
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and study of donor–acceptor conjugated polymers based on isoindigo units via a metal-free aldol polymerization strategy
Prerak R.
Patel
a,
Mayur J.
Patel
c,
Parameswar K.
Iyer
 *bc,
Sanjio S.
Zade
*d and
Arun L.
Patel
*a
*bc,
Sanjio S.
Zade
*d and
Arun L.
Patel
*a
aDepartment of Chemistry, Faculty of Science, The Maharaja Sayajirao University of Baroda, Vadodara-390002, Gujarat, India. E-mail: arunpatel_5376@yahoo.co.in
bCentre of Nanotechnology, Indian Institute of Technology Guwahati, Guwahati, Assam-781039, India
cDepartment of Chemistry, Indian Institute of Technology Guwahati, Guwahati, Assam-781039, India. E-mail: pki@iitg.ac.in
dDepartment of Chemical Sciences, Indian Institute of Science Education and Research (IISER) Kolkata, Mohanpur-741246, India. E-mail: sanjiozade@iiserkol.ac.in
First published on 30th December 2025
Abstract
The synthesis of donor–acceptor conjugated polymers is challenging due to the dependence on expensive transition-metal catalysts and potentially hazardous reagents, which can have negative environmental effects. Aldol polycondensation provides a promising, metal-free alternative for polymerizing such materials. In this study, we report the synthesis of three polymers, namely, AIRI, AICRI and AITRI, through an aldol condensation reaction between a diformylisoindigo compound and reduced bis-isatins. Photophysical, electrochemical, thermogravimetric, atomic force microscopy (AFM), gel permeation chromatography (GPC), powder X-ray diffraction (PXRD), density functional theory (DFT) calculations, and space charge limited current (SCLC) hole mobility measurement studies were carried out for these polymers. Photophysical and electrochemical studies showed high visible light absorption, with HOMO energy levels around −5.7 eV and LUMO energy levels near −3.7 eV for all polymers. Thermogravimetric analysis (TGA) indicated that the polymers are stable up to approximately 300 °C. The SCLC hole mobilities measured for polymers AIRI, AICRI and AITRI were 7.13 × 10−2, 8.15 × 10−2, and 9.38 × 10−2 cm2 V−1 s−1, respectively.
Introduction
The advancement of organic electronics, including organic photovoltaics (OPVs),1 organic field-effect transistors (OFETs),2,3 organic light-emitting devices (OLEDs)4 and organic electrochromics (OECs),5 has been propelled by continuous developments in the synthesis of conjugated molecules and polymers. Structural designs have evolved based on a deeper comprehension of the critical parameters dictating high performance in these devices. Parameters like ionization potentials (IPs), electron affinities (EAs), optical gaps and absorption profiles have been specifically tailored by integrating electron-rich (donor) and electron-deficient (acceptor) arenes within the electroactive core of conjugated materials.6 This strategic approach enables precise control over electronic properties, thereby enhancing the overall efficiency and performance of these organic electronic devices. This progress is attributed to advancements in photoactive materials, especially electron-donating polymers, as well as device optimization.Recently, donor–acceptor (D–A) polymers have attracted particular interest due to their in-chain donor–acceptor interactions, which lower the band gap and make them important light-harvesting systems for solar cells.7 This class of polymers offers advantages because their photoelectronic properties, including absorption spectra and energy levels, can be easily adjusted by combining different electron-rich and electron-deficient units, as well as by introducing various side groups. Numerous electron-rich groups have been identified, such as fluorene,1 silafluorene,8 carbazole,9 indolo[3,2-b]carbazole,10 cyclopenta[2,1-b:3,4-b′]dithiophene,11 dithieno[3,2-b:2′,3′-d]silole,12 indacenodithiophene,13 thieno[3,4-b]thiophene,14 and benzo[1,2-b:4,5-b′]dithiophene (BDT).15,16 However, the identification of successful electron-deficient units has been less extensive, including 2,1,3-benzothiadiazole,1 quinoxaline,17 diketopyrrolo[3,4-c]-pyrrole-1,4-dione (DPP),18 and thieno[3,4-c]pyrrole-4,6-dione (TPD).19
Isoindigo, a structural isomer of the well-known pigment indigo, is a naturally occurring indigoid pigment found in plants like Isatis tinctoria (Woad).20,21 Although the synthesis of isoindigo dates back to 1988,22 and some isoindigo-containing polymers and small molecules were reported in 2008, it was not recognized as a building block for creating conjugated p-type materials for solar cells until Reynolds et al.23 reported two small molecules with isoindigo as an electron-donating unit in 2010. Since then, isoindigo has garnered significant attention in the scientific community as an ideal electron-deficient unit for constructing donor–acceptor (D–A) polymers for solar cells. Isoindigo possesses strong electron-withdrawing characteristics due to its two lactam rings, and it also adjusts the LUMO levels of the resulting polymers to an ideal range. Furthermore, there is a report where an isoindigo-containing material served as an electron acceptor, replacing conventional fullerene derivatives in bulk heterojunction (BHJ) solar cells.24,25 Moreover, high-performance field-effect transistors (FETs) based on isoindigo polymers have also been achieved, demonstrating hole mobility as high as 3.62 cm2 V−1 s−1.26–28 Because of their distinctive characteristics, several polymers incorporating isoindigo have been documented in the scientific literature, showcasing donor–acceptor (D–A) structures (Fig. 1(a)).24–28
 | ||
| Fig. 1 (a) Structures of the reported isoindigo-based conjugated polymers; (b) some reported donor–acceptor aldol polycondensation polymers. | ||
Conjugated polymers have traditionally relied on expensive transition metal catalysts for synthesis, posing environmental and economic challenges.29,30 The demand for environmentally sustainable synthetic methods is growing due to the use of hazardous monomers and the generation of harmful byproducts in traditional polymerization techniques like Stille coupling.31–33 To address these issues, researchers are exploring alternative methods such as direct heteroarylation polymerization (DHAP) and other metal-free polymerizations, such as aldol condensation. DHAP offers atom efficiency; however, it may suffer from regiospecificity limitations.34 In contrast, the aldol condensation reaction presents advantages including the formation of water as a byproduct, the use of non-halogenated solvents, and catalysis by simple acids or bases.35,36 The aldol condensation reaction generates α,β-unsaturated carbonyl structures, facilitating the synthesis of novel ladder-type conjugated polymers that are challenging to achieve through traditional cross-coupling techniques. For instance, Zhang et al. utilized aldol condensation to synthesize donor–acceptor copolymers with bithiophene donors for organic thin-film transistor (OTFT) applications.37 Several other examples of donor–acceptor conjugated polymers synthesized via aldol condensation are also reported (Fig. 1(b)).28,38–40 In summary, there is a pressing need for environmentally friendly synthetic approaches for the synthesis of conjugated polymers. The aldol condensation reaction emerges as a promising alternative, offering distinct advantages and enabling the creation of innovative polymers with desirable properties.
