DOI:
10.1039/D5MA01046B
(Paper)
Mater. Adv., 2026,
7, 2225-2240
Nature-inspired α-MnMoO4 nanocubes from Arachis hypogaea for next-generation wastewater treatment and organic pollutant catalysis
Received
11th September 2025
, Accepted 4th January 2026
First published on 23rd January 2026
Abstract
In this study, bimetallic α-MnMoO4 nanoparticles (NPs) were successfully synthesized via a one-step solution combustion method using Arachis hypogaea (peanut) seed powder as a green fuel. This eco-friendly route was adopted to explore the adsorption, photocatalytic, and catalytic properties of the resulting NPs. The structural, morphological, and optical characteristics of α-MnMoO4 NPs were systematically characterized using XRD, FTIR, UV-vis, and PL spectroscopy, SEM, and EDX techniques. Notably, α-MnMoO4 NPs demonstrated excellent adsorption capability toward methylene blue (MB) dye, achieving a removal efficiency of 86.70%, which was primarily attributed to their negatively charged surface. Moreover, the nanoparticles demonstrated a remarkable photocatalytic activity, achieving 81.55% degradation of MB through the photo-oxidation of water into hydroxyl (ȮH) radicals by photogenerated holes. Beyond dye remediation, the study further explored the catalytic capabilities of o-phenylenediamine via oxidative condensation with substituted aromatic aldehydes to synthesize benzimidazole derivatives, achieving yields ranging from 35% to 85%, depending on the substituents used. This integrated approach highlights the potential of α-MnMoO4 NPs not only in pollutant removal but also in facilitating the green synthesis of high-value chemical products, demonstrating promising applications in environmental remediation and fine chemical industries.
1. Introduction
In the present scenario, natural resources are being exhausted due to rapid urbanization and an increase in population growth. Urbanization has resulted in the increased discharge of organic effluents from industry and agriculture.1 These effluents are the primary source of water pollution, which poses a threat to human health and has a severe negative impact on aquatic life.2 In particular, during the past few decades, dyes and the dyeing industries have experienced tremendous growth. Numerous potentially hazardous substances that are detrimental and carcinogenic to the flora and fauna have been released into the environment as effluents by these industries.3 The organic effluents cause various human disorders like ulceration of the skin, nausea, and haemorrhage.4 This raises more serious concerns regarding organic effluent discharges and the significant difficulty in degrading these organic compounds for the well-being of mankind.5 Various techniques have been proposed for the degradation of organic effluents from water systems; they include physical methods such as adsorption,6 coagulation/flocculation,7 ultrafiltration,8 and ion exchange techniques.9 These techniques have the limitations of high expenditure and restricted versatility.10 Biological methods, involving various microbes and fungi, have been explored as alternatives due to their environmentally friendly nature. However, they are often not cost-effective and show limited efficiency in degrading the complex structures and stable chromophores of organic effluents. Additionally, these methods typically require relatively long treatment times, are sensitive to environmental conditions (e.g., pH and temperature), and may not fully mineralize persistent organic pollutants, making them less suitable for large-scale or rapid remediation processes.11 Chemical methods enable the complete degradation of highly stable organic compounds utilizing chemical oxidation and catalytic degradation processes.12 However, on an operational scale, the usage of these methods has been hindered due to high operational costs, secondary pollutant generation, and the requirement for controlled reaction conditions. Additionally, the use of toxic or non-renewable reagents in some chemical processes raises environmental and safety concerns, limiting their large-scale applicability.13
The new realm of utilizing nanotechnology for the degradation of organic effluents has gained significant attention due to the photocatalytic properties exhibited by transition-metal oxides; they absorb photons under light irradiation and facilitate redox reactions that break down complex chromophores in organic structures into simple, less harmful compounds.14 Various transition-metal oxides, such as titanium dioxide (TiO2),15 zinc oxide (ZnO),16 tungsten trioxide (WO3),17 vanadium pentoxide (V2O5),18 zirconium dioxide (ZrO2),19 tin dioxide (SnO2),20 and cerium dioxide (CeO2),21 have been extensively employed in photocatalytic degradation processes owing to their large surface area, strong photochemical stability, and chemical inertness.22 The application and practical utility of these monometal oxides are impeded due to their wide bandgap.23 To narrow down the bandgap, researchers have extensively worked on doping a foreign transition metal in the monometal oxide, which tunes the bandgap, thereby altering the optical absorption properties of the nanomaterials, leading to the formation of mixed bimetallic transition metal oxides.3 Among these, molybdenum-based metal oxides have attracted considerable research interest owing to their high efficiency in photocatalytic degradation and excellent photostability against organic cationic dyes.24 In addition, they have been widely applied in the oxidation of various organic pollutants, such as toluene.25 These bimetallic oxides have also been extensively investigated for applications in supercapacitors,26 electrode materials for lithium-ion batteries,27 and other energy storage devices.28 However, the synthesis of such bimetallic transition-metal oxides has traditionally relied on chemical methods involving toxic and hazardous solvents.29 Thus, the employment of biological materials reduces the formation of harmful by-products as they contain various phytochemicals, which include polyphenols, alkaloids, flavonoids, proteins, and polysaccharides.30 These biomolecules facilitate the reduction and capping of metal ions while simultaneously preventing agglomeration, thereby promoting the formation of diverse morphological structures.31 Such a sustainable synthesis strategy holds significant potential for the development of eco-friendly nanocatalysts aimed at the degradation and conversion of organic pollutants.
In recent decades, increasing attention has been directed toward the green synthesis of bimetallic transition-metal oxides, owing to their narrow bandgaps and superior efficiency in degrading organic pollutants through oxidation–reduction processes.32 In this study, we report the green combustion synthesis of bimetallic manganese molybdate (α-MnMoO4) nanoparticles using peanut powder, as a sustainable fuel source, enriched with sucrose and protein content. The structural, morphological, and optical characteristics of the synthesized α-MnMoO4 NPs are systematically analyzed using XRD, FTIR, UV-vis, PL, SEM, and EDX techniques. Furthermore, their catalytic performance is evaluated through the photocatalytic degradation of water-soluble methylene blue (MB) dye and the catalytic conversion of o-phenylenediamine into benzimidazole derivatives. The novelty of this work lies in the dual-functional application of green-synthesized α-MnMoO4 NPs—not only as efficient agents for dye removal and photocatalysis, but also as catalysts enabling the sustainable synthesis of benzimidazole derivatives from industrial effluents. These findings highlight the potential of the α-MnMoO4 nanoparticles for practical applications in textile industries, both for dye degradation and the conversion of organic pollutants into value-added products.
2. Experimental procedure
2.1. Synthesis of MnMoO4 nanoparticles
Based on reported methods,33 α-MnMoO4 NPs were synthesized by the combustion method using peanut powder as fuel. The raw peanuts were initially ground into a fine powder using a blender. Thereafter, 0.1 M manganese nitrate and 0.1 M ammonium heptamolybdate, at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric molar ratio, were dissolved in 10 mL of deionized water, along with half the calculated amount of peanut powder based on the total precursor weight. The solution was continuously stirred until it became a homogeneous suspension. Then, the solution was kept in a muffle furnace at 500 °C for about 10 min. After that, a greyish-black foam-like product was formed. The above experiment was repeated for different amounts (double and triple) of fuel with the same molar ratio of precursors. The greyish-black foam obtained was ground into fine powder and calcinated in a muffle furnace at 600 °C for 3 h at a heating rate of 5 °C min−1, and the calcined NPs were used for further characterization and application.
