DOI:
10.1039/D5LC00851D
(Paper)
Lab Chip, 2026,
26, 89-105
Use of poly(glycerol sebacate) as an alternative to poly(dimethylsiloxane) as a platform for making micro-fluidic channels
Received
7th September 2025
, Accepted 8th November 2025
First published on 13th November 2025
Abstract
Poly(dimethylsiloxane) (PDMS) has been used extensively for making microfluidic devices because of several favorable features: optical transparency, bio-compatibility, easy moldability and its inertness towards most materials. However, PDMS is non-biodegradable and swells when in contact with most solvents. It is also an expensive material. As an alternative, here we have presented the application of poly(glycerol sebacate) (PGS), a thermoset elastomer for fabrication of microchannels. PGS is synthesized via the condensation reaction of sebacic acid, a non-edible vegetable oil, and glycerol which results in a flexible, biocompatible and optically transparent bio-elastomer; its Young's modulus has been varied over a wide range: 0.125–1.4 MPa. We have shown that controlled etching with an aqueous solution of NaOH yields microchannels of a variety of shapes, sizes and complexities: straight and spiral channels and ones with patterned surfaces of the channel wall and depth. The PGS surface shows hydrophobicity initially but turns hydrophilic over time, with minimal swelling effect, thereby highlighting its suitability for both aqueous and non-aqueous media. With acidic and neutral buffers, PGS shows negligible leaching, thereby suggesting usability of microfluidic channels under physiological conditions. PGS channels were used also in multiple layers with a membrane in-between, to explore controlled transport of drug and nutrient molecules and to determine possible bioanalytical applications.
1. Introduction
Elastomers have drawn significant attention because of their unique combination of properties: elasticity, durability, chemical stability and processability which have led to their applications in diverse fields like soft robotics, microfluidics, flexible electronics, adhesives, sealants, food storage, and several others. Among elastomers, poly(dimethylsiloxane) (PDMS) possesses excellent properties like high flexibility, optical transparency, chemical inertness, biocompatibility, and ease of fabrication, which make it the preferred material for fabrication of microfluidic channels for many scientific and engineering applications, e.g. in bio-analytical devices, disease diagnostics, and point-of-care equipment. Microfluidic channels made of PDMS are primarily made by soft-lithography,1–3 and/or by crosslinking against a template which can be either two dimensional planar or three-dimensional4–6 or by laser ablation.7,8 In addition to conventional top-down fabrication methods, bottom-up strategies have also been explored, wherein channels are generated spontaneously, such as through hydrophilic–hydrophobic phase separation leading to self-assembled microchannels,9 or strain-induced self-rolling of PDMS films forming microcapillaries.10 The channels are generally straight, but can also be spiral,11,12 helical,5,13 super-helical5 or any other orientation as desired; the channels are generally rectangular in cross-section but can also be circular,5,6 trapezoidal12 or elliptic type;14,15 the channel surfaces are generally smooth, but can also be decorated with ridges16 and pillars of different shapes and size.17,18 One important aspect of PDMS being used as a microfluidic platform is that it is benign to many materials, and it doesn't involve in non-specific interaction with biological molecules like proteins and amino acids and biological cells. In contrast, PDMS is amenable to a variety of surface modification methods e.g. ligand immobilization,19,20 plasma treatment,21–23 UV-ozone treatment,24,25 silanization,25–27 graft polymerization,28,29 sol–gel approaches30,31 and so on. To summarize, PDMS has proved to be an excellent material, considering almost all aspects of soft microfluidics.
Despite its many uses, PDMS has few notable limitations. It is non-biodegradable; it is expensive, not easily available; it is made from non-renewable feedstock; it also swells when in contact with many solvents which poses operational difficulties. Since microfluidic devices are being used on a large scale, they need to be inexpensive both in terms of cost and sustainability. Microfluidic platforms are also expected to be compatible with fluids of different types: aqueous and non-aqueous, polar and non-polar. To meet these needs, as well as to address the deficiencies of PDMS, we have explored here poly(glycerol sebacate) (PGS) as a potential alternative to PDMS. PGS is inexpensive, biodegradable, biocompatible and importantly it is a material approved by the Food and Drug Administration (USA).32,33 Several other biodegradable polