Dictating the CuO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h2_char_2009.gif) :Cu(OH)2 ratio and microstructural evolution by varying the temperature of anodization of Cu for the OER-masking GOR
:Cu(OH)2 ratio and microstructural evolution by varying the temperature of anodization of Cu for the OER-masking GOR
Received
12th December 2025
, Accepted 20th February 2026
First published on 23rd February 2026
Abstract
This study investigates the anodization of a Cu foam substrate at various temperatures and examines its influence on the surface composition and the subsequent electrocatalytic glucose oxidation reaction (GOR). It has been found that anodizing Cu at 28 °C results in a higher proportion of Cu(OH)2 compared to CuO with better control over the typical nanoneedles obtained while anodizing Cu. Increasing the temperature of anodization increases the CuO proportion while shortening the average length of the nanoneedles from ∼3 μm at 28 °C to ∼50 nm at 46 °C. When all these were tested with a 0.15 M glucose solution in 1 M KOH, the one anodized at 28 °C achieved the highest current density of approximately 500 mA cm−2 at 1.924 V vs. RHE. The superior performance is attributed to the unique surface chemistry featuring a relatively higher proportion of Cu(OH)2, which facilitates a more efficient electron transfer during the glucose oxidation process with the lattice OH groups, vital for glucose activation. With CuO, glucose oxidation demands the in situ conversion of CuO into Cu(OH)2 or CuOOH for the same and tends to be slower. Moreover, the Cu electrode anodized at 28 °C demonstrates the critical advantage of effectively masking the oxygen evolution reaction (OER), and exceptional durability under operational conditions. These insights into anodization of Cu electrodes are crucial for the practical application of Cu-based electrodes in co-electrolysis-based sustainable hydrogen production.
Introduction
Hydrogen has emerged as a key player in the context of erratic climate change stemming from the uncontrolled exploitation of fossil fuels and increased atmospheric CO2 contents. It has been garnering increasing attention due to its potential as a clean energy carrier capable of significantly reducing carbon emissions. Among the most promising methods for producing green hydrogen is water electrolysis powered by renewable electricity. This process offers a particularly environmentally friendly approach, as it can be powered using renewables and be operated under ambient conditions unlike steam reforming of methane, ensuring minimal environmental impact. The theoretical cell voltage required to split water into hydrogen and oxygen with hydrogen being produced at the cathode and oxygen evolving at the anode is 1.23 V versus the reversible hydrogen electrode (RHE). However, the overall efficiency of this process is heavily influenced by the electrocatalysts used. Electrocatalysts are essential for facilitating the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER), which are key to effective water splitting. Despite their importance, the OER suffers from sluggish kinetics meaning that a higher voltage is necessary to drive the overall water splitting reaction efficiently. As a result, the practical voltage required for efficient water electrolysis rises to about 1.48 V vs. RHE.1–4 Using small molecules such as methanol, urea, and glucose as alternative anodic reactants to be oxidized instead of the OER can significantly lower the voltage required for efficient water electrolysis and makes the overall process water–small molecule co-electrolysis.5–7 This strategy, in which the energy demanding OER is masked by a more favourable anodic oxidation reaction such as the glucose oxidation reaction (GOR), is commonly referred to as the OER-masking GOR.
Although precious metals, such as Ir, Ru, and Pt, exhibit the lowest overpotential as their oxides and are among the most efficient electrocatalysts for the OER and for most of these small molecule oxidation reactions, their high cost and limited availability have prompted researchers to explore alternative, more sustainable options. As a result, non-noble metal catalysts such as Ni, Cu, Fe, and Co have gained significant attention. These transition metals are not only abundant and cost-effective but also exhibit high catalytic activity, especially in alkaline environments. The unique properties of these metals, including the presence of 3d electrons, their ability to adopt multiple oxidation states, and their diverse morphological characteristics, have made them increasingly attractive alternatives for noble metals in electrocatalytic applications. Specifically, Cu has been found to promote and mediate the glucose oxidation reaction (GOR) efficiently on par with other 3d metals.8–12 In general, three-dimensional (3D) CuO-based electrocatalysts have garnered significant interest due to their earth abundance, cost effectiveness, and favourable Cu2+/Cu+ redox characteristics. Moreover, the 3D architecture offers a large electrochemically active surface area and improved stability which are crucial for efficient electrochemical water splitting.13,14 In our recent study, we found that the GOR on anodized copper scrap outperformed both the methanol oxidation reaction (MOR) and the urea oxidation reaction (UOR). Notably, the GOR required a voltage that was 38 mV lower than the equilibrium potential limit of 1.23 V vs. RHE.15 Recent studies have demonstrated that metallic Cu, Cu2O, and Cu(OH)2 exhibit excellent catalytic activity for the oxidation of glucose. It has been shown that pre-anodized Cu in alkaline media significantly enhances electron transfer and provides long term stability to the electrode, which is particularly beneficial for the GOR.16,17 The formation of an oxide/hydroxide layer on the Cu surface is a key factor in improving catalytic performance which can be generated through various methods, such as annealing Cu substrates to form an air-oxidized layer or using electrochemical techniques. Annealing methods often require elevated temperatures and extended periods (sometimes up to several days).18–21 In contrast, electrochemical methods allow for the formation of an oxide/hydroxide layer on Cu much more quickly, making them a more efficient approach for optimizing the electrocatalytic properties of Cu for the GOR. Previously, electrochemical anodization of Cu surfaces has been performed using higher potentials and extended durations.22–25 For instance, Ganzha and co-workers reported that anodizing Cu at a potential of 0.10 V for 15 min yielded the best results compared to other potentials they tested.26 Similarly, Scherzer and co-workers found that Cu could be anodized by using chronoamperometry (CA) at −0.26 V (vs. Ag/AgCl) for 20 h.27 In our recent study, we introduced an oxidation-potential guided electrochemical method that uses CA for anodizing Cu which achieved complete anodization in just 87 s, with noticeable changes occurring within the first 20 s. The selected potential for this rapid anodization technique was −0.06 V (vs. Hg/HgO) demonstrating a significantly faster approach compared to other traditional methods.28 In another recent study from our group, we revealed an intriguing finding that the size, morphology, surface chemistry, and electrochemical accessibility of active Cu sites can be effectively tuned by adjusting the concentration of KOH used as the electrolyte during the anodization process. We observed that increasing the strength of the KOH solution significantly reduced the anodization time on the Cu surface to just 6 s at the highest pH. Additionally, it was observed that as the pH of the electrolyte increased, the formation of Cu(OH)2 on the Cu foam after anodization was enhanced as compared to the formation of CuO, which favoured the methanol oxidation reaction (MOR).29 In a related study by Honnappa and co-workers, our approach was extended with additional structure directing agents like urea and acetic acid, and significant changes were found occurring on the morphologies of the resultant anodized Cu substrate leading to different activity trends in the MOR and OER.30 In this work, we applied a potential of −0.06 V (vs. Hg/HgO) in 1 M KOH, based on our previously optimized conditions for anodizing Cu, to achieve efficient and rapid surface modification. Furthermore, we sought to investigate the impact of temperature on the anodization process of Cu and how it subsequently influences the GOR activity. We conducted temperature-dependent experiments to assess these effects. We initially hypothesized that with increasing temperature, the rate of reaction for anodizing Cu foam would accelerate, in line with the Arrhenius theory. However, we found no such uniform trend as thermal oxidation competed for the same surface sites with electrochemical oxidation. In addition, at lower temperatures, more copper hydroxide (Cu(OH)2) forms on the Cu surface, which promotes a faster GOR. The presence of Cu(OH)2 significantly enhances the catalytic activity, promoting swift glucose oxidation. In contrast, as the temperature increased, some of the Cu(OH)2 was converted to CuO, which negatively impacted the GOR activity. Notably, the electrode anodized at 28 °C (room temperature) was rich in Cu(OH)2 which favoured the GOR, aligning with our previous pH study where a similar trend was observed for the methanol oxidation reaction (MOR). This study, thus, provides insights into the effects of temperature on the anodization of Cu substrates and how it can influence the surface chemistry of CuO and Cu(OH)2 affecting the GOR activity eventually.
