
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEco-friendly cyanation strategies of aryl halides using recyclable nickel nanocatalysts with promising antibacterial and antioxidant activities
Asit
Kumar Das
 *a,
Md Sattar
Ali
a,
Arindam
Misra
a,
Md Sultan
Saikh
a,
Subhendu
Dhibar
b,
Sumit Kumar
Panja
c,
Aniruddha
Das
d,
Gourav
Ghatak
e,
Lokesh Kumar
Rathore
f,
Ashok
Bera
f,
Sanjay
Bhar
g and
Smritikana
Biswas
*e
*a,
Md Sattar
Ali
a,
Arindam
Misra
a,
Md Sultan
Saikh
a,
Subhendu
Dhibar
b,
Sumit Kumar
Panja
c,
Aniruddha
Das
d,
Gourav
Ghatak
e,
Lokesh Kumar
Rathore
f,
Ashok
Bera
f,
Sanjay
Bhar
g and
Smritikana
Biswas
*e
aDepartment of Chemistry, Murshidabad University, Berhampore, 742101, India. E-mail: akdche@msduniv.ac.in
bDepartment of Physics, Indian Institute of Technology Patna, Bihar, 801106, India
cTarsadia Institute of Chemical Science, Uka Tarsadia University, Surat, 394350, India
dDepartment of Chemistry, Shiv Nadar Institution of Eminence Deemed to be University, Uttar Pradesh, 201314, India
eDepartment of Physiology, Murshidabad University, Berhampore, 742101, India. E-mail: physio.smriti2005@gmail.com
fDepartment of Physics, Indian Institute of Technology Jammu, Jammu, J & K 181221, India
gDepartment of Chemistry, Jadavpur University, Kolkata-700032, India
First published on 15th January 2026
Abstract
Recyclable nickel nanoparticles have been utilized as an efficient, stable, heterogeneous catalyst for the synthesis of aryl nitriles using commercially available and less toxic K4[Fe(CN)6]·3H2O as an environmentally benign cyanide source. The reactions are not dependent on an inert atmosphere or a ligand. Several aryl chlorides, aryl bromides, and aryl iodides survived well and were associated with high yield in the aforesaid method. The synthesized Ni-γAl2O3 nanocatalysts could be recovered and recycled again without significantly reducing their efficacy. Moreover, “Sheldon's test (hot filtration method)” was carried out to establish the heterogeneity of the catalyst. The significant benefits of this catalytic methodology align with green chemistry principles, making this process potentially applicable in industrial chemistry. The synthesized Ni-γAl2O3 nanocatalysts exhibited moderate antioxidant activity, with maximum antioxidant activity (68.17%) at 200 mg mL−1 concentration. Ni-γAl2O3 nanocatalysts were found to be effective against Staphylococcus aureus ATCC25923 (Gram-positive) and Escherichia coli (Gram-negative) with zones of inhibition of 10 ± 0.25 mm and 12 ± 0, respectively. MIC values against Escherichia coli (Gram-negative) and Staphylococcus aureus ATCC25923 (Gram-positive) were 200 mg mL−1 and 205 mg mL−1, respectively, while MBC values were 220 and 230 mg mL−1 for Staphylococcus aureus ATCC25923 and Escherichia coli, respectively. This study is provided to demonstrate the dual applicability of the recyclable Ni-γAl2O3 nanocatalyst for a green synthetic route to aryl nitriles, and to exhibit potential antibacterial and antioxidant activity.
1. Introduction
Nanomaterials have emerged as one of the most promising tools for biomedical and pharmaceutical applications.1 It is necessary to stop the risk of pathogenic bacteria in terms of community health safety. Numerous types of antibacterial agents are emerging. However, adverse effects and the consecutive use of antibacterial agents lead to antibiotic resistance.2–6 Concerning the recent trend in the substitute antimicrobial strategy, metal nanoparticles have promising dynamic and potential applications due to their unique chemical and physical properties.2,7 Although nickel nanoparticles (Ni NPs), a metal nanoparticle, exhibit significant and effective antimicrobial properties against several kinds of bacterial strains, including pathogenic ones.8 Larger surface area, high reactivity of Ni NPs boosts and induces their antibacterial property.Aryl nitriles represent an important structural framework in organic chemistry because of their multifarious application in diverse disciplines, including polymers, materials, pharmaceuticals, agrochemicals, dyes, pigments, natural products, and biologically active compounds.9 In addition, nitriles are straightforwardly converted into a range of several efficient scaffolds such as amidines, amides, oximes, aldehydes, ketones, esters, and carboxylic acids.10 Benzonitriles are key intermediates in organic synthesis and are used as industrially important solvents for several significant organic transformations.11 Furthermore, nitrile moieties are essential building blocks in pharmaceutically potent drugs, such as Letrozole®, Finrozole®, Citalopram®, Etravirine®, Periciazine®, Bicalutamide®, and 5-lipoxygenase inhibitors have been recognised (Fig. 1).12 Historically, two conventional methods have been employed to synthesize aryl nitriles: diazotization of aromatic amines followed by a Sandmeyer reaction13a–d and Rosenmund–von Braun reaction of aromatic halides.13e However, these reactions are associated with serious disadvantages, such as high temperature, requirement of a super-stoichiometric amount of extremely poisonous CuCN as cyanating agent, and generation of a major amount of heavy metal waste, leading to unavoidable environmental complications.
 | ||
| Fig. 1 Some pharmaceutically potential organonitrile drugs. | ||
Therefore, the development of highly efficient and eco-friendly methodologies for the synthesis of important organic molecules is of significant importance.14 In this context, the transition metal-mediated cyanation of aryl halides15 for synthesizing the corresponding nitriles has gained immense attention from the scientific community. In 1973, Takagi et al. first developed the Pd-catalyzed cyanation reaction of aryl bromides and aryl iodides using KCN as a cyanide source at 140–150 °C.16 Later on, numerous transition metals, including Pd,17a Rh,17b Ir,17c and Cu17d were used in the synthesis of nitriles in the presence of toxic cyanating agents, such as Zn(CN)2,18a CuCN,18b TMSCN,18c KCN,18d and NaCN.17a,18e Some less toxic nonmetallic agents such as aliphatic nitriles,19a cyanohydrins from acetone,19b benzyl thiocyanate,19c phenyl cyanate,19d and N-cyanobenzimidazole19e were also utilized with the employment of various hazardous and expensive nitrogen and phosphorus ligands. However, most of these protocols were highly toxic to humans and the environment and posed a high-risk during handling and workup procedures, which limited their industrial application. Therefore, the situation demanded environmentally benign chemical processes20 for the synthesis of aryl nitriles using a non-hazardous, economically safe cyanating agent. Beller and co-workers17a first reported the use of a commercially available eco-friendly K4[Fe(CN)6] as a cyanating agent for the cyanation of aryl halides with Pd(OAc)2 as the metal precursor with dppf as the ligand. Recently, nickel-catalyzed21 cyanation reactions (Scheme 1) have drawn significant attention due to their ease of accessibility, lower toxicity, and their inexpensive, eco-friendly nature compared to reported metal-mediated reactions. Although the nickel-catalysed developed procedures (Scheme 1) are quite acceptable, their limitations are significant because of the involvement of expensive cyanide sources, perilous ligands, laborious catalyst preparation, necessity of additives, generation of metal waste, recyclability problem of the catalysts, and difficult work-up methods that are less eco-friendly from the perspective of sustainability.22
