fig. 2(a) and (b) indicate that NCL possesses a nanoflower morphology composed of a large number of self-assembled ultrathin nanoflakes with smooth surfaces. Fig. 3(a) (enlarged image inserted) displays the low- and high-magnification TEM images of the pristine NCL, illustrating its flower-like hierarchical nanostructure, which is consistent with the FESEM observation.39 These arbitrarily aligned nanoflakes were interwoven with each other and perpendicular to the CTAB-micelle, forming 3D hierarchical nanoflower structures. From Fig. 2(a) and (c), the diameter of the nanoflower and the thickness of the nanoflakes are found to be 5 µm and 75 nm, respectively. After one-pot phospho-selenisation treatment, the surface of the derived NCP–Se/NF is rough, but it still almost retains its size and nanoflower morphology (Fig. 2(d)). It is interesting that the solid flakes of the nanoflower architecture created an internal porous structure, as represented in Fig. 2(e). In addition, the TEM image of the Se-doped bimetal phosphide in Fig. 3(b) and (c) reveals that many pores are distributed uniformly throughout the catalyst, which was also evidenced by BET analysis. The development of this internal porous framework facilitates efficient penetration of electrolyte anions, thereby promoting the electrochemical reaction at a reduced applied potential. The HRTEM image of the NCP–Se catalyst in Fig. 3(d) demonstrates that the interplanar spacings of 0.22 nm and 0.20 nm can be ascribed to the (111) and (102) crystal planes of NCP and (NiCo)2Se, respectively. Moreover, the yellow circles in Fig. 3(e) indicate the plentiful regional lattice defects at the grain boundaries of bimetal phosphide in the NCP–Se material, which are mainly attributed to the P vacancies (proved by XPS and EPR analyses). This P-vacancy optimized electronic structure around the Ni/Co sites leads to an increase in the intrinsic catalytic activity. Furthermore, the selected area electron diffraction (SAED) pattern of NCP–Se, as portrayed in Fig. 3(f), presents a series of concentric diffraction rings, which are attributed to the crystal planes of NCP [(111), (210)] and (NiCo)Se2 [(101), (102)], respectively. This pattern is well consistent with the XRD results, confirming the polycrystalline nature of the NCP–Se nanoflower structure. The elemental confirmation was characterized by energy-dispersive X-ray spectroscopy (EDX) and colour mapping analysis, as shown in Fig. S3(c) and Fig. 3(g), respectively. This result demonstrates that elements Ni, Co, P, and Se are uniformly distributed throughout the catalyst.
 |
| | Fig. 2 FESEM images of the (a)–(c) NCL/NF and (d) and (e) NCP–Se/NF catalysts at different magnifications. | |
 |
| | Fig. 3 TEM images of the (a) NCL (a high magnification image was inserted) and (b) and (c) NCP–Se catalysts. (d) and (e) HRTEM image, (f) SAED pattern, and (g) colour mapping images of the NCP–Se material. | |
The chemical compositions and elemental oxidation states of the as-prepared catalysts (NCL, NCP and NCP–Se) are analyzed by the XPS technique. As demonstrated in Fig. S4(e), the wide-scan survey spectra exhibit the most prominent signals of C, O, Ni, and Co, which are present in the NCL electrocatalyst. Fig. 4(e) represents the survey spectra of both the NCP and NCP–Se samples, revealing the presence of Ni, Co, and P elements, whereas Se is utterly detected in NCP–Se, confirming the successful introduction of Se into the structure of NCP–Se. Furthermore, no additional signal was detected in the wide scan, indicating the high purity of the specimen, as confirmed by the XRD results. As observed in Fig. S49(a), the C 1s of NCL displays three distinctive signals at 284.8 and 288.1 eV, corresponding to the adventitious sp2 hybridized C–C/CC and CO bonds, respectively.21,40 Similarly, the O 1s spectrum is fitted into a single peak positioned at 530.8 eV, indicating the lattice oxygen and metal oxygen species (Fig. S4(b)).35,41 For the NCL sample, the Ni 2p spectrum displays two distinctive peaks at 855.1 and 873.1 eV, which are associated with the 2p3/2 and 2p1/2 electron orbital signals, respectively, with an energy gap of 18 eV, indicating the presence of Ni in +2 and +3 oxidation states (Fig. S4(c)). Thereafter, the two characteristic signals were further fitted into several additional peaks, which were discussed. The signals with binding energies located at 855.1 eV are related to Ni2+, whereas the peaks at 856.3 and 873.1 eV correspond to the Ni3+ species, respectively. Moreover, Ni2+ and Ni3+ are accompanied by three satellite peaks positioned at 861.2, 866.1, and 879.2 eV.42–46 The Co 2p spectrum also exhibits two