Experimental section
Materials
Cu foam (99.9%) was procured from Nanoshell, India and was pretreated with 3 M HCl solution to remove the surface oxides and to expose the fresh metallic surface free from existing oxides or hydroxides. A standard three electrode setup was used, where the acid etched Cu foam was used as the working electrode, with large Pt foil as the counter electrode and Hg/HgO as the reference electrode. All anodization studies and electrochemical experiments were carried out in a 1 M KOH (Qualigens) solution. Various characterization techniques including diffractometric, spectroscopic, microscopic, and electroanalytical methods were employed to examine the anodized electrodes at different bath temperatures and to evaluate their activity towards oxidation of glucose which included concentration optimization, scan rate studies, multi-potential electrochemical impedance spectroscopy (EIS), and long-term stability studies.
Anodizing Cu foam with different bath temperatures
One of the most efficient methods to grow an oxide/hydroxide layer on Cu foam is electrochemical anodization, which is not only rapid but also highly effective as introduced earlier. The Cu foam was anodized at various temperatures starting from 28 °C to 31 °C, 36 °C, 41 °C, and 46 °C using CA in a 1 M KOH solution. The set potential for the oxidation of Cu was −0.06 V vs. Hg/HgO, which was optimized in our previous study.28 The anodized electrodes were then characterized for morphological and compositional changes using different analytical techniques.
Glucose electrooxidation and electrochemical characterization
To optimize the glucose concentration, electrooxidation experiments were conducted with varying concentrations of glucose of 0.10, 0.15, 0.20, 0.25, and 0.30 M in a 1 M KOH base solution for all the anodized electrodes at different temperatures and compared with that of bare Cu foam. Linear sweep voltammetry (LSV) was performed at a scan rate of 10 mV s−1 to determine the optimal concentration for the anodized electrodes. Additionally, the effect of the scan rate on the GOR was studied by varying the same (10, 50, 100, 150, and 200 mV s−1, respectively). A detailed EIS analysis was carried out for all the electrodes with a frequency range from 1 MHz to 0.1 Hz and an AC perturbation of 10 mV to understand the mechanism and charge transfer kinetics of the GOR; the applied DC potential of EIS was varied regularly from 1.10 V to 1.80 V vs. RHE with an interval of 50 mV. Finally, to evaluate the stability of all the electrodes, CA was performed at 0.5 V vs. Hg/HgO. The obtained results are comprehensively interpreted and discussed below by correlating the same with the observations made in other characterization studies.
Results and discussion
Temperature-dependent electrochemical anodization of Cu foam
Based on our recent findings, the potential for anodizing each Cu foam was set at −0.06 V vs. Hg/HgO. This is the potential where metallic Cu is oxidized to Cu2+ to form a Cu(OH)2/CuO layer on the Cu foam as identified in the respective cyclic voltammogram (CV). The anodization CA was conducted for 1200 s to ensure complete anodization of all the electrodes, although anodization was apparently completed as indicated by the flatlined anodization current in Fig. 1 well before the duration. The current density was seen to decrease exponentially, as expected for a diffusion-controlled reaction. The completion of anodization was indicated by a constant near-zero current density (in mA cm−2).
 |
| | Fig. 1 The current density–time curves of the temperature-dependent anodization of Cu foam at −0.06 V vs. Hg/HgO in 1.0 M KOH. | |
The anodization of Cu foam was carried out at various temperatures such as 28 °C, 31 °C, 36 °C, 41 °C, and 46 °C to investigate the effect of temperature on the anodization time and electrocatalytic GOR activity. As shown in Fig. 1, there was no clear trend in the current density–time curves across the different anodization temperatures employed. It was also observed that the surface of the Cu foam anodized at 28 °C appeared greenish, while the Cu foam anodized at higher temperatures had a more blackish hue, indicating a higher proportion of CuO compared to Cu(OH)2. This irregular trend may be attributed to a combination of factors, including subtle changes in the surface area of the electrodes and the competition between thermal oxidation and electrochemical oxidation as the temperature increases. Moreover, the elevated temperature could also dehydrate initially formed Cu(OH)2 by electrochemical oxidation to form CuO in situ. The topographical differences commonly found in three-dimensional substrates like Cu foam could also play a role behind not seeing any such expected trend in the time of anodization with increasing temperature. Properties of metal foam substrates such as changes in pore size, volume, and density could have also likely affected the extent of electrolyte diffusion, further influencing the anodization process causing irregularities in the time of anodization with respect to the temperature of anodization. A similar irregular trend in the current density–time curve could be reproduced as shown in Fig. S1. These observations hint that it might not be possible to see a direct relationship between the temperature of anodization and the time it takes to completely oxidize the Cu surface. With this understanding, the electrodes were thoroughly characterized with various analytical techniques before proceeding to GOR studies.