:1 stoichiometric molar ratio, were dissolved in 10 mL of deionized water, along with half the calculated amount of peanut powder based on the total precursor weight. The solution was continuously stirred until it became a homogeneous suspension. Then, the solution was kept in a muffle furnace at 500 °C for about 10 min. After that, a greyish-black foam-like product was formed. The above experiment was repeated for different amounts (double and triple) of fuel with the same molar ratio of precursors. The greyish-black foam obtained was ground into fine powder and calcinated in a muffle furnace at 600 °C for 3 h at a heating rate of 5 °C min−1, and the calcined NPs were used for further characterization and application.
2.2. Characterisation and instrumentations
A Shimadzu 7000 X-ray diffractometer (XRD) with monochromatized CuKα radiation (1.5418 Å) was used to analyse the structure and size of the synthesized α-MnMoO4 NPs in the 2θ range of 10°–70°. The Fourier transform infrared (FTIR) spectroscopy of α-MnMoO4 NPs was conducted using a Bruker Alpha P-spectrophotometer in the wavenumber range from 350 to 4000 cm−1. The reflectance spectrum was evaluated to determine the optical properties of the nanoparticles using an Agilent Technology Cary-60 UV-vis diffuse reflectance spectrophotometer (UV-vis DRS) in the range of 200–800 nm. The photoluminescence spectrum of the α-MnMoO4 nanoparticles was measured using an Agilent Technology Cary Eclipse fluorescence spectrophotometer at an excitation wavelength of 387 nm. Using an Agilent Technology Cary-60 UV-vis absorption spectrophotometer, the degradation of dyes was monitored. The 1H NMR spectrum was recorded in DMSO-d6 using a Bruker Avance 400 MHz FT-NMR spectrometer.
2.3. Adsorption studies
To evaluate the adsorption property of the synthesized α-MnMoO4 NPs, a specific amount of absorbent was introduced into 20 mL of an MB dye solution at room temperature. Contact time and dye concentration factors were optimized for efficient adsorption. The adsorption experiments were performed in different ppm dye concentrations (25–100 ppm) at varying time intervals (0–18 min). The adsorption of the MB dye solution was monitored by observing the absorbance peak of MB at λmax of 665 nm using UV-vis absorption spectroscopy, and the adsorption percentage was examined using eqn (1).34| | 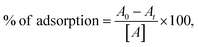 | (1) |
where A0 is the initial concentration of the MB dye solution, and At is the concentration of the MB dye solution.
2.4. Photo-catalytic studies
The degradation of organic dyes, such as methylene blue (MB), was performed to assess the photo-catalytic activity of the synthesized α-MnMoO4 NPs. To evaluate the photo-catalytic activity, 5–20 mg of MnMoO4 NPs was weighed and evenly distributed in the dye solution (5–20 ppm). Then, to attain adsorption–desorption equilibrium between the NPs and the dye solution, it was kept in the dark for 30 min. Once a homogenous mixture was achieved, it was irradiated with a UV light source. The dye solution was collected at an interval of 30 min and centrifuged to get rid of the nanoparticles. The degradation of the MB dye solution was monitored by observing the absorbance peak of MB at λmax of 665 nm using UV-vis absorption spectroscopy, and the percentage degradation was examined using eqn (2).13| | 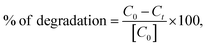 | (2) |
where C0 is the initial concentration of the MB dye solution, and Ct is the concentration of the MB dye solution collected at an interval of 30 min.
2.5. General procedure for the synthesis of 2-substituted benzimidazole derivatives
Typically, a 50 mL round-bottom flask was charged with a mixture of substituted benzaldehyde (1.0 mmol) and o-phenylenediamine (1.0 mmol) in 5 mL of ethanol, along with 20 mg of the α-MnMoO4 nanocatalyst. The reaction mixture was subjected to continuous magnetic stirring and refluxed at room temperature for 12 h. Using TLC (EtOAc:hexane = 1:5), the reaction's advancement was regularly monitored. Once the reaction was completed, it was quenched with water and extracted using ethyl acetate. The nanocatalyst was separated from the mixture using a Whatman filter paper. The filtrate was then dried over sodium sulfate; the solvent was removed under a vacuum, and the resulting reaction mixture was purified through column chromatography.35
3. Results and discussions
3.1 Powder X-ray diffraction (XRD) analysis
All MnMoO4 NPs formed through the solution combustion method with varying green fuel concentrations were confirmed using an X-ray diffractometer. The synthesized MnMoO4 NP XRD patterns are depicted in Fig. 1a. For all synthesized MnMoO4 NPs (1:0.5, 1:1, and 1:1.5), peaks at 2θ = 12.95°, 18.85°, 22.81°, 25.81°, 26.87°, 27.84°, 31.27°, 33.10°, 35.77°, 37.81°, 40.58°, 42.61°, 45.83°, 51.29°, 52.79°, 56.40°, 59.43°, 65.16° were observed, which were indexed to planes (1 1 0), (−2 0 1), (0 2 1), (2 2 0), (−1 1 2), (−3 1 1), (1 1 2), (−2 2 2), (4 0 0), (0 4 0), (4 2 0), (−4 2 2), (2 4 1), (−2 0 4), (4 4 0), (4 4 1), (−4 2 4), (−2 4 4), respectively. The peaks matched JCPDS (Joint Committee on Powder Diffraction Standards) Card number: 01-072-0285.36 The presence of a major (2 2 0) reflection peak indicated the presence of an alpha phase, and it was observed that upon increasing the fuel concentration, the intensity of this XRD peak increased. This could be attributed to the small crystallite size of MnMoO4 NPs.
 |
| | Fig. 1 (a) XRD patterns of MnMoO4 NPs synthesized at different ratios. (b) FTIR spectra of MnMoO4 NPs prepared at different ratios. | |
The average crystal sizes of the synthesized MnMoO4 NPs were calculated using Debye–Scherrer's eqn (3):
| | 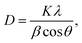 | (3) |
where
D is the crystallite size,
λ is the wavelength of the X-ray (1.54 Å) used,
β is the full-width at half maximum (FWHM), and
θ is the diffraction angle. Additionally, the lattice parameter of MnMoO
4 NPs was calculated using Bragg's
eqn (4):
| | 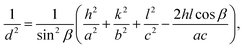 | (4) |
where
a,
b,
c and
β represent the lattice parameters, and
d is the distance between parallel lattice planes with Miller indices (
h k l), as represented in Table S1, which match the JCPDS Card number: 01-072-0285. Further, the strain of the synthesized MnMoO
4 NPs was evaluated using
eqn (5).
| | 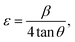 | (5) |
where
ε represents the micro strain,
β is the full-width at half maximum (FWHM), and
θ is the diffraction angle. The crystallite size, lattice parameter, and strain are represented in
Table 1.