In this article, we describe the synthesis of donor–acceptor copolymers by integrating non-fused bis(indolinone)-based monomers with monomers containing diformyl dialkylated isoindigo moieties. The synthesized monomers were thoroughly characterized using IR, 1H & 13C NMR, and HRMS techniques. The synthesized polymers underwent a comprehensive characterization process employing various analytical methods. Structural analysis of the polymers was carried out using 1H NMR spectroscopy, while gel permeation chromatography (GPC) determined their molecular weights. UV-visible spectroscopy was utilized to examine electronic transitions, and thermogravimetric analysis (TGA) provided insights into thermal stability. Furthermore, atomic force microscopy (AFM) and powder X-ray diffraction (PXRD) studies were conducted to analyze morphology and crystallinity. Density functional theory (DFT) calculations were performed to understand the molecular properties better. Moreover, the hole mobilities of polymers AIRI, AITRI, and AICRI were investigated through space charge limited current (SCLC) measurements, offering insights into their charge transport properties.
Results and discussion
Synthesis of monomers and polymers
Compound 2 was synthesized from compound 1. The precursor compound 1 was synthesized following a literature procedure.35,38 The reduction of compound 1 was achieved using hydrazine hydrate, resulting in the desired compound 2 (Scheme 1).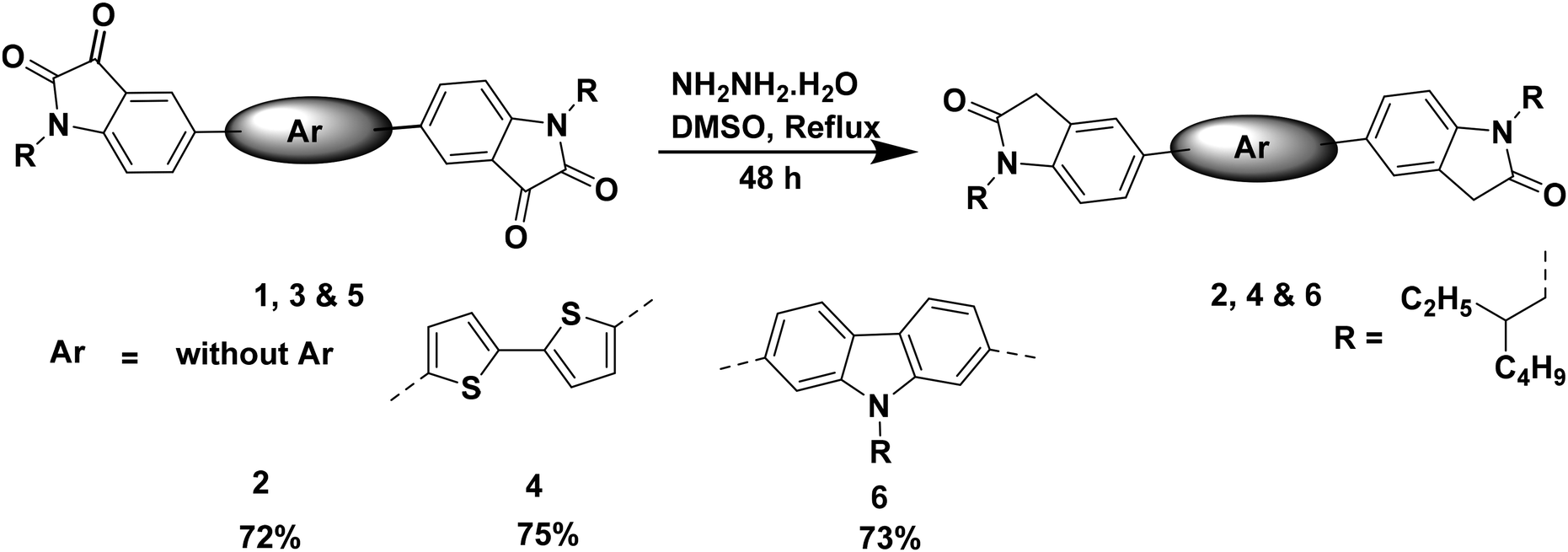 | ||
| Scheme 1 Synthesis of compounds 2, 4 and 6. | ||
The precursor compound 3 was synthesized following a literature procedure.41 The reduction of compound 3 was achieved using hydrazine hydrate, resulting in the desired compound 4. The precursor compound 5 was synthesized following a literature procedure.42 The reduction of compound 5 was achieved using hydrazine hydrate, resulting in the desired compound 6 (Scheme 1).
