
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Localized ionomer degradation analysis of sulfonated poly(phenylene sulfones) in fuel cell applications using confocal Raman microscopy
Julian
Stiegeler†
 ab,
Didem
Yazili-Marini†
c,
Christian
Piesold
ad,
Dennis
Rusitov
a,
Giorgi
Titvinidze
de,
Susanne
Koch
ad,
Nodar
Dumbadze
e,
Nertila
Joachimsen
ab,
Andreas
Münchinger
ad,
Tym
de Wild
*ad and
Carolin
Klose
*ad
ab,
Didem
Yazili-Marini†
c,
Christian
Piesold
ad,
Dennis
Rusitov
a,
Giorgi
Titvinidze
de,
Susanne
Koch
ad,
Nodar
Dumbadze
e,
Nertila
Joachimsen
ab,
Andreas
Münchinger
ad,
Tym
de Wild
*ad and
Carolin
Klose
*ad
aElectrochemical Energy Systems, IMTEK – Department of Microsystems Engineering, University of Freiburg, Georges-Koehler-Allee 103, 79110 Freiburg, Germany. E-mail: carolin.klose@imtek.uni-freiburg.de; tym.de.wild@imtek.uni-freiburg.de
bUniversity of Freiburg, Institute and FIT – Freiburg Center for Interactive Materials and Bioinspired Technologies (FIT), Georges-Koehler-Allee 105, 79110 Freiburg, Germany
cZentrum für Sonnenenergie- und Wasserstoff-Forschung Baden-Württemberg (ZSW), Helmholtzstraße 8, 89081, Ulm, Germany
dHahn-Schickard, Georges-Koehler-Allee 103, 79110 Freiburg, Germany
eDepartment of Natural Sciences and Biotechnology Engineering Polymers Laboratory, Agricultural University of Georgia, 240 David Aghmashenebeli Alley, Tbilisi, 0131, Georgia
First published on 3rd December 2025
Abstract
The development of highly conductive and stable fluorine-free polymer materials is critical for transitioning from perfluorosulfonic acid-based membranes to non-fluorinated alternatives for proton exchange membrane fuel cells and water electrolyzers. Among these, sulfonated poly(phenylene sulfones) (sPPS) are a promising class of polymers. However, little is known about their stability when used for these applications. To gain deeper insight into the aging mechanisms of sPPS membranes, confocal Raman microscopy was employed as a non-destructive, contact-free technique to determine membrane thickness and local equivalent weight. In this study, two sandwiched sPPS membranes were aged in situ using an accelerated stress test under open-circuit voltage (OCV) conditions in a fuel cell setup. Confocal Raman microscopy revealed that after the OCV-hold test, the combined thickness of sPPS membranes decreased from 27 µm to 15 µm, confirming chemical degradation. 60% of this reduction occurs on the membrane facing the anode side of the cell, indicating localized acceleration of degradation processes near the anode. Notably, despite the observed degradation, the local EW remained unchanged at the end of the test. This hints at a mechanism where chain scission is prevalent over desulfonation. Complementary techniques, nuclear magnetic resonance spectroscopy and gel permeation chromatography, were used on aged polymers to further validate these findings. It was found that despite no clear chemical changes (e.g. the degree of sulfonation or EW), the molecular weight decreased by 50%.
Introduction
Ionomer membranes are critical for electrochemical devices such as fuel cells and electrolyzers. State-of-the-art membranes have been based on perfluorosulfonic acid (PFSA), which, although effective, pose significant drawbacks such as high hydrogen crossover,1 loss of mechanical strength at high temperatures,2 and environmental concerns due to their persistence and accumulation in ecosystems.3–5 These concerns have driven the search for fluorine-free alternatives,6 with sulfonated polyphenylene sulfone (sPPS) emerging as a promising candidate due to the reported high proton conductivity at low gas permeability and good thermooxidative and hydrolytic stability7–10 while avoiding environmental concerns associated with perfluorinated materials.8 Despite these advantages, the long-term durability of sPPS membranes, particularly under operational stress, remains an open question.For effective fuel cell operation, the membrane must exhibit three key properties: (1) it must be impermeable to gases, preventing mixing of oxygen and hydrogen from the cathode and anode compartments; (2) exhibit good proton conduction to facilitate electrochemical reactions, and (3) maintain mechanical stability during operation as mechanical failure would lead to loss of its function as a barrier. During fuel cell operation, the membrane is exposed to harsh environments where fluctuating temperatures, relative humidity, and mechanical stress challenge its integrity. Additionally, the acidic environment, high electrochemical potentials, and reactive radicals can chemically degrade the membrane over time.11–15
Chemical degradation of ionomer membranes primarily occurs during fuel cell operation by the formation of radical species. These radicals can form in situ from oxygen and hydrogen, e.g. catalyzed by Pt to form H2O2,16 which can further decompose into reactive species such as H˙, HO˙, and HOO˙.17 There is still no consensus on whether degradation occurs predominantly on the cathode or anode side of the membrane electrode assembly (MEA).17–24 Various studies suggest that membrane degradation is highly influenced by factors such as operating conditions (e.g. cell voltage), the materials and loading of MEA components (e.g. platinum catalyst), and the overall cell design (e.g. flow field material impurities).21,25 This is especially true for non-PFSA-based membrane materials as they might have different gas transport resistances and degradation behaviors. Therefore, a reliable experimental approach is needed to characterize the chemical degradation mechanisms of materials independent of specific cell designs.
Chemical membrane degradation typically manifests in two effects: (1) loss of functional groups and (2) cleavage of the polymer backbone.11,24,26 The loss of functional groups increases the equivalent weight (EW) of the membrane and consequently reduces the proton conductivity of the membrane.11 Polymer backbone degradation reduces molecular weight, which results in membrane thinning and eventually leads to mechanical failure, e.g. pin-hole formation.11,27–29 In the case of sPPS membranes, there is limited understanding of whether functional group loss or backbone degradation is the primary degradation pathway. Understanding these mechanisms is crucial for developing strategies to mitigate chemical degradation in sPPS-based systems.
Various methods can be used to analyze the chemical and structural changes in ionomers. For EW determination, titration is the most common technique, though it requires precise sample drying and large quantities of materials for a reliable measurement – larger than typically available for end-of-life evaluation. Additionally, reinforced membranes cannot be measured as the weight of reinforcement would artificially increase the measured EW. Other methods include nuclear magnetic resonance (NMR) spectroscopy and energy-dispersive X-ray (EDX) spectroscopy, though both have limitations. NMR provides a global measurement that lacks spatial resolution, while EDX is surface-sensitive and requires cross-sectioning to examine material thickness and chemical composition across the material depth.30 Other X-ray-based methods, such as X-ray fluorescence (XRF) and X-ray tomography (XRT), can be used to get 3-dimensional data of samples but have limitations for membrane research. XRF provides limited resolution with good confocal XRF setups reaching resolutions of 10 µm. This is in many cases only slightly lower than the full membrane thickness, which starts at thicknesses of about 10 µm in modern electrochemical applications.31 XRT can achieve the required resolution to locally analyze membranes but lacks the ability to measure small changes in chemical compositions in the membrane.32
Confocal Raman microscopy (CRM) presents a promising alternative. CRM allows for non-destructive, spatially resolved analysis of local chemical changes within the membrane. Its confocal setup enables the acquisition of information from within the membrane, providing 3D measurements without physically sectioning the material.25,33,34 Thus, CRM offers a comprehensive view of the entire membrane volume. Previous studies by Böhm et al.25 demonstrated the effectiveness of CRM in analyzing degradation in Nafion-based membranes. Additionally, CRM was shown to be feasible to investigate water management33 and transport properties of different chemical compounds through PFSA-based membranes.35 Nishiyama et al. used a similar method, coherent anti-Stokes Raman scattering spectroscopy, to characterize the water molecule's state in a fuel cell during operation.36
In this study, we apply CRM to examine local chemical changes in sPPS membranes caused by chemical degradation during fuel cell operation. We demonstrate the feasibility of CRM to measure the thickness and equivalent weight of both pristine and degraded sPPS membranes, providing new insights into the degradation processes of these membranes during an open-circuit voltage (OCV) hold accelerated stress test (AST). Our findings are further supported by nuclear magnetic resonance (NMR) spectroscopy and gel permeation chromatography (GPC), validating the CRM data and confirming the degradation mechanisms in sPPS-based systems.
Experimental
sPPS ionomers
All sPPS ionomers were provided and synthesized by an optimized synthesis method first published by Schuster et al.9Reference sPPS film preparation
For Raman EW calibration, sPPS samples with different EWs are needed, labelled as sPPS-XX with XX being the EW.Non-reinforced membranes with different EWs were obtained by solvent casting sPPS-360, -390, -400 and -420 using the following procedure. The different sPPS polymers were prepared previously.8,9
Polymers were dissolved in DMSO under constant stirring at 120 °C overnight (10 wt% for sPPS-360, sPPS-390, and sPPS-400 and 5 wt% for sPPS-420). The resulting polymer solutions were filtered through a 3.1 µm glass microfiber syringe filter and degassed prior to membrane casting. Membranes were cast via doctor blading onto a glass plate and dried on the glass plate in the oven at 80 °C. After drying, the membranes were removed from the glass plate and immersed in 2 M HCl and left in the acid overnight to ensure full protonation. The membranes were washed in deionized water for 24 hours to remove residual acid. Finally, the membranes were dried at room temperature overnight.