Table 1 Crystallite sizes and microstrain of MnMoO4 NPs
| Type of nanoparticle |
Crystallite size (nm) ± SD |
Microstrain (× 10−3) ± SD |
| MnMoO4 (1:0.5) |
35 ± 1 |
0.4 ± 0.01 |
| MnMoO4 (1:1) |
31 ± 1 |
0.5 ± 0.02 |
| MnMoO4 (1:1.5) |
23 ± 0.4 |
0.7 ± 0.02 |
3.2. Fourier transform infrared spectroscopy (FTIR) analysis
Fig. 1b and Fig. S1 show the FTIR spectra of MnMoO4 NPs in the range of 350–4000 cm−1. The characteristic peaks at 728, 798, 869, and 943 cm−1 confirmed the formation of MnMoO4. Specifically, the strong peak at 943 cm−1 was attributed to the stretching vibration of the Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group, while the peaks at 728, 798, and 869 cm−1 corresponded to the bending modes of Mo–O–Mo bonds. In addition, the peaks observed at 618 cm−1 and 410 cm−1 were assigned to the vibrational modes of the Mo–O and Mn–O bonds, respectively. Notably, the intensity of these characteristic MnMoO4 peaks increased with increased fuel concentration, as summarized in Table S2. Furthermore, narrow peaks at 1110 cm−1 and 1270 cm−1 were attributed to C–O stretching and CC vibrational modes, respectively, while the broad peaks at 1484 cm−1 and 3443 cm−1 were ascribed to C–H bending and O–H stretching vibrations, respectively. A shoulder peak at 1632 cm−1 corresponded to the CO stretching mode.
O group, while the peaks at 728, 798, and 869 cm−1 corresponded to the bending modes of Mo–O–Mo bonds. In addition, the peaks observed at 618 cm−1 and 410 cm−1 were assigned to the vibrational modes of the Mo–O and Mn–O bonds, respectively. Notably, the intensity of these characteristic MnMoO4 peaks increased with increased fuel concentration, as summarized in Table S2. Furthermore, narrow peaks at 1110 cm−1 and 1270 cm−1 were attributed to C–O stretching and CC vibrational modes, respectively, while the broad peaks at 1484 cm−1 and 3443 cm−1 were ascribed to C–H bending and O–H stretching vibrations, respectively. A shoulder peak at 1632 cm−1 corresponded to the CO stretching mode.
The UV-vis DRS spectra of the synthesized MnMoO4 NPs were recorded from 200 to 800 nm to examine the optical properties and energy bandgap of the NPs. The spectral details revealed that a peak was observed in the 200-to-600-nm region of the visible spectrum, and on increasing the fuel concentration, the peak shifted towards a relatively low wavelength (hypsochromic shift). Fig. 2a shows the reflectance spectra of the synthesized NPs, and the optical bandgap energy was obtained by analysing the modified tau plot called the Kubelka–Munk relation, as given in eqn (6).
| | 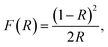 | (6) |
where
R is the reflectance value. The energy corresponding to the wavelength was calculated using
eqn (7).
| | 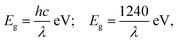 | (7) |
where
Eg is the bandgap energy (eV),
h is the Planck's constant (6.626 × 10
−34 J s),
c is the light velocity (3 × 10
8 m s
−1), and
λ is the wavelength (nm). The optical bandgap of the NPs was revealed
via extrapolating the linear portion of the plot [
F(
R)
hν]
1/2vs. hν, which is represented in
Fig. 2b. The results indicated that increasing the fuel concentration led to variations in the bandgaps of the synthesized NPs. The crystallite size of the semiconductor nanoparticles played a significant role in governing their bandgaps. For MnMoO
4 synthesized with a fuel concentration ratio of 1
:
1.5, the relatively small crystallite size induced a quantum confinement effect, resulting in an increased bandgap. In contrast, MnMoO
4 NPs synthesized with a 1
:
1 fuel concentration ratio exhibited a relatively low bandgap of 4.5 eV, which was favorable for the enhanced photocatalytic degradation of dyes. Additionally, the positions of the valence band (VB) and conduction band (CB) strongly influenced the photocatalytic efficiency, as the bandgap energy corresponded to the minimum energy required for an electron to transition from the VB to the CB. These band edge positions could be estimated using the Mulliken electronegativity approach, as described in
eqn (8) and (9).
where
χ, the absolute electronegativity of MnMoO
4, was found to be 5.92, as calculated using
eqn (10).
| | | χ = [x(A)ax(B)bx(C)c]1/(a+b+c). | (10) |
 |
| | Fig. 2 (a) UV-Visible spectra, (b) Kubelka–Munk plots, (c) excitation spectra (λemission = 287 nm), and (d) emission spectra (λexcitation = 258 nm) of MnMoO4 NPs (1:0.5, 1:1, and 1:1.5). | |
E° is the constant value of the free electron energy on the hydrogen scale (4.5), and Eg is the optical bandgap of the material. The obtained bandgap, valence band, and conduction band of the NPs are listed in Table S3.
3.3. Photoluminescence analysis
PL spectra were recorded to monitor the light released by the synthesized NPs when the photogenerated electron/hole recombined. The charge separation efficiency and fundamental radiative characteristics of the synthesized MnMoO4 NPs were assessed using these spectra. The photoluminescence emission and excitation spectra of all the synthesized MnMoO4 NPs are shown in Fig. 2c and d. The excitation wavelength of the sample was found to be 258 nm, whereas the main emission peak was observed at 287 nm. Among the synthesized NPs, MnMoO4 (1:1) NPs showed a lower emission intensity, which could be ascribed to their greater charge separation than those in other produced NPs. Therefore, MnMoO4 (1:1) NPs could enable a relatively high photodegradation percentage. Fig. S2 represents the CIE 1931 x–y chromaticity diagram (Commission Internationale de l'Elcairage) for the detection of the light colour produced by the NPs. The chromaticity points facilitated the observation of the color purity. The corresponding MnMoO4 (1:0.5, 1:1, and 1:1.5) NP chromaticity points were (0.17, 0.634), (0.144, 0.667), and (0.145, 0.678), respectively. The distinct x and y coordinates revealed that the ionic liquid had a major impact on the crystallization process, in addition to the light's high color and purity.
3.4. Scanning electron microscopy (SEM) and energy-dispersive X-ray (EDX) analysis
The SEM micrographs of the synthesized MnMoO4 (1:1) NPs at different magnifications are shown in Fig. 3a–d. The nano range of the NPs was clearly observed in the figure depicted. The rhombohedron-like morphology of MnMoO4 (1:1) NPs with the C2/m space group was observed. A similar morphology was reported for MnMoO4 NPs prepared by hydrothermal and precipitation methods. The agglomeration of the NPs was also observed, which was due to the high surface area energy that led to the NPs forming bundles and the subsequent decrease in temperature to room temperature after the growth, causing the enthalpy to become negative. This indicated that in order to maintain equilibrium, the size of the NPs changed and agglomerated. The values of the weight % and atom % of MnMoO4 (1:1) NPs were recorded using EDX measurements. The EDX spectra of the synthesized NPs are shown in Fig. 3e. It revealed that the sample contained Mn, Mo, and O with a definite stoichiometry expected (Mn:Mo:O = 1:1:4).
 |
| | Fig. 3 SEM micrographs of MnMoO4 NPs at magnifications of (a) and (b) 5 µm and (c) and (d) 1 µm. (e) EDX spectra of MnMoO4 NPs. | |
3.5. Adsorption property
3.5.1. Contact time.
Fig. 4a represents the impact of contact time on the adsorption of the MB dye (50 ppm) at various time intervals (18 min) using 10 mg of MnMoO4 NPs. It can be noted from the adsorption percentage plot given in Fig. 4b that as time increases, the adsorption rate also increases and reaches the saturation point. Maximum adsorption was observed at 18 min. This could be ascribed to the availability of more active sites for the adsorption at the beginning. As the process continued further, there was a reduction in the availability of the binding sites, which were occupied by the dye molecules. A phase time of 18 min was observed, in which the availability of the active site vanished, a saturation point was obtained, and further adsorption was hindered. Thus, 18 min was the optimum time for the maximum adsorption of the MB dye by MnMoO4 NPs.