The synthesis of compound 7 was conducted using a modified literature procedure.43
Compound 9 was synthesized using a modified reported literature process.27 5-Bromoindolin-2-one (7) was obtained by the reduction of 5-bromoisatin in the presence of NH2NH2·H2O. Compound 8 was obtained by the condensation of 5-bromoisatin and 5-bromo oxindole. The N-alkylation of compound 8 was carried out by using 7-(bromomethyl)pentadecane to obtain the alkylated compound 9. A Stille coupling reaction was carried out between compound 9 and 2-(tributylstannyl)thiophene to obtain compound 10, which was further formylated under Vilsmeier–Haack reaction conditions to obtain the desired compound 11 (Scheme 2).
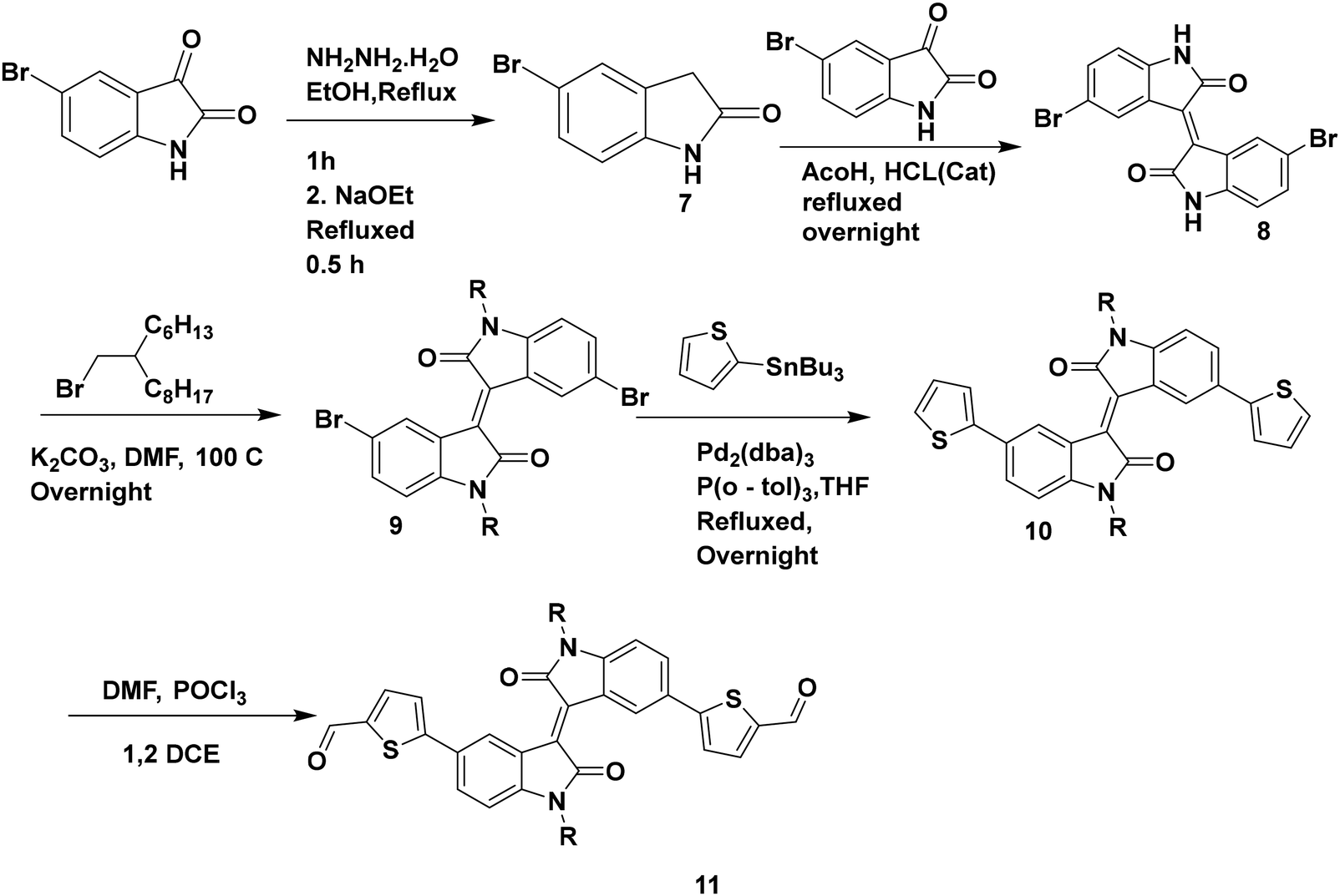 | ||
| Scheme 2 Synthesis of compound 11. | ||
The synthesized compound 11 was subjected to polymerization under aldol polycondensation conditions with compounds 2, 4 and 6 in the presence of pTSA under a nitrogen atmosphere (Scheme 3). The synthesized polymers were further purified by Soxhlet extraction using methanol, hexane, toluene, and CHCl3. The chloroform fraction is used for further studies.