prominent signals at 780.4 and 796.3 eV, corresponding to the 2p3/2 and 2p1/2 electron orbital signals, respectively, with a spin-energy gap of 15.9 eV. In the Co 2p spectrum, the signals at 782.5 and 796.4 eV are assigned to the +2 oxidation state of Co. The peak positioned at 780.4 corresponds to the Co3+ states, and the remaining two peaks at 786.5 and 802.1 eV are linked to the satellite signals (Fig. S4(d)).44–46 These observations suggest that the Ni2+ and Co2+ ions are partially oxidized to Ni3+ and Co3+ during the course of the solvothermal reaction, respectively. However, after phosphidation, the Ni 2p spectrum of NCP displays two new signals at 852.6 and 869.1 eV, corresponding to the Ni–P bonds.47 For the Co 2p spectrum of NCP, the signals located at 781 and 797 eV are associated with the Co–P bond, along with the appearance of Co2+ and satellite peaks. The formation of metal–phosphide bonds suggests good interaction among the Ni and Co atoms with that of the P atoms. In comparison to NCP, the binding energy of Ni–P in NCP–Se exhibits a positive shift towards a higher region, which can be attributed to the stronger Ni–Se bond compared to the Ni–P bond and also explains the electronic interaction between the NCP and Se elements (Fig. 4(a)). Similarly, the binding energy of the Co–P bond in NCP–Se exhibits a positive shift towards higher binding energy compared to NCP, which again explains the stronger Co–Se bond compared to the Co–P bond (Fig. 4(b)). Further, these outcomes suggest that NCP participates in the electrochemical reaction.21 In addition to Ni and Co, the P 2p spectrum of NCP exhibits a signal at 133.8 eV, indicating the existence of P–O bonds.46 After the selenization process, the P 2p spectrum of NCP–Se exhibits two additional signals at 130.1 and 137.4 eV, corresponding to the appearance of the P–O bond (Fig. 4(c)). The XPS signal with binding energies at 130.1 and 134.2 eV is associated with the M–P and P–O bonds because of surface oxidation. In contrast, the signal at 137.4 eV corresponds to the P–Se bonds.16,43 It has been assumed that Se doping may have weakened the strength of the M–P bonds. In the Se 3d spectrum of NCP–Se, the signals around 54.2 and 55.1 eV correspond to the 3d5/2 and 3d3/2 of selenium, respectively (Fig. 4(d)).21 The information obtained from the XPS analysis suggests that the Se atoms were introduced successfully, and the doping of Se results in the generation of P vacancies. Moreover, the quantitative analysis of the Ni3+/Ni2+ and Co3+/Co2 ratios in the deconvoluted Ni 2p and Co 2p spectra of NCL, NCP, and NCP–Se materials, respectively, is presented in Table S1. This result reveals that the NCP–Se exhibits higher concentrations of Ni3+/Ni2+ and Co3+/Co2+ compared to NCP and NCL, indicating electron transfer from the metal centre to the surrounding anions. This valence-state evolution is also consistent with the positive binding energy changes observed in the Ni 2p and Co 2p spectra of NCP–Se, confirming strong metal–Se interactions and electronic-structure modulation. These high valence states of the metal (Ni3+ and Co3+) in NCP–Se facilitate OH− adsorption, promote the formation of M–OOH intermediates, and lower the activation energy, subsequently improving the OER activity.
 |
| | Fig. 4 (a) Ni 2p, (b) Co 2p, and (c) P 2p spectra of the NCP and NCP–Se catalysts. (d) Se 3d spectrum of NCP–Se. (e) Survey scan of NCP and NCP–Se XPS spectra. | |
To verify the existence of P vacancies again, the electron paramagnetic resonance (EPR) of NCP, NCP–Se3, NCP–Se, and NCP–Se5 is characterized. As illustrated in Fig. S5, NCP–Se displays the highest-intensity EPR signal with a g value of 2.003 compared to the weak signal obtained from NCP having the same g value, corresponding to the existence of unpaired electrons that have been trapped by the P vacancies.19 This implies that during the pyrolysis-based phospho-selenisation process, some of the Se atoms may have entered and distorted the lattice of NCP by partially substituting the P atoms because of their larger electronegativity and atomic radius. However, the EPR signal intensity increases when the Se-doping amount improves from NCP–Se3 to NCP–Se and then decreases again for the NCP–Se5 material. This outcome demonstrates that employing the optimum amount of Se doping results in a higher P vacancy in NCP–Se (Se powder to NCL weight ratio of 4:1) compared to the NCP–Se3 and NCP–Se5 materials. EPR results support the XPS analysis, and the highest concentration of P vacancy plays a crucial role in creating additional active sites in NCP–Se, which in turn significantly boosts its electrocatalytic performance.