 |
| | Fig. 4 (a) Adsorption of the MB dye at varying times (0–18 min), (b) adsorption percentage of the MB dye with respect to time, (c) UV spectra of the adsorption of the MB dye at varying concentrations, and (d) adsorption percentage of the MB dye at varying concentrations. | |
3.5.2. Dye concentration.
Dye concentration is a factor influencing the adsorption capacity of the absorbent (MnMoO4), which is illustrated in Fig. 4c and d. Various dye concentrations of 25 ppm, 50 ppm, 75 ppm, and 100 ppm were used along with 10 mg of MnMoO4 NPs to perform this experiment. It was observed that the maximum adsorption percentage was obtained in the 25 ppm MB dye concentration, and on increasing the MB dye concentration, the adsorption percentage reduced. This could be attributed to the greater number of active sites than the MB dye molecules in a 25 ppm concentration solution. Therefore, a high adsorption percentage was observed at a low MB dye concentration. Consequently, when the dye concentration increased due to the limited number of active sites, competition between the dye molecules occurred, which lowered the adsorption percentage. The saturation point was achieved when the dye molecules covered the monolayer of the absorbent, and further adsorption was restricted.
3.5.3. Adsorption kinetics.
To evaluate the influence of contact time on the adsorption of the MB dye by MnMoO4 NPs, two kinetic models were applied: the pseudo-first-order model proposed by Lagergren and the pseudo-second-order model developed by Ho and McKay. The pseudo-first-order kinetic model is commonly used to describe adsorption processes in solid–liquid systems, whereas the pseudo-second-order model assumes that the adsorption rate is dependent on the adsorption equilibrium capacity. The linearized forms of the two models are expressed in eqn (11) and (12):| | 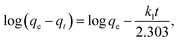 | (11) |
| | 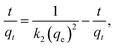 | (12) |
where qt is the adsorption capacity at time t, k1 is the pseudo-first-order rate constant (min−1), and k2 is the pseudo-second-order rate constant (g mg−1 min−1). The correlation coefficient (R2) obtained for the pseudo-first-order model was 0.97954, compared to 0.93398 for the pseudo-second-order model, as depicted in Fig. 5a and b. The relatively low R2 value for the pseudo-second-order model indicated limited simultaneous interactions between the MB dye molecules and multiple adsorption sites. In contrast, the relatively high R2 value for the pseudo-first-order model suggested that the adsorption of the MB dye onto MnMoO4 NPs predominantly occurred through interactions with single active sites. Hence, the adsorption kinetics of the MB dye onto MnMoO4 NPs were best described by the pseudo-first-order model. Moreover, the high rate constant (k1) obtained from the pseudo-first-order plot further confirmed the relatively fast reaction kinetics under this model. The calculated parameters (R2, k1, and k2) are summarized in Table 2.
 |
| | Fig. 5 Adsorption kinetics plots: (a) pseudo-first order and (b) pseudo-second order. Adsorption isotherm plots: (c) Langmuir and (d) Freundlich plots for the adsorption of the MB dye. | |
Table 2 Kinetic parameters for the MB dye adsorption by MnMoO4 NPs
| Concentration (mg L−1) |
Pseudo-first order |
Pseudo-second order |
|
R
2
|
k
1 (1 min−1) |
R
2
|
k
2 (g mg−1 min−1) |
| 50 |
0.98 |
0.112 |
0.93 |
0.003 |
3.5.4. Adsorption isotherm.
The adsorption behavior of MnMoO4 NPs toward the MB dye was further evaluated using two commonly applied isotherm models: the Langmuir and the Freundlich eqn (13) and (14).| |  | (13) |
| |  | (14) |
where qm (mg g−1) and b (L mg−1) are the Langmuir constants, representing the maximum adsorption capacity and adsorption energy, respectively. Kf and n are the Freundlich constants, indicating the adsorption capacity and adsorption intensity, respectively. qe (mg g−1) is the equilibrium adsorption capacity, which can be calculated using eqn (15):| |  | (15) |
where C0 (mg L−1) and Ce (mg L−1) are the initial and equilibrium dye concentrations, V (L) is the volume of the dye solution, and m (g) is the mass of the adsorbent.
As per the Langmuir model, the dye molecules were adsorbed as monolayers on the energy sites of MnMoO4 NPs. Fig. 5c depicts the plot of Ce/qe against Ce, which provides the Langmuir coefficients (qm and b). This coefficient was used to measure the adsorption ability of MnMoO4 NPs, where qm and b were found to be 413.22 and 0.265, respectively. The R2 value (0.95308) obtained for the Langmuir plot was relatively high, which indicated that the monolayer adsorption was favoured. The Freundlich plot is illustrated in Fig. 5d, divulging Freundlich constants Kf and n, which describe the heterogeneous sorption phenomenon of adsorption. The Kf and 1/nf values obtained were 3.66 and 0.60146. These constants were the essential parameters for examining the extent of favorability of the process. Based on the magnitude of 1/nf, the adsorption processes were classified as reversible (1/nf = 0), favorable (0 < 1/nf < 1), and unfavorable (1/nf = 1). The process was favorable in this case, as the magnitude applied to the condition. Simultaneously, nf can also be used to evaluate whether the adsorption is physical (nf > 1) or chemical (nf < 1). Here, the value of nf was greater than 1. Therefore, it implied that the process was a favorable physical adsorption. The low R2 value (0.93347) indicated that multilayer adsorption on heterogeneous sorption sites was hindered. The data are computed in Table 3.
Table 3 Isotherm parameters for the MB adsorption by MnMoO4 NPs
| Isotherm |
Isotherm parameters |
Values |
| Langmuir |
q
m (mg g−1) |
413.22 |
|
b (L mg−1) |
0.27 |
|
R
2
|
0.95 |
| Freundlich |
K
f (mg g−1) (L mg−1)1/nf |
3.66 |
| 1/n |
0.61 |
|
R
2
|
0.93 |
3.5.5. Post-adsorption studies.
After the adsorption of methylene blue, notable changes were observed in the FTIR spectrum of the adsorbent, as shown in Fig. S3a. The broad O–H stretching band around 3400 cm−1 became weak and slightly shifted, indicating possible hydrogen bonding or electrostatic interactions between the surface functional groups and methylene blue molecules. A new or intensified band appeared near 1600 cm−1, corresponding to the CC and CN stretching vibrations of methylene blue's aromatic thiazine ring. Additionally, enhanced peaks in the 1300–1200 cm−1 region were attributed to the C–N and C–S stretching modes of the dye. The reduction in the intensity of the bands around 1000–1100 cm−1 suggested that these surface groups were actively involved in the adsorption process.
Complementary evidence from the XRD patterns, as shown in Fig. S3b, clearly reveals significant structural modifications in the material before and after adsorption. Before adsorption, the sharp and intense diffraction peaks indicated a highly crystalline structure with well-defined phases. The most prominent peak appeared at 2θ = 25.81°, representing the dominant crystalline phase of the material. After adsorption, the diffraction peaks become relatively broad and less intense, and the major peak at 25.81° showed a clear bifurcation, indicating a distortion of the corresponding crystal planes. This bifurcation and overall reduction in the peak intensity suggested a lattice strain, surface interaction, and partial phase transformation caused by the adsorption of molecules onto the material surface. The broadening and decreased intensity of the peaks confirmed a reduction in crystallinity and partial amorphization, resulting from surface modifications. The absence of new diffraction peaks further confirmed that no new crystalline phases were formed, implying that adsorption primarily altered the surface structure and order of the existing crystalline phases while the bulk crystal framework remained largely intact.
3.6. Photocatalytic property
3.6.1. Degradation of organic dye.
Under a UV light source, the photo-catalytic performance of MnMoO4 NPs was assessed by degrading various organic dyes at a constant concentration of 5 ppm (pH 7). Fig. 6a depicts the percentage degradation values of methylene blue (MB), rhodamine B (RB), indigo carmine (IC), and methyl orange (MO). In the presence of 5–20 mg of MnMoO4 NPs, a highly selective degradation of 81.55% was observed for the methylene blue dye. To monitor the degradation of the MB dye, the corresponding absorption spectra were observed. The reduction of λmax at 665 nm was attributed to the breakdown of the MB dye, and the degradation capacity was attributed to the location of aromatic rings in the structure. The above figure illustrates the MB dye degradation of the synthesized MnMoO4 NPs with varying fuel concentrations. MnMoO4 (1:1) NPs showed a higher amount of degradation than the other NPs, as presented in Fig. 6b. This enhanced photocatalytic activity could be attributed to the low bandgap of MnMoO4, which facilitated the excitation of more electrons into the conduction band while generating corresponding holes in the valence band. The resulting reactive species, namely hydroxyl radicals (˙OH) and superoxide anions (O2˙−), actively participated in accelerating the photodegradation of the MB dye. To further maximize the degradation efficiency, key operational parameters, such as catalyst loading, initial dye concentration, and solution pH, were systematically optimized. Moreover, the photostability and recyclability of MnMoO4 NPs were also investigated to evaluate their potential for repeated practical applications.
 |
| | Fig. 6 (a) Photo-catalytic activities of MnMoO4 NPs over different organic dyes. (b) Degradation percentages of the MB dye in the presence of all MnMoO4 NPs. Optimisation of the catalyst dosage (5–20 mg) for the degradation of the MB dye in the presence of MnMoO4 NPs at neutral pH: (c) photocatalytic activity and (d) rate constant. | |
3.6.2. Optimization of photocatalytic degradation by various factors.
3.6.2.1. Catalyst dosage.
Catalyst dosage was optimized for the degradation of the MB dye by varying the amount of MnMoO4 NPs from 5 to 20 mg in the MB dye solution (100 mL, 5 ppm) under UV-light radiation at a neutral pH. Fig. 6c illustrates that the degradation efficiency increases till 10 mg of catalyst dosage, and upon increasing the amount of the catalyst, the efficiency drops. This reduction in efficiency could be attributed to the accumulation of the MB dye molecules on the surface of the NPs, which reduced the absorption capacity of the NPs. This suggested that a minimal amount of catalyst dosage was required for the efficient degradation of the MB dye. Thus, the optimized catalyst dosage of 10 mg was used for further degradation.
3.6.2.2. Dye concentration.
To enhance the photodegradation, the dye concentration also has a crucial role. The photodegradation of the MB dye solution was conducted in the presence of MnMoO4 NPs (5 mg), along with varying dye concentrations from 5–20 ppm at a neutral pH under UV light radiation. Fig. 7a shows the decrease in percentage degradation with the increase in the concentration of the dye. This can be interpreted through Beer–Lambert's law, which elucidates that on increasing the dye concentration, the photons absorbed by the NPs have a relatively short path length, leading to a reduction in the amount of active radical (ȮH and O2˙−) produced. This signifies that a low concentration of dye is required for the enhanced degradation process. Therefore, the optimum 5 ppm concentration is provided for further experiments.
 |
| | Fig. 7 Optimisation of the dye concentration (5–20 ppm) for the degradation of the MB dye in the presence of MnMoO4 NPs at neutral pH: (a) photocatalytic activity and (b) rate constant. Optimisation of the pH values for the degradation of the MB dye in the presence of MnMoO4 NPs at neutral pH: (c) photocatalytic activity and (d) rate constant. | |
3.6.3. Kinetics of the photocatalytic properties.
In order to support the experimental results, kinetic studies were conducted. Fig. 6d and 7b depict the dye degradation process following the pseudo-first-order reaction, with the slope indicating the rate of reaction (k) calculated from the  versus reaction time described by the Langmuir and Hinshelwood model, defined as eqn (16) and (17).
versus reaction time described by the Langmuir and Hinshelwood model, defined as eqn (16) and (17).| |  | (17) |
The k values for the respective catalyst dosage, dye concentrations, and pH medium are represented in Table S4. An increased k value was noted for 10 mg of catalyst, 5 ppm concentration, and basic medium in the kinetic studies. This high k value determined the optimum condition for the enhanced degradation of the MB dye under a UV light source. Therefore, the photocatalytic degradation of the MB dye was performed with 10 mg of MnMoO4 NPs in 5 ppm of MB dye concentration under a UV light source at a neutral pH to enhance the degradation, as presented in Fig. S4. The results of a literature survey of the photodegradation of the methylene blue dye are compiled in Table 4.
Table 4 Literature survey for the photodegradation of the methylene blue dye by NPs
| S. no. |
Nanoparticles |
Synthesis method |
Green source |
Degradation percentage (%) |
Ref. |
| 1 |
ZnO |
Co-precipitation method |
Croton macrostachyus
|
66 |
37
|
| 2 |
α-Fe2O3 |
Co-precipitation method |
Mentha pulegium
|
78 |
38
|
| 3 |
Ag |
Co-precipitation method |
Camellia sinensis
|
58.3 |
39
|
| 4 |
ZnMn2O4 |
Green combustion method |
Arachis hypogaea
|
79.45 |
33
|
| 5 |
α-MnMoO4 |
Green combustion method |
Arachis hypogaea
|
81.55 |
Present |
3.6.4. Mechanism of the photodegradation.
To ascertain the mechanism of the photodegradation by MnMoO4 NPs, a simple radical trapping experiment was conducted. Scavengers, like ascorbic acid (10 mM), potassium dichromate (10 mM), EDTA (10 mM), and tert-butyl alcohol (1:20 v/v), were added to various MB dye solution mixtures to trap O2˙−, e−, h+, and ȮH radicals. It was found that, as the accompanying Fig. 9a illustrates, EDTA and tert-butyl alcohol inhibited and reduced the photodegradation of the MB dye solution to 55.90% and 48.03%, respectively. Therefore, it revealed that the h+ and ȮH radicals were more actively engaged in the photodegradation of the MB dye solution by MnMoO4 NPs than the e− and O2˙− radicals.