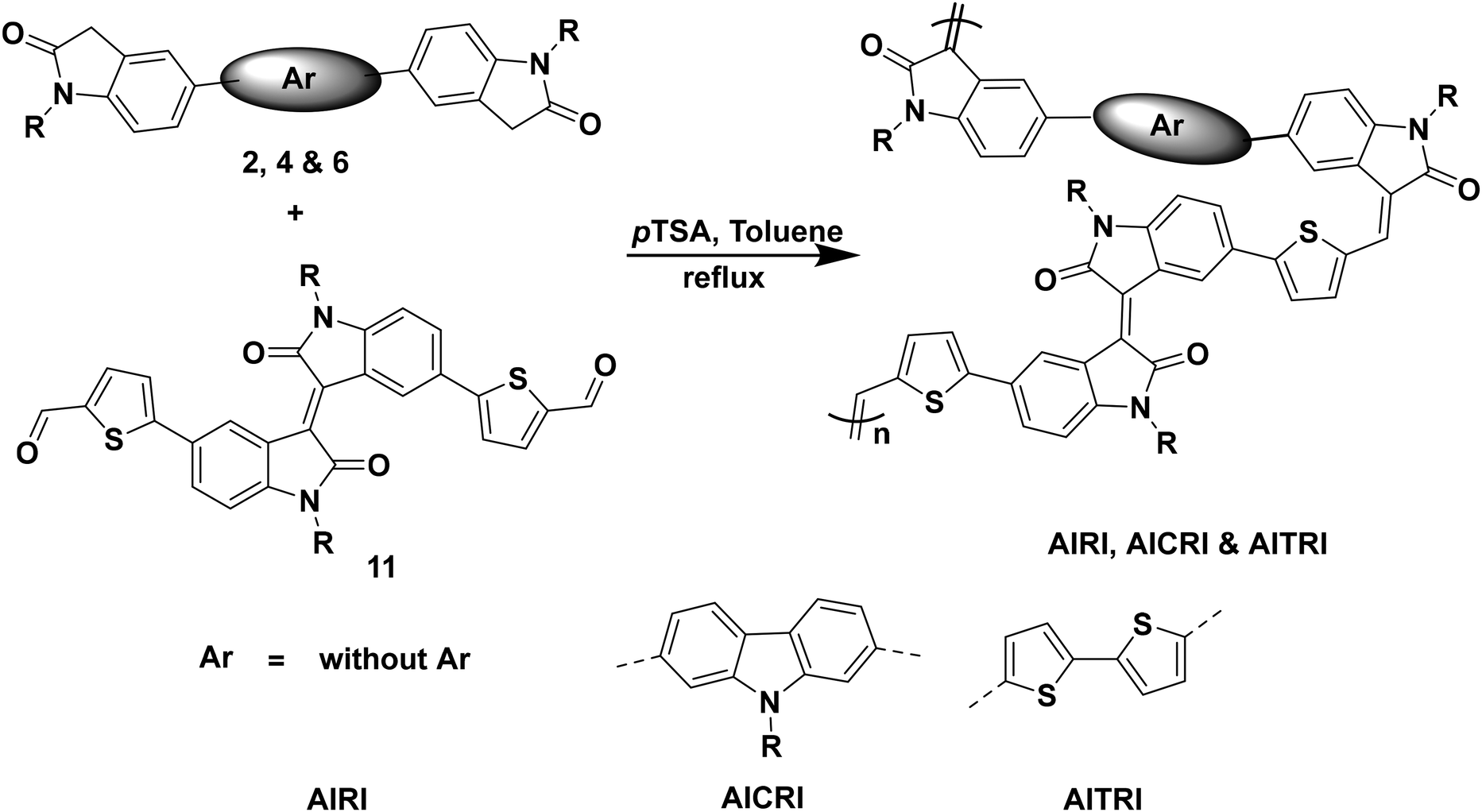 | ||
| Scheme 3 Synthesis of polymers AIRI, AICRI and AITRI. | ||
Thermal and molecular weight properties of polymers
The thermal characteristics of all the synthesized conjugated polymers are investigated by thermogravimetric analysis (TGA) under a nitrogen atmosphere at a heating rate of 10 °C min−1 (Fig. 2). The polymer's breakdown temperature (Td) is the temperature at which it loses 5% of its weight. The polymers AIRI, AICRI and AITRI exhibited decomposition temperatures of 373 °C, 390 °C and 340 °C, respectively. The synthesized polymers exhibit decomposition temperatures above 340 °C, which demonstrates that the thermal stability of the polymer is suitable for optoelectronic applications. | ||
| Fig. 2 Thermogravimetric analysis of the polymers. | ||
The weight average molecular weight (Mw), poly-dispersity index (Đ) and decomposition temperatures (Td) of the polymers are summarized in Table 1. The molecular weights of the polymers were determined by GPC in tetrahydrofuran (THF) solution relative to polystyrene standards. The molecular weights of the polymers AIRI, AICRI and AITRI were found to be 18.73 kDa, 39.12 kDa and 30.01 kDa, with poly-dispersity indices of 2.04, 2.25 and 1.75, respectively (Table 1).
Photophysical properties of polymers
The photophysical studies of aldol polymers AIRI, AICRI and AITRI were carried out by UV-visible spectroscopy in 1 × 10−6 M chloroform solutions (Fig. 3). Polymers AIRI, AICRI and AITRI show broad charge transfer (CT) bands ranging from 350 nm to 550 nm, having absorption maxima (λmax) at 438 nm, 434 nm and 412 nm, respectively. The optical band gaps (Eoptg) of polymers, AIRI, AICRI and AITRI, are found to be 2.14 eV, 2.13 eV and 2.25 eV, respectively. The photophysical properties of the polymers AIRI, AICRI and AITRI are summarized in Table 1. | ||
| Fig. 3 Absorption spectra of polymers in chloroform solution: AIRI (black line), AICRI (red line) and AITRI (blue line). | ||
Electrochemical properties of polymers
The frontier molecular orbital energy levels of the aldol polymers are measured using cyclic voltammetry (CV). CV experiments are carried out in dry acetonitrile using tetra-n-butylammonium hexafluorophosphate (TBAPF6) as the supporting electrolyte using a three-electrode system: a Pt disc electrode as the working electrode, a Pt wire electrode as the counter electrode and Ag/Ag+ as the reference electrode.All three aldol polymers show irreversible oxidation and reduction in the CV (Fig. 4). The oxidation potentials of polymers AIRI, AICRI and AITRI were found at +1.54 V, +1.52 V and +1.56 V, respectively. The corresponding onset oxidation potentials for AIRI, AICRI and AITRI were found to be +1.36 V, +1.29 V and +1.34 V, respectively. The HOMO energy levels were calculated from the onset oxidation potentials and found to be −5.78 eV, −5.71 eV and −5.76 eV for polymers AIRI, AICRI and AITRI, respectively. The reduction potentials of polymers AIRI, AICRI and AITRI were found to be −1.04 V, −1.08 V and −1.10 V with onset reduction potentials of −0.68 eV, −0.70 eV and −0.71 eV, respectively (Fig. 4). The equation ELUMO = EHOMO + Eoptg was used to calculate the LUMO energy levels. The calculated LUMO energy levels were found to be −3.64 eV, −3.58 eV and −3.51 eV for polymers AIRI, AICRI and AITRI, respectively. The electrochemical properties of the synthesized aldol polymers are summarized in Table 2.