3. Electrochemical activity towards the OER
The NCL and its derived nanohybrid electrocatalysts (NCP, NCP–Se) in situ grown on an NF substrate were evaluated for their electrocatalytic OER activities in an alkaline medium using a three-electrode system. The 1 cm2 effective surface area of the modified NF was used as the working electrode. First, all the working electrodes were activated through CV measurement with 50 mV s−1 for 20 cycles in the potential range of 1.01 to 1.71 V vs. RHE. The iR-compensated LSV curve was measured at 2 mV s−1. The polarization outcome given in Fig. 5(a) shows that Se-doped bimetal phosphide (NCP–Se/NF) displayed the highest OER activity among all the catalysts. A broad oxidation peak is observed at the potential region of 1.3–1.4 V vs. RHE in Fig. 5(a), attributed to the Ni(II)–Ni(III) redox transformation.48 The overpotential values at different current densities of all as-prepared electrodes are presented using a bar diagram in Fig. 5(b). NCP–Se/NF requires the lowest overpotential values of 260, 320 and 346 mV to achieve 10, 50, and 100 mA cm−2 current densities, respectively. Meanwhile, NCP/NF (307, 350, 367 mV) and NCL (332, 381, 401 mV) require relatively high overpotential values for the same current densities. The derived bimetal phosphide (NCP/NF) exhibits metallic properties, leading to superconductivity, and it provides abundant reactive sites for OER, which support better activity than the parent NCL/NF. Furthermore, the enhanced OER activity over that of NCP–Se/NF might be due to the presence of simultaneous P vacancies and electron-rich Se2− (as evident from XPS), which accelerates the desorption of the O2 molecules by reducing the kinetic barrier.21,45 The formation of mesopores in the flakes of the nanoflower architecture facilitates effective electron transport. Furthermore, the OER results of NCP–Se/NF were compared to those of the benchmarking commercial RuO2 material. As depicted in Fig. 5(a) and (b), RuO2 required 298 mV to attain the 10 mA cm−2 current density, which was also higher than that required by the present Se-substituted NCP system. Moreover, to gain a deeper understanding of the kinetics of the OER activity, Tafel plots are analyzed, extracted from iR-corrected polarisation curve conversion, and presented in Fig. 5(c). The Tafel slope can be obtained by fitting the linear region of the Tafel curve according to the Tafel formula (eqn (S3)). NCP–Se/NF delivered the lowest Tafel slope value of 28.17 mV dec−1, which signifies that it exhibits quicker charge-transfer kinetics at the electrode–electrolyte interface in comparison to RuO2 (29.4 mV dec−1), NCP/NF (33 mV dec−1), and NCL/NF (50.64 mV dec−1). The above-mentioned comparison of performance further demonstrates the effective enhancement of the OER activity by the modulation of the charge density of Ni and Co through Se doping, which causes a regional defect near the P site in the NCP–Se/NF material. Furthermore, certain advantages in terms of the OER performance of NCP–Se/NF are comparable to those of the recently reported materials (Table S2). Fig. 5(d) encloses the EIS plots and their corresponding equivalent circuit diagram (inserted). The charge-transfer resistance of the catalyst can be determined by simulating the equivalent circuit diagram through the ZView software. In a circuit diagram, Rs is the solution resistance, and Rct is the charge-transfer resistance of the interphase between the electrode and electrolytic solution. NF supported all electrocatalysts, which exhibited low charge-transfer resistance, as represented by the Nyquist plot in Fig. 5(d). In particular, the smallest Nyquist semicircle of NCP–Se/NF reveals its lower charge-transfer resistance of Rct = 0.78 Ω in comparison to those of NCP/NF (Rct = 1.55 Ω), RuO2 (Rct = 2.85 Ω), NCL/NF (Rct = 2.92 Ω), and bare NF (Rct = 8 Ω). This result indicates that after the phosphidation of NCL/NF, the charge-transfer resistance decreased with increasing conductivity of the NF-supported bimetal phosphide mesoporous nanosheet, which is coincident with the value of the Tafel slope. Se doping and P vacancy improve the electron-transfer rates and OER kinetics.14,19
 |
| | Fig. 5 (a) LSV, (b) overpotential, (c) Tafel plot, and (d) EIS of the as-synthesized NF, NCL/NF, NCP/NF, NCP–Se/NF and RuO2/NF catalysts. | |
The electrochemically active surface area (ECSA) is analyzed to determine the number of real active sites available for the OER, which can be calculated by performing cyclic voltammetry (CV) in the non-Faraday region (Fig. S6). The electrical double-layer capacitance (Cdl) is the slope value of the linear fit difference of the cathodic and anodic current density from the measured CV cycles at various scan rates. The Cdl values of NCL/NF, NCP/NF and NCP–Se/NF are found to be 4.51, 5.59, and 9.24 µF cm−2, respectively, as displayed in Fig. 6(a). The Cdl is directly proportional to the ECSA value, and it is calculated by eqn (S5), as represented in SI. As depicted in Fig. 6(b), the determined ECSA value of NCP–Se/NF (0.23 cm2) is 1.6 and 2 times larger than those of NCP/NF (0.14 cm2) and NCL/NF (0.11 cm2). These outcomes demonstrate that the higher electronic conductivity of the derived bimetal phosphide, NCP/NF, compared to that of the parent hydroxides, NCL/NF, results in a larger ECSA value. More specifically, the significantly increased ECSA value of the NCP–Se/NF catalyst indicates the