As the ȮH radical prevails as the active species that governs the overall photodegradation process, the determination of the ȮH radicals is important. Therefore, a simple experiment of converting coumarin to 7-hydroxycoumarin using the ȮH radicals released during the photo oxidation of water was conducted by mixing 5 mg of MnMoO4 (1:1) NPs in 100 mL of 10 mM coumarin solution and irradiating the mixture under UV light. At an interval of 30 min, the solution was collected, and at excitation of 440 nm, the fluorescence spectra were recorded, as shown in Fig. S5a. It divulged the increasing intensity confirms the formation of 7-hydroxycoumarin. This confirmed that MnMoO4 NPs generated ȮH radicals during the photo oxidation with water molecules, which could contribute to the photodegradation. Additionally, it was observed that the fluorescence intensity was quenched in the presence of the MB dye, as shown in Fig. S5b, suggesting photo-induced electron transfer from the excited MnMoO4 nanocubes to molecular oxygen. This electron transfer facilitated the formation of superoxide radicals (O2˙−), which also contributed to the photocatalytic degradation process. The thermodynamic feasibility of electron transfer from the MB dye to photoexcited MnMoO4 NPs was evaluated using the Rehm–Weller eqn (18).
| | | ΔG0 = ED/D+ − EA/A− − E00, | (18) |
where
ED/D+ is the oxidation potential of the donor,
EA/A− is the reduction potential of the acceptor, and
E00 is the excited state energy, approximated by the bandgap. The conduction band reduction potential of α-MnMoO
4 NPs was experimentally determined as −0.86 eV, while the bandgap energy was measured to be 4.56 eV through UV-vis diffuse reflectance spectroscopy. Considering the oxidation potential of the MB dye at +0.94 eV, the Gibbs free energy change (Δ
G0) for the electron transfer from the MB dye to the excited α-MnMoO
4 NPs was calculated as −2.76 eV, indicating a highly favorable and spontaneous electron transfer process. This thermodynamic evidence supported the proposed mechanism in which the photoexcited α-MnMoO
4 nanoparticles efficiently accepted electrons from the MB dye molecules, initiating their degradation. Consequently, the generated charge carriers participated in redox reactions, leading to the breakdown of the MB dye.
The photocatalytic degradation efficiency of nanoparticles primarily depends on their bandgap energy, surface area, and electron–hole (e−/h+) separation efficiency. A schematic of the proposed degradation mechanism is presented in Fig. 8. MnMoO4 (1:1) NPs possessed a relatively large surface area and enhanced charge carrier separation, which favoured improved photocatalytic activity. As discussed earlier, the h+ and ˙OH radicals play a dominant role in the degradation of the MB dye. Upon UV irradiation, electrons (e−) were excited from the valence band to the conduction band, leaving behind holes (h+). These charge carriers drove redox reactions: h+ oxidized H2O to generate hydroxyl radicals (˙OH), while e− reduced O2 to produce superoxide anions (O2˙−). These reactive oxygen species (ROS) accumulated on the nanoparticle surface and attacked the MB dye molecules, ultimately decomposing the organic structure into environmentally benign end products, such as CO2 and H2O.
 |
| | Fig. 8 Mechanism of the photodegradation by MnMoO4 NPs. | |
3.6.5. Recyclability and photostability of MnMoO4 NPs.
For the commercialization of the synthesized nanoparticles, recyclability and photostability are important parameters. In order to estimate these parameters, centrifugation was performed to remove the nanoparticles from the MB dye solution, and the resulting solids were then washed with methanol and distilled water and dried at 100 °C for 3 h. They were subjected to 4 degradation cycles to monitor the percentage of the MB dye degradation. It was found that the change in the degradation percentage was almost similar, with a slight decrease in the 4th cycle, as shown in Fig. 9b. This stability could be attributed to their tetragonal structure. Thus, the synthesised MnMoO4 NPs could be used consecutively for four cycles of the MB dye degradation with a reduction in the percentage of degradation.
 |
| | Fig. 9 (a) Effects of different scavengers during the degradation of the MB dye. (b) Photostability and reusability of MnMoO4 (1:1) NPs over four successive cycles. | |
3.7. Catalytic property of MnMoO4 NPs as a nano-catalyst
The catalytic activity of the MnMoO4 nanoparticles was investigated in the synthesis of 2-substituted benzimidazoles via the oxidative condensation of substituted benzaldehydes with o-phenylenediamine. As a model reaction, the condensation of 5-bromosalicylaldehyde with o-phenylenediamine was selected. Reaction optimization was carried out by varying the solvents, oxidants, and catalyst loadings, with the results summarized in Table 4. Among the tested solvents (DMF, DMSO, ACN, H2O, and EtOH), ethanol was identified as the most effective medium. Hydrogen peroxide (H2O2) served as the oxidant, while reactions carried out in its absence yielded only trace amounts of the product. As presented in Table 5, the optimum yield was achieved with 20 mg of MnMoO4 NPs. Under these optimized conditions (Table 5, entry 5), the substarte scope of the oxidative condensation reaction was further examined using various substituted benzaldehyde derivatives with o-phenylenediamine. The results, as shown in Table 6, revealed that the desired benzimidazole products were obtained in yields ranging from 47% to 85%. Product identity was confirmed by melting point analysis, and selected compounds (3c, 3d, 3i, and 3j) were further characterized by 1H NMR spectroscopy (SI).
Table 5 Optimization of 2-substituted benzimidazole derivativesa
| Entry |
Catalyst (mg) |
Solvent |
Oxidant |
Time (h) |
Temp (°C) |
Isolated yield (%) |
| DMF-dimethylformamide, ACN-acetonitrile, DMSO-dimethyl sulfoxide. Reaction condition: 5-bromosalicylaldehyde (1.0 mmol), o-phenylenediamine (1.0 mmol), MnMoO4 catalyst (20 mg), oxidant (0.1 equiv.), and solvent (5 mL). Conversion was determined by thin-layer chromatography (TLC). |
| 1 |
20 |
DMF |
H2O2 |
6 |
60 |
34 |
| 2 |
20 |
DMSO |
H2O2 |
6 |
60 |
Traceb |
| 3 |
20 |
ACN |
H2O2 |
6 |
60 |
42 |
| 4 |
20 |
H2O |
H2O2 |
6 |
60 |
74 |
|
5
|
20
|
EtOH
|
H
2
O
2
|
6
|
60
|
85
|
| 6 |
20 |
EtOH |
— |
6 |
60 |
No reactionb |
| 7 |
20 |
EtOH |
H2O2 |
4 |
60 |
81 |
| 8 |
20 |
EtOH |
H2O2 |
6 |
40 |
79 |
| 9 |
— |
EtOH |
H2O2 |
6 |
60 |
No reactionb |
| 10 |
30 |
EtOH |
H2O2 |
6 |
60 |
83 |
| 11 |
10 |
EtOH |
H2O2 |
6 |
60 |
69 |
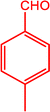
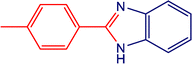
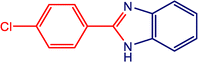
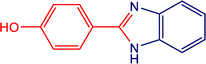
Table 6 Oxidative condensation of o-phenylenediamine with substituted-benzaldehydes and its substrate scopea
|
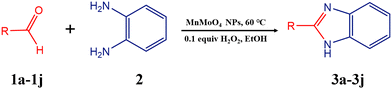
|
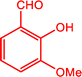
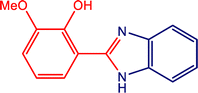
| Entry |
–R (aldehyde) |
Product |
Yieldb (%) |
M
p (°C) |
|
Reaction conditions: substituted benzaldehyde (1.0 mmol), o-phenylenediamine (1.0 mmol), MnMoO4 catalyst (20 mg), oxidant (0.1 equiv.), and solvent (5 mL).
Yield (%): isolated yield.
3a, 3b, 3e, 3f, and 3g were characterized by comparing the melting points with those of samples of reported methods and by comparison with TLC results.