Fuel cell testing and chemical membrane degradation AST
The MEA was composed of two layers of sPPS-390 both reinforced with a 5 µm porous polyethylene (PE) layer (Tejin Limited, MIRAIM®, 5 μm) (cross-section shown in Fig. S1). The MEAs were assembled in the dry state with commercial gas diffusion electrodes (GDEs) (Fuel Cells Etc®). The GDEs were based on a gas diffusion layer (GDL) from Freudenberg H23C8 and had a Nafion-based catalyst- layer with a loading of 0.5 mg Pt per cm2 (Pt/C: 60 wt%). The GDEs incorporated a Nafion interlayer to enhance the membrane-to-catalyst contact interface.The electrochemical testing was conducted using a PEM single-cell (Fuel Cell Technologies, Inc., New Mexico), featuring an active area of 5 × 5 cm2. Graphite flow fields with a triple serpentine design were utilized within the cell housing. The MEAs were assembled using a polytetrafluoroethylene (PTFE) gasket and sub-gasket to achieve approximately 15% compression of the GDE on each side. The cell was sealed by applying a torque of 7 Nm to the eight bolts surrounding the perimeter of the assembly. A G40 fuel cell test station (Greenlight Innovation, Canada) coupled with a potentiostat (BioLogic, France) was used.
Electrochemical conditioning
Before starting the electrochemical measurements, a pressure drop test was conducted by sealing the cathode outlet and introducing nitrogen through the cathode inlet, followed by sealing the anode outlet and introducing nitrogen through the anode inlet. This process ensured that there were no leaks either from the cell housing or through the membrane. Testing proceeded only when the pressure drop rate was below 1 kPa min−1. Following the pressure drop test, MEA conditioning was performed at a cell temperature of 80 °C with fully humidified (100% RH) H2 and O2 supplied to the anode and the cathode, respectively. The backpressure was set to 150 kPag, with gas flow rates of H2/O2 at 0.2/0.2 nlpm. The current density was maintained at 0.5 A cm−2 for 105 minutes to complete the conditioning process.Polarization (I–V) measurement
After conditioning, the cell was kept at 80 °C, 100% RH and 150 kPag. Reactant flows were fixed to a stoichiometry of 1.5/1.5 (H2/O2) with a minimum flow of 0.2/0.2 nlpm. Once the open-circuit voltage stabilised, the current density was stepped from 0 to 3.50 A cm−2. Between 0 and 0.25 A cm−2 the step size was 0.012 A cm−2 with a 60 s hold; above 0.25 A cm−2 it was 0.25 A cm−2 with a 180 s hold. Voltage was recorded every 2 s. For each current, the polarization curve was constructed from the mean of the final ten data points, corresponding to the last 20 s of data.OCV hold test
Subsequently, a long-term stability test was carried out by maintaining the fuel cell at OCV under the same conditions (80 °C, 100% RH, 150 kPag backpressure, and gas flow rates of H2/O2 at 0.2/0.2 nlpm). Cell voltage was logged every 2 s, and the high-frequency resistance (HFR) check was triggered every 5 min; the HFR reported is 1 kHz. The termination criterion of the test is a voltage drop of 20% from the initial OCV value.Hydrogen crossover (LSV)
Linear-sweep voltammograms were recorded at 80 °C and 100% RH. The anode and cathode were supplied with 0.2 nlpm H2 and 0.2 nlpm N2, respectively. The cell voltage was swept from 0.20 to 0.50 V at 2 mV s−1.MEA preparation for ex situ analysis
Removing all traces of the electrode material before conducting Raman microscopy is essential, as carbon black at the electrodes is highly sensitive to thermal degradation induced by laser irradiation. This decomposition can also damage the surrounding ionomer. Therefore, for ex situ characterization of the membrane, the MEA was disassembled. First, the GDEs were carefully removed from the membrane. Secondly, in order to remove the remaining catalyst layer on the membrane, the membrane was cleaned with an adapted wet process proposed by Bas et al.37 Specifically, the sample was immersed in an isopropyl alcohol (IPA) and water mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 weight ratio) at 60 °C and sonicated for ten seconds. This step removed the majority of the catalyst layers and allowed the two halves of the CCM to be separated, facilitating a better understanding of degradation mechanisms on the anode and cathode sides. To track membrane orientation, each half of the membrane had the side facing the catalyst layer marked and the orientation was carefully tracked throughout all future preparation and analysis. The remaining catalyst on the two half-CCMs was removed by two additional IPA-water baths, each conducted at 60 °C in an ultrasonic bath for ten seconds with 5 minutes of drying in between each step.
:1 weight ratio) at 60 °C and sonicated for ten seconds. This step removed the majority of the catalyst layers and allowed the two halves of the CCM to be separated, facilitating a better understanding of degradation mechanisms on the anode and cathode sides. To track membrane orientation, each half of the membrane had the side facing the catalyst layer marked and the orientation was carefully tracked throughout all future preparation and analysis. The remaining catalyst on the two half-CCMs was removed by two additional IPA-water baths, each conducted at 60 °C in an ultrasonic bath for ten seconds with 5 minutes of drying in between each step.
Raman microscopy
For quantitative Raman analysis, it is important to keep the measurement conditions as constant as possible. For Nafion, it was shown in the literature that the intensity of the SO3− Raman peak is sensitive to the hydration state of the system.38,39 To keep the hydration constant through all measurements, all samples were measured submerged in water. To ensure equal hydration, the samples were placed in DI water at least 18 h before the measurement. Raman measurements of the submerged samples were performed using a custom sample holder (shown in SI Fig. S2) with a water immersion objective (Zeiss W Achroplan/W N-Achroplan 63×/0.9).Raman measurements were conducted using a WITec alpha 300 RA (WITec, Germany) confocal Raman microscope. The microscope featured a 785 nm laser operating at 70 mW power. Spectra were obtained using a WITec UHTS 300 VIS-NIR spectrometer equipped with a Peltier-cooled, back-illuminated CCD camera (1024 pixels) and a 300 grooves per mm optical grating, providing a spectral resolution of approximately 2.1 cm−1. For depth scans, individual spectra were acquired for five seconds with six accumulations at 0.5 μm intervals through fully hydrated membranes to avoid changes in hydration. For calibration, five depth scans per reference sample were measured. For the beginning of life (BOL) membrane, 16 depth scans at different locations were performed and for the aged membranes, depth scans at ten different locations per sample were conducted.
Data processing
First, cosmic ray removal (CRR) and background subtraction were performed using WITec Project 5+ software. Filter settings were kept the same for all spectra. For the CCR algorithm, a filter size of 4 and a dynamic factor of 5 were used. Background subtraction was done using the shape-based algorithm with a shape size of 100.For all further evaluations, custom Python scripts were used. A visual guide for data processing is given in Fig. S3. And all used scripts can be found in the SI.
In the first script, the raw data were transferred to a 3D dataset (Fig. S3a). To achieve this, an average spectrum is calculated from all spectra recorded for a sample at the same z-height. Subsequently, for every wavenumber of interest, a 2D dataset (intensity vs. z-height) is generated by first fitting a Gaussian40 at the specified wavenumber and then calculating the area of the Gaussian between ±3σ for every z-height (Fig. S3b).
In the second script, the intensity vs. z-height data is smoothed, and the EW is calculated (Fig. S3c). First, two filters are applied to the data sequentially: a wavenumber filter and an intensity outlier filter. The wavenumber filter removes all data points where the wavenumber of the Gaussian peak center deviates by more than ±3 cm−1 from the reference value. This filtration step is crucial for eliminating incorrectly fitted peaks, which may result from cosmic rays or other artifacts that were not sufficiently removed by previous filters.
Following this, the intensity outlier filter is applied. Here, a moving average with a window size of three is computed. Any data points deviating by more than 30% from this average are considered outliers and subsequently removed from the dataset.
The cleaned data were then fitted using a cubic B-spline. The beginning and end of the membrane within this fitted dataset are identified by locating the points where the intensity is equal to or greater than 50% of the maximum intensity (Fig. S3d). This allows for the calculation of the membrane thickness. The position data are then normalized relative to the calculated membrane thickness, resulting in a dataset of intensity vs. relative membrane depth (ranging from 0 to 1) (Fig. S3e).
With these datasets, EW can be calculated for each measurement point from the peak ratios using eqn (1):41
 | (1) |
With m being the number ratio of backbone units per sulfonic acid group, IBB being the intensity of the backbone signal, IFG being the intensity of the functional group (sulfonic acid signal at 1022 cm−1) and a being the slope of the linear correlation obtained by Raman calibration data. This calculation is repeated for all relevant wavenumbers across all samples. Subsequently, the median EW and the standard deviation across samples are computed (Fig. S3f). Finally, the positional data are converted back to an absolute scale by multiplying them by the mean membrane thickness.
For the calibration samples, the same evaluation was used. But instead of calculating EW, only the peak ratio is calculated followed by calculating the mean peak ratio over the whole membrane thickness as well as the sample-to-sample standard deviation.
We want to mention that Maier et al. recently published an improved method to determine the thickness of a membrane in Raman z-scans by fitting a sigmoidal curve to the beginning and end of the spectra to account for changes in refractive index between the sample and the surrounding medium as well as the absorption of the material.42 We did not apply this method as the intensity did not change strongly over the scan depth, only introducing a small error in the thickness evaluation, and the refractive indices of the membrane and water are very close so that the mismatch can be neglected. Additionally, the computational effort would be significantly higher as we work with a three-layer setup with a mixed area since we analyzed PE reinforced membranes.
NMR measurements
After Raman analysis, parts of the samples were dried at 125 °C in a vacuum for four hours and then placed in DMSO-d6 and heated to 100 °C for 24 hours in a sealed glass vial to dissolve all the ionomer in the membrane. The solution was then transferred to an NMR tube using a syringe. Since in the case of reinforced samples, the reinforcement was not soluble in DMSO-d6, the NMR sample remained unaffected by the reinforcement, which remained in the vial. 1H spectra with 128 scans were acquired on a Bruker Avance IV Neo 400 MHz NMR-spectrometer.For evaluation, MestreNova 14.2.1 was used. Before analyzing the spectra, all spectra were auto-phase corrected and auto-background corrected. The chemical shift was referenced to the DMSO peak at 2.50 ppm.
EW-analysis by NMR
EW can be determined by NMR by calculating the number ratio of sulfonated monomer units to total monomer units, denoted as m−1, using eqn (1).41 An exemplary spectrum of sPPS-390 is shown in Fig. 1c. For the evaluation of m−1, the “peak” integration method in MestReNova was used. All relevant peaks were first assigned (1H NMR (400 MHz, DMSO-d6): δ 8.59 (d, H1), 8.54 (d, H2), 8.26 (d, H3), 8.20 (m, H4), 8.06 (d, H5), 7.98 (d, H6)). Peaks 1, 2, and 3, corresponding to three protons total per phenyl group, were used as proxies for the functional group. Peaks 4, 5, and 6, representing four protons total per phenyl group, were used for the unsulfonated repeat units. | ||
| Fig. 1 (a) Chemical structure of sPPS with X giving the ratio of the two dyads. (b) Normalized Raman spectrum of sPPS-360 (black), sPPS-390 (yellow), sPPS-400 (green), and sPPS-420 (blue). Colored areas correspond to the colored chemical groups in (a), gray and red marked peaks could not be assigned to one specific chemical group (see the Raman spectrum of sPPS section). (c) NMR spectrum of sPPS-390, where numbers 1–6 correspond to the assigned protons in the chemical structure (a). | ||
The ratio m−1 is calculated using eqn (2), where I is the integral of proton peaks 1 to 6 according to Fig. 1.
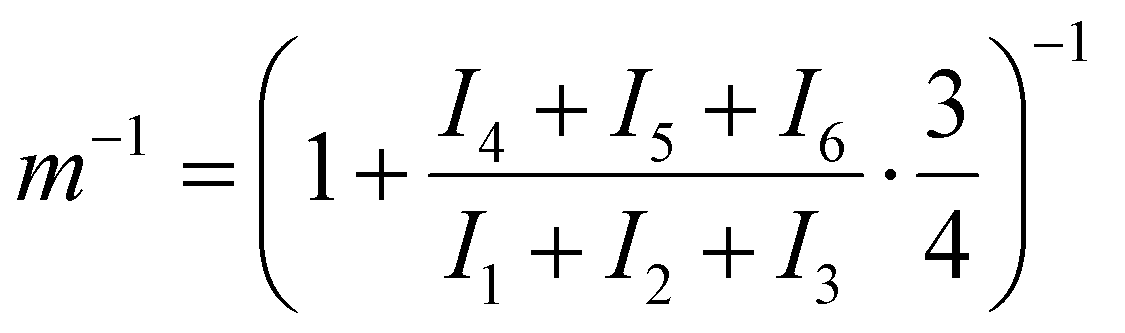 | (2) |
Gel permeation chromatography (GPC)
GPC samples were prepared by drying the NMR samples and dissolving 2 mg of dry polymer in 4 ml of DMSO–LiBr solution (WLiBr = 0.5 wt%) at 100 °C for 4 hours and subsequently filtered through a 1.0 µm glass fibre filter. The measurement was performed on a PSS 1200 system equipped with a PSS GRAM Precolumn 10 μm (8 × 50 mm) and three analytical columns (1× PSS GRAM 100Å and 2× PSS GRAM 3000Å, each 8 × 300 mm). The system was calibrated before the experiment using pullulan standards (polymer standard services GmbH) using an RI-detector (PSS G1362A, 1260-Series) and a differential viscosity detector (PSS DVD 1260). DMSO with LiBr (0.5 wt%) was used as a mobile phase at 70 °C. Measurements were performed with an injection volume of 100 µl and a flow rate of 0.8 ml min−1.Results
Raman spectrum of sPPS
To investigate sPPS by CRM, the sPPS-Raman modes of the aromatic ring (Fig. 1a: blue) and the SO2-bridge (Fig. 1a: green) of the backbone and the sulfonic acid functional group (Fig. 1a: yellow) were assigned to the respective Raman peaks (Fig. 1b; full spectrum: Fig. S4a). For the peak assignment, literature data of bisphenol-S43 and benzenesulfonic acid were used44 as they are good model compounds for the sulfonated phenylene groups in sPPS. Bisphenol-S resembles the unsulfonated backbone, and benzenesulfonic acid serves as the model for the sulfonated phenyl ring. Raman spectra of the sulfonated and unsulfonated monomers (see Fig. S4b) and a series of Raman spectra of four different types of sPPS (Fig. 1b) were recorded. The sPPS samples comprised sPPS-360 (EW ≈ 360 g mol−1, X = 0), sPPS-390 (EW ≈ 390 g mol−1, X ≈ 0.11), sPPS-400 (EW ≈ 400 g mol−1, X ≈ 0.14), and sPPS-420 (EW ≈ 420 g mol−1, X ≈ 0.21). The samples were measured while submerged in water to avoid signal changes due to changes in hydration.The Raman peak of sPPS observed at 1581 cm−1 is assigned to the C–C-stretching vibration in the phenyl ring (blue mark, Fig. 1a), as supported by the literature studies on bisphenol-S and benzenesulfonic acid.43,44 Due to its high intensity and clear assignment to the backbone, this peak will be used as a reference for all spectra.
The peak observed at 1155 cm−1 is assigned to an S–O stretching vibration mode arising from the sulfone bridge in the backbone (green mark, Fig. 1a) since a similar peak is present in bisphenol-S and the Raman spectra of the unsulfonated monomer (Fig. S4b). Additionally, its intensity remains unchanged for sPPS ionomers with different EW (Fig. 1b).43
The two peaks at 1172 cm−1 and 1022 cm−1 are assigned to the S–O stretching vibration mode arising from the sulfonic acid group (yellow mark, Fig. 1a) since both peaks varied with the EW of the sPPS ionomer (inset Fig. 1b). The assumption is supported by the occurrence of both peaks in the Raman spectrum of the functionalized monomer (Fig. S4b) and benzenesulfonic acid.44 As the peak at 1172 cm−1 overlaps the peak at 1155 cm−1 it is only observable as a shoulder.43,44
The peak at 1090 cm−1 is attributed to the backbone (grey mark, Fig. 1b), as it does not appear in any other spectra used for peak assignment and shows no dependency on EW.
The 1070 cm−1 peak (red mark, Fig. 1b) is observed at a similar intensity for all measured sPPS samples, was also observed in bisphenol-S and was assigned by Ullah et al.43 to either a CC stretching vibration mode or an SO stretch vibration. Since this peak is only present in the unsulfonated monomer and does not vary with EW, it is likely attributed to the backbone.
Other peaks were not assigned due to their low intensities, making them unsuitable for evaluation because of higher error margins. A summary of the assigned peaks is provided in Table 1.