larger active surface area, revealing a greater number of catalytically active sites on the electrode surface, which is mainly ascribed to the one-step introduction of P vacancy by Se doping in bimetal phosphide and the formation of many mesopores on the large lateral sheet of the NCP–Se/NF nanoflower structure.49 To again explore the intrinsic OER activity of a single site on the catalyst, the specific activity was calculated. Upon normalization of the geometrical LSV polarisation curve by the resulting ECSA value, the particular activity was obtained (eqn (S6)). As shown in Fig. 6(b), the specific activity is calculated by normalizing the ECSA current density at a potential of 1.56 V vs. RHE. Interestingly, the NCP–Se/NF electrode exhibits the highest specific activity of 333.73 mA cm−2, which is approximately 1.8 and 2.7 times higher than those of NCP/NF and NCL/NF, respectively. Moreover, the surface-metal active sites were determined by measuring the reduction surface area of the catalyst (Fig. S7). As portrayed in Fig. 6(c), the estimated value indicates that NCP–Se/NF (9.9875 × 1017) exhibits the most exposed metal active sites compared to NCP/NF (8.7890 × 1017) and parent NCL/NF (6.7400 × 1017).45,50,51 The electrochemical examination of the surface area and active sites is evidenced by the BET measurement, which confirms that NCP–Se/NF has a greater number of active sites, which are beneficial for effective O2 evolution at low applied potentials. Furthermore, the intrinsic catalytic performance towards the OER was investigated based on the turnover frequency (TOF), which signifies the number of O2 molecules generated per second per active site.52 TOF is calculated through the measured reduction surface area (Fig. S7) at a potential of 1.66 V vs. RHE. As displayed in Fig. 6(c), the TOF values of NCL/NF, NCP/NF and NCP–Se/NF are 0.08, 0.10 and 0.12 s−1, respectively. Therefore, by comparing the TOF results, it can be concluded that the Se-doped induced P-vacancy mesoporous bimetal phosphide nanoflower catalyst delivers a larger specific activity, as well as greater intrinsic activity, with a very high concentration of O2 gas per unit active site per second, compared to the other two remaining catalysts.51 Besides, the multi-step current CP analysis of NCP–Se/NF in Fig. 6(d) was performed using 1 M KOH electrolyte for measuring the stability in terms of the potential response upon a rapid change in the current density. The stability was measured by increasing the current density from 10 to 50 mA cm−2, with an increment of 10 mA cm−2, followed by a further decrease from 50 to 10 mA cm−2, with a 1 h hold at each step. At every step of the current density, the potential remains constant and instantly levels off at the next current density value. Similar phenomena were observed for the downward steps of the current density. This result indicates the superior conductivity, mass-transport properties, and mechanical robustness of the NCP–Se/NF catalyst.49
 |
| | Fig. 6 (a) Cdl, (b) ESCA and specific activity and (c) surface active sites and TOF value of the NCL/NF, NCP/NF and NCP–Se/NF catalysts. (d) Multi-step current CP plot of the NCP–Se/NF catalyst. | |
Further, the long-term stability of the NCP–Se/NF electrocatalyst towards the OER was verified by chronoamperometry (CA) at 1.65 V vs. RHE. The resultant CA plot, portrayed in Fig. 7(a), illustrates that the optimum catalyst, NCP–Se/NF, maintains 84.1% of its original activity even after 50 hours of exposure to a harsh anodic reaction, which strongly validates its excellent stability. A dynamic stability, i.e., accelerated durability (AD), study of NCP–Se/NF was also analyzed with 1000 constant CV cycles at a scan rate of 50 mV s−1. When the resulting backwards polarisation information curve was compared with the polarisation information before cycling (Fig. 7(b)), a moderate overpotential deviation was observed at 10, 50 and 100 mA cm−2, due to the accumulation of O2 gas on the electrode surface. In addition, the EIS analysis in Fig. 7(c) revealed the same with a minor rise in resistance after 1000 CV cycles. The post-dynamic LSV and EIS results indicated the long-term durability of the electrocatalyst towards the OER in the alkali medium. The CV analysis of the NCP–Se/NF catalyst in Fig. S7(d) shows a slight decrease in the onset potential after 1000 CV cycles, indicating improved desorption of O2 molecules due to the occurrence of Se-doping and P-vacancy. Its long-term stability and high current operation indicate superior charge transport, fast reaction kinetics, and facile gas removal, all of which are crucial for continuous and scalable oxygen production. Additionally, achieving a low overpotential at a high current density suggests a direct correlation with enhanced energy efficiency and marketable capability. In addition, the structural and morphological stabilities of NCP–Se/NF after prolonged electrocatalytic OER operation were evaluated using XRD, FESEM and HRTEM analyses. The XRD patterns of NCP–Se/NF before and after electrochemical analysis are nearly identical, with only a slight intensity reduction (Fig. S8(a)). The low- and high-magnification FESEM and TEM images in Fig. S8(b) and (c), respectively, confirm the retention of mesoporous nanoflower morphology of the NCP–Se/NF catalyst. Further, the post-OER colour mapping (Fig. S8(d)) and EDAX (Fig. S9(e)) study illustrate the presence of all the elements (Ni, Co, O, P and Se) and their uniform elemental distribution. The presence of oxygen indicates the in situ electrochemical transformation of the Ni/Co-based hydroxide/(oxy)hydroxide layer on the catalyst surface, which serves as the true active species for the OER.51 Furthermore, the post-OER XPS was performed to determine the chemical-state stability of the NCP–Se nanohybrid. As displayed in Fig. S9(a), a slight attenuation was observed for Ni 2p, which was consistent with the reduction of the Ni diffraction signal in the XRD outcomes. In the Ni 2p spectrum of NCP–Se after the prolonged OER experiment, distinctive signals corresponding to the Ni–P bond are still evident, suggesting the structural integrity of the NCP–Se catalyst (Fig. S9(a)). Moreover, the signals appearing at 855.5 and 873.3 with a spin splitting of 17.8 eV are consistent with the standard splitting value of NiOOH. This indicates that the generation of NiOOH during OER has been confirmed as the real active species catalyzing the reaction. Similarly, a reduction in the peak area for Co 2p was observed for the used NCP–Se nanohybrid (Fig. S9(b)). In the P 2p spectrum, the M–P, P–O and P–Se bonds are still present in the catalyst after the OER operation, which again indicates the stability of the nanohybrid (Fig. S9(c)). The existence of Se 3d signals in Fig. S9(d) verifies the presence of Se in the −2 oxidation state in the reused catalyst. At the end, all elements present in the NCP–Se nanohybrid display a negative shifting binding energy after the OER operation, indicating that the reconfiguration of the catalyst over a long time period under the reaction condition led to changes in the elemental content and valence states, which affected the local electron cloud distribution and was one of the reasons for the slight decrease in the electrocatalytic activity. This post-OER characterization result demonstrates the durable and stable nature of the mesoporous NCP–Se/NF nanoflower catalyst despite its exposure to stringent anodic polarisation.
 |
| | Fig. 7 (a) CA plot, (b) polarization curves and (c) EIS plot of NCP–Se/NF before and after 1000 CV cycles. | |
The proposed mechanism for the electrocatalytic OER on the NCP–Se/NF surface in an alkaline medium is presented in Scheme 2. The OER progresses through a four-electron transfer pathway involving O*, OH*, and OOH* intermediates (eqn (1)–(5)). “M” signifies the catalytically active sites in the NCP–Se/NF nanohybrid. In the OER, the adsorption process takes place in the first step. Still, the activity depends on the entire four-step process, as established in earlier literature. Usually, the third step, i.e., the transformation of oxide (M–O) to oxyhydroxide (M–OOH), is the rate-determining step (RDS), as more energy is required to complete the process.50,53 Here, Se doping and P vacancies help in reducing the activation energy of the RDS step, as it is highly polarisable and can markedly stabilize the high-oxidation state Ni–OOH compound, making Ni an active site for the whole OER process.
| | | M − OH + OH− → M − O + e− + H2O | (2) |
| | | M − O + OH− → M − OOH + e− | (3) |
| | | M − OOH + OH− → M − O2 + e− + H2O | (4) |
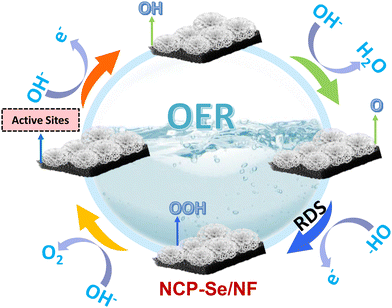 |
| | Scheme 2 Schematic of the proposed OER mechanism over the NCP–Se/NF nanohybrid. | |
The enhanced OER activity and prolong stability of NCP–Se/NF in comparison to those of the NCP/NF and NCL/NF catalyst can be associated with the co-operative effect of the simultaneous formation of P vacancies and Se doping, unique mesoporous flakes of the nanoflower architecture and synergistic impact of heteroatoms (P and Se) and metal components (Ni and Co), which can be summarized as follows: (i) first, the binder-free active electrocatalyst in situ grown on the 3D network-based NF increased the contact area between the catalytic sites and electrolyte while maintaining the current collector properties, resulting in efficient charge transfer. Moreover, the close electronic interaction between the conductive substrate and catalyst reveals excellent mechanical strength for long-term water electrolysis. (ii) Second, the derived bimetal phosphide exhibits better OER activity in comparison to the parent NCL/NF material because it possesses metallic character, leading to better electronic conductivity, faster mass transport, copious active centres and high structural stability. (iii) Then, after the one-pot phospho-selenization synthesis process, Se doping is implemented and P vacancies are created simultaneously in the lattice of bimetal phosphide, which induce electronic redistribution around the Ni/Co sites. This optimizes the d-band centre, subsequently providing a more exposed active site and enhancing the adsorption/desorption of the OER intermediates (*OH, *O, *OOH). The P vacancy hastens the rebuilding of active species throughout the OER process, increasing the intrinsic catalytic activity. At the same time, Se incorporation increases the electrical conductivity of bimetal phosphide by altering orbital overlap, which minimizes the diffusion path length and reduces the kinetic energy barrier. (iv) CTAB-integrated NCL/NF nanoflower under high temperature gives a mesoporous framework in the NCP–Se/NF catalyst. These unique mesoporous flakes with a nanoflower structure increase the specific surface area, facilitate electrolyte penetration, and accelerate the formation/liberation of O2 gas bubbles.