3c, 3d, 3h, 3i, and 3j were characterized by 1H NMR spectral analysis.
|
|
3a
|

|
|
83 |
291–292c (ref. 40) |
|
3b
|
|
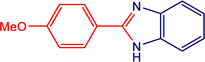
|
68 |
224–225c (ref. 40) |
|
3c
|

|
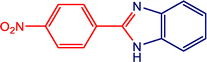
|
59 |
320–322d (ref. 40) |
|
3d
|
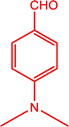
|
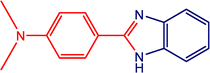
|
66 |
272–274d (ref. 40) |
|
3e
|
|
|
69 |
269–270c (ref. 40) |
|
3f
|

|
|
47 |
294–295c (ref. 40) |
|
3g
|

|
|
60 |
286–287c (ref. 41) |
|
3h
|
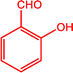
|
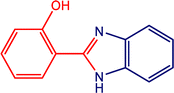
|
62 |
181–182c (ref. 41) |
|
3i
|
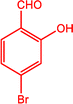
|
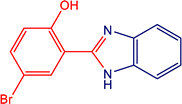
|
85 |
256–257d (ref. 41) |
|
3j
|
|
|
73 |
95–97d (ref. 41) |
3.7.1. Plausible reaction mechanism.
A plausible mechanism for the MnMoO4-catalyzed synthesis of 2-substituted benzimidazoles is illustrated in Scheme 1. Initially, MnMoO4 activated the carbonyl group of the aromatic aldehyde, thereby enhancing the electrophilicity of the carbonyl carbon. This activation facilitated its condensation with one of the –NH2 groups of o-phenylenediamine to generate a Schiff base intermediate. Subsequently, the second –NH2 group of o-phenylenediamine nucleophilically attacked the carbon of the imine intermediate, leading to intramolecular cyclization and the formation of a five-membered heterocyclic ring. Finally, under the oxidative conditions provided by H2O2, the intermediate underwent aromatization to afford the desired 2-substituted benzimidazole.
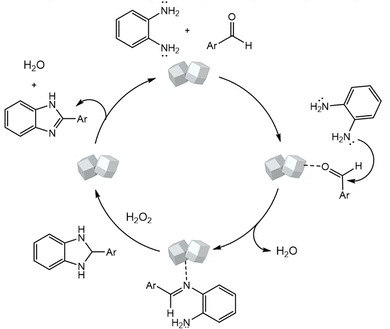 |
| | Scheme 1 Plausible mechanism for the synthesis of the benzimidazole derivatives. | |
3.7.2. Green chemistry parameters.
The green chemistry metrics for the synthesis of the model benzimidazole product were systematically evaluated to assess the environmental sustainability of the developed protocol. Key parameters, including the E-factor, atom economy, reaction mass efficiency, and Eco-scale, were calculated under optimized conditions, and the results are summarized in Table 7. A radar chart, as shown in Fig. 10, was constructed to visualize these metrics, revealing a synergistic relationship that underscored the overall greenness of the synthetic process. Detailed step-by-step calculations for each parameter are provided in the SI.
Table 7 Evaluation of green chemistry metrics
| Yield (%) |
E-factor |
Reaction mass efficiency |
Atom economy |
Eco-scale |
| 85 |
0.34 |
74.43 |
93.28 |
81 |
 |
| | Fig. 10 Radar chart plot of the green chemistry metrics calculated for the synthesis of model reaction product 3i. | |
3.7.3. Reusability of the MnMoO4 nano-catalyst.
Considering the principles of green chemistry and industrial scalability, catalyst recovery and reusability are essential factors for practical applications. To assess these, the recyclability of the MnMoO4 nano-catalyst was examined using o-phenylenediamine and 5-bromosalicylaldehyde as model substrates. Upon the completion of the reaction, the catalyst was recovered by simple filtration, thoroughly washed with ethanol, and dried at 60 °C under a vacuum. The recycled catalyst was subsequently reused in consecutive runs of the same model reaction. As shown in Fig. S11, the catalyst maintained high efficiency for up to four successive cycles without significant loss of activity. The slight decrease in performance observed in later cycles may be attributed to partial pore blockage by adsorbed reactants or products under identical reaction conditions. These results demonstrated that the MnMoO4 nano-catalyst was not only highly effective but also readily recoverable and reusable, underscoring its promise for sustainable catalytic processes.
4. Conclusion and future prospects
In this study, bimetallic α-MnMoO4 nanoparticles were successfully synthesized through a facile and eco-friendly one-step solution combustion method using Arachis hypogaea seed powder as a sustainable green fuel. Comprehensive characterization using XRD, FTIR, UV-vis, PL, SEM, and EDX techniques confirmed that the as-prepared α-MnMoO4 nanoparticles possessed well-defined structural, morphological, and optical properties. Owing to their highly negative surface charge, the nanoparticles demonstrated excellent adsorption performance toward methylene blue (MB), achieving an adsorption efficiency of 86.70%. Additionally, the α-MnMoO4 nanoparticles exhibited strong photocatalytic activities, enabling 81.55% degradation of MB through a mechanism dominated by the generation of reactive hydroxyl radicals via photo-oxidation processes. Beyond dye remediation, the multifunctionality of the nanoparticles was further explored using o-phenylenediamine (OPD), a toxic aromatic diamine commonly found in industrial effluents from dye, pharmaceutical, and fine-chemical manufacturing. Rather than arising as a dye degradation product, OPD was intentionally used as a model pollutant and was subsequently upgraded through oxidative condensation with a series of substituted aromatic aldehydes to synthesize benzimidazole derivatives. The reaction proceeded efficiently in the presence of α-MnMoO4 NPs, producing benzimidazoles in yields ranging from 35% to 85%, depending on the electronic characteristics of the substituents. This integrated strategy highlighted the dual role of the α-MnMoO4 nanoparticles in environmental remediation and green synthesis: effectively removing hazardous dyes from aqueous media while catalyzing the transformation of industrial effluents into high-value chemical products. The demonstrated stability, reusability, and photostability of the nanoparticles further strengthened their suitability for practical deployment in wastewater treatment and sustainable fine-chemical production. Future work may expand the utility of the α-MnMoO4-based nanocatalysts as cost-effective, environmentally benign, and multifunctional materials for broad applications in environmental protection and green chemistry.
Conflicts of interest
There are no conflicts to declare.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5ma01046b.
Acknowledgements
The authors would like to thank Christ University, Bangalore, and Energy Materials Research Laboratory, Siddaganga Institute of Technology, Tumakuru, for their constant support and encouragement for this research.
References
- Y. Wang, X. Sun, T. Xian, G. Liu and H. Yang, Opt. Mater., 2021, 113, 110853, DOI:10.1016/j.optmat.2021.110853.
- A. R. Khataee, M. N. Pons and O. Zahraa, J. Hazard. Mater., 2009, 168, 451–457, DOI:10.1016/j.jhazmat.2009.02.052.
- P. Ahuja, S. K. Ujjain, R. Kanojia and P. Attri, J. Compos. Sci., 2021, 5, 82, DOI:10.3390/jcs5030082.
- R. Kishor, D. Purchase, G. D. Saratale, R. G. Saratale, L. F. R. Ferreira, M. Bilal, R. Chandra and R. N. Bharagava, J. Environ. Chem. Eng., 2021, 9, 105012, DOI:10.1016/j.jece.2020.105012.
- K. S. Rajmohan, R. Chandrasekaran and S. Varjani, Indian J. Microbiol., 2020, 60, 125–138, DOI:10.1007/s12088-019-00841-x.
- S. Wong, N. A. Ghafar, N. Ngadi, F. A. Razmi, I. M. Inuwa, R. Mat and N. A. S. Amin, Sci. Rep., 2020, 10, 2928, DOI:10.1038/s41598-020-60021-6.
- M. R. Gadekar and M. M. Ahammed, Desalin. Water Treat., 2016, 57, 26392–26400, DOI:10.1080/19443994.2016.1165150.