 | ||
| Scheme 1 Ni-catalysed different approaches for the cyanation of aryl halides. | ||
Therefore, considering the present environmental scenario, there is an enormous demand to develop highly effective approaches23 for the synthesis of nitriles that refrain from utilizing expensive and harmful metal catalysts and rather employ less hazardous and less expensive reagents. However, in continuing on our previous work utilizing nickel nanocatalysts,24 we have reported here the excellent catalytic attributes of Ni-γAl2O3 nanocatalyst (Scheme 1) for the efficient synthesis of aryl nitriles from the cyanation reaction of aryl halides using innocuous K4[Fe(CN)6]·3H2O as an eco-friendly cyanide source.
2. Experimental
2.1. Preparation of nickel nanocatalysts
Nickel acetylacetonate (Merck), hydrazine hydrate (Sigma Aldrich), sodium hydroxide (Merck), PEG-400 (Loba Chemie), and ethanol (Merck) have been used as received. Every experiment was conducted using deionized water. The synthetic method involves dissolving 5 mmol of Ni(acac)2 (nickel acetylacetonate) in 20 mL of PEG-400 solvent, and it was magnetically stirred for 20 minutes at room temperature. Then, 2 mL of 0.1 M NaOH was added, followed by 2 mL of hydrazine hydrate. Here, hydrazine hydrate serves as a reducing agent, and NaOH maintains the reaction alkaline. By this time, the reaction mixture had changed color from green to deep blue. The reaction mixture was stirred magnetically at 80 °C for 2 h, and a black precipitate was obtained, which indicates the reduction of Ni2+ to Ni0. Then, γ-Al2O3 (5 g) was added to the aforesaid mixture containing nickel nanoparticles, and the reaction mixture was magnetically stirred for 1 h. The reaction mixture was then cooled to room temperature, washed successively with ethanol and water. The resultant residue was dried in an oven at 120 °C for 24 h. Then it was stored under ambient conditions for further investigation. Fig. 2 presents the schematic presentation used in this work for synthesizing heterogeneous nickel nanocatalysts. | ||
| Fig. 2 Schematic representation for the synthesis of Ni-γAl2O3 nanocatalyst. | ||
2.2. Ni-γAl2O3 catalysed general experimental procedure for the ligand-free cyanation of aryl halides
A solution of appropriate aryl halides 1 (1 mmol), K4[Fe(CN)6]·3H2O (0.2 mmol) and K2CO3 (1.2 mmol) in DMF (3 mL), Ni-γAl2O3 (6 mol%) was added and stirred at 120 °C for the specified time. TLC was used to monitor the reaction. The reaction mixture was cooled to room temperature upon completion. The product was then dissolved by adding 15 mL of ethyl acetate. The catalyst was removed by simple filtration. The recovered catalyst was thoroughly washed with ethyl acetate, followed by H2O. Ethyl acetate was used several times to extract the aqueous part. The organic extracts were washed using water and dried on anhydrous Na2SO4. Product 2 was produced by evaporating the solvent at lowered pressure. Then it was further purified by column chromatography on silica gel using EtOAc-hexane as eluent.2.3. Antioxidant activity of Ni-γAl2O3
Antioxidant activity of the synthesized Ni-γAl2O3 nanocatalysts was assessed using the DPPH free radical scavenging assay.25 First, 200 µL of a DPPH methanol solution was mixed with a different concentration of the samples (200, 100, 80, 40, 10 mg mL−1) followed by incubation at 37 °C for 30 min, and absorbance was noted at the wavelength of 517 nm using a UV-VIS dual beam spectrophotometer. The reduction capacity was estimated in terms of ascorbic acid per mg. DPPH free radical scavenging potential (in percentage) was estimated by using the following equation.| DPPH free radical scavenging activity (%) = (Acontrol − Asample)/Acontrol × 100 |
2.4. Antibacterial assay of Ni-γAl2O3
2.5. Minimum inhibitory (MIC) and minimum bactericidal concentrations (MBC) determination
The minimum inhibitory concentration was determined using the dilution method. Ni-γAl2O3 NPs were diluted to attain the concentrations of 250, 240, 230, 220, 210, 205, 200, 195, 190 mg mL−1, respectively. Then, 100 µL of the respective bacterial suspension (105 CFU mL−1) was spread over a Mueller–Hinton agar plate, followed by the addition of 40 µL of each dilution of Ni-γAl2O3 NPs to the respective wells in the plate. After that, these plates were incubated at 37 °C for 24–48 h. The lowest concentration of Ni-γAl2O3 NPs that produced a clearing zone was denoted as MIC value, whereas the concentration that killed all bacterial cells was considered as MBC (minimum bactericidal concentration).3. Results and discussion
3.1. Catalyst characterization
The FTIR spectra of γAl2O3 and the synthesized Ni-γAl2O3 nanocatalyst are shown in Fig. 3. The FTIR spectra of γAl2O3 displayed the broad bands in the 3620–3032 cm−1 region, because of the existence of several hydroxyl groups on the γAl2O3 surfaces. Moreover, the FTIR spectra of γAl2O3 showed absorption peaks at 2090, 1966, and 1639 cm−1, which are assigned to the bending mode of vibrations of adsorbed water molecules.27 However, the intensities of these bands were reduced in the FTIR spectrum of the synthesized Ni-γAl2O3 nanocatalyst. These findings suggested the effective encapsulation of the metal into the pores of γAl2O3. The similar IR peaks at 1448 cm−1, 1159 cm−1, 1072 cm−1 in the γAl2O3 and 1460 cm−1, 1162 cm−1, 1078 cm−1 in the synthesized Ni-γAl2O3 nanocatalyst were also observed. This observation showed the effective formation of Ni-γAl2O3 nanocatalyst through the complexation and stabilization of nanoparticles within the pores of γAl2O3. The FTIR spectra of Ni-γAl2O3 nanocatalyst exhibited the absorption peaks at 736 cm−1, 619 cm−1, and 495 cm−1 due to the stretching and bending vibrations between Ni–O, Ni–Al, and Al–O bonds.28 | ||
| Fig. 3 FTIR analysis of γAl2O3 and synthesized Ni-γAl2O3 nanocatalyst. | ||
We performed powder X-ray diffraction analysis of the prepared Ni-γAl2O3 nanocatalyst to determine its crystallinity. The X-ray diffraction patterns of the prepared Ni-γAl2O3 catalysts are displayed in Fig. 4. The diffraction peaks exhibit that Al2O3 exists in gamma (γ) crystalline phase. The peak intensities confirm that the particles are crystalline. The appearance of diffraction peaks at 2θ = 44.5° and 2θ = 51.7° can be ascribed to the Ni(111) and Ni(200) crystalline planes, respectively. These characteristic peaks also indicate the metallic nickel phase with fcc structure. This is also consistent with the reported literature.29 The characteristic 2θ values of the nickel also revealed that the Ni2+ ions were completely reduced to Ni0, and the synthesized Ni0 was incorporated within the pores of γAl2O3 support.