Morphological and chemical characterisation after anodization
To monitor the changes that occurred before and after anodization, X-ray diffraction (XRD) analysis was performed for all the electrodes anodized at different temperatures and compared with bare Cu foam (Fig. 2a). Both the bare Cu foam and the temperature-dependent anodized electrodes displayed similar peaks that would correspond to the metallic Cu base. This indicates that the surface oxides or any hydroxides formed during anodization were thin enough to allow X-rays to penetrate and diffract from the underlying metallic Cu base. Besides these sharp metallic Cu peaks, some low intensity peaks could be observed in the range of 10° to 40°. Based on previous literature reports, the observed low-angle peaks (at 17.1°, 23.5°, and 34° corresponding to the (020), (103) and (002) crystal planes) were identified and assigned to Cu(OH)2, especially in the electrodes anodized at lower temperatures.31–34 As the temperature of anodization was increased to 41 °C and 46 °C, only the (002) phase of Cu(OH)2 was retained. In the meantime, the (020) and (103) planes were diminished, indicating a decrease in the amount of Cu(OH)2 formed at higher temperatures. Instead, peaks associated with CuO, particularly those at 38° and 39° corresponding to the (−111) and (111) planes, became more dominant as the temperature of anodization was increased.
 |
| | Fig. 2 (a) XRD patterns of the bare Cu foam and anodized electrodes at 28 °C, 31 °C, 36 °C, 41 °C, and 46 °C, where the asterisk symbol indicates the phases of Cu(OH)2 and the number symbol indicates the CuO phases. (b) Raman spectra of the same, where the asterisk symbol in the Raman spectrum of the Cu electrode anodized at 28 °C shows the presence of Cu(OH)2. Both XRD patterns and Raman spectra reveal that Cu(OH)2 phases are more prominent in Cu foam anodized at 28 °C. | |
This suggests that as the temperature increased during anodization, Cu(OH)2 underwent partial conversion into CuO through the removal of water from it, as CuO is thermodynamically more stable at elevated temperatures. This observed trend strongly indicates that at lower temperatures, the proportion of Cu(OH)2 is more as compared to CuO, and the formation of Cu(OH)2 decreases as the temperature for anodization is increased. This presence of a higher proportion of the Cu(OH)2 layer at the surfaces of the electrodes anodized at lower temperatures further supports the idea that temperature plays a crucial role in determining the phase structure and morphology of the anodized Cu, possibly influencing its electrochemical properties in the GOR to be screened. To further investigate the surface changes imparted by temperature-dependent anodization, the Raman spectra of the bare Cu foam and electrodes anodized at different temperatures were recorded (Fig. 2b). The bare Cu electrode was pre-cleaned with 3 M HCl and hence, showed no stretching vibrations corresponding to copper oxide or hydroxide layers proving that the Cu substrate was free from oxides and hydroxides before anodization. All the anodized electrodes exhibited various characteristic stretching vibrations that corresponded to the Cu(OH)2 and CuO entities, confirming successful anodization of Cu at all applied temperatures.15,35,36 Notably, the Raman spectral results revealed that the intensity of the Cu(OH)2 peak was most prominent for the electrode anodized at 28 °C, indicating a higher proportion of Cu(OH)2 on the surface. As the anodization temperature increased, the Cu(OH)2 peak intensity began lowering, whereas the intensity of peaks corresponding to CuO was seen to become prominent as the temperature of anodization increased, implying more CuO formation at high anodization temperatures. This trend resonates well with the XRD results, further supporting the observation that the proportion of Cu(OH)2 decreases with increasing anodization temperature, while CuO becomes the predominant phase at higher temperatures. Overall, the Raman analysis confirmed the phase changes observed in the XRD analysis and complemented the former to cement the fact that anodization can be used to control the surface oxide–hydroxide ratio of anodized Cu substrates. To know if and how morphological changes were brought out by the temperature-dependent anodization adapted, scanning electron microscopy (SEM) images were obtained initially. At lower anodization temperatures (28 °C (Fig. 3a and b), 31 °C (Fig. 3d and e), and 36 °C (Fig. 3g and h)), dense nanoneedle structures were observed on the surface with decreasing density as the temperature of anodization increased. This suggested a reduction in surface area in the resultant anodized Cu electrode as the temperature of anodization increased. At higher temperatures such as 41 °C (Fig. 3j and k) and 46 °C (Fig. 3m and n), visible cracks began to appear on the surface hinting at temperature-driven disintegration of oxide and hydroxide layers formed initially by the electrochemical anodization.
 |
| | Fig. 3 SEM images of the anodized Cu electrodes at 28 °C (a and b), 31 °C (d and e), 36 °C (g and h), 41 °C (j and k), and 46 °C (m and n) along with their corresponding ED spectra given in (c), (f), (i), (l) and (o), respectively. SEM images reveal that morphological integrity deteriorates with increasing anodization temperature with well-developed nanoneedles at lower temperatures. | |
These cracks seemed to widen and become more pronounced, which might compromise the anodized electrode's electrochemical properties. Additionally, at the anodization temperature of 46 °C, the nanoneedle structures' length was observed to be ∼50 nm. This contrasts with the well-developed nanoneedles observed at 28 °C, which were about 2–3 μm in length. The nanoneedles of the Cu electrode anodized at 28 °C are clearly observed in FESEM images, too (Fig. S2). The loss of the nanoneedle structure at higher temperatures suggests a notable change in morphology, primarily driven by the thermal dehydration and competing thermal oxidation of Cu. From the SEM and FESEM results, it can be safely concluded here that the temperature of anodization had a clear impact on the morphology of the electrodes. Firstly, the density of the nanoneedles decreased as the temperature increased. Secondly, the length of the nanoneedles became much smaller at higher temperatures. These morphological changes could significantly influence the electrochemical behaviour of the Cu electrodes anodized at different temperatures in terms of GOR activity.
The corresponding energy dispersive X-ray spectroscopy (EDS) results were used to analyse the surface composition of the anodized electrodes. These spectra showed that all electrodes contained Cu and O with no other impurities, confirming that their surfaces were made of the oxides and hydroxides of Cu only. To compare the composition of Cu and O better, a bar diagram was plotted based on the EDS observations (Fig. 4), which shows the mass percentage ratio of Cu and O for the different temperature anodized electrodes. As the anodization temperature increased, the proportion of Cu also increased, while the oxygen mass percentage decreased. This trend can be explained directly by the difference in the Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O ratio between Cu(OH)2 and CuO. Moreover, Cu(OH)2 has an orthorhombic crystal structure, which is less tightly packed compared to CuO, which has a monoclinic structure. As every Cu in Cu(OH)2 has two O, the higher proportion of O at lower temperatures of anodization directly implies high Cu(OH)2 content. As the temperature was increased to 41 °C and 46 °C, the mass% of Cu takes over the mass% of O indicating a sharp composition change in the surface of the anodized Cu electrodes as hinted by the XRD, Raman, and SEM results, as well. This observation is crucial here because it has just been proved that by simply varying the temperature of anodization, not just the morphological outcomes, but also the chemical composition of the surface of the anodized Cu electrodes can be tuned, controlled, and dictated for the very first time to the best of our knowledge. Even though we cannot say how this can affect the GOR activity trend yet, we can call it a success in the materials aspect as our method let us have explicit control over both the chemical composition and morphology. This ability might prove beneficial in so many other areas and not just in the GOR or other related electrocatalytic reactions. For further evaluation of the nanoneedles seen in SEM analysis, transmission electron microscopy (TEM) analysis was performed by taking the electrode anodized at 28 °C which showed the best results toward electrocatalytic GOR activity in this study, which is discussed later (Fig. 5a). The nanoneedles were scratched off from the Cu electrode anodized at 28 °C and extracted into a glass vial containing a few drops of deionised (DI) water. Further, these particles were sonicated and homogenized. Drop casting about 3 μL of the same solution on a carbon-coated Cu grid resulted in the specimen for TEM. It was seen from the TEM analysis that the nanoneedles seen in SEM are now shattered but preserved almost the morphology. The inset of Fig. 5a shows the selected area electron diffraction (SAED) pattern of the same. The pattern exhibited dot-plus-ring features, indicative of a polycrystalline material with varying crystallite sizes. The observed pattern matched well with the (170) and (042) planes of Cu(OH)2. Another specimen was also prepared in the same way for high-resolution transmission electron microscopy (HRTEM) analysis (Fig. 5b–d). The lattice fringes observed in the HRTEM images corresponded to the (021) plane of Cu(OH)2, confirming the presence of the Cu(OH)2 phase primarily at the nanoscale. The results of TEM and HRTEM analyses were complementary to the observations made with SEM and EDS compositional analyses that at 28 °C, the primary component is Cu(OH)2.
:O ratio between Cu(OH)2 and CuO. Moreover, Cu(OH)2 has an orthorhombic crystal structure, which is less tightly packed compared to CuO, which has a monoclinic structure. As every Cu in Cu(OH)2 has two O, the higher proportion of O at lower temperatures of anodization directly implies high Cu(OH)2 content. As the temperature was increased to 41 °C and 46 °C, the mass% of Cu takes over the mass% of O indicating a sharp composition change in the surface of the anodized Cu electrodes as hinted by the XRD, Raman, and SEM results, as well. This observation is crucial here because it has just been proved that by simply varying the temperature of anodization, not just the morphological outcomes, but also the chemical composition of the surface of the anodized Cu electrodes can be tuned, controlled, and dictated for the very first time to the best of our knowledge. Even though we cannot say how this can affect the GOR activity trend yet, we can call it a success in the materials aspect as our method let us have explicit control over both the chemical composition and morphology. This ability might prove beneficial in so many other areas and not just in the GOR or other related electrocatalytic reactions. For further evaluation of the nanoneedles seen in SEM analysis, transmission electron microscopy (TEM) analysis was performed by taking the electrode anodized at 28 °C which showed the best results toward electrocatalytic GOR activity in this study, which is discussed later (Fig. 5a). The nanoneedles were scratched off from the Cu electrode anodized at 28 °C and extracted into a glass vial containing a few drops of deionised (DI) water. Further, these particles were sonicated and homogenized. Drop casting about 3 μL of the same solution on a carbon-coated Cu grid resulted in the specimen for TEM. It was seen from the TEM analysis that the nanoneedles seen in SEM are now shattered but preserved almost the morphology. The inset of Fig. 5a shows the selected area electron diffraction (SAED) pattern of the same. The pattern exhibited dot-plus-ring features, indicative of a polycrystalline material with varying crystallite sizes. The observed pattern matched well with the (170) and (042) planes of Cu(OH)2. Another specimen was also prepared in the same way for high-resolution transmission electron microscopy (HRTEM) analysis (Fig. 5b–d). The lattice fringes observed in the HRTEM images corresponded to the (021) plane of Cu(OH)2, confirming the presence of the Cu(OH)2 phase primarily at the nanoscale. The results of TEM and HRTEM analyses were complementary to the observations made with SEM and EDS compositional analyses that at 28 °C, the primary component is Cu(OH)2.
 |
| | Fig. 4 Plot of mass% of Cu (blue) and O (orange) against the temperature of anodization. | |
 |
| | Fig. 5 (a) TEM image of surface segregates of the Cu electrode anodized at 28 °C. The inset of (a) is the SAED pattern of the same. (b–d) HRTEM images of the surface segregates of the Cu electrode anodized at 28 °C. | |
To gain the chemical oxidation state information of Cu in the anodized electrode, we utilized X-ray photoelectron spectroscopy (XPS), which is a more surface-sensitive analytical technique. The survey scan (Fig. 6a) revealed the presence of all the expected elements with no impurities. The C 1s spectrum was used for calibration (Fig. 6b). On the other hand, Fig. 6c presents the O 1s XP spectrum of the Cu electrode anodized at 28 °C, which reveals the presence of both M–O (530.6 eV) and M–OH (531.7 eV) moieties.15,37 These peaks confirm the successful anodization process and formation of both copper hydroxide and copper oxide on the electrode surface. Fig. 6d shows the Cu 2p XP spectrum, which was deconvoluted to reveal two peaks for both Cu 2p3/2 (932.3 eV and 934.1 eV) and Cu 2p1/2 (953.8 eV and 956.1 eV), which are consistent with our previous observations.
 |
| | Fig. 6 XPS survey scan (a) of the Cu electrode anodized at 28 °C. XPS narrow scans of C 1s (b), O 1s (c), and Cu 2p (d) of the same. | |
The peak found at 932.3 eV indicates the presence of Cu while the one at 934.1 eV indicates the presence of the Cu2+ state which could be due to both CuO and Cu(OH)2. From the Cu 2p XP spectrum alone, the identified Cu2+ cannot be assigned conclusively to CuO or Cu(OH)2. However, as our earlier analyses revealed that at 28 °C, the anodized Cu electrode featured Cu(OH)2, we attribute that this peak could also be dominantly from Cu(OH)2. The results of XPS analysis resonated well with the observations made using XRD, SEM, EDS, and Raman analyses.