 | ||
Fig. 4 Oxidation–reduction curves of the polymers, (a) AIRI (black line), (b) AICRI (red line) and (c) AITRI (blue line), obtained by cyclic voltammetry at 50 mV s−1 in dry acetonitrile–chloroform (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) using TBAPF6 as a supporting electrolyte; :3) using TBAPF6 as a supporting electrolyte; 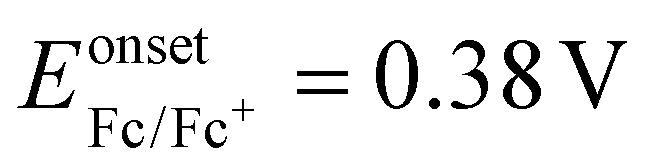 . . | ||
| Polymers |
E
oxia (eV) |
E
onsetoxa (eV) |
E
HOMOb (eV) |
E
reda (V) |
E
onsetreda (V) |
E
LUMOc (eV) |
|---|---|---|---|---|---|---|
a Potential vs. Ag/Ag+.
b Calculated from the equation 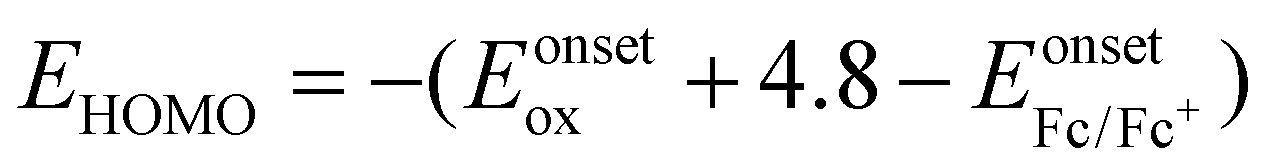 .
c Calculated from the equation ELUMO = EHOMO + Eoptg. .
c Calculated from the equation ELUMO = EHOMO + Eoptg.
|
||||||
| AIRI | +1.54 | +1.36 | −5.78 | −1.04 | −0.68 | −3.64 |
| AICRI | +1.52 | +1.29 | −5.71 | −1.08 | −0.70 | −3.58 |
| AITRI | +1.56 | +1.34 | −5.76 | −1.10 | −0.71 | −3.51 |
X-ray diffraction studies of polymers
Fig. 5 shows the results of the powder X-ray diffraction (PXRD) analysis for the polymer films of AIRI, AICRI and AITRI. Each film exhibits a significant broad peak at approximately 2θ = 22.15°, which is indicative of their amorphous structure. Additionally, these polymers exhibit a uniform d spacing of 4.011 Å, highlighting a consistent structural feature among them. | ||
| Fig. 5 Thin film X-ray diffraction pattern spectra of aldol polymers AIRI, AICRI and AITRI. | ||
Atomic force microscopy (AFM) analysis
Tapping mode atomic force microscopy (AFM) was employed to examine a polymer/NiOx/FTO film, focusing on the morphological characteristics of the polymer films. As shown in Fig. 6, the polymer AITRI has the lowest root mean square (RMS) roughness value of 6.99 nm, setting it apart from the other polymers. In contrast, polymers AIRI and AICRI exhibit RMS roughness values of 21.23 nm and 17.84 nm, respectively. | ||
| Fig. 6 AFM images of aldol polymers (a) AIRI, (b) AICRI and (c) AITRI. | ||
The higher surface smoothness is thought to enhance the efficiency of charge transfer pathways at the interfaces, thereby improving charge transfer dynamics across the different layers. This observation highlights the crucial role of surface morphology in determining charge transport properties within device architectures, providing important insights into the mechanisms underlying the enhancement of charge carrier mobility in polymer-based electronic systems.
Space charge limited current (SCLC) hole mobility measurements of aldol polymers
The assessment of hole mobilities in the polymers was achieved using the Mott–Gurney equation,27,43 which correlates the current density (J) with the applied voltage (V) and film thickness (L) based on the material's dielectric properties.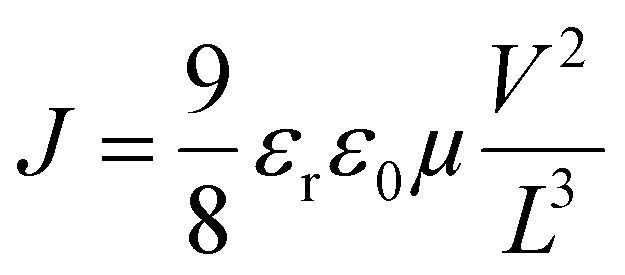
In this formula, ε0 represents the vacuum permittivity (8.85 × 10−12 F m−1), while εr signifies the dielectric constant, typically assumed to be 3 for organic semiconductors. By calculating the hole mobility (µ), we gain a quantitative understanding of the charge carrier transport properties within the material. This method, rooted in fundamental electrostatic principles, highlights the thorough scientific methodology used to study charge carrier dynamics in polymer-based electronic systems.