 | ||
| Fig. 4 X-ray diffraction analysis of the synthesized Ni-γAl2O3 nanocatalyst. | ||
The TEM and HRTEM images of the synthesized Ni-γAl2O3 nanocatalyst are illustrated in Fig. 5. The TEM image of the synthesized Ni-γAl2O3 nanocatalyst reveals a layered structure, suggesting a well-defined architecture (Fig. 5a). Moreover, a good dispersion of metallic Ni on the surfaces of γAl2O3 support was observed in the structure of Ni-γAl2O3 nanocatalyst. Fig. 5b and c indicate that the Ni nanoparticles have an average size of 4–7 nm, demonstrating the nanoscale diameter of the active metal component. Remarkably, the HRTEM image in Fig. 5c revealed the clear lattice fringes, further confirming the good dispersion and crystallinity of the synthesized nanomaterials on the surfaces of γAl2O3 support. Fig. 5c also exhibits the lattice fringe distances of 0.20 and 0.17 nm, which can be attributed to the d-spacing values of the metallic Ni(111) and Ni(200), respectively.30 Besides, the SAED (selected area electron diffraction) pattern substantiated the crystalline structure (Fig. 5d), showing distinct diffraction spots, which further established the well-ordered arrangement and high crystallinity of the Ni-γAl2O3 nanocatalyst.
 | ||
| Fig. 5 TEM and HRTEM images (a)–(c) and SAED pattern (d) of synthesized Ni-γAl2O3 nanocatalysts. | ||
We next recorded the scanning electron microscopy (SEM) images of the prepared Ni-γAl2O3 catalyst, which are depicted in Fig. 6(a–d). The results showed that the synthesized Ni-γAl2O3 catalyst displays a distinctly crystalline structure. Furthermore, the nanoparticles were irregularly distributed on the γAl2O3 support, indicating a non-uniform arrangement. This irregularity could affect the catalytic performance and efficiency of nanocatalysts across a variety of catalytic applications. This finding is also corroborated by the aforesaid TEM images.
 | ||
| Fig. 6 Scanning electron microscopy (SEM) images (a)–(d) of synthesized Ni-γAl2O3 nanocatalysts. | ||
To investigate the elemental composition of the prepared Ni-γAl2O3 nanocatalysts, an EDX (energy dispersive X-ray spectroscopy) study was also conducted. Fig. 7 shows the elemental peaks, which correspond to Ni, Al, and O in the synthesized Ni-γAl2O3 catalyst. Besides, the elemental mapping images of the synthesized Ni-γAl2O3 catalyst were also carried out. The distribution of Ni, Al, and O elements was also examined by mapping, and well-ordered distributions of each element were observed in the synthesized Ni-γAl2O3 nanocatalyst (Fig. 8).
 | ||
| Fig. 7 Energy dispersive X-ray spectroscopy analysis of Ni-γAl2O3 nanocatalyst. | ||
 | ||
| Fig. 8 Elemental mapping images of the synthesized Ni-γAl2O3 nanocatalyst. | ||
The N2 adsorption–desorption study of the synthesized Ni-γAl2O3 catalyst (Fig. 9a) demonstrates a type IV isotherm, supporting the presence of a mesoporous structure. BET analysis showed a high specific surface area of 173 m2 g−1, confirming the porous γAl2O3 support and homogeneous distribution of Ni species. The BJH desorption pore-size distribution (Fig. 9b) reveals a predominant mesopore population centered at around 4 nm as well as a secondary broader distribution around 7–8 nm, confirming the bimodal mesoporosity of the Ni-γAl2O3 catalyst. Such a large, high-surface-area and accessible mesopores are expected to facilitate the effective diffusion of aryl halides and cyanide released from K4[Fe(CN)6]·3H2O to the active Ni sites, thereby corroborating the observed catalytic efficiency in the cyanation of aryl halides.
 | ||
| Fig. 9 N2 adsorption/desorption isotherm (a) and pore size distribution (b) of synthesized Ni-γAl2O3 nanocatalyst. | ||
Thermogravimetric analysis (TGA) was conducted to examine the thermal behavior of the as-synthesized Ni-γAl2O3 catalyst. The TGA curve (Fig. S1, SI) shows a gradual multi-step weight loss (∼12 wt% total) between 25 °C and 600 °C. The first weight loss (4.26%) up to ∼200 °C is associated with the elimination of physically adsorbed moisture and residual water desorption from the high-surface-area of γAl2O3 support. The second weight decrease (4.17%) observed between ∼200 °C and 385 °C is assigned to the decomposition of residual organic species originating from acetylacetonate ligand, PEG-400, and hydrazine used during synthesis. The last weight loss (3.71%) between ∼385 °C and 520 °C is attributed to the oxidation of metallic Ni(0) nanoparticles to NiO on the γAl2O3 surface. Above 520 °C, the TGA curve remains essentially stable, demonstrating the complete elimination of volatile species and indicating the high thermal stability of the γ-Al2O3 support. Overall, the TGA analysis validates the effective formation of Ni-γAl2O3 catalyst, successful elimination of most organic residues, and the excellent thermal stability of the Ni-γAl2O3 catalyst.
These detailed characterizations highlight the unique applicability of the synthesized Ni-γAl2O3 nanocatalyst across a range of catalytic applications, owing to its unusual crystalline characteristics.
3.2. Catalytic activity of the Ni-γAl2O3 nanocatalyst
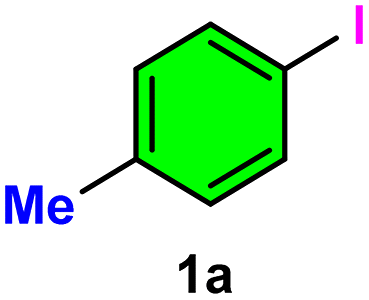
The catalytic efficiency of Ni-γAl2O3 was investigated in the cyanation of aryl halides (such as aryl iodides, aryl bromides, and aryl chlorides) using readily available, non-toxic, and cost-effective K4[Fe(CN)6]·3H2O as a green cyanide source. To achieve optimal conditions, several control experiments were investigated using 4-iodotoluene 1a (1 mmol) as the model substrate and K4[Fe(CN)6]·3H2O as the safe cyanide source in the presence of different bases in several solvents at various temperatures with the alteration of catalyst amount and reaction time (Table 1).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Base (mmol) | Solvent (mL) | Temperature (°C) | Time (h) | Yieldb (%) |
| a Reaction conditions: 1a (1.0 mmol), K4[Fe(CN)6]·3H2O (0.2 mmol); base (1.2 mmol), solvent (3 mL), catalyst, and temperature (as indicated) under ambient condition. b Isolated yield. | ||||||
| 1 | — | — | DMF | Reflux | 14 | — |
| 2 | — | K2CO3 | DMF | Reflux | 14 | — |
| 3 | 2 | — | DMF | Reflux | 14 | Trace |
| 4 | 4 | K2CO3 | DMF | 120 | 12 | 68 |
| 5 | 5 | K2CO3 | DMF | 120 | 12 | 74 |
| 6 | 6 | K 2 CO 3 | DMF | 120 | 12 | 92 |
| 7 | 7 | K2CO3 | DMF | 120 | 12 | 93 |
| 8 | 6 | Na2CO3 | DMF | 120 | 12 | 85 |
| 9 | 6 | NaOH | DMF | 120 | 12 | 22 |
| 10 | 6 | KOH | DMF | 120 | 12 | 27 |
| 11 | 6 | Et3N | DMF | 120 | 12 | Trace |
| 12 | 6 | Pyridine | DMF | 120 | 12 | Trace |
| 13 | 6 | K2CO3 | DMSO | Reflux | 12 | 68 |
| 14 | 6 | K2CO3 | Toluene | Reflux | 12 | — |
| 15 | 6 | K2CO3 | Xylene | Reflux | 12 | — |
| 16 | 6 | K2CO3 | H2O | Reflux | 12 | — |
| 17 | 6 | K2CO3 | CH3CN | Reflux | 12 | 29 |
| 18 | 6 | K2CO3 | EtOAc | Reflux | 12 | 35 |

The reaction initially failed without using Ni-γAl2O3 and base (entry 1), and in the presence of base (entry 2), no nitrile product 2a was isolated. However, in the absence of base, only a small amount of 2a was observed after 14 h when the reaction was examined with 2 mol% of catalyst under reflux conditions in DMF solvent (entry 3). Using 4 mol% of Ni-γAl2O3 catalyst and 1.2 mmol of K2CO3 base (entry 4), the conversion reached 68% within 12 h at 120 °C. After increasing the catalyst concentration (5 mol%), the conversion was improved to 74% after 12 h (entry 5). Satisfactory outcome was found after 12 h using Ni-γAl2O3 (6 mol%) and K2CO3 (1.2 mmol) (entry 6). Excess catalyst (>6 mol%) showed no further increase in conversion rate (entries 6 and 7). When K2CO3 was replaced with Na2CO3, the reaction yield was slightly reduced to 85% (entry 8). Strong bases such as NaOH (entry 9) and KOH (entry 10) reduced the yield drastically. Treatment with organic bases such as Et3N (entry 11) and pyridine (entry 12) yielded only trace amounts of product under similar reaction conditions. Finally, K2CO3 proved to be the most effective base in DMF medium at 120 °C for the cyanation reaction. Moreover, different solvents were screened. The reaction efficiency slightly decreases when DMSO is used in place of DMF (entry 13). Other solvents, such as toluene, xylene, and water, showed negative results (entries 14–16). Inferior performance was obtained when CH3CN and EtOAc were used as the reaction medium in the cyanation of aryl halides (entries 17 and 18). Therefore, the conditions delineated in entry 6 were selected as the optimized reaction conditions to investigate the substrate scope and ensure the practical applicability of this reaction. The aforesaid protocol did not occur with γAl2O3 alone nor with Ni nano without γAl2O3, nor with Ni(acac)2-γAl2O3. The importance of Ni-γAl2O3 for this cyanation reaction is highly crucial due to its high stability and better catalytic activity.
To establish the general applicability of this protocol, the optimized reaction conditions have been employed for the cyanation of aryl iodides/bromides/chlorides bearing several electron-withdrawing and electron-donating substituents at various places of the benzene. The corresponding nitriles were produced with good to high yields. The results are presented in Table 2.
| Entry | Substrate | Product | Time (h) | Yieldb (%) |
|---|---|---|---|---|
| a Reaction conditions: 1a (1 mmol), K4[Fe(CN)6]·3H2O (0.2 mmol), K2CO3 (0.5 mmol), Ni-γAl2O3 (6 mol%), DMF (3 mL) at 120 °C. b Yield states to the isolated pure product. c In presence of an additive (KI) (1.0 mmol) for bromo and chloro compounds. d In absence of an additive (KI) (1.0 mmol) for bromo and chloro compounds. | ||||
| Cyanation of aryl iodides | ||||
| 1 |
|