- H. M. Diallo, H. Ayyoub, F. Elazhar, M. Tahaikt, A. Elmidaoui and M. Taky, Phys. Chem. Earth, Parts A/B/C, 2024, 135, 103672, DOI:10.1016/j.pce.2024.103672.
- P. Thiripelu, J. Manjunathan, M. Revathi and P. Ramasamy, J. Water Process Eng., 2024, 58, 104815, DOI:10.1016/j.jwpe.2024.104815.
- J. Ayach, W. El Malti, L. Duma, J. Lalevée, M. Al Ajami, H. Hamad and A. Hijazi, Polymers, 2024, 16, 1959, DOI:10.3390/polym16141959.
-
A. Mohanty, S. Sharma and S. S. Meena, Microbial Metagenomics in Effluent Treatment Plant, 2024, pp. 167–183 DOI:10.1016/B978-0-443-13531-6.00010-0.
- M. M. El-Sadaawy and N. S. Agib, Blue Econ., 2024, 2, 1, DOI:10.57241/2805-2994.1023.
- Y. Khan, M. N. Khan, A. Salam, H. Sadia, M. F. Ullah, M. I. Khan, B. S. Abdullaeva, F. A. Awwad and E. A. Ismail, Open Chem., 2024, 22, 20240026, DOI:10.1515/chem-2024-0026.
- F. Mohamadpour and A. M. Amani, RSC Adv., 2024, 14, 20609–20645, 10.1039/D4RA03259D.
- A. Mishra, A. Mehta and S. Basu, J. Environ. Chem. Eng., 2018, 6, 6088–6107, DOI:10.1016/j.jece.2018.09.029.
- S. Vasantharaj, S. Sathiyavimal, P. Senthilkumar, V. N. Kalpana, G. Rajalakshmi, M. Alsehli, A. Elfasakhany and A. Pugazhendhi, J. Environ. Chem. Eng., 2021, 9, 105772, DOI:10.1016/j.jece.2021.105772.
- A. Fakhri and S. Behrouz, Sol. Energy, 2015, 112, 163–168, DOI:10.1016/j.solener.2014.11.014.
- S. Alghool, H. F. Abd El-Halim and A. M. Mostafa, J. Inorg. Organomet. Polym. Mater., 2019, 29, 1324–1330, DOI:10.1007/s10904-019-01096-1.
- P. Bansal, G. R. Chaudhary and S. K. Mehta, Chem. Eng. J., 2015, 280, 475–485, DOI:10.1016/j.cej.2015.06.039.
- G. Elango and S. M. Roopan, J. Photochem. Photobiol., B, 2016, 155, 34–38, DOI:10.1016/j.jphotobiol.2015.12.010.
- R. A. Amoresi, R. C. Oliveira, N. L. Marana, P. B. De Almeida, P. S. Prata, M. A. Zaghete, E. Longo, J. R. Sambrano and A. Z. Simoes, ACS Appl. Nano Mater., 2019, 2, 6513–6526, DOI:10.1021/acsanm.9b01452.
- T. S. Anirudhan, F. Shainy, V. C. Sekhar and V. S. Athira, J. Photochem. Photobiol., A, 2021, 418, 113333, DOI:10.1016/j.jphotochem.2021.113333.
- G. A. R. Bari, M. Islam and J. H. Jeong, Metals, 2024, 14, 423, DOI:10.3390/met14040423.
- A. Di Mauro, M. M. Natile, A. Landström, I. Concina, M. Ferroni, V. Privitera, G. Impellizzeri and M. Epifani, J. Photochem. Photobiol., A, 2021, 413, 113258, DOI:10.1016/j.jphotochem.2021.113258.
- H. Shoukat, A. A. Altaf, M. Hamayun, S. Ullah, S. Kausar, M. Hamza, S. Muhammad, A. Badshah, N. Rasool and M. Imran, ACS Omega, 2021, 6, 19606–19615, DOI:10.1021/acsomega.1c02163.
- T. E. Balaji, H. Tanaya Das and T. Maiyalagan, ChemElectroChem, 2021, 8, 1723–1746, DOI:10.1002/celc.202100098.
- Y. Fu, J. Zhang, H. Wang, L. Tao, S. Liu, Y. Wang, S. Wang and J. Liu, J. Electroanal. Chem., 2023, 942, 117561, DOI:10.1016/j.jelechem.2023.117561.
- N. Devi, S. K. Ghosh, V. K. Perla and K. Mallick, ACS Omega, 2020, 5, 18693–18699, DOI:10.1021/acsomega.0c01576.
- A. Nyabadza, É. McCarthy, M. Makhesana, S. Heidarinassab, A. Plouze, M. Vazquez and D. Brabazon, Adv. Colloid Interface Sci., 2023, 103010, DOI:10.1016/j.cis.2023.103010.
- B. V. N. Sahithi and V. V. Lakshmaiah, RSC Adv., 2025, 15, 28721–28729, 10.1039/D5RA04728E.
- S. Hegde, A. Venkatesan, A. Nizam, V. V. Lakshmaiah and S. B. N. Krishna, Tetrahedron, 2025, 185, 134813, DOI:10.1016/j.tet.2025.134813.
- U. O. Aigbe and A. O. Osibote, J. Hazard. Mater. Adv., 2024, 100401, DOI:10.1016/j.hazadv.2024.100401.
- A. Venkatesan, A. Nizam, A. R. Cherian, R. Patel, J. Xavier, K. R. P and G. N, RSC Adv., 2025, 15, 32638–32653, 10.1039/D5RA05696A.
- Hisana, A. Shahzaib, N. Nishat, S. M. Alshehri, T. Ahamad and Z. Haque, Hybrid Adv., 2024, 5, 100145, DOI:10.1016/j.hybadv.2024.100145.
- P. Yadav, P. Kakati, P. Singh and S. K. Awasthi, Appl. Catal., A, 2021, 612, 118005, DOI:10.1016/j.apcata.2021.118005.
- Y. Zhang, G. Du, X. Dong, H. Li, S. Zeng, C. Cui, C. Fu and L. Wang, J. Alloys Compd., 2023, 944, 169105, DOI:10.1016/j.jallcom.2023.169105.
- A. Negash, S. Mohammed, H. D. Weldekirstos, A. D. Ambaye and M. Gashu, Sci. Rep., 2023, 13(1), 22234, DOI:10.1038/s41598-023-48826-7.
- M. B. Goudjil, H. Dali, S. Zighmi, Z. Mahcene and S. E. Bencheikh, Desalin. Water Treat., 2024, 317, 100079, DOI:10.1016/j.dwt.2024.100079.
- S. Ali, H. Ijaz, M. U. Ahmad, Rukhma, N. Ullah, A. Sarwar, M. J. Khan, T. Aziz, A. Shami and F. Al-Asmari, Sci. Rep., 2025, 15, 1851, DOI:10.1038/s41598-025-85894-3.
- K. Bahrami and Z. Karami, J. Exp. Nanosci., 2018, 13, 272–283, DOI:10.1080/17458080.2018.1542511.
- X. He, Y. Wu, W. Jin, X. Wang, C. Wu and Y. Shang, Appl. Organomet. Chem., 2018, 32, e4284, DOI:10.1002/aoc.4284.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Aatika
Nizam
a,
Aatika
Nizam



















 versus reaction time described by the Langmuir and Hinshelwood model, defined as eqn (16) and (17).
versus reaction time described by the Langmuir and Hinshelwood model, defined as eqn (16) and (17).
