Hole mobility evaluation of the synthesized polymers was carried out using the SCLC technique in a hole-only device configuration described as FTO/NiOx/polymer/MoO3/Al, with an active area of 0.12 cm2 (Fig. 7). The FTO glass slides were meticulously cleaned through sonication in a soap solution, followed by sequential rinsing with deionized water, acetone, and isopropyl alcohol, each for 15 minutes. Post-cleaning, the slides were treated under UV-ozone. The NiOx precursor solution was then spin-coated onto the FTO glass surface at 3500 rpm for 45 seconds and subsequently annealed at 300 °C for 1 hour to form a hole transport layer (HTL) approximately 120 nm thick. The polymer solution, dissolved in chloroform, was spin-coated onto the substrate at 1000 rpm for 60 seconds and then thermally annealed at 80 °C for 10 minutes to ensure proper film formation. A molybdenum trioxide (MoO3) layer, 20 nm thick, was thermally deposited as a buffer layer. Finally, aluminium (Al) metal electrodes, 100 nm thick, were deposited using a shadow mask. The current density–voltage characteristics were recorded using a CHI660E instrument at a scan rate of 100 mV s−1, enabling a thorough characterization of device performance.
 | ||
| Fig. 7 (a) The energy levels of the hole-only device; (b) schematic diagram of the device. | ||
The hole mobilities were determined by analyzing data from the linear region of the logJ versus logV curves, as shown in Fig. 8. The measured hole mobilities for the polymers AIRI, AICRI and AITRI were observed to be 7.13 × 10−2, 8.15 × 10−2 and 9.38 × 10−2 cm2 V−1 s−1, respectively. This precise analytical method allowed for accurate quantification of charge carrier mobility within the prepared devices, providing valuable insights into the electronic transport properties of the respective polymer materials.
 | ||
| Fig. 8 SCLC hole mobilities of polymers, (a) AIRI, (b) AICRI and (c) AITRI. | ||
Computational studies
To gain insights into the structural characteristics, frontier molecular orbitals, and band gaps of the polymers, DFT calculations were performed on polymer models (with N-alkyl chains simplified to N-ethyl groups) using Gaussian09 at the PBC/B3LYP/6-31G(d) level44 (Fig. 9). The polymer unit cells were constructed by taking the comonomer unit as the repeating unit. The calculated band gap values were 2.05 eV, 2.05 eV and 1.82 eV for AIRI, AICRI and AITRI, respectively. These results are consistent with the experimental band gaps (Table 1), except for AITRI, where the calculated value was underestimated. The isoindigo unit is nearly planar, with a torsion angle of about 10° between the two indolinone units, while the dihedral angles between the indolinone units and the carbazole and bithiophene spacers are approximately 40° and 28°, respectively. The HOMOs of all polymers are delocalized over the thienomethylene-capped di(indolinone) with the spacer, whereas the LUMOs are delocalized on the isoindigo units. Since the HOMO and LUMO coefficients are spatially disjoint, none of the polymers exhibited a significant charge-transfer band. Instead, all polymers exhibited π→π* transition bands with only minimal contributions from charge-transfer transitions in their absorption spectra.
 | ||
| Fig. 9 DFT-calculated frontier molecular orbitals of model polymers AIRI, AICRI and AITRI. | ||
Conclusion
In summary, acid-catalyzed aldol polycondensation provides a practical and safe alternative to cross-coupling reactions for the synthesis of conjugated polymers. This method was used to synthesize three polymers, namely, AIRI, AICRI and AITRI, which were subsequently purified and characterized using NMR analysis. These polymers demonstrated moderate to excellent solubility in common organic solvents and exhibited significant thermal stability. In UV-visible absorption spectra, AIRI, AICRI and AITRI showed broad absorption peaks with maxima at 438 nm, 434 nm, and 412 nm, respectively. Each polymer displayed reversible oxidation with onset potentials Eonsetox of +1.36 V, +1.29 V and +1.34 V, respectively. The band gap values were determined to be 2.14 eV for AIRI, 2.13 eV for AICRI and 2.25 eV for AITRI. Powder X-ray diffraction studies revealed a consistent d-spacing of 4.011 Å across all polymers, indicating similar structural characteristics. The measured values of SCLC hole mobilities were 7.13 × 10−2, 8.15 × 10−2 and 9.38 × 10−2 cm2 V−1 s−1 for polymers AIRI, AICRI and AITRI, respectively. The high hole mobilities suggest that these polymers could be effective p-type semiconductors for use in organic photovoltaic devices.Experimental section
General procedures
All chemicals used were of reagent grade and employed without additional purification. Reactions sensitive to moisture were conducted under an anhydrous nitrogen atmosphere using dry solvents. Reaction progress was monitored via thin-layer chromatography (TLC) on Merck 60 F254 aluminium-coated plates, with spots visualized under UV light. Silica gel column chromatography was used for purification. Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance-III 400 spectrometer, using CDCl3 as a solvent. High-resolution mass spectrometry (HRMS) analysis was carried out on an Xevo G2-XS QTOF mass spectrometer. UV-visible absorption spectra were recorded using a Jasco V-630 spectrophotometer with quartz cuvettes.The molecular weights of the polymer samples were determined using an Agilent 1260 Infinity gel permeation chromatography (GPC) system, equipped with a refractive index (RI) detector and calibrated with polystyrene standards. Approximately 5 mg of each polymer sample was dissolved in 5 mL of tetrahydrofuran (THF), filtered through a 0.2 µm filter and analyzed with THF as the eluent at a flow rate of 1–2 mL min−1. Thermogravimetric analysis (TGA) was performed on an Exstar SII TG/DTA 6300 instrument under a nitrogen atmosphere. Electrochemical measurements were conducted using a BioLogic SP-200 electrochemical analyzer. The current–voltage (I–V) characteristics of the devices were measured using a Keithley 2450 source meter.