4. Conclusion
In summary, an active and robust NCP–Se/NF catalyst was successfully fabricated for efficient OER in an alkaline electrolyte. First, NCL was grown on an NF-conductive substrate through a solvothermal reaction. Then, it was used as a precursor for constructing the Se-doped NCP nanoflower architecture with high porosity. During pyrolysis, Se doping and P vacancy are simultaneously introduced in bimetallic phosphide to form an NCP–Se/NF hybrid through a one-pot phospho-selenization technique. The optimized NCP–Se/NF hybrid exhibited enhanced OER activity compared to NCP/NF and NCL/NF. It exhibited an overpotential of 260 mV to attain a current density of 10 mA cm−2, with the lowest Tafel slope of 28.17 mV dec−1 and the smallest Rct value (0.78 Ω). The calculated TOF value was approximately 0.12 s−1, which is nearly 1.5 times higher than that of the parent material. Moreover, NCP–Se/NF exhibited outstanding long-term stability for over 50 hours with negligible degradation under the OER conditions. The excellent performance of the NCP–Se/NF electrode is attributed to the synergistic effect of both Se doping and P vacancies, which modulates the electronic structure of bimetal phosphide, thereby accelerating electron/mass transfer and optimizing the adsorption/desorption of intermediates. Moreover, the formation of mesopores on the flakes of the NCP–Se/NF nanoflower permits the exposure of more active sites, facilitates ion diffusion and electrolyte penetration, and enhances reaction kinetics. This research involves a simple, cost-effective, and feasible preparation process, providing a new direction for regulating the electronic structure through doping and vacancy engineering to create an efficient bimetallic phosphide catalyst for the OER in an alkaline medium.
Conflicts of interest
The authors declare no conflicts of interest regarding the conduct of this study and the publication of its results.
Data availability
Additional data have been provided in the supplementary information (SI). Supplementary information: the fabrication methods of the electrocatalyst; physicochemical characterization technique; electrocatalytic measurement of the OER; calculation of the overpotential, Tafel slope, ESCA, specific activity and TOF; XRD plots of NCP–Se3 and NCP–Se5; LSV and overpotential plots of NCP–Se3, NCP–Se5 and NCP–Se(WC); N2 adsorption–desorption curves and pore-size distribution curves of NCP and NCP–Se(WC); FESEM images of the bare NF and NCL/NF and EDAX image of the NCP–Se/NF catalyst; C 1s, O 1s, Ni 2p, Co 2p, and survey scan XPS spectra of NCL; quantitative analysis ratios of Ni2+/Ni3+, Co2+/Co3+, Ni3+/Ni2+ and Co3+/Co2+ in the Ni 2p and Co 2p XPS spectra of NCL, NCP and NCP–Se nanohybrid presented in a table; EPR spectra of NCP, NCP–Se3, NCP–Se and NCP–Se5; cyclic voltammetry features of NCL/NF, NCP/NF, and NCP–Se/NF with different applied scan rates for obtaining the Cdl information; reduction area corresponding to the Ni3+ → Ni2+ conversion of NCL/NF, NCP/NF, NCP–Se/NF and the first and 1000th CV cycles of NCP–Se/NF; post-OER characterisation of XRD, FESEM, TEM, elemental colour mapping and EDAX of the NCP–Se catalyst; post-OER XPS analysis of NCP–Se (Ni 2p, Co 2p, P 2p and Se 3d spectra); and the electrocatalytic OER activities of NCP–Se/NF compared with those of similar types of catalysts reported in the table. See DOI: https://doi.org/10.1039/d5ma01125f.
Acknowledgements
The authors are very grateful to the Siksha ‘O’ Anusandhan (Deemed to be University) management for their support and encouragement in publishing this work. Upali Aparajita Mohanty is grateful to UGC for the award of a SJSGC fellowship (UGCES-22-GE-ORI-F-SJSGC-5637). Ritik Mohanty is thankful to CSIR New Delhi for awarding CSIR-SRF (File No. 09/0969 (18687)/2024-EMR-I).
References
- Y. Wang, D. Yan, S. El Hankari, Y. Zou and S. Wang, Adv. Sci., 2018, 5, 1800064 CrossRef PubMed.
- D. P. Sahoo, K. K. Das, S. Mansingh, S. Sultana and K. Parida, Coord. Chem. Rev., 2022, 469, 214666 Search PubMed.
- A. Karmakar, K. Karthick, S. S. Sankar, S. Kumaravel, R. Madhu and S. Kundu, J. Mater. Chem. A, 2021, 9, 1314–1352 RSC.