Raman EW calibration curve
Raman spectra are mostly used for qualitative analysis as the absolute intensity of a measurement not only depends on the sample but also on the laser wavelength and intensity used, the detector, and the optics. Additionally, quantitative analysis is further complicated as each Raman-active group has its own Raman activity. To circumvent most of these issues, normalization is necessary. For Nafion, Böhm et al. used the relative peak ratio of a backbone peak and the sidechain peak to determine the EW of Nafion-based membranes.25,45 The same approach is used in this study. In short, the Raman peak intensity of a chemical group is proportional to its number of groups in the measured volume46 and its Raman activity and laser intensity.46 With this approach, one can use the following equation to calculate the ratios of backbone units per sulfonic acid group m−1. | (3) |
With the Raman measurement constant K (laser wavelength, intensity, sensor, etc.), the number of sample molecules n, the specific Raman activity of the molecule fragment vibration mode of the sulfonic acid group aFG and the backbone aBB and the respective intensities IFG and IBB. As n and K are identical for both peaks, they cancel each other out. The activities aFG and aBB can be simplified to an effective activity a, which can be experimentally measured by a concentration study (cf. section: Data processing). For further details, we refer to Böhm et al.25
In the following, we relate to m−1 to refer to the ratio of functional groups to backbone units instead of the EW, which refers to the backbone mass. To establish a calibration curve for m−1 of sPPS ionomers, four non-reinforced sPPS sample membranes with different but known m−1 were measured. m−1 for sPPS-360, sPPS-390, sPPS-400, and sPPS-420 has been determined by NMR to be m−1 = 0.498, m−1 = 0.443, m−1 = 0.426, and m−1 = 0.408, respectively (Table 2).
| Sample | Target EW [g molSO3H−1] | EW by NMR [g molSO3H−1] | m −1 by NMR |
|---|---|---|---|
| sPPS-360 | 360 | 360.4 | 0.498 |
| sPPS-390 | 390 | 394.9 | 0.443 |
| sPPS-400 | 400 | 407.6 | 0.426 |
| sPPS-420 | 420 | 421.7 | 0.408 |
To counteract any influence of chromatic aberration, we performed full depth scans and calculated the peak ratio with the normalized datasets. Afterward, the peak ratio was averaged over the full membrane depth.
Fig. 2 displays the intensity ratio of the sulfonic acid mode (1022 cm−1) divided by the three most significant backbone modes: 1090 cm−1 (blue), 1155 cm−1 (green), and 1581 cm−1 (yellow) over m−1. The data show some scattering but follow, as expected, a linear trend. For the linear correlation, the y-intercept was defined as y = 0, since there should be no sulfonic acid signal for an unfunctionalized polymer. The parameters obtained by linear fitting of the different intensity ratios are given in Table S5. The scattering for the peak ratio of 1090 cm−1 to 1022 cm−1 is higher than that for the other peaks, since the intensity of the 1090 cm−1 mode is much lower compared to the intensity of the 1155 cm−1 and 1181 cm−1 modes, as can be seen by the greater slope. Lower intensity means that the absolute error due to random noise is higher. The small shoulder of the 1055 cm−1 peak should not influence the results greatly, as we do a Gaussian fitting before evaluating the area of the fit, which should neglect the additional signal. However, there might still be a small contribution from the additional signal. Thus, in principle, all evaluated peak ratios can be used for the determination of m−1 as the linear fits demonstrate excellent agreement with the data, as indicated by the high coefficient of determination (R2 > 0.99). However, the 1581 cm−1 peak is the most suitable for EW evaluation as it exhibits the second highest intensity and shows no contribution from other signals. The high intensity leads to smaller errors due to noise. The peak at 1155 cm−1 has higher intensity but is close to a very strong signal of the backbone (see Fig. S8a), risking overlap or wrong fitting by the algorithm.
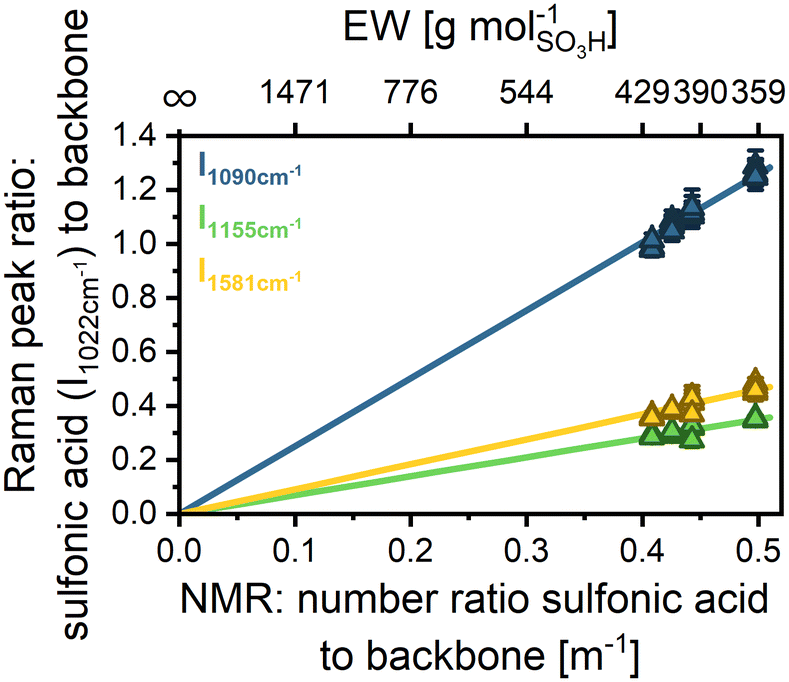 | ||
| Fig. 2 Raman calibration: correlation between the Raman peak ratio and the NMR number ratio of sulfonic acid groups per backbone repeat unit and respective EW for 4 samples (sPPS-360, sPPS-390, sPPS-400, and sPPS-420). Error bars indicate standard deviation within a single depth-scan. The lines are error weighted linear fits with a defined y-intercept at (0|0). Fit parameters are given in Table S5. | ||
Chromatic aberration in confocal Raman microscopy
When analyzing the normalized intensity vs. scan depth for a pristine membrane at the 1022 cm−1 and 1581 cm−1 peaks, a positional shift in the intensity data is apparent (Fig. 3a). Since the backbone and sulfonic acid group are covalently bonded, the signal shift of about 1 µm cannot be explained by the ionomer structure or conformation. Since the evaluation of the EW, given by the ratio of the two peaks, is significantly altered by the observed shift (Fig. S6 blue), particularly at the membrane edges, the respective peak intensities need to be position corrected. | ||
| Fig. 3 (a) Peak shift: normalized peak intensity with respect to scan-depth for a pure sPPS-390 membrane. (b) Positional shift of the measured start and end of the membrane for the BOL reinforced SPPS-390 sample with 16 measurements and a concatenated weighted linear fit over all data points and its 95% prediction band (yellow). The reference point (marked with gray dashed lines) is the sulfonic acid peak at 1022 cm−1. | ||
An analysis of the positional shifts of other peaks relative to the 1022 cm−1 peak revealed a linear trend of peak shift versus wavenumber (Fig. 3b). The occurrence of this shift is most likely caused by chromatic aberration in the optical setup, as the confocal microscope is optimized for the visible range, and the utilized 785 nm laser operates at the edge of the device's operational window. Chromatic aberration would lead to different focal planes depending on the light wavelength, and with that to the observed positional shifts.47,48
The chromatic aberration does not affect the measured membrane thickness, as can be seen by evaluating the membrane thickness of different peaks (1089 cm−1, 1155 cm−1, and 1581 cm−1) with respect to the membrane thickness evaluated from the sulfonic acid signal (1022 cm−1) (Fig. S7). This finding allows us to normalize the peak location separately with regard to the membrane thickness, creating an intensity vs. relative membrane thickness dataset (ranging from 0 to 1 with 0 = beginning of the membrane and 1 = end of the membrane). The thickness corrected dataset allows us to determine the signal ratio of the sulfonic acid peak to the backbone peaks while avoiding errors due to chromatic aberration. Comparing EW evaluation by absolute scan depth and thickness-normalized location for a pristine membrane shows a clear improvement in data consistency (cf. Fig. S6).
EW measurement of reinforced membranes
Reinforcements might affect the EW measurement if the reinforcement material exhibits a chemically similar structure to the ionomer. This is, e.g. often the case for PFSA membranes, which are typically reinforced by an expanded PTFE substrate.25 However, as long as there is a backbone peak and a functional group peak that does not overlap with the reinforcement spectra, the EW should remain accessible by CRM.33,45 For hydrocarbon membranes still in the R&D state, different reinforcement materials are being tested.49–51 In this study, polyethylene (PE)-reinforced sPPS membranes are used. For PE and sPPS, only the peak at 1070 cm−1 overlaps (Fig. S8a), leaving a sufficient number of peaks for accurate EW evaluation by CRM.Thus, while the signal intensity of sPPS-390 decreases in absolute terms, when PE is present as reinforcement, due to the reduced local concentration of sPPS-390 (Fig. S8b), the peak ratios of the sulfonic acid group and the backbone peaks (1090 cm−1, 1155 cm−1, and 1581 cm−1) remain constant. This can be seen in a depth scan of a PE-reinforced sPPS membrane (Fig. S8c). The determined EW across the thickness of the membrane (using the signal at 1581 cm−1) remains constant and unaffected by the integrated reinforcement over the whole membrane depth, indicating no interaction between the reinforcement and the ionomer that would influence the Raman peak ratio. To locate the reinforcement, one can examine the intensity of the sPPS-390 peaks, as the local concentration of sPPS-390 is reduced where the reinforcement is present, leading to lower signal intensities but a similar intensity ratio (see Fig. S8b and c). Alternatively, the reinforcement position can be determined by analyzing its specific Raman signals. Although this method uses absolute location data and is subject to a small error due to chromatic aberration, it is more accurate in identifying the reinforcement boundaries, as changes in the sPPS-390 signals are minimal. In Fig. S8d, the depth scan for sPPS-390 is shown, along with the reinforcement location determined by the Raman signals of PE at 1296 cm−1. We corrected the PE location by applying a factor obtained from the linear correlation when analyzing chromatic aberration.
Membrane degradation
Fig. 4 shows the relationship between the cell voltage (OCV, blue line) and the high-frequency resistance (HFR, green line) over time during the OCV hold. The cell voltage and the rate of voltage loss provide insights into the membrane degradation process, as the OCV is largely governed by hydrogen permeation from the anode to the cathode.
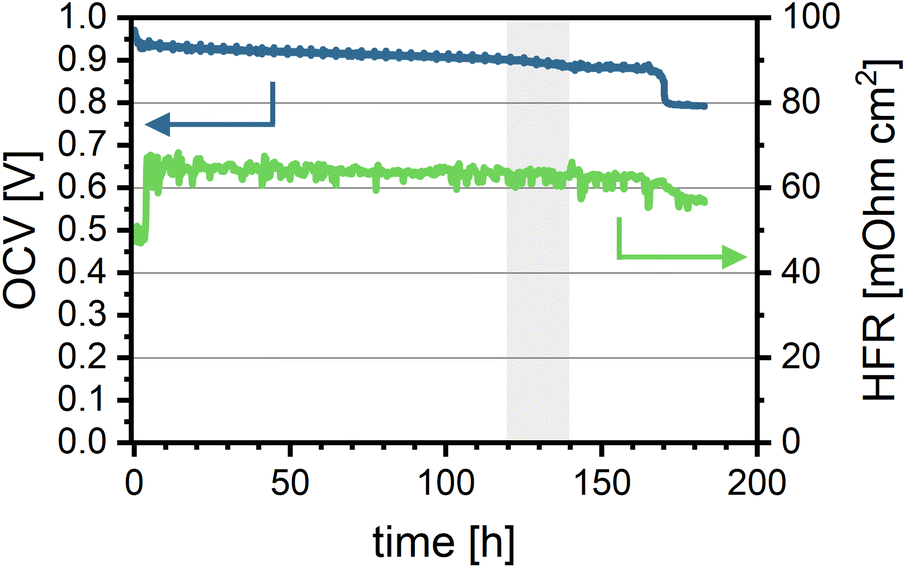 | ||
| Fig. 4 Cell in situ aging data: OCV over aging time (blue, left axis) and HFR over aging time (green, right axis). The grey area marks the timeframe between 120 and 140 hours. Measured at 80 °C, 100% RH, 150 kPag backpressure, and gas flow rates of H2/O2 at 0.2/0.2 nlpm. | ||
At the beginning of the test, the cell voltage is the highest (971 mV) and gradually decreases at a rate of approximately 20 mV h−1, with slightly more pronounced degradation observed between 120 and 140 hours. At 170 hours, a sharp drop of 100 mV in OCV is observed. This sudden voltage loss is likely due to hole formation and/or loss of mechanical integrity in the polymer membrane caused by oxidative damage, which increases hydrogen crossover. After 183 hours, the cell voltage falls below the minimum threshold (20% OCV loss), and the test is terminated. The overall loss in OCV suggests membrane thinning and the formation of pinholes, leading to increased gas crossover and potentially contributing to local performance degradation. This increased crossover is also evident from LSV measurements comparing the BOL and EOL states (see Fig. S9a). Hydrogen crossover current density increases from 0.21 mA cm−2 (1.08 × 10−6 mmol cm−2 s−1) to 2.92 mA cm−2 (15.14 × 10−6 mmol cm−2 s−1).
The HFR decreases overall by 16%, from 68 to 57 mΩ cm2, with a more significant reduction coinciding with the voltage drop after around 170 hours. The slight decrease in the HFR may indicate improved proton transport through the membrane over time, potentially due to a decrease in membrane thickness while retaining proton conductivity. However, it is important to note that the decrease in HFR can also be caused by changes in electrical or contact resistances in the cell. Nevertheless, it does align reasonably well with the later measured reduction in membrane thickness (see S10).54 The relatively stable HFR implies that the membrane's EW remains nearly constant throughout the test duration.
Overall performance degradation was not evaluated for this system as EOL performance measurements were not performed due to low OCV as well as high crossover and shunt resistance. BOL performance (Fig. S9b) is comparable to or better than the performance of other hydrocarbon-based cells reported in the literature.52,55,56
Membrane thickness
Using CRM allows for virtual cross-sections, providing localized chemical data of the membrane structure. When performing depth scans with CRM, the thickness of different layers can be extracted as well. As expected from the decrease in OCV and HFR, the membrane likely undergoes thinning during the accelerated stress test. This finding is supported by the CRM virtual cross-section (Table 3 and Fig. 5a). Comparing the end-of-life (EOL) thickness of the anode-half and the cathode-half to the BOL thickness of the PE-reinforced sPPS membrane by CRM, a reduction of approximately 44% from 27 µm down to 15 µm is observed. While significant, this thinning is less severe than that typically reported for a PFSA membrane (Nafion NR-211 with 30 µm original thickness) by Böhm et al., who observed a 64% reduction in the membrane thickness after 100 hours of OCV-hold. This observation aligns with the slow loss in OCV over time, which is likely caused by increasing gas crossover due to continuous membrane thinning. Similar to Böhm et al., we observe more pronounced degradation on the anode, with the EOL thickness of the anode being 2 µm (or 25%) lower than that of the cathode.25 The statistical significance of this observation in the thickness difference is confirmed by the Mann–Whitney U test (U = 92.0, p = 0.0017). It should be noted that a change in the molecular weight of the polymer could change the swelling properties of the material. Meaning that with a decrease in molecular weight, the membrane swells more.57 This would lead to CRM underestimating the change in membrane thickness between BOT and EOT compared to the dry state, as it is measured in a fully hydrated state. | ||
| Fig. 5 (a) Membrane thickness at the beginning and end of life (BOL and EOL). The box gives the 25th to 75th percentile of all measured thicknesses; whiskers show the min and max of the dataset. The horizontal line in the box gives the median and the black diamonds mark the average thickness. Next to the barplot is the smoothed distribution function with every single data point marked as a circle. The black dotted line indicates the nominal thickness of the PE-reinforcement. (b) EW with respect to membrane depth. The shown depth is the average of all measured membrane thicknesses (cf. the Data processing section). | ||
| Thickness [µm] | Thickness [% BOL] | M n [kDa] | M w [kDa] | Đ M (Mw/Mn) | M n [% BOL] | M w [% BOL] | Đ M [% BOL] | EW by Raman [g molSO3H−1] | EW by NMR [g molSO3H−1] | |
|---|---|---|---|---|---|---|---|---|---|---|
| Comparison of global EW measured by Raman and NMR. | ||||||||||
| BOL | 13.3 ± 1.7 | 100 | 18.7 | 58.1 | 3.1 | 100 | 100 | 100 | 382 | 382 |
| EOL-cathode | 8.6 ± 1.3 | 64.7 | 12.4 | 34.1 | 2.8 | 66.2 | 58.8 | 88.7 | 378 | 393 |
| EOL-anode | 6.4 ± 1.5 | 48.1 | 9.3 | 19.3 | 2.1 | 49.6 | 33.3 | 67.0 | 383 | 381 |
This conclusion is further supported by HFR measurements, which show a slight decrease during AST, indicating that proton transport dynamics remain stable despite aging with the slight decrease of HFR due to membrane thinning. To validate the Raman data, we compare the global EW measured by Raman (average EW over the full membrane thickness) to the EW determined by NMR (see Table 3). NMR results confirm the Raman measurements, as both methods show only insignificant differences between BOL and EOL EW. Additionally, the data demonstrate excellent agreement between NMR and Raman, further reinforcing the reliability of the findings.
To better understand the degradation mechanism of sPPS, it is crucial to consider possible reaction pathways. As Schuster et al.9 demonstrated excellent hydrolytic stability for sPPS-type polymers, we exclude hydrolytic degradation as a major pathway, focusing instead on radical induced polymer degradation.