|
12 | 92 |
| 2 |

|

|
12 | 88 |
| 3 |

|
|
12 | 91 |
| 4 |

|

|
10 | 94 |
| 5 |

|

|
12 | 93 |
| 6 |

|

|
12 | 92 |
| Cyanation of aryl bromides | ||||
| 7 |

|

|
12c/18d | 88c/32d |
| 8 |

|

|
12c/18d | 90c/35d |
| 9 |

|

|
12c/18d | 88c/30d |
| 10 |

|

|
12c/18d | 92c/33d |
| 11 |

|

|
12c/18d | 91c/30d |
| 12 |

|

|
12c/18d | 80c/25d |
| Cyanation of aryl chlorides | ||||
| 13 |

|

|
12c/22d | 76c/22d |
| 14 |

|

|
12c/22d | 73c/19d |
| 15 |

|

|
12c/22d | 78c/23d |
| 16 |

|

|
12c/22d | 76c/22d |
As evident from Table 2, a wide variety of aryl iodides (1a–1f) bearing electron-donating and electron-withdrawing substituents were transformed into the products (2a–2f) in good to excellent yield. Cyanation of ortho-substituted compounds such as 2-iodotoluene (1b) afforded the corresponding nitrile (2b) under the optimized reaction conditions with slightly lower yield due to steric reasons. 3-Nitroiodobenzene (1d) smoothly cyanated into their corresponding nitrile (2d) with 94% yield within a shorter reaction time without interfering –NO2 groups. The aforesaid protocol displayed selectivity for the cyanation of 4-bromoiodobenzene (1e), producing 4-bromobenzonitrile (1e) with excellent yield without cleaving the C–Br bond. This could be due to the lower bond dissociation energy of the C–I bond compared to the C–Br bond. This reaction is also highly effective for the substrate bearing a highly reducible group. 4-Iodobenzaldehyde (1f) also responded efficiently, providing the corresponding nitrile 2f in good yield within 12 h. The formation of 2f was confirmed by the 1H and 13C NMR spectroscopy. The 1H NMR spectrum of 2f showed the existence of a –CHO group, which appeared as a singlet at δ 10.07 and two doublets at δ 7.975 because of the aromatic ortho protons with respect to the –CHO group and at δ 7.825 because of the aromatic ortho protons with respect to the –CN group. The 13C NMR spectrum of 2f displayed the appearance of two main signals at δ 190.7 and δ 117.8, demonstrating the existence of both –CHO and –CN groups, respectively. We next extended the optimized reaction conditions for a wide variety of aryl bromides and aryl chlorides. The corresponding nitrile was observed with a lower yield even after a longer reaction time. The bond dissociation energy of C–X (X = Cl, Br, I) is C–Cl > C–Br > C–I. Therefore, the oxidative addition of Ni on aryl bromides as well as aryl chlorides is complicated when compared with aryl iodides. Hence, the cyanation of aryl bromides (1g–1l) and aryl chlorides (1m–1p) showed lower yield. However, the reaction yield was comparatively high when the reaction was used as the oxidant. The main role of iodide ion is to catalyze the construction of aryl iodide from aryl bromides and aryl chlorides, and therefore, the in situ cyanation reaction takes place, as reported by Buchwald et al.31 We observed that 1.0 mmol of KI is necessary to endorse the incorporation of iodide into aryl bromides as well as aryl chlorides. Therefore, both differently substituted aryl bromides and aryl chlorides readily furnished their cyanated products with moderate to good yield in the presence of 1.0 mmol of KI as an additive. This is an immensely essential feature of the present method in comparison to reported methods, where no such additional reactivity of aryl bromides and aryl chlorides was observed.17a–d
The plausible reaction pathway for this Ni-γAl2O3 catalysed cyanation of aryl halides is presented in Scheme 2.32a–c The oxidative addition of aryl halides to the nickel metal seems to be the initiation step of this catalytic reaction, and therefore metallic nickel oxidised to Ni(II) species (A). Then the exchange of ligand from the inside coordination sphere of K4[Fe(CN)6] to the Ni(II) species (B) of the catalyst occurs via a transmetallation process. Finally, the reductive removal stage resulted in the formation of arylnitriles with the regeneration of the metallic nickel catalyst.
 | ||
| Scheme 2 Plausible mechanism for the Ni-γAl2O3 catalyzed cyanation of aryl halides. | ||
To design a sustainable protocol, the recoverability and reusability33a–d of the catalyst are highly essential from a green perspective. Therefore, the recycling test of our prepared Ni-γAl2O3 catalyst was performed using 4-iodotoluene 1a (1 mmol), K4[Fe(CN)6]·3H2O (0.2 mmol), K2CO3 (1.2 mmol), Ni-γAl2O3 (6 mol%), DMF (3 mL) at 120 °C. We separated the Ni-γAl2O3 catalyst by simple filtration after the end of the reaction. The crude product was extracted from the filtrate using EtOAc solvent. The recovered catalyst was thoroughly washed using ethyl acetate, then with H2O. The recovered Ni-γAl2O3 catalyst was then dried at 120 °C for 1 hour. The recovered Ni-γAl2O3 catalyst was then applied for a series of catalytic reactions, with little variation in yield (Fig. 10a). The significant recyclability of this Ni-γAl2O3 catalyst prompted us to further analyse the characterization study of the recycled Ni-γAl2O3 to confirm its stability. Therefore, SEM and TEM analyses of the recycled Ni-γAl2O3 were investigated (Fig. 10b and c). The aforesaid studies demonstrate that the structural features of the Ni-γAl2O3 catalyst were relatively stable during this investigation. Moreover, ICP-OES analysis was performed to determine the actual Ni loading and investigate the stability of the Ni-γAl2O3 catalyst. The fresh catalyst contained 5.1 wt% Ni, whereas the recycled catalyst (after the 5th catalytic run) retained 5.04 wt% Ni, showing only 0.06% metal leaching. This negligible loss indicates that Ni remains well dispersed and firmly anchored on the γ-Al2O3 support.
 | ||
| Fig. 10 (a) Recycling test of Ni-γAl2O3 catalyst, (b) SEM image of Ni-γAl2O3 (after recycled 5 times), and (c) TEM image of Ni-γAl2O3 (after recycled 5 times). | ||
3.3. Hot filtration experiment (Sheldon's test) of the Ni-γAl2O3 nanocatalyst
Furthermore, the hot filtration test34a,b (Sheldon's test) was carried out to establish the heterogeneous nature of Ni-γAl2O3 catalyst with 4-iodotoluene (1a) as the model substrate. When 32% conversion of the 1a was observed after 3 h, the separation of Ni-γAl2O3 catalyst was carried out through filtration under hot conditions. The reaction without a catalyst was continued for 14 h. No more transformation of 1a occurred (Fig. 11). This experiment revealed that no catalytically active species remained in the reaction mixture. Therefore, the heterogeneous nature of Ni-γAl2O3 was successfully proven. | ||
| Fig. 11 Hot-filtration test of Ni-γAl2O3 catalyst using 1a (1 mmol), K4[Fe(CN)6]·3H2O (0.2 mmol), K2CO3 (1.2 mmol), Ni-γAl2O3 (6 mol%), DMF (3 mL) at 120 °C under ambient conditions. | ||
3.4. Comparative efficiency of Ni-γAl2O3 catalyst with other reported Ni-based catalysts