- Z. Cai, X. Bu, P. Wang, J. C. Ho, J. Yang and X. Wang, J. Mater. Chem. A, 2019, 7, 5069–5089 RSC.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 Search PubMed.
- S. Anantharaj, S. R. Ede, K. Karthick, S. Sam Sankar, K. Sangeetha, P. E. Karthik and S. Kundu, Energy Environ. Sci., 2018, 11, 744–771 RSC.
- T. M.-J. Choi, L. Wang, K. A. Stoerzinger, S.-Y. Chung, S. A. Chambers and Y. Du, Adv. Energy Mater., 2023, 13, 2300239 Search PubMed.
- Z. Lang, G.-L. Song, P. Wu and D. Zheng, Nano Res., 2022, 16, 2224 CrossRef.
- H. Tan, B. Tang, Y. Lu, Q. Ji, L. Lv, H. Duan, N. Li, Y. Wang, S. Feng, Z. Li, C. Wang, F. Hu, Z. Sun and W. Yan, Nat. Commun., 2022, 13, 2024 Search PubMed.
- M. Wang, X. Liu and X. Wu, Nano Energy, 2023, 114, 108681 Search PubMed.
- L. Wu, F. Zhang, S. Song, M. Ning, Q. Zhu, J. Zhou, G. Gao, Z. Chen, Q. Zhou, X. Xing, T. Tong, Y. Yao, J. Bao, L. Yu, S. Chen and Z. Ren, Adv. Mater., 2022, 34, 2201774 CrossRef CAS PubMed.
- A. Ray, S. Sultana, L. Paramanik and K. M. Parida, J. Mater. Chem. A, 2020, 8, 19196–19245 RSC.
- L.-A. Stern, L. Feng, F. Song and X. Hu, Energy Environ. Sci., 2015, 8, 2347 RSC.
- X. Wang, X. Liu, S. Wu, K. Liu, X. Meng, B. Li, J. Lai, L. Wang and S. Feng, Nano Energy, 2023, 109, 108292 Search PubMed.
- X. Wei, Y. Jiao, X. Zou, Y. Guo, W. Li and T. Ai, Inorg. Chem. Front., 2025, 12, 2678–2690 Search PubMed.
- Z. Zhan, P. Gao, T. Lei, S. Zhang and Y. Du, ACS Sustainable Chem. Eng., 2023, 11, 4980–4989 CrossRef CAS.
- L. Chen, H. Chen, L. Wu, G. Li, K. Tao and L. Han, ACS Appl. Mater. Interfaces, 2024, 16, 8751–8762 Search PubMed.
- M. Jeong, S. Park, T. Kwon, M. Kwon, S. Yuk, S. Kim, C. Yeon, C.-W. Lee and D. Lee, ACS Appl. Mater. Interfaces, 2024, 16, 34798–34808 CrossRef CAS PubMed.
- K. Wang, X. Liu, Q. Yu, X. Wang, J. Zhu, Y. Li, J. Chi, H. Lin and L. Wang, Small, 2024, 20, 2308613 CrossRef CAS PubMed.
- B. Zhang, S.-l Xu, J. Li, X.-X. Zhao, R. Zhao, M. Zhang, D. Zhao and L. Miao, CrystEngComm, 2025, 27, 5485–5500 RSC.
- S. Cui, Y. Tang, W. Cui, G. Li, X. Xiao and L. Han, ACS Appl. Mater. Interfaces, 2024, 16, 42230–42241 CrossRef CAS PubMed.
- A. Qiu, D. Qiu, X. Ye, W. Chen and Y. Yu, Int. J. Hydrogen Energy, 2025, 105, 545–555 CrossRef CAS.
- Q. T. T. Le, T. T. Nguyen, K. D. Tran, Q. P. Ngo, D. T. Tran, N. H. Kim and J. H. Lee, Chem. Eng. J., 2025, 166378 CrossRef CAS.
- M. Zhang, H. Xu, H. Yang, X. Shang, M. Yuan, Y. Fu, Y. Xiao, S. Wang, X. Wang, B. Jia and S. Li, Small, 2025, 2504837 Search PubMed.
- R. He, J. Li and L. Feng, Catal. Commun., 2022, 163 Search PubMed.
- Y. Yan, J. Lin, J. Cao, S. Guo, X. Zheng, J. Feng and J. Qi, J. Mater. Chem. A, 2019, 7, 24486–24492 Search PubMed.
- J. Su, N. Jiang, Y. Wang, B. Jiang, X. Wang and H. Song, Energy, 2025, 318, 134905 CrossRef CAS.
- L. Zhang, Y. T. Ma, J. J. Duan, Y. Q. Yao, J. J. Feng and A. J. Wang, J. Colloid Interface Sci., 2022, 611, 205–214 Search PubMed.
- M. Liu, Q. He, S. Huang, W. Zou, J. Cong, X. Xiao, P. Li, J. Cai and L. Hou, ACS Appl. Mater. Interfaces, 2021, 13, 9932–9941 Search PubMed.