While there are many reaction pathways suggested for sulfonated arylene-based polymers with C–C bonds in the backbone,62,64 there is no mechanistic study of arylene groups linked only by sulfone groups, and only a few studies on heteroatom linked arylenes such as sulfonated polyether(ether)ketone (sPEEK)65 and sulfonated polyethersulfone (sPSU),66 making suggestions about reaction pathways for the analyzed sPPS highly speculative. In the few studies analyzing arylene-based polymers with C–C bonds in the backbone, neither Holmes et al. nor Dockheer et al. proposed an attack on the C–S bond in sulfonated phenylated polyphenylenes64 or poly(sodium styrene sulfonate), respectively.62
For sPEEK, it was demonstrated that chain scission is possible by cleaving the hetero C–O bond.65 Using density functional theory (DFT), Panchenko calculated that in sPSU, the C–S bond between the backbone and the sulfonic acid can be split. Although the reaction is unlikely to occur in the gas phase due to high reaction free energies, it can occur in the presence of water.66 The reason this is not observed in measurable amounts might be the slightly different chemical structures: sPPS only contains sulfone bridges, whereas sPSU features alternating sulfone and ether bridges, leading to different electron densities in the arylene backbone.
However, based on the mechanism described by Holmes, Dockheer and Panchenko, it is likely that hydroxyl radicals rapidly react with the aromatic rings to form cyclohexadienyl radical adducts, which react further (with e.g. oxygen), eventually leading to chain scission or desulfonation. The observation of stable EW suggests that desulfonation is not the major pathway.
For a better understanding of the chemical and structural degradation mechanism of sPPS, additional dedicated experiments will be required. Particularly, analyzing degradation products exiting the fuel cells might provide additional insights into the exact mechanism.
Conclusion
In this work, the capabilities of confocal Raman microscopy (CRM) for analyzing sulfonated poly(phenylene sulfone) (sPPS) membranes have been demonstrated. Our results show that CRM is a reliable method for measuring equivalent weight (EW), with a strong agreement with the NMR results. In addition to NMR, CRM not only provides the global EW as an average over the whole membrane area but also enables the evaluation of the local EW throughout the depth of the membrane. A correction of the measurement data has been provided to avoid errors due to chromatic aberration. This allows us to implement the method for different set-ups not necessarily optimized for the utilized wavelength.To show the feasibility of CRM for the analysis of membrane aging, a sPPS membrane was aged for 180 h under in situ OCV-hold conditions. While some local EW variations were observed after the test, the values remained closely aligned with the overall EW with only slight changes to the EW at the beginning of the test. Thus, for sPPS, the sulfonic acid groups directly attached to the polymer backbone are not prone to chemical degradation, and the C–S bonds between the backbone and sulfonic acid groups are highly stable. The significant decrease in sPPS membrane thickness after the AST, down to 56% of the original value, was attributed to an attack on the backbone of the arylene polymer, leading to a reduction in molecular mass and the likely removal of equal fragments of sulfonated and non-sulfonated polymers. This degradation mechanism is fundamentally different from that of PFSAs, where functional group or side-chain degradation occurs more rapidly than main-chain scission. In sPPS, by contrast, degradation is driven primarily by an attack on the polymer backbone, while the sulfonic acid groups remain largely intact.
Additionally, we showed that the integration of a membrane reinforcement material does not interfere with the EW measurements by CRM, provided that carefully selected peaks are used to avoid signal overlap. Since the reinforcement structure can be localized, future studies can leverage this feature to differentiate between the anode and cathode sides of a membrane using the reinforcement as a marker. This technique enables the use of a single reinforced membrane for degradation studies, maintaining spatial resolution between the two sides.
Looking ahead, this method holds potential for investigating degradation not only in sPPS-based fuel cell membranes but also in other membrane materials. Furthermore, the influence of operating conditions, including operation under electrolysis conditions, on degradation as well as on different degradation modes of various hydrocarbon-based membranes can be analyzed with the presented method. However, since the method is limited to the material that remains after testing, we would suggest that future studies should include effluent water analysis, as degradation products are likely present and could provide additional insights into the underlying degradation mechanisms.
Author contributions
Julian Stiegeler: conceptualization, methodology, software, formal analysis, investigation, data curation, writing – original draft, writing – review & editing, visualization, supervision, and project administration. Didem Yazili-Marini: methodology, investigation, resources, writing – original draft, writing – review & editing, and funding acquisition. Christian Piesold: validation, resources, formal analysis, investigation, and writing – review & editing. Dennis Rusitov: resources and writing – review & editing. Giorgi Titvinidze: validation, resources, and writing – review & editing. Susanne Koch: software, validation, formal analysis, data curation, and writing – review & editing. Nodar Dumbadze: resources and writing – review & editing. Nertila Joachimsen: resources and investigation. Andreas Münchinger: investigation and writing – review & editing. Tym de Wild: validation, writing – review & editing, supervision, and project administration. Carolin Klose: validation, writing – review & editing, supervision, project administration, project lead, and funding acquisition.Conflicts of interest
There are no conflicts of interest to declare.Data availability
Data for this article, including Raman Spectra, Raman EW-calibration, peak shift data, cell durability, and Raman depth-scans, as well as the used evaluation scripts are available. See DOI: https://doi.org/10.60493/yktz3-67524.Supplementary information (SI): Full Raman spectra of sPPS-390, Raman spectra of the monomers, linear fit data for the EW calibration, data to show independence of measured membrane thickness with the used Raman peak for evaluation, and further data supporting the manuscript. See DOI: https://doi.org/10.1039/d5lp00284b.
Acknowledgements
The authors acknowledge funding from the Federal Ministry of Research, Technology and Space of Germany (BMFTR) through the projects FC-RAT (grant 03EW0011B) and Beyond-PFSA (grant 03SF0643A). We also acknowledge Dr. Victor Hugo Pacheco Torres and Ivonne Knauer (Institute for Macromolecular Chemistry, University Freiburg) for measuring GPC and NMR spectroscopy. We thank Alexander Kohushölter (IMTEK, University of Freiburg) for providing SEM cross-sections. During the preparation of this work, the authors used ChatGPT in order to improve text clarity and readability. After using this tool/service, the authors have reviewed and edited the content as needed and take full responsibility for the content of the publication.References
- H. Ito, T. Maeda, A. Nakano and H. Takenaka, Properties of Nafion membranes under PEM water electrolysis conditions, Int. J. Hydrogen Energy, 2011, 36, 10527–10540, DOI:10.1016/j.ijhydene.2011.05.127.
- K.-D. Kreuer, The role of internal pressure for the hydration and transport properties of ionomers and polyelectrolytes, Solid State Ionics, 2013, 252, 93–101, DOI:10.1016/j.ssi.2013.04.018.
- M. Feng, R. Qu, Z. Wei, L. Wang, P. Sun and Z. Wang, Characterization of the thermolysis products of Nafion membrane: A potential source of perfluorinated compounds in the environment, Sci. Rep., 2015, 5, 9859, DOI:10.1038/srep09859.
- W. Wang, G. Rhodes, J. Ge, X. Yu and H. Li, Uptake and accumulation of per- and polyfluoroalkyl substances in plants, Chemosphere, 2020, 261, 127584, DOI:10.1016/j.chemosphere.2020.127584.
- P. Casal, B. González-Gaya, Y. Zhang, A. J. F. Reardon, J. W. Martin, B. Jiménez and J. Dachs, Accumulation of Perfluoroalkylated Substances in Oceanic Plankton, Environ. Sci. Technol., 2017, 51, 2766–2775, DOI:10.1021/acs.est.6b05821.
- H. Nguyen, C. Klose, L. Metzler, S. Vierrath and M. Breitwieser, Fully Hydrocarbon Membrane Electrode Assemblies for Proton Exchange Membrane Fuel Cells and Electrolyzers: An Engineering Perspective, Adv. Energy Mater., 2022, 12, 2103559, DOI:10.1002/aenm.202103559.
- G. Titvinidze, K.-D. Kreuer, M. Schuster, C. C. de Araujo, J. P. Melchior and W. H. Meyer, Proton Conducting Phase-Separated Multiblock Copolymers with Sulfonated Poly(phenylene sulfone) Blocks for Electrochemical Applications: Preparation, Morphology, Hydration Behavior, and Transport, Adv. Funct. Mater., 2012, 22, 4456–4470, DOI:10.1002/adfm.201200811.
- Z. Katcharava, T. Saatkamp, A. Muenchinger, N. Dumbadze, K.-D. Kreuer, M. Schuster and G. Titvinidze, Optimized step–growth polymerization of water–insoluble, highly sulfonated poly(phenylene sulfone), Polym. Adv. Technol., 2022, 33, 2336–2343, DOI:10.1002/pat.5688.
- M. Schuster, K.-D. Kreuer, H. T. Andersen and J. Maier, Sulfonated Poly(phenylene sulfone) Polymers as Hydrolytically and Thermooxidatively Stable Proton Conducting Ionomers, Macromolecules, 2007, 40, 598–607, DOI:10.1021/ma062324z.