Table 3 provides a comparative overview of previously reported Ni-catalyzed cyanation reactions (entries 1–5) and compares the efficiency of our synthesized Ni-γAl2O3 nanocatalyst (entry 6). The earlier reported methods (entries 1–4) employ homogeneous nickel catalytic procedures in combination with harmful cyanating agents, such as tBuCN,35a BrCN,35b or the relatively less hazardous 1,4-dicyanobenzene.35c Despite the potential efficiency of these homogeneous systems, the necessity of expensive reagents and strong additives,35c,d requirements of an inert atmosphere and photochemical activation,35c laborious preparation35d and poor recyclability of the catalysts35a–d were the serious limitations of these methods. Although the NiFe2O4 catalyst demonstrated good catalytic activity and recyclability (entry 5), its scope is limited by the employment of highly toxic NaCN as the cyanating agent.35e In this context, our developed Ni-γAl2O3 nanocatalyst represents an economically efficient and operationally simple catalytic system for the cyanation reactions with the utilization of K4[Fe(CN)6]·3H2O as a safe and eco-friendly cyanide source. The excellent catalytic efficiency of the Ni-γAl2O3 nanocatalyst indicates good dispersion of nickel species on the γAl2O3 support, suggesting the strong metal-support interaction. Importantly, the Ni-γAl2O3 catalyst can be efficiently recycled and reused for five successive runs without substantial loss of activity, indicating greater stability than previously reported Ni-based catalytic systems. Moreover, the attractive features, such as the new catalyst design, green cyanation reaction, high efficiency, and excellent recyclability, clearly distinguish the present catalytic methodology from earlier Ni-based catalytic systems.| Entry | Catalyst | Cyanating agent | Reaction conditions | Yield (%) | Catalyst type & recyclability | Ref. |
|---|---|---|---|---|---|---|
| a Iodobenzene was used as the model substrate. b Yield of 4-methoxyiodobenzene. c Yield of 4-methoxybromobenzene. d Yield of 3-bromo benzaldehyde. | ||||||
| 1 | NiBr2/PCy3/Mn | t BuCN | NaHCO3, toluene, 150 °C, 22 h | 56 | Homogeneous, not reported | 35a |
| 2 | NiCl2·1,10-phen/Zn | BrCN | Dioxane, 50 °C, 12 h | 84b | Homogeneous, not reported | 35b |
| 3 | NiI2, dtbbpy, purple light (390–395 nm) | 1,4-Dicyano benzene | DBU, TMSBr, (TMS)SiH3, toluene, Ar atm., 50 °C, 24 h | 79c | Homogeneous, not reported | 35c |
| 4 | Ni(PPh3)2(1-Naph)Cl, JosiPhos | K4[Fe(CN)6]·3H2O | TBAHS, DIPEA, nBuOAc:H2O, 95 °C | 83d | Homogeneous, not reported | 35d |
| 5 | NiFe2O4 | NaCN | K2CO3, DMF, 100 °C, 17 min | 92 | Heterogeneous, 5 cycles | 35e |
| 6 | Ni-γAl2O3 | K4[Fe(CN)6]·3H2O | K2CO3, DMF, 120 °C, 12 h | 91 | Heterogeneous, 5 cycles | This work |
3.5. Antioxidant activity of Ni-γAl2O3 nanocatalysts
In the present study, Ni-γAl2O3 nanoparticles showed moderate antioxidant activity with a maximum antioxidant activity (68.17%) attained at 200 mg mL−1 concentration (Fig. 12). Previous literature has also shown their antioxidant activity.36a,b Antioxidant property exhibited by Ni-γAl2O3 nanoparticles might be due to their free radical scavenging capacity to reduce oxidative stress. This might be achieved by donating electrons or hydrogen atoms to neutralize free radicals, or by using redox-active sites on nickel nanoparticles. In addition, alumina also exhibits a high surface area, which plays an important role in free radical scavenging capacity (antioxidant activity) by promoting adsorption and interaction with free radicals.36a,b IC50 of ascorbic acid was 22.73 µg mL−1 while it was 146.69 mg mL−1 for Ni-γAl2O3 nanoparticles. This observation suggests that DPPH reduction capacity is lower than that of ascorbic acid, as reported earlier.36b | ||
| Fig. 12 DPPH radical scavenging activity of Ni-γAl2O3 nanoparticles at different concentration. | ||
3.6. Antibacterial activities of Ni-γAl2O3 nanocatalysts
The antibacterial activities of the Ni-γAl2O3 nanoparticles were tested against Staphylococcus aureus ATCC25923 (Gram-positive) and Escherichia coli (Gram-negative). The aforementioned Ni-γAl2O3 nanoparticles were found to be effective against both Staphylococcus aureus ATCC25923 and Escherichia coli (Fig. 13). The zone of inhibition against Escherichia coli was 12 ± 0.31 while it was 10 ± 0.25mm in the case of Staphylococcus aureus ATCC25923 (Table 4). Inhibitory effect of Ni nanoparticles against both Gram-negative and Gram-positive bacteria was also reported earlier by Angel Ezhilarasi et al.,37a Prabhu et al.,37b Rajith Kumar et al.37c Larger surface area, high reactivity of Ni NPs might boost and promote their antibacterial property. MIC values against Escherichia coli and Staphylococcus aureus ATCC25923 were 200 mg mL−1 and 205 mg mL−1, respectively, while MBC values were 220 and 230 mg mL−1, respectively (Table 4). Lower MIC and MBC values of Ni nanoparticles against Escherichia coli than against Staphylococcus aureus ATCC25923 were also reported earlier.38 Besides, previous literature also reported that inhibition of the growth of E. coli by Ni–Al2O3 nanoparticles was found to require 0.01 g mL−1 of Ni–Al2O3 nanoparticles.38d | ||
| Fig. 13 (A) Antibacterial activity of Ni-γAl2O3 nanoparticles against Escherichia coli and Staphylococcus aureus ATCC25923, which were grown on Mueller–Hinton agar plate at 37 °C. DMSO was used as a negative control. (B) The plausible mechanism of action of Ni-γAl2O3 nanoparticles exhibiting antibacterial activity. | ||
| Nanoparticle | Bacterial strain | Zone of inhibition (mm) | MIC values (mg mL−1) | MBC values (mg mL−1) |
|---|---|---|---|---|
| Ni-γAl2O3 | Escherichia coli | 12 ± 0.31 | 200 | 220 |
| Staphylococcus aureus ATCC25923 | 10 ± 0.25 | 205 | 230 |
Additionally, a comparative table (Table S1, SI) has been presented to compare the antibacterial performance of our synthesized Ni-γAl2O3 nanocatalysts with those of previously reported other metal-based nanocatalysts. However, the Ni-γAl2O3 nanocatalysts exhibited higher MIC and MBC values than those of conventional antibiotics. The absence of specific bioactive groups in the Ni-γAl2O3 nanocatalysts resulted in high MIC/MBC values, indicating poor membrane permeability and less interaction with bacterial cell surfaces. The present study demonstrates that the pristine Ni-γAl2O3 catalyst displays baseline antibacterial efficacy. However, future progress could be accomplished through surface modification with bioactive phytochemicals, the introduction of synergistic metallic dopants, and the integration of the nanocatalyst with traditional antibiotics to improve membrane penetration and offer synergistic bactericidal properties.1a,b These approaches will serve to substantially lower MIC/MBC values and extend catalyst utility in subsequent biological applications.
Antibacterial activity may be attributed to nickel nanoparticles, which induce gaps and pits that fragment the bacterial cell membrane. This fragmentation of the bacterial membrane had also been reported by other researchers.38a,38b,38c Furthermore, the metals such as Ni effectively affect the transport system of the bacteria by interacting with proteins to block regulated transport through the plasma and thus cause their death.39 Besides this hyperactive property of Ni NPs due to unpaired electrons on the surface of Ni NPs underscores complex interactions with bacterial cellular components. Thus, Ni NPs disrupt metabolic pathways, leading to bacterial cell death.40 Moreover, Ni NPs have been found to generate oxidative stress in bacteria, which is a pivotal mechanism for their antimicrobial property by damaging DNA, oxidizing proteins, and finally disrupting the cell membrane as well.41a,41b For instance, destruction of bacterial cells confers the release of cellular content and cell death.41b Inhibitory effect of Ni-γAl2O3 nanoparticles against Gram-negative bacteria Escherichia coli was higher than the Gram-positive Staphylococcus aureus ATCC25923, which might be due to differences in the cell wall structure of Gram-negative and Gram-positive bacteria. Similar findings were also shown by Angel Ezhilarasi et al.,37a Prabhu et al.,37b Rajith Kumar et al.37c Gram-negative bacteria had an outer layer of lipopolysaccharides and thin peptidoglycans that made it easy for nanoparticles to enter inside the cell. However, Gram-positive bacteria have a thick peptidoglycan layer covalently bonded to teichoic and teichuronic acids, acting as a protective layer.42 A schematic diagram regarding the mechanism of action is given in Fig. 13B.