Synthesis of monomers
Detailed synthetic procedures for intermediate compounds 1, 3, 5 and 7–9 are given in the SI.General procedure for the synthesis of compounds 2, 4 and 6
In a clean dry round bottom flask, compound 1 or 3 or 5 and hydrazine monohydrate [for compound 2: compound 1 (0.500 g, 0.96 mmol), hydrazine monohydrate (12 mL, 246 mmol) and DMSO (12 mL); for compound 4: compound 3 (0.400 g, 0.586 mmol), hydrazine monohydrate (8 mL, 150 mmol) and DMSO (8 mL); for compound 6: compound 5 (0.400 g, 0.504 mmol), hydrazine monohydrate (7 mL, 128 mmol) and DMSO (7 mL)] were dissolved in DMSO under a nitrogen atmosphere. The reaction mixture was refluxed for 30 hours at 140 °C. After the completion of the reaction, the reaction mixture was poured into the brine solution and extracted with ethyl acetate three times. The combined organic fractions were dried on anhydrous sodium sulfate and purified by column chromatography using 50% ethyl acetate in petroleum ether as the eluent to afford the desired product.General procedure for the synthesis of aldol polymers
In a clean dry round bottom flask, equimolar amounts of both monomers (1 equivalent, 0.0001 mmol) and p-TSA (0.6 eq., 0.00006 mmol) [for polymer AIRI, compound 2 (0.150 g), compound 11 (0.188 g) and p-TSA (0.031 g); for polymer AICRI, compound 4 (0.125 g), compound 11 (0.099 g) and p-TSA (0.0184 g) and for polymer AITRI, compound 6 (0.1 g), compound 11 (0.090 g) and p-TSA (0.016 g)] were taken in a 20 mL toluene under a nitrogen atmosphere. The reaction mixture was refluxed for 48 hours. After the completion of the reaction, the solvent was evaporated, the crude product was poured into methanol, and the solid polymer was separated by filtration. The polymer was further purified by Soxhlet extraction using acetone, hexane and chloroform. The chloroform fraction was separated, dried and used for further characterization.Conflicts of interest
The authors declare no conflicts of interest.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: detailed synthetic procedures for monomers, intermediate compounds and polymers and characterization data of the synthesized compounds and polymers. See DOI: https://doi.org/10.1039/d5lp00341e.Acknowledgements
The authors would like to acknowledge the DST-FIST program for funding the NMR facility at the Department of Chemistry, Faculty of Science, The Maharaja Sayajirao University of Baroda, Vadodara, Gujarat (India). The authors wish to thank Mr Gunvant Patel, Managing Director of Rhodium Master, for providing Pd-based catalysts.References
- A. W. Hains, Z. Liang, M. A. Woodhouse and B. A. Gregg, Chem. Rev., 2010, 110, 6689–6735 CrossRef CAS PubMed.
- H. Bronstein, C. B. Nielsen, B. C. Schroeder and I. McCulloch, Nat. Rev. Chem., 2020, 4, 66–77 CrossRef CAS PubMed.
- Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS PubMed.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- C. Zhong, C. Duan, F. Huang, H. Wu and Y. Cao, Chem. Mater., 2011, 23, 326–340 Search PubMed.
- P. M. Beaujuge, C. M. Amb and J. R. Reynolds, Acc. Chem. Res., 2010, 43, 1396–1407 CrossRef CAS PubMed.
- K. Müllen and W. Pisula, J. Am. Chem. Soc., 2015, 137, 9503–9505 CrossRef PubMed.
- G. Li, C. Kang, X. Gong, J. Zhang, W. Li, C. Li, H. Dong, W. Hu and Z. Bo, J. Mater. Chem. C, 2014, 2, 5116–5123 RSC.
- N. Blouin, A. Michaud and M. Leclerc, Adv. Mater., 2007, 19, 2295–2300 CrossRef CAS.
- E. Zhou, S. Yamakawa, Y. Zhang, K. Tajima, C. Yang and K. Hashimoto, J. Mater. Chem., 2009, 19, 7730–7737 RSC.
- Z. Zhu, D. Waller, R. Gaudiana, M. Morana, D. Mühlbacher, M. Scharber and C. Brabec, Macromolecules, 2007, 40, 1981–1986 CrossRef CAS.
- L. Liao, L. Dai, A. Smith, M. Durstock, J. Lu, J. Ding and Y. Tao, Macromolecules, 2007, 40, 9406–9412 CrossRef CAS.
- Y. Zhang, S. K. Hau, H.-L. Yip, Y. Sun, O. Acton and A. K.-Y. Jen, Chem. Mater., 2010, 22, 2696–2698 CrossRef CAS.
- J. Hou, H.-Y. Chen, S. Zhang, R. I. Chen, Y. Yang, Y. Wu and G. Li, J. Am. Chem. Soc., 2009, 131, 15586–15587 CrossRef CAS PubMed.
- Y. Liang, Y. Wu, D. Feng, S.-T. Tsai, H.-J. Son, G. Li and L. Yu, J. Am. Chem. Soc., 2009, 131, 56–57 CrossRef CAS PubMed.
- Y. Liang, Z. Xu, J. Xia, S.-T. Tsai, Y. Wu, G. Li, C. Ray and L. Yu, Adv. Mater., 2010, 22, E135–E138 CrossRef CAS PubMed.
- E. Wang, L. Hou, Z. Wang, S. Hellström, F. Zhang, O. Inganäs and M. R. Andersson, Adv. Mater., 2010, 22, 5240–5244 CrossRef CAS PubMed.