- C. Klose, T. Saatkamp, A. Münchinger, L. Bohn, G. Titvinidze, M. Breitwieser, K.-D. Kreuer and S. Vierrath, All–Hydrocarbon MEA for PEM Water Electrolysis Combining Low Hydrogen Crossover and High Efficiency, Adv. Energy Mater., 2020, 10, 1903995, DOI:10.1002/aenm.201903995.
- L. Dubau, L. Castanheira, F. Maillard, M. Chatenet, O. Lottin, G. Maranzana, J. Dillet, A. Lamibrac, J.-C. Perrin, E. Moukheiber, A. ElKaddouri, G. de Moor, C. Bas, L. Flandin and N. Caqué, A review of PEM fuel cell durability: materials degradation, local heterogeneities of aging and possible mitigation strategies, Wiley Interdiscip. Rev.: Energy Environ., 2014, 3, 540–560, DOI:10.1002/wene.113.
- W. Vielstich, H. A. Gasteiger and H. Yokokawa, Advances in electrocatalysis, materials, diagnostics and durability; part 2. Advances in Electrocatalysis, Materials, Diagnostics and Durability, Volumes 5 and 6, Wiley, Chichester West Sussex England, Hoboken NJ, 2009 Search PubMed.
- W. Vielstich, A. Lamm and H. A. Gasteiger, Handbook of fuel cells. Fundamentals, technology, and applications, Wiley, Chichester England, Hoboken N.J., 2003 Search PubMed.
- V. Di Noto, T. A. Zawodzinski, A. M. Herring, G. A. Giffin, E. Negro and S. Lavina, Polymer electrolytes for a hydrogen economy, Int. J. Hydrogen Energy, 2012, 37, 6120–6131, DOI:10.1016/j.ijhydene.2012.01.080.
- G. de Moor, C. Bas, N. Charvin, E. Moukheiber, F. Niepceron, N. Breilly, J. André, E. Rossinot, E. Claude, N. D. Albérola and L. Flandin, Understanding Membrane Failure in PEMFC: Comparison of Diagnostic Tools at Different Observation Scales, Fuel Cells, 2012, 12, 356–364, DOI:10.1002/fuce.201100161.
- W. Liu and D. Zuckerbrod, In Situ Detection of Hydrogen Peroxide in PEM Fuel Cells, J. Electrochem. Soc., 2005, 152, A1165, DOI:10.1149/1.1904988.
- L. Gubler, S. M. Dockheer and W. H. Koppenol, Radical (HO•, H• and HOO•) Formation and Ionomer Degradation in Polymer Electrolyte Fuel Cells, J. Electrochem. Soc., 2011, 158, B755, DOI:10.1149/1.3581040.
- M. Chandesris, R. Vincent, L. Guetaz, J.-S. Roch, D. Thoby and M. Quinaud, Membrane degradation in PEM fuel cells: From experimental results to semi-empirical degradation laws, Int. J. Hydrogen Energy, 2017, 42, 8139–8149, DOI:10.1016/j.ijhydene.2017.02.116.
- H. Ericson, T. Kallio, T. Lehtinen, B. Mattsson, G. Sundholm, F. Sundholm and P. Jacobsson, Confocal Raman Spectroscopic Investigations of Fuel Cell Tested Sulfonated Styrene Grafted Poly(vinylidene fluoride) Membranes, J. Electrochem. Soc., 2002, 149, A206, DOI:10.1149/1.1431964.
- C. Huang, K. S. Tan, J. Lin and K. L. Tan, XRD and XPS analysis of the degradation of the polymer electrolyte in H2–O2 fuel cell, Chem. Phys. Lett., 2003, 371, 80–85, DOI:10.1016/s0009-2614(03)00259-8.
- N. Ohguri, A. Y. Nosaka and Y. Nosaka, Detection of OH radicals as the effect of Pt particles in the membrane of polymer electrolyte fuel cells, J. Power Sources, 2010, 195, 4647–4652, DOI:10.1016/j.jpowsour.2010.02.010.
- A. Pozio, R. F. Silva, M. de Francesco and L. Giorgi, Nafion degradation in PEFCs from end plate iron contamination, Electrochim. Acta, 2003, 48, 1543–1549, DOI:10.1016/s0013-4686(03)00026-4.
- L. Zhang and S. Mukerjee, Investigation of Durability Issues of Selected Nonfluorinated Proton Exchange Membranes for Fuel Cell Application, J. Electrochem. Soc., 2006, 153, A1062, DOI:10.1149/1.2180715.
- S. Kundu, M. W. Fowler, L. C. Simon, R. Abouatallah and N. Beydokhti, Degradation analysis and modeling of reinforced catalyst coated membranes operated under OCV conditions, J. Power Sources, 2008, 183, 619–628, DOI:10.1016/j.jpowsour.2008.05.074.
- T. Böhm, R. Moroni, M. Breitwieser, S. Thiele and S. Vierrath, Spatially Resolved Quantification of Ionomer Degradation in Fuel Cells by Confocal Raman Microscopy, J. Electrochem. Soc., 2019, 166, F3044–F3051, DOI:10.1149/2.0051907jes.
- P. C. Okonkwo, I. B. Belgacem, W. Emori and P. C. Uzoma, Nafion degradation mechanisms in proton exchange membrane fuel cell (PEMFC) system: A review, Int. J. Hydrogen Energy, 2021, 46, 27956–27973, DOI:10.1016/j.ijhydene.2021.06.032.
- J. Wu, X. Z. Yuan, J. J. Martin, H. Wang, J. Zhang, J. Shen, S. Wu and W. Merida, A review of PEM fuel cell durability: Degradation mechanisms and mitigation strategies, J. Power Sources, 2008, 184, 104–119, DOI:10.1016/j.jpowsour.2008.06.006.
- W. Shi and L. A. Baker, Imaging heterogeneity and transport of degraded Nafion membranes, RSC Adv., 2015, 5, 99284–99290, 10.1039/c5ra20291d.
- R. Shimizu, J. Tsuji, N. Sato, J. Takano, S. Itami, M. Kusakabe, K. Miyatake, A. Iiyama and M. Uchida, Durability and degradation analysis of hydrocarbon ionomer membranes in polymer electrolyte fuel cells accelerated stress evaluation, J. Power Sources, 2017, 367, 63–71, DOI:10.1016/j.jpowsour.2017.09.025.
- E. Moukheiber, G. de Moor, L. Flandin and C. Bas, Investigation of ionomer structure through its dependence on ion exchange capacity (IEC), J. Membr. Sci., 2012, 389, 294–304, DOI:10.1016/j.memsci.2011.10.041.
- K. Heimler, C. Gottschalk and C. Vogt, Confocal micro X-ray fluorescence analysis for the non-destructive investigation of structured and inhomogeneous samples, Anal. Bioanal. Chem., 2023, 415, 5083–5100, DOI:10.1007/s00216-023-04829-x.
- J. T. Lang, D. Kulkarni, C. W. Foster, Y. Huang, M. A. Sepe, S. Shimpalee, D. Y. Parkinson and I. V. Zenyuk, X-ray Tomography Applied to Electrochemical Devices and Electrocatalysis, Chem. Rev., 2023, 123, 9880–9914, DOI:10.1021/acs.chemrev.2c00873.
- M. S. Mu'min, T. Böhm, R. Moroni, R. Zengerle, S. Thiele, S. Vierrath and M. Breitwieser, Local hydration in ionomer composite membranes determined with confocal Raman microscopy, J. Membr. Sci., 2019, 585, 126–135, DOI:10.1016/j.memsci.2019.05.032.
- A. Gruger, A. Régis, T. Schmatko and P. Colomban, Nanostructure of Nafion® membranes at different states of hydration: An IR and Raman study, Vib. Spectrosc., 2001, 26, 215–225, DOI:10.1016/S0924-2031(01)00116-3.
- C. Götz, M. Komma, D. Dworschak, S. Thiele and T. Böhm, Quantification of Organic Solvent Concentration Profiles in Ion Exchange Membranes Via Confocal Raman Microscopy, Adv. Mater. Interfaces, 2024, 11, 2300887, DOI:10.1002/admi.202300887.
- H. Nishiyama, S. Takamuku, K. Oshikawa, S. Lacher, A. Iiyama and J. Inukai, Chemical States of Water Molecules Distributed Inside a Proton Exchange Membrane of a Running Fuel Cell Studied by Operando Coherent Anti-Stokes Raman Scattering Spectroscopy, J. Phys. Chem. C, 2020, 124, 9703–9711, DOI:10.1021/acs.jpcc.0c00347.
- C. Bas, L. Flandin, A.-S. Danerol, E. Claude, E. Rossinot and N. D. Alberola, Changes in the chemical structure and properties of a perfluorosulfonated acid membrane induced by fuel–cell operation, J. Appl. Polym. Sci., 2010, 117, 2121–2132, DOI:10.1002/app.31386.