Electrocatalytic oxidation of glucose
After the successful comparative preliminary characterisation (XRD, Raman, SEM, and EDS) of all the anodized electrodes, we proceeded to evaluate their GOR activity to assess how the changing anodization temperature and its subsequent consequences on the morphology and surface chemistry influence the oxidation of glucose. By introducing small molecules like glucose at the anode, where the OER takes place, we aimed to reduce the overall cell voltage required for water splitting electrocatalytically though it would be technically called water–glucose co-electrolysis. First, the concentration of glucose was optimized for each anodized electrode using LSV at a scan rate of 10 mV s−1 (Fig. 7a, c, e, g and i). To facilitate better comparison, we plotted the current density against the glucose concentration as a bar diagram next to the corresponding LSVs in Fig. 7b, d, f, h and j. Almost all of them exhibited the highest current density at a glucose concentration of 0.15 M in 1 M KOH compared to other (0.10 M, 0.20 M, 0.25 M, and 0.30 M) concentrations of glucose studied. Therefore, we selected 0.15 M glucose as the optimized concentration for subsequent electrochemical studies.
 |
| | Fig. 7 (a, c, e, g and i) GOR LSVs recorded at 10 mV s−1 for electrodes anodized at 28 °C, 31 °C, 36 °C, 41 °C, and 46 °C in 1 M KOH. (b, d, f, h and j) The respective current density–concentration bar diagrams for the same. From the LSV analysis, 0.15 M glucose in 1 M KOH was chosen as the optimized concentration for further electrochemical studies. | |
Importantly, the Cu electrode anodized at room temperature (featuring more Cu(OH)2) appears to have outperformed all the other electrodes making it the best catalytic electrode for the GOR of them all. To see the interfacial behaviour GOR with the anodized electrodes, a series of CVs were recorded to evaluate the glucose oxidation reaction (GOR) at various scan rates such as 10, 50, 100, 150, and 200 mV s−1 using 0.15 M glucose in 1 M KOH for all the anodized electrodes at various bath temperatures (Fig. 8a–f). At all scan rates, we observed that the current at the maximum anodic potential (1.924 V vs. RHE) remained nearly constant, suggesting that the glucose oxidation reaction is a kinetically controlled reaction within the chosen potential window.
 |
| | Fig. 8 GOR CVs with increasing scan rates in 1 M KOH containing 0.15 M glucose for bare (a) and anodized electrodes at 28 °C (b), 31 °C (c), 36 °C (d), 41 °C (e), and 46 °C (f), respectively. | |
Additionally, we noticed a subtle but noticeable increase in current density of the diffusion controlled Cu2+/Cu3+ redox peak appearing prior to the GOR. This scan rate-dependent rise in the current densities and increasing peak-to-peak separation of this redox couple indicate that this is indeed a diffusion-controlled process, and quasi-reversible too in nature. These observations are mechanistically significant as this emphasizes the need for the oxidation of Cu2+ to Cu3+ before glucose oxidation can even begin. To see if the electrochemically accessible sites (ECAS) of Cu were behind the observed activity trend, we have integrated the area under the reduction peak of the CV curves recorded at 200 mV s−1 for Cu foam anodized at different temperatures (Fig. 9a–e) and the corresponding ECASCu values are shown in Fig. 9f. The observed trend is completely random, suggesting almost no relationship to the observed GOR trend. This might also mean that the observed GOR trend was mainly driven by the chemical composition rather than ECASCu or the surface area (driven by morphology changes).
 |
| | Fig. 9 (a–e) Area integrated reduction of peaks of GOR CVs (recorded at 200 mV s−1) for Cu foam electrodes anodized at 28 °C, 31 °C, 36 °C, 41 °C, and 46 °C, respectively. (f) Corresponding ECASCu bar diagram displaying no specific trend. | |
Further, to assess the stability of all the electrodes, CA analysis was conducted at a constant potential of 0.5 V vs. Hg/HgO using a 0.15 M glucose solution in 1 M KOH (Fig. 10). Over the first 3 h of the experiment, a rapid decrease in the current density was observed in the first 20 min indicating the concurrent occurrence of surface reconstruction and/or ongoing further Cu oxidation alongside the GOR. However, as the time of the analysis increased, a very stable performance could be observed which matched reasonably with the LSV results. It should be noted here that this gradual decrease could also be due to the rapidly decreasing interfacial glucose concentration initially before achieving an equilibrium between the rate of diffusion of glucose and the rate of the GOR. Nevertheless, this can safely be ignored as the GOR under the chosen potential is kinetically controlled. To further confirm the same, the glucose concentration in the electrolyte was replenished. As soon as the replenishment was done, the GOR current density shot up swiftly only to decrease to the stable performance seen earlier within a few minutes. This behaviour, while indicating the excellent stability of all the electrodes during the GOR, also hints at ongoing surface reconstruction by further Cu oxidation though it was relatively shorter after the glucose replenishment manoeuvre.
 |
| | Fig. 10 GOR stability study using CA at a set potential of 0.5 V vs. Hg/HgO in 1 M KOH containing 0.15 M glucose for 3 h and for 2 more hours after replenishing the concentration of glucose. | |
Importantly, the observed decrease in current density was not due to catalytic degradation of the electrodes but rather due to the gradual consumption of glucose in the electrolyte and the ongoing surface reconstruction. These observations confirmed the excellent stability of all the anodized electrodes. To understand the mechanism of the GOR on the surface of these anodized Cu foam electrodes, a multi-potential EIS analysis was performed in the range of 1.10 to 1.80 V vs. RHE in the frequency range of 1 MHz to 0.1 Hz with 10 mV AC perturbation with 0.15 M added glucose (Fig. 11a) and without any added glucose (Fig. 11b). This potential window was chosen in such a way that all the electron transfer processes (i.e., Cu2+/Cu3+ redox, OER, GOR, and the adsorption of active intermediates thereof) can be accommodated and compared reasonably.
 |
| | Fig. 11 (a and b) Bode phase angle plot of the 28 °C anodized electrode for the GOR and OER from 1.1 to 1.8 V vs. RHE. | |
When the solution is 0.15 M in 1.0 M KOH, the Bode phase angle plots showed completely unique features (Fig. 11b) compared to when there was just 1.0 M KOH, indicating a complete change in the mechanism between the GOR and OER. For instance, with the GOR, at lower potentials where Cu2+/Cu3+ redox was still predominant, the peak phase angle observed was closer to 50° (especially at 1.10 V vs. RHE) indicating the diffusion-controlled nature of the same. However, as soon as the set potential was increased beyond 1.10 V vs. RHE, the phase angle of the same decreased notably below 45° hinting more charge transfer control of Cu2+ oxidation. This means that the Cu2+, whichever is getting oxidized, is getting depleted swiftly by the concurrent GOR making the resultant Cu2+ available for faster oxidation. In the meantime, the peak also shifts towards the higher frequency range hinting even faster consumption of Cu3+ formed and a faster GOR. The subtle resistive phase angle peak seen even at 1.10 V vs. RHE can be attributed to adsorbed OH* on the surface of the electrode which is also getting scavenged by the GOR alongside Cu3+. At higher applied potentials, we can notice that this low intensity peak merges with the GOR peak indicating an even faster GOR at the highest anodic applied potential. In contrast, with just KOH, the phase angle corresponding to Cu2+ oxidation was closer to 80° and was also located at the lowest frequency of the study indicating that no significant charge transfer occurred except for the adsorption of OH. Interestingly, the high frequency peak is more prominent in this case indicating a longer lifetime of adsorbed OH* on the surface even before the OER was begun. As the set potential was increased beyond the onset of the OER, the low frequency shifted even further to the lower frequency side while the intensity was reduced below 45°, indicating poor consumption of formed Cu3+ during the OER with charge transfer control after the OER onset. Unlike the GOR, no merging of the phase angle peaks of Cu3+ oxidation and OH adsorption was observed in the case of just the OER. This implies that the adsorbed OH* and OER are loosely interconnected (kinetically) in the mechanism, whereas, with the GOR, they were synergistic in enhancing the GOR rate on the surface of the anodized Cu electrode. These observations are consistent with the recent results reported in the literature. Based on these observations, a probable mechanism is proposed below. At anodic potentials, Cu(OH)2 is oxidized to a higher valent surface oxyhydroxide which is the active oxidant (eqn (1)).
| | 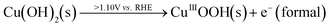 | (1) |
Glucose is mostly cyclic in neutral water, but in strongly alkaline media, it is present mostly as open-chain and/or deprotonated forms (alkoxide/enediolate). Adsorption of the same to the Cu surface occurs
via oxygen (–OH, aldehyde/ketone oxygen) and it may involve coordination of the C1 aldehyde (open-chain) or multiple vicinal hydroxyls to surface Cu sites (
eqn (2)).
| | | Glc (aq) → Glc: (CuIIIOOH(s)) (adsorption) | (2) |
First 2e
−/2OH
− oxidation is assumed to convert the aldehydic C1 (or anomeric centre) into the corresponding carboxylate (gluconate) or its surface-bound intermediate. This step is common in metal-catalysed alkaline sugar oxidation (C
1 → carboxylate). The active oxidant is the surface Cu
3+ species, which is reduced back to Cu
2+ (
eqn (3)).
| | | Glc: (CuIIIOOH(s)) + 2OH− → Gluconate: (CuII(s)) + e− + 2H2O | (3) |
Once oxidized to an aldonic acid (or activated enediolate), retro-aldol or β-elimination is assumed to occur on the surface, possibly yielding C
1 fragments (formate precursors) and other C
n fragments (
e.g., glycolate, glycerate,
etc.). However, we could not observe the latter in the reaction mixture indicating that these C
n fragments would have been rapidly fragmented further by C–C scission and oxidation to formate (the only product witnessed). An example simplified step (one of the many possible cleavage routes) is given below (
eqn (4)).
| |  | (4) |
These fragments will continue to undergo oxidation until the C
2 fragment formed at the end is also scissored and oxidized to formate. At the end of every oxidation step, Cu
3+ is reduced back to Cu
2+ which is re-oxidized electrochemically at the anode (
eqn (1)). The overall reaction would be the one given in
eqn (5). In this, the Cu
2+/Cu
3+ couple is the redox mediator.
| | | C6H12O6 + 18OH− → 6HCOO− + 12e− + 12H2O | (5) |
The overall mechanism discussed above is given as a catalytic cycle in
Scheme 1. This mechanism is only a plausible hypothesis from the EIS analysis and other electrochemical characterization studies and is not a confirmed pathway from the fact that the only product identified was formate. The actual glucose-to-formate oxidation route on Cu(OH)
2 may differ as well, and experimental validation is required before any of these steps can be considered definitive.
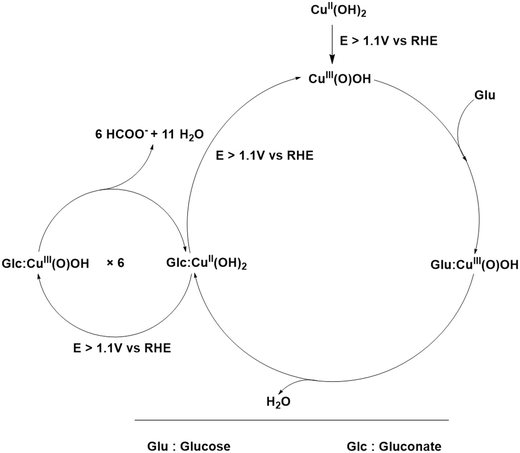 |
| | Scheme 1 Possible electrocatalytic mechanism of the GOR on anodized Cu electrodes. | |
Post stability analysis
We observed that at lower temperatures of anodization, where the proportion of Cu(OH)2 is higher, the corresponding electrodes performed the GOR more swiftly as compared to the electrodes which were anodized at higher temperatures. Following this, a 12 h stability study was conducted using CA at 0.5 V vs. Hg/HgO in 0.5 M glucose solution along with 1 M KOH (Fig. S3) by taking all the electrodes anodized at various temperatures. Among them, the one anodized at 28 °C with a higher Cu(OH)2 proportion delivered both high activity and better stability. To assess any changes (in morphology and chemical composition) that could have occurred during such a long-term stability test, post-stability XRD analysis was carried out for all the electrodes and is depicted in Fig. S4a. The resultant pattern demonstrated that while the XRD peaks remained largely similar, subtle changes were observed in the intensities of a few peaks. Notably, a decrease in peak intensity was observed in the 2θ range of 30° to 40°. These observations hinted at possible surface changes during the GOR as anticipated. This is because the GOR is a reaction that demands the extensive and progressive oxidation and reduction of surface sites of Cu, and such changes after a long-term stability analysis are common. To further investigate the changes, Raman analysis was also conducted for all of them, and the resultant spectra are shown in Fig. S4b.
These spectra showed almost similar signatures of Cu–O and Cu–OH moieties. To further investigate the morphological changes of the electrode anodized at 28 °C which showed better activity and better endurance in the stability study, FESEM images of the same were obtained (Fig. S5a–d) from which we could observe notable surface reconstruction of the electrode as anticipated. This observation matched well with recent reports.15,28,29 Following this, the TEM (Fig. S6a) and HRTEM (Fig. S6b–d) images were also obtained for the same electrode to further study the morphological changes of the nanostructures present on its surface. The same sample preparation methodology described previously was applied. Despite the observed surface reconstruction, the HRTEM analysis revealed a d-spacing of 0.226 nm, which corresponds to the (130) plane of Cu(OH)2, indicating that despite significant structural changes, the chemical composition remained predominantly to be Cu(OH)2 after the stability test. This finding was again proved by the SAED pattern shown as the inset of Fig. S6a. The SAED pattern exhibited characteristic ring plus dot features, suggesting that the material retained its polycrystalline nature, with variations in crystallite size. The diffraction planes observed in the SAED pattern were identified as (131) and (221), which are characteristic of Cu(OH)2 and CuO, respectively. Given that SAED is particularly sensitive to the spatial resolution of crystalline domains, the presence of these diffraction planes strongly supports the coexistence of both Cu(OH)2 and CuO phases on the electrode surface. The elemental composition was mapped using EDS and the corresponding elemental colour mapping (Fig. S7b–d), which clearly revealed the uniform presence of Cu and O on the surface. The EDS results (Fig. S7b) showed an atomic percentage of Cu at 86% and O at 14%, further supporting that the observed phases are surface confined. To further investigate the chemical oxidation states, XPS analysis was performed (Fig. S8a–d). The survey scan (Fig. S8a) revealed that the electrode was not contaminated with anything else other than K from the electrolyte used. The C 1s scan revealing sp3 carbon (Fig. S8b) was used to calibrate the narrow scans of other elements. Interestingly, the O 1s narrow scan (Fig. S8c) could be largely deconvoluted to the M–OH moiety compared to M–O indicating the increased Cu(OH)2 population after the stability. The XPS narrow scan of Cu 2p shown in Fig. S8d revealed that the Cu 2p1/2 and Cu 2p3/2 peaks were nearly identical to those obtained prior to the stability test, indicating that the chemical states of Cu did not undergo significant changes during the 12 h stability period. These XPS results provide valuable insight into the surface chemistry of the electrode, confirming that the stability study did not significantly alter the predominant chemical states at the surface. The overall post-stability characterization results unanimously concluded that though there were significant structural changes during the stability analysis, the chemical characteristics remained largely favourable by preserving most of the surface to be in the Cu(OH)2 state. This confirms synergistic evolution of the catalytic electrode.
Conclusions
This study explores the anodization of Cu foam electrodes at various temperatures and its impact on the electrocatalytic GOR. Our results demonstrate that the anodization of Cu electrodes at 28 °C significantly enhances the electrocatalytic activity for glucose oxidation, exhibiting faster anodic reaction kinetics. Specifically, when 0.15 M glucose in 1 M KOH was used, the Cu electrode showed the highest current density (∼500 mA cm−2 at 1.924 V vs. RHE) compared to other glucose concentrations. Moreover, the onset potential for anodic oxidation was reduced, indicating that room-temperature anodization of Cu can serve as an efficient alternative for water–glucose co-electrolysis. This process offers a promising energy-efficient pathway, positioning it as a viable alternative to the traditional OER for water splitting. Additionally, CA tests revealed excellent stability of the anodized Cu electrode, with the ability to restore its catalytic performance after adding glucose solution. The electrode's robustness under continuous operation is noteworthy, further highlighting its potential for long-term use in energy conversion systems. Another important outcome of this study is the precise control that one can gain over the morphological outcome and the chemical composition of the anodized surface. By performing the anodization at 28 °C, one can obtain Cu(OH)2-rich longer (∼2–3 μm) nanoneedle arrays. On the other hand, by performing the anodization at 46 C, one can reduce the length of the nanoneedles to just ∼50 nm and increase the proportion of CuO. This versatility makes this method applicable potentially in other areas of research where the chemical composition, surface area, and morphology of nanoneedle arrays are crucial. On top of this, given its high catalytic efficiency, ability to mask the OER, and overall durability, this system holds promise for integrating into practical electrolyzer systems for sustainable hydrogen production. Beyond water–glucose co-electrolysis, such Cu-based anodized electrodes may find broader application in electrochemical sensing and hybrid energy conversion devices, where simultaneous chemical upgrading and hydrogen generation are desirable. The ability to operate at reduced cell voltages while utilizing abundant, low cost materials makes this approach attractive for decentralized hydrogen production and alkaline electrolyzers coupled with renewable electricity sources.
Author contributions
N. C. M.: conceptualization, methodology, validation, formal analysis, investigation, data curation, writing – original draft, and visualization. B. S. D. and C. P. K.: investigation, formal analysis, data curation, and visualization. B. K.: methodology, validation, formal analysis, investigation, and data curation. A. S.: conceptualization, methodology, validation, formal analysis, investigation, resources, data curation, writing – original draft, writing – review & editing, visualization, supervision, project administration, and funding acquisition.
Conflicts of interest
The authors declare no competing interests.
Data availability
All the data discussed in this work are provided in full within the main manuscript and the supplementary information (SI). The access to the unprocessed data will be given upon a proper request made via an email addressed to the corresponding author.
Supplementary information: SEM-ED spectra, anodization CA, and stability studies are provided. See DOI: https://doi.org/10.1039/d5lf00387c.
Acknowledgements
This work is supported by the PMECR grant from ANRF, India (ANRF/ECRG/2024/000063/CS).
References
-
Y. Naimi and A. Antar, in Advances In Hydrogen Generation Technologies, InTech, 2018 Search PubMed.
- S. Shiva Kumar and H. Lim, Energy Rep., 2022, 8, 13793–13813 CrossRef.
- S. Wang, A. Lu and C.-J. Zhong, Nano Convergence, 2021, 8, 4 CrossRef CAS PubMed.
- M. El-Shafie, Results Eng., 2023, 20, 101426 CrossRef CAS.