4. Conclusion
The cyanation of aryl halides to aryl nitriles has been developed using economically supportable and operationally simple Ni-γAl2O3 as a heterogeneous catalyst and commercially available K4[Fe(CN)6] as an eco-friendly cyanating agent. The reactions are not dependent on an inert atmosphere or a ligand. Several aryl iodides, bromides, and chlorides bearing various functional moieties were well-survived and associated with good to high yield in the aforesaid method. The synthesized Ni-γAl2O3 nanocatalysts could be recovered and recycled again without significantly reducing their efficacy. Moreover, the “hot filtration method (Sheldon's test)” was carried out to establish the heterogeneity of the catalyst. The present method attracts attention for its procedural simplicity, significant catalytic durability and productivity, simple recovery, high recyclability, and survival of diverse functional groups in the reaction. In the present study, Ni-γAl2O3 nanocatalysts exhibited moderate antioxidant activity, with a maximum antioxidant activity (68.17%) at a concentration of 200 mg mL−1. Ni-γAl2O3 nanocatalysts of the present study were found to inhibit the growth of Staphylococcus aureus ATCC25923 (Gram-positive) and Escherichia coli (Gram-negative). However, the zone of inhibition (12 ± 0.31 mm) against Escherichia coli was higher than that (10 ± 0.25 mm) against Staphylococcus aureus ATCC25923. Larger surface area, unique physiochemical property, and high reactivity of Ni-γAl2O3 nanocatalysts might be responsible for their inhibitory effect against both Gram-negative and Gram-positive bacteria.Author contributions
Asit Kumar Das: conceptualization, project administration, writing-original draft preparation, supervision. Md Sattar Ali: methodology, investigation, data curation, formal analysis. Arindam Misra: methodology, investigation, data curation, formal analysis. Md Sultan Saikh: methodology, investigation, data curation, formal analysis. Subhendu Dhibar: resources, data curation. Sumit Kumar Panja: resources, data curation. Aniruddha Das: data curation, validation, formal analysis. Gourav Ghatak: investigation, biological study. Lokesh Kumar Rathore: resources, data curation. Ashok Bera: resources, data curation. Sanjay Bhar: editing, writing, and review. Smritikana Biswas: investigation, biological study, writing – original draft preparation, supervision.Conflicts of interest
There is no conflict of interest to declare.Data availability
The data that support the findings of this study are available in the supplementary information (SI) of this article. Supplementary information is available. See DOI: https://doi.org/10.1039/d5ma00879d.Acknowledgements
A. K. Das gratefully acknowledges the Department of Science & Technology and Biotechnology, Govt. of West Bengal, for financial grant (Grant No. 2147(Sanc.)/STBT-11012(25)/1/2024-ST SEC). S. Biswas gratefully acknowledges DST-SERB for a financial grant (File No. SUR/2022/001437 dated 04.10.2023). Special thanks to Dr M. Maji and Dr S. Mondal for their assistance.Notes and references
- (a) M. Binandeh, Eur. J. Med. Chem. Rep., 2022, 6, 100072 Search PubMed; (b) M. Binandeh, Bioinspired, Biomimetic Nanobiomater., 2023, 11, 121–127 Search PubMed; (c) A. Raghunath and E. Perumal, Int. J. Antimicrob. Agents, 2017, 49, 137–152 Search PubMed.
- M. Rai, A. Yadav and A. Gade, Biotechnol. Adv., 2009, 27, 76–83 CrossRef CAS PubMed.
- J. J. Lin, W. C. Lin, S. D. Li, C. Y. Lin and S. H. Hsu, ACS Appl. Mater. Interfaces, 2013, 5, 433–443 Search PubMed.
- B. Le Ouay and F. Stellacci, Nano Today, 2015, 10, 339–354 Search PubMed.
- P. Huo, J. Ind. Eng. Chem., 2018, 57, 125–133 Search PubMed.
- R. Dadi, R. Azouani, M. Traore, C. Mielcarek and A. Kanaev, Mater. Sci. Eng., C, 2019, 104, 109968 Search PubMed.
- R. A. Ulwali, H. Kh Abbas, N. Yasoob and H. A. Alwally, Neuro Quantol., 2021, 19, 42–52 Search PubMed.
- S. Debnath, S. Ghosh, S. Das, P. Das, S. Sarkar and S. M. Mandal, J. Basic Microbiol., 2016, 56, 614–625 Search PubMed.
- (a) R. C. Larock, Comprehensive Organic Transformations: a Guide to Functional Group Preparations, Wiley-VCH, Weinheim, Germany, 1989, p. 819 Search PubMed; (b) A. Kleemann, J. Engel, B. Kutscher and D. Reichert, Pharmaceutical Substances: Syntheses, Patents, Applications, Georg Thieme, Stuttgart, 4th edn, 2001 Search PubMed.
- R. C. Larock, Comprehensive Organic Transformations, Wiley, New York, 2nd edn, 1999 Search PubMed.
- P. Anbarasan, T. Schareina and M. Beller, Chem. Soc. Rev., 2011, 40, 5049–5067 Search PubMed.
- (a) M. N. Janakirman, K. D. Watenpaugh and K. T. Chong, Bioorg. Med. Chem. Lett., 1998, 8, 1237 CrossRef PubMed; (b) S. I. Murahashi, Sci. Synth., 2004, 19, 345 Search PubMed; (c) L. H. Jones, N. W. Summerhill, N. A. Swain and J. E. Mills, Med. Chem. Commun., 2010, 1, 309 RSC; (d) A. M. Sweeney, P. Grosche, D. Ellis, K. Combrink, P. Erbel, N. Hughes, F. Sirockin, S. Melkko, A. Bernardi, P. Ramage, N. Jarousse and E. Altmann, ACS Med. Chem. Lett., 2014, 5, 937 Search PubMed.
- (a) T. Sandmeyer, Ber. Dtsch. Chem. Ges., 1884, 17, 1633 CrossRef; (b) T. Sandmeyer, Chem. Ber., 1884, 17, 2650 CrossRef; (c) T. Sandmeyer, Chem. Ber., 1885, 18, 1492 CrossRef; (d) T. Sandmeyer, Chem. Ber., 1885, 18, 1946 Search PubMed; (e) K. W. Rosenmund and E. Struck, Chem. Ber., 1919, 2, 1749 Search PubMed.
- (a) A. K. Das and K. Sarkar, New J. Chem., 2025, 49, 12898–12930 RSC; (b) A. Pramanik, A. K. Das, S. Bhar and A. Ghatak, ChemistrySelect, 2025, 10, e202500035 Search PubMed; (c) S. Nandy, A. K. Das and S. Bhar, Synth. Commun., 2020, 50, 3326–3336 CrossRef CAS; (d) A. Ghatak, A. K. Das, S. Bhar and A. Pramanik, J. Het. Chem., 2025, 62, 760–780 CrossRef CAS.
- (a) M. Sundermeier, A. Zapf and M. Beller, Eur. J. Inorg. Chem., 2003, 3513–3526 CrossRef CAS; (b) G. P. Ellis and T. M. Romney-Alexander, Chem. Rev., 1987, 87, 779 CrossRef CAS; (c) V. V. Grushin and H. Alper, Chem. Rev., 1994, 94, 1047 CrossRef CAS.
- T. Kentaro, O. Tadashi, S. Yasumasa and O. Shinzaburo, Chem. Lett., 1973, 471–474 Search PubMed.
- (a) T. Schareina, A. Zapf and M. Beller, Chem. Commun., 2004, 1388 Search PubMed; (b) A. B. Khemnar, D. N. Sawant and B. M. Bhanage, Tetrahedron Lett., 2013, 54, 2682–2684 Search PubMed; (c) C. W. Liskey, X. Liao and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 11389–11391 CrossRef CAS PubMed; (d) A. B. Khemnar and B. M. Bhanage, RSC Adv., 2014, 4, 13405–13408 RSC.