- R. Buzzoni, S. Bordiga, G. Ricchiardi, G. Spoto and A. Zecchina, Interaction of H2O, CH3OH, (CH3)2O, CH3CN, and Pyridine with the Superacid Perfluorosulfonic Membrane Nafion: An IR and Raman Study, J. Phys. Chem., 1995, 99, 11937–11951, DOI:10.1021/j100031a023.
- S. R. Lowry and K. A. Mauritz, An investigation of ionic hydration effects in perfluorosulfonate ionomers by Fourier transform infrared spectroscopy, J. Am. Chem. Soc., 1980, 102, 4665–4667, DOI:10.1021/ja00534a017.
- M. Bradley, Curve fitting in Raman and IR spectroscopy: basic theory of line shapes and applications, Thermo Fisher Scientific, Madison, WI, Application Note: 50733, 2007, https://documents.thermofisher.com/TFS-Assets/CAD/Application-Notes/AN50733_E.pdf (accessed December 11, 2025).
- A. Kusoglu and A. Z. Weber, New Insights into Perfluorinated Sulfonic-Acid Ionomers, Chem. Rev., 2017, 117, 987–1104, DOI:10.1021/acs.chemrev.6b00159.
- M. Maier and T. Böhm, Modeling the Depth Resolution of Translucent Layers in Confocal Microscopy, Small Sci., 2024, 4, 2400120, DOI:10.1002/smsc.202400120.
- R. Ullah and X. Wang, Raman spectroscopy of Bisphenol ‘S’ and its analogy with Bisphenol ‘A′ uncovered with a dimensionality reduction technique, J. Mol. Struct., 2019, 1175, 927–934, DOI:10.1016/j.molstruc.2018.08.025.
- H. Edwards, D. R. Brown, J. A. Dale and S. Plant, Raman spectroscopy of sulfonated polystyrene resins, Vib. Spectrosc., 2000, 24, 213–224, DOI:10.1016/S0924-2031(00)00070-9.
- M. Maier, D. Abbas, M. Komma, M. S. Mu'min, S. Thiele and T. Böhm, A comprehensive study on the ionomer properties of PFSA membranes with confocal Raman microscopy, J. Membr. Sci., 2023, 669, 121244, DOI:10.1016/j.memsci.2022.121244.
- G. Turrell and J. Corset, Raman microscopy: developments and applications, Academic Press, 1996 Search PubMed.
- K. W. Dunn and E. Wang, Optical aberrations and objective choice in multicolor confocal microscopy, BioTechniques, 2000, 28, 542–544, DOI:10.2144/00283rr03 , 546, 548–50.
- H. J. Tiziani and H. M. Uhde, Three-dimensional image sensing by chromatic confocal microscopy, Appl. Opt., 1994, 33, 1838–1843, DOI:10.1364/AO.33.001838.
- J. Miyake, M. Kusakabe, A. Tsutsumida and K. Miyatake, Remarkable Reinforcement Effect in Sulfonated Aromatic Polymers as Fuel Cell Membrane, ACS Appl. Energy Mater., 2018, 1, 1233–1238, DOI:10.1021/acsaem.7b00349.
- Y. S. Noh, H. Y. Jeong, T.-H. Kim, J. Choi, J. Y. Lee, S. So and D. M. Yu, Sulfonated poly(p-phenylene)-based ionomer/PTFE composite membrane with enhanced performance and durability for energy conversion devices, J. Power Sources, 2023, 580, 233422, DOI:10.1016/j.jpowsour.2023.233422.
- R. Qelibari, E. C. Ortiz, N. van Treel, F. Lombeck, C. Schare, A. Münchinger, N. Dumbadze, G. Titvinidze, C. Klose and S. Vierrath, 74 µm PEEK–Reinforced Sulfonated Poly(phenylene sulfone)–Membrane for Stable Water Electrolysis with Lower Gas Crossover and Lower Resistance than Nafion N115, Adv. Energy Mater., 2024, 14, 2303271, DOI:10.1002/aenm.202303271.
- D. Yazili, E. Marini, T. Saatkamp, A. Münchinger, T. de Wild, L. Gubler, G. Titvinidze, M. Schuster, C. Schare, L. Jörissen and K.-D. Kreuer, Sulfonated Poly(Phenylene sulfone) blend membranes finding their way into proton exchange membrane fuel cells, J. Power Sources, 2023, 563, 232791, DOI:10.1016/j.jpowsour.2023.232791.
- Z. Han, T. Nemeth, M. Yandrasits, H. Ren, W. Bangay, T. Saatkamp and L. Gubler, Hydrocarbon Proton Exchange Membranes for Fuel Cells: Do We Need New Chemical Durability Testing Protocols?, ACS Electrochem., 2024, 1(5), 588–598, DOI:10.1021/acselectrochem.4c00123.
- C. Piesold, C. Schare, C. Klose, M. Viviani and G. Titvinidze, Novel branched sulfonated poly(phenylene sulfone)s for PEM- water electrolysis application, Polym. Bull., 2026, 83, 129, DOI:10.1007/s00289-025-06098-3.
- H. Nguyen, F. Lombeck, C. Schwarz, P. A. Heizmann, M. Adamski, H.-F. Lee, B. Britton, S. Holdcroft, S. Vierrath and M. Breitwieser, Hydrocarbon-based Pemion™ proton exchange membrane fuel cells with state-of-the-art performance, Sustainable Energy Fuels, 2021, 5, 3687–3699, 10.1039/D1SE00556A.
- H. Liepold, A. Bird, P. A. Heizmann, H. Fadlullah, H. Nguyen, C. Klose, S. Holdcroft, A. Kusoglu, S. Vierrath and A. Münchinger, High protonic resistance of hydrocarbon-based cathodes in PEM fuel cells under low humidity conditions: Origin, implication, and mitigation, J. Power Sources, 2024, 624, 235537, DOI:10.1016/j.jpowsour.2024.235537.
- Y. Li, R. Jiang and C. Gittleman, Effects of melt flow index and equivalent weight on the dimensional stability and mechanical behavior of perfluorosulfonic acid ionomer membranes, J. Power Sources, 2020, 478, 228734, DOI:10.1016/j.jpowsour.2020.228734.
- A. A. Martens, N. A. M. Besseling, S. Rueb, E. J. R. Sudhölter, H. P. Spaink and L. C. P. M. de Smet, Random Scission of Polymers: Numerical Simulations, and Experiments on Hyaluronan Hydrolosis, Macromolecules, 2011, 44, 2559–2567, DOI:10.1021/ma200009y.
- V. J. Triacca, P. E. Gloor, S. Zhu, A. N. Hrymak and A. E. Hamielec, Free radical degradation of polypropylene: Random chain scission, Polym. Eng. Sci., 1993, 33, 445–454, DOI:10.1002/pen.760330802.
- P. Gloor, Y. Tang, A. Kostanska and A. Hamielec, Chemical modification of polyolefins by free radical mechanisms: a modelling and experimental study of simultaneous random scission, branching and crosslinking, Polymer, 1994, 35, 1012–1030, DOI:10.1016/0032-3861(94)90946-6.
- V. Lohmann, G. R. Jones, N. P. Truong and A. Anastasaki, The thermodynamics and kinetics of depolymerization: what makes vinyl monomer regeneration feasible?, Chem. Sci., 2024, 15, 832–853, 10.1039/D3SC05143A.
- S. M. Dockheer, L. Gubler, P. L. Bounds, A. S. Domazou, G. G. Scherer, A. Wokaun and W. H. Koppenol, Damage to fuel cell membranes. Reaction of HO* with an oligomer of poly(sodium styrene sulfonate) and subsequent reaction with O(2), Phys. Chem. Chem. Phys., 2010, 12, 11609–11616, 10.1039/c0cp00082e.
- M. Zatoń, J. Rozière and D. J. Jones, Current understanding of chemical degradation mechanisms of perfluorosulfonic acid membranes and their mitigation strategies: a review, Sustainable Energy Fuels, 2017, 1, 409–438, 10.1039/C7SE00038C.
- T. Holmes, T. J. G. Skalski, M. Adamski and S. Holdcroft, Stability of Hydrocarbon Fuel Cell Membranes: Reaction of Hydroxyl Radicals with Sulfonated Phenylated Polyphenylenes, Chem. Mater., 2019, 31, 1441–1449, DOI:10.1021/acs.chemmater.8b05302.
- M. Pinteala and S. Schlick, Direct ESR detection and spin trapping of radicals generated by reaction of oxygen radicals with sulfonated poly(ether ether ketone) (SPEEK) membranes, Polym. Degrad. Stab., 2009, 94, 1779–1787, DOI:10.1016/j.polymdegradstab.2009.06.009.
- A. Panchenko, DFT investigation of the polymer electrolyte membrane degradation caused by OH radicals in fuel cells, J. Membr. Sci., 2006, 278, 269–278, DOI:10.1016/j.memsci.2005.11.010.
Footnote |
| † Equally contributing authors. |
| This journal is © The Royal Society of Chemistry 2026 |