- X. Zhang, J. Wang, K. Zong, Z. Chen, X. Yang, L. Yang, X. Wang and Z. Chen, Carbon Energy, 2025, 7, e679 CrossRef CAS.
- C. Lin, H. Li, P. Zhang, C. Deng, L. Meng, Q. Zhou, S. Wang, J. Wu, C. Liu, J. Tian and Y. Qian, J. Electroanal. Chem., 2020, 861, 113946 CrossRef CAS.
- W. Liu, X. Niu, J. Tang, Q. Liu, J. Luo, X. Liu and Y. Zhou, Chem. Synth., 2023, 3, 44 CAS.
- M. A. Khan, H. Zhao, W. Zou, Z. Chen, W. Cao, J. Fang, J. Xu, L. Zhang and J. Zhang, Electrochem. Energy Rev., 2018, 1, 483–530 CrossRef CAS.
- X. Chen, Z. Zhang, Y. Yang, B. Hu, Q. Wu, W. Fan, J. Hao and W. Shi, Chem. Eng. Sci., 2024, 291, 119937 CrossRef CAS.
- M. Tominaga, Y. Taema and I. Taniguchi, J. Electroanal. Chem., 2008, 624, 1–8 CrossRef CAS.
- L. Chen and J. Shi, Sci. China Mater., 2022, 65, 1–9 CrossRef CAS.
- J.-E. Lim, S. H. Ahn, S. G. Pyo, H. Son, J. H. Jang and S.-K. Kim, Bull. Korean Chem. Soc., 2013, 34, 2685–2690 CrossRef CAS.
- A. P. Tiwari, M. S. Rahman and W. J. Scheideler, Adv. Mater. Technol., 2024, 9(13), 2400160 CrossRef CAS.
- A. P. Tiwari, J. E. Patterson, M. S. Rahman and W. J. Scheideler, Adv. Mater. Technol., 2025, 10(15), e00122 CrossRef CAS.
- N. C. Minj, B. Kamaraj, S. Mittal, S. Yadav, P. Trivedi and A. Sengeni, ACS Sustainable Chem. Eng., 2025, 13, 7824–7836 CrossRef CAS.
- I.-H. Yeo and D. C. Johnson, J. Electroanal. Chem., 2000, 484, 157–163 CrossRef CAS.
- Y. Wang, M. Xu, X. Wang, R. Ge, Y.-Q. Zhu, A.-Z. Li, H. Zhou, F. Chen, L. Zheng and H. Duan, Sci. Bull., 2023, 68, 2982–2992 CrossRef CAS PubMed.
- S. Choudhary, J. V. N. Sarma, S. Pande, S. Ababou-Girard, P. Turban, B. Lepine and S. Gangopadhyay, AIP Adv., 2018, 8(5), 055114 CrossRef.
- E. Touzé and C. Cougnon, Electrochim. Acta, 2018, 262, 206–213 CrossRef.
- F. I. Barbaccia, T. de Caro, F. Federici, A. Mezzi, L. Sansone, M. Giordano and A. Macchia, Materials, 2025, 18, 1487 CrossRef CAS PubMed.
- M. Košiček, J. Zavašnik, O. Baranov, B. Šetina Batič and U. Cvelbar, Cryst. Growth Des., 2022, 22, 6656–6666 CrossRef.
- D.-D. La, S.-Y. Park, Y.-W. Choi and Y.-S. Kim, Bull. Korean Chem. Soc., 2010, 31, 2283–2288 CrossRef CAS.
- D. Giziński, A. Brudzisz, J. S. Santos, F. Trivinho-Strixino, W. J. Stępniowski and T. Czujko, Catalysts, 2020, 10, 1338 CrossRef.
- L. Jiang, P. Li, S. Wang, R. Liu, X. Zhu, Y. Song and T.-V. Ree, Energy Mater., 2022, 2, 200038 CrossRef CAS.
- N. K. Allam and C. A. Grimes, Mater. Lett., 2011, 65, 1949–1955 CrossRef CAS.
- S. V. Ganzha, S. N. Maksimova, S. N. Grushevskaya and A. V. Vvedenskii, Prot. Met. Phys. Chem. Surf., 2011, 47, 191–202 CrossRef CAS.
- M. Scherzer, F. Girgsdies, E. Stotz, M.-G. Willinger, E. Frei, R. Schlögl, U. Pietsch and T. Lunkenbein, J. Phys. Chem. C, 2019, 123, 13253–13262 CrossRef CAS.
- S. Anantharaj, H. Sugime and S. Noda, ACS Appl. Mater. Interfaces, 2020, 12, 27327–27338 CrossRef CAS PubMed.
- S. Anantharaj, H. Sugime, S. Yamaoka and S. Noda, ACS Appl. Energy Mater., 2021, 4, 899–912 CrossRef CAS.
- B. Honnappa, T. R. N. Kumar, P. J. J. Sagayaraj, S. Shenoy, C. Chuaicham, M. S. Yesupatham, A. Sengeni, B. Neppolian, K. Sasaki and K. Sekar, J. Mater. Chem. A, 2023, 11, 25854–25858 RSC.
- H. K. Ayask, J. V. Khaki and M. H. Sabzevar, J. Ultrafine Grained Nanostruct. Mater., 2015, 48(1), 37–44 Search PubMed.
- P. Marathey, S. Khanna, R. Pati, I. Mukhopadhyay and A. Ray, J. Mater. Res., 2019, 34, 3173–3185 CrossRef CAS.
- A. M. Awwad and B. Albiss, Adv. Mater. Lett., 2015, 6, 51–54 CrossRef.
- L. Wang, K. Zhang, Z. Hu, W. Duan, F. Cheng and J. Chen, Nano Res., 2014, 7, 199–208 CrossRef CAS.
- C.-Y. Peng, C.-C. Hou, Q.-Q. Chen, C.-J. Wang, X.-J. Lv, J. Zhong, W.-F. Fu, C.-M. Che and Y. Chen, Sci. Bull., 2018, 63, 1583–1590 CrossRef CAS PubMed.
- J. Wang, L. Zhu, L. Ji and Z. Chen, J. Mater. Res., 2018, 33, 581–589 CrossRef CAS.
- T. M. F. Marques, R. N. Morais, F. X. Nobre, J. M. Rocha, A. Ghosh, T. A. S. Soares, B. C. Viana, G. Machado, J. C. S. Costa and J. M. E. De Matos, An. Acad. Bras. Cienc., 2019, 91(04), e20190082 CrossRef.
Footnote |
| † These authors have contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a