- (a) M. T. Martin, B. Liu, B. E. Cooley and J. F. Eaddy, Tetrahedron Lett., 2007, 48, 2555 CrossRef CAS; (b) L. Cai, X. Liu, X. Tao and D. Shen, Synth. Commun., 2004, 34, 1215 Search PubMed; (c) M. Sundermeier, S. Mutyala, A. Zapf, A. Spannenberg and M. Beller, J. Organomet. Chem., 2003, 684, 50 Search PubMed; (d) Y. Ren, Z. Liu, S. Zhao, X. Tian, J. Wang, W. Yin and S. He, Catal. Commun., 2009, 10, 768 CrossRef CAS; (e) H. Yu, R. N. Richey, W. D. Miller, J. Xu and S. A. May, J. Org. Chem., 2011, 76, 665 CrossRef CAS PubMed.
- (a) F. H. Luo, C. I. Chu and C. H. Cheng, Organometallics, 1998, 17, 1025 Search PubMed; (b) M. Sundermeier, A. Zapf and M. Beller, Angew. Chem., Int. Ed., 2003, 42, 1661 Search PubMed; (c) Z. Zhang and L. S. Liebeskind, Org. Lett., 2006, 8, 4331 Search PubMed; (d) N. Sato and Q. Yue, Tetrahedron, 2003, 59, 5831 Search PubMed; (e) P. Anbarasan, H. Neumann and M. Beller, Chem. – Eur. J., 2010, 16, 4725 CrossRef CAS PubMed.
- (a) A. K. Das, S. Nandy and S. Bhar, RSC Adv., 2022, 12, 4605–4614 RSC; (b) B. N. Patra, A. K. Das, S. Misra, P. P. Jana, P. Brandao, M. Afzal, A. Alarifi, T. Saha, D. Bera, S. Halder, D. Mal and N. Sepay, J. Mol. Struct., 2024, 1300, 137229 CrossRef CAS; (c) A. K. Das, S. Ali, A. Misra, S. Islam, B. Kar, S. Biswas, G. Ghatak, D. Mal, M. Shit, M. Dolai and A. Das, Appl. Organomet. Chem., 2025, 39, e7796 CrossRef CAS.
- (a) P. Yu and B. Morandi, Angew. Chem., Int. Ed., 2017, 56, 15693 Search PubMed; (b) Y. Ueda, N. Tsujimoto, T. Yurino, H. Tsurugi and K. Mashima, Chem. Sci., 2019, 10, 994–999 RSC; (c) F. H. Luo, C. I. Chu and C. H. Chen, Organometallics, 1998, 17, 1025 Search PubMed; (d) L. R. Mills, J. M. Graham, P. Patel and S. A. L. Rousseaux, J. Am. Chem. Soc., 2019, 49, 19257–19262 CrossRef PubMed; (e) D. D. Beattie, T. Schareina and M. Beller, Org. Biomol. Chem., 2017, 15, 4291–4294 Search PubMed.
- (a) A. Sadhukhan, B. N. Patra, T. Maity, A. K. Das, C. K. Ghosh, P. Brandao, D. M. Gil, D. Bera, C. Roy Choudhury, P. K. Bhaumik, D. Mal and A. Frontera, Eur. J. Inorg. Chem., 2024, e202400459 CrossRef CAS; (b) A. Ghatak, A. Pramanik, A. K. Das and S. Bhar, Tetrahedron, 2022, 127, 133090 Search PubMed; (c) A. K. Das, S. Saikh, A. Misra, S. Ali, P. Pradhan, N. Sepay, S. Dhibar, M. Afzal, J. Abbas and N. Sepay, J. Mol. Struct., 2026, 1352, 144012 Search PubMed; (d) A. K. Das, A. Misra, S. Ali, S. Saikh, S. Dhibar, K. K. Banerjee, G. Ghatak, D. Mal, M. Shit and S. Biswas, RSC Adv., 2025, 15, 35844–35858 Search PubMed.
- (a) D. Saha, A. K. Das, M. Raish and N. Sepay, J. Mol. Struct., 2025, 1329, 141363 CrossRef CAS; (b) S. Nandy, A. Ghatak, A. K. Das and S. Bhar, Synlett, 2018, 2208–2212 Search PubMed.
- A. K. Das, S. Nandy and S. Bhar, Appl. Organomet. Chem., 2021, 35, e6282 CrossRef CAS.
- Y. Wang, Y. Gao, H. Ding, S. Liu, X. Han, J. Gui and D. Liu, Food Chem., 2017, 218, 152–158 CrossRef CAS PubMed.
- A. M. Al-Dbass, S. A. Daihan, A. A. Al-Nasser, L. S. Al-Suhaibani, J. Almusallam, B. I. Alnwisser, S. Saloum, R. S. Alotaibi, L. A. Alessa and R. S. Bhat, Molecules, 2022, 27, 7656 Search PubMed.
- P. Adamou, E. Harkou, A. Bumajdad, X. De Jong, M. Van Haute, A. Constantinou and S. M. Al-Salem, ACS Omega, 2024, 9(17), 19057–19062 Search PubMed.
- B. Djebarri, F. Touahra, N. Aider, F. Bali, M. Sehailia, R. Chebout, K. Bachari and D. Halliche, Bull. Chem. React. Eng. Catal., 2020, 15, 331–347 Search PubMed.
- P. Srimara, T. Chevapruk, P. Kumnorkaew, T. Muangnapoh and P. Vas-Umnuay, Mater. Today: Proc., 2020, 23, 720–725 CAS.
- A. Bustinza, M. Frías, Y. Liu and E. García-Bordejé, Catal. Sci. Technol., 2020, 10, 4061–4071 Search PubMed.
- J. Zanon, A. Klapars and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 2890–2891 Search PubMed.
- (a) A. Schareina, A. Zapf and M. Beller, J. Organomet. Chem., 2004, 689, 4576–4583 Search PubMed; (b) A.-R. Hajipour, F. Abrisham and G. Tavakoli, Transition Met. Chem., 2011, 36, 725–730 Search PubMed; (c) B. S. Kumar, A. J. Amali and K. Pitchumani, ACS Appl. Mater. Interfaces, 2015, 7, 22907–22917 Search PubMed.
- (a) M. Binandeh, J. Mater. Sci: Mater. Eng., 2025, 20, 17 Search PubMed; (b) A. Das, D. Chavda, M. Manna and A. K. Das, New J. Chem., 2024, 48, 18249–18260 RSC; (c) A. Das and A. K. Das, New J. Chem., 2023, 47, 5347–5355 RSC; (d) A. K. Das, N. Sepay, S. Nandy, A. Ghatak and S. Bhar, Tetrahedron Lett., 2020, 61, 152231 CrossRef CAS.
- (a) H. E. Lempers and R. A. Sheldon, J. Catal., 1998, 175, 62 CrossRef CAS; (b) M. Binandeh, Nano Trends, 2025, 11, 100138 Search PubMed.
- (a) Y. L. Zhang, Z. G. Zhang, Y. Y. Hu, Y. K. Liu, H. W. Jin and B. W. Zhou, Org. Biomol. Chem., 2022, 20, 8049–8053 RSC; (b) Y. J. Wu, C. Ma, M. Bilal and Y. F. Liang, Molecules, 2024, 29, 6016 Search PubMed; (c) Y. Yan, J. Sun, G. Li, L. Yang, W. Zhang, R. Cao, C. Wang, J. Xiao and D. Xue, Org. Lett., 2022, 24, 2271–2275 CrossRef CAS PubMed; (d) N. A. Wilson, W. M. Palmer, M. K. Slimp, E. M. Simmons, M. V. Joannou, J. Albaneze-Walker, J. M. Ganley and D. E. Frantz, ACS Catal., 2025, 15, 6459–6465 CrossRef CAS PubMed; (e) F. M. Moghaddam, G. Tavakoli and H. R. Rezvani, Appl. Organomet. Chem., 2014, 28, 750–755 CrossRef.
- (a) M. Zamani, A. M. Delfani and M. Jabbari, Spectrochim. Acta, Part A, 2018, 201, 288–299 CrossRef CAS PubMed; (b) A. Chahardoli, N. Karimi, X. Ma and F. Qalekhani, Sci. Rep., 2020, 10, 3847 CrossRef CAS PubMed.
- (a) A. Angel Ezhilarasi, J. Judith Vijaya, K. Kaviyarasu, L. John Kennedy, R. Ramalingam and H. A. Al-Lohedan, J. Photochem. Photobiol. B Biol., 2018, 180, 39–50 CrossRef CAS PubMed; (b) S. Prabhu, T. D. Thangadurai, P. V. Bharathy and P. Kalugasalam, Results Chem., 2022, 4, 100285 CrossRef CAS; (c) C. R. Rajith Kumar, V. S. Betageri, G. Nagaraj, G. H. Pujar, B. P. Suma and M. S. Latha, J. Sci. Adv. Mater. Dev., 2020, 5, 48–55 Search PubMed.
- (a) K. Ishaq, A. A. Saka, A. O. Kamardeen, A. Ahmed, M. I. H. Alhassan and H. Abdullahi, Eng., Sci. Technol., 2017, 20, 563–569 Search PubMed; (b) N. A. Amro, L. P. Kotra, K. Wadu-Mesthrige, A. Bulychev, S. Mobashery and G. Y. Liu, Langmuir, 2000, 16, 2789–2796 Search PubMed; (c) Y. Hyosuk, D. K. Ji, C. C. Hyun and W. L. Chul, Bull. Korean Chem. Soc., 2013, 34, 3261–3264 Search PubMed; (d) M. R. Ahghari, V. Soltaninejad and A. Maleki, Sci. Rep., 2020, 10, 12627 Search PubMed.
- (a) H. El Ghandoor, H. M. Zidan, M. M. Khalil and M. I. M. Ismail, Int. J. Electrochem. Sci., 2012, 7, 5734–5745 Search PubMed; (b) I. M. Obaidat, Nanomaterials, 2017, 7, 415 Search PubMed; (c) J. Chaudhary, G. Tailor, B. L. Yadav and O. Michael, Heliyon, 2019, 5, 2405–8440 CrossRef PubMed; (d) S. B. Park, Sci. Rep., 2019, 9, 1–11 Search PubMed.
- (a) S. Mukherjee, S. Dasgupta, S. Sengupta, P. Patra, K. Das and A. Ghosh, Langmuir, 2008, 24, 9817–9826 Search PubMed; (b) W. R. Li, X. B. Xie, Q. S. Shi, H. Y. Zeng, O. Y. You-Sheng and Y. B. Chen, Appl. Microbiol. Biotechnol., 2010, 85, 1115–1122 CrossRef CAS PubMed.
- (a) B. H. Shnawa, P. J. Jalil, S. M. Hamad and M. H. Ahmed, BioNanoSci, 2022, 12, 1264–1278 CrossRef; (b) Y. Li, P. Leung, L. Yao, Q. W. Song and E. Newton, J. Hosp. Infect., 2006, 1, 58–63 CrossRef PubMed.
- Y. N. Slavin, J. Asnis, U. O. Häfeli and H. Bach, J. Nanobiotechnol., 2017, 15, 65 Search PubMed.
| This journal is © The Royal Society of Chemistry 2026 |