- H. Liang, A. N. Gandi, D. H. Anjum, X. Wang, U. Schwingenschlögl and H. N. Alshareef, Nano Lett., 2016, 16, 7718–7725 Search PubMed.
- K. Z. Cai, Y. Q. Huo, Y. Teng, X. Liu and H. Y. Chen, J. Solid State Chem., 2021, 299, 122173 Search PubMed.
- D. V. Wellia, A. Syafawi and Y. E. Putri, RSC Adv., 2023, 13, 29645–29656 RSC.
- X. Xu, H. Liang, F. Ming, Z. Qi, Y. Xie and Z. Wang, ACS Catal., 2017, 7, 6394–6399 CrossRef CAS.
- S. Khan, T. Ali, X. Wang, W. Iqbal, T. Bashir, W. Chao, H. Sun, H. Lu, C. Yan and R. M. Irfan, Chem. Eng. Sci., 2022, 247, 117020 Search PubMed.
- D. P. Sahoo, K. K. Das, S. Patnaik and K. M. Parida, Inorg. Chem. Front., 2020, 7, 3695–3717 Search PubMed.
- R. Zhang, P. A. Russo, M. Feist, P. Amsalem, N. Koch and N. Pinna, ACS Appl. Mater. Interfaces, 2017, 9, 14013–14022 CrossRef CAS PubMed.
- Y. Pei, L. Huang, L. Han, H. Zhang, L. Dong, Q. Jia and S. Zhang, Green Energy Environ., 2022, 7, 467–476 CrossRef CAS.
- N. Ali, S. Ahmad, A. Khan, S. Khan, M. Bilal, S. Ud Din, N. Ali, H. M. Iqbal and H. Khan, Chem. – Asian J., 2020, 15, 2660–2673 Search PubMed.
- M. Li, L. Fang, H. Zhou, F. Wu, Y. Lu, H. Luo, Y. Zhang and B. Hu, Appl. Surf. Sci., 2019, 495, 143554 CrossRef CAS.
- K. K. Das, U. A. Mohanty, R. Mohanty, P. P. Sarangi, D. P. Sahoo and K. M. Parida, ACS Appl. Energy Mater., 2024, 7, 6360–6375 Search PubMed.
- K. K. Das, S. Mansingh, R. Mohanty, D. P. Sahoo, N. Priyadarshini and K. M. Parida, J. Phys. Chem. C, 2022, 127, 22–40 Search PubMed.
- C. Lv, Y. Jing, Z. Lu, X. Wang, K. Zhu, J. Yin, Y. Xie and K. Ye, Chem. Eng. J., 2025, 510, 161709 CrossRef CAS.
- P. Yue, M. Wang, J. Chen, T. Song, Y. Zhao and X. Wu, Adv. Sustainable Sys., 2025, 9, 2400539 CrossRef CAS.
- W. Sun, Z. Liu, D. Liu, B. Zhang, Y. Li, C. Wang, X. Liu, X. Wang, X. Z. Song and Z. Tan, J. Mater. Chem. A, 2025, 13, 22792–22803 Search PubMed.
- R. Madhu, J. Muthukumar, P. Arunachalam, P. Gudlur and S. Kundu, J. Phys. Chem. C, 2024, 128, 12891–12902 CrossRef CAS.
- H. Zhao, M. Liu, X. Du and X. Zhang, Inorg. Chem. Front., 2025, 12, 2103–2112 Search PubMed.
- M. Shi, F. Sultana, X. Qin, P. Zhang, K. Qian, T. Wei, Y. Duan, T. Li, J. Bai and R. Li, Appl. Catal., B, 2025, 371, 125210 CrossRef CAS.
- L. Zhang, Z. Hu, H. Li, Q. Ren, Y. Qiu, J. Qu and S. Hu, Chem. Phys. Chem., 2021, 22, 1785–1791 Search PubMed.
- D. Li, Y. Xing, R. Yang, T. Wen, D. Jiang, W. Shi and S. Yuan, ACS Appl. Mater. Interfaces, 2020, 12, 29253–29263 Search PubMed.
- A. De, R. Madhu, K. Bera, H. N. Dhandapani, S. Nagappan, S. S. Roy and S. Kundu, J. Mater. Chem. A, 2023, 11, 25055–25071 Search PubMed.
- A. Karmakar, A. V. Krishnan, R. Jayan, R. Madhu, M. M. Islam and S. Kundu, J. Mater. Chem. A, 2023, 11, 10684–10698 RSC.
- S. Nagappan, R. Jayan, N. Rajagopal, A. V. Krishnan, M. M. Islam and S. Kundu, Small, 2024, 20, 2403908 CrossRef CAS PubMed.
- G. Rajeshkhanna, S. Kandula, K. R. Shrestha, N. H. Kim and J. H. Lee, Small, 2018, 14, 1803638 CrossRef PubMed.
Footnote |
| † Equal contribution. |
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Kulamani
Parida
and
Kulamani
Parida