- J. C. Bijleveld, V. S. Gevaerts, D. Di Nuzzo, M. Turbiez, S. G. J. Mathijssen, D. M. de Leeuw, M. M. Wienk and R. A. J. Janssen, Adv. Mater., 2010, 22, E242–E246 CrossRef CAS PubMed.
- C. Piliego, T. W. Holcombe, J. D. Douglas, C. H. Woo, P. M. Beaujuge and J. M. J. Fréchet, J. Am. Chem. Soc., 2010, 132, 7595–7597 CrossRef CAS PubMed.
- M. Puchalska, K. Połeć-Pawlak, I. Zadrożna, H. Hryszko and M. Jarosz, J. Mass Spectrom., 2004, 39, 1441–1449 CrossRef CAS PubMed.
- T. Mohn, I. Plitzko and M. Hamburger, Phytochemistry, 2009, 70, 924–934 CrossRef CAS PubMed.
- C. Papageorgiou and X. Borer, Helv. Chim. Acta, 1988, 71, 1079–1083 CrossRef CAS.
- J. Mei, K. R. Graham, R. Stalder and J. R. Reynolds, Org. Lett., 2010, 12, 660–663 CrossRef CAS PubMed.
- T. Lei, J.-H. Dou, Z.-J. Ma, C.-H. Yao, C.-J. Liu, J.-Y. Wang and J. Pei, J. Am. Chem. Soc., 2012, 134, 20025–20028 CrossRef CAS PubMed.
- J. Mei, D. H. Kim, A. L. Ayzner, M. F. Toney and Z. Bao, J. Am. Chem. Soc., 2011, 133, 20130–20133 CrossRef CAS PubMed.
- T. Lei, J.-H. Dou and J. Pei, Adv. Mater., 2012, 24, 6457–6461 CrossRef CAS PubMed.
- T. Lei, Y. Cao, Y. Fan, C.-J. Liu, S.-C. Yuan and J. Pei, J. Am. Chem. Soc., 2011, 133, 6099–6101 CrossRef CAS PubMed.
- A. Ganguly, K. He, A. D. Hendsbee, M. Abdelsamie, R. N. Bennett, Y. Li, M. F. Toney and T. L. Kelly, ACS Appl. Mater. Interfaces, 2020, 12, 14265–14271 CrossRef CAS PubMed.
- Y. Jiang, X. Zheng, Y. Deng, H. Tian, J. Ding, Z. Xie, Y. Geng and F. Wang, Angew. Chem., Int. Ed., 2018, 57, 10283–10287 CrossRef CAS PubMed.
- L. Yao, H. Liao, M. K. Ravva, Y. Guo, J. Duan, Y. Wang, Y. Yu, Z. Li, I. McCulloch and W. Yue, Polym. Chem., 2020, 11, 3695–3700 RSC.
- B. Carsten, F. He, H. J. Son, T. Xu and L. Yu, Chem. Rev., 2011, 111, 1493–1528 CrossRef CAS PubMed.
- B. S. Nehls, U. Asawapirom, S. Füldner, E. Preis, T. Farrell and U. Scherf, Adv. Funct. Mater., 2004, 14, 352–356 CrossRef CAS.
- M. Mooney, A. Nyayachavadi and S. Rondeau-Gagné, J. Mater. Chem. C, 2020, 8, 14645–14664 RSC.
- C. Sun, F. Pan, H. Bin, J. Zhang, L. Xue, B. Qiu, Z. Wei, Z. G. Zhang and Y. Li, Nat. Commun., 2018, 9, 743 CrossRef PubMed.
- Y. W. Huang, Y. C. Lin, J. S. Li, W. C. Chen and C. C. Chueh, J. Mater. Chem. C, 2021, 9, 9473–9483 RSC.
- A. Onwubiko, W. Yue, C. Jellett, M. Xiao, H. Y. Chen, M. K. Ravva, D. A. Hanifi, A. C. Knall, B. Purushothaman, M. Nikolka, J. C. Flores, A. Salleo, J. L. Bredas, H. Sirringhaus, P. Hayoz and I. McCulloch, Nat. Commun., 2018, 9, 1–9 CAS.
- W. Yang, M. Sun, Y. Wang, H. Yan, G. Zhang and Q. Zhang, Polym. Chem., 2021, 12, 2317–2324 RSC.
- P. R. Patel, Y. G. Kapdi, S. S. Soni, S. S. Zade and A. L. Patel, New J. Chem., 2024, 48, 7007–7019 RSC.
- D. Wang, Y. Hou, K. Yang, Y. Wang, Q. Liao, S. Y. Jeong, J. Li, H. Y. Woo, X. Deng and X. Guo, Dyes Pigm., 2023, 212, 111138 CrossRef CAS.
- G. Zhang, Y. Dai, Y. Liu, J. Liu, H. Lu, L. Qiu and K. Cho, Polym. Chem., 2017, 8, 3448–3456 RSC.
- Y. Jiang, J. Chen, Y. Sun, Q. Li, Z. Cai, J. Li, Y. Guo, W. Hu and Y. Liu, Adv. Mater., 2019, 31, 1900342 CrossRef PubMed.
- Y. Yu, N. Xue, C. Xiao, M. K. Ravva, Y. Guo, L. Wu, L. Zhang, Z. Li, W. Yue and Z. Wang, J. Mater. Chem. C, 2019, 7, 12263–12269 RSC.
- V. S. Kadam, P. A. Bhatt, H. K. Machhi, S. S. Soni, S. S. Zade and A. L. Patel, Nano Sel., 2020, 1, 491–498 CrossRef.
- W. Koch and M. C. Holthausen, A Chemist's Guide to Density Functional Theory, 2001 Search PubMed.
| This journal is © The Royal Society of Chemistry 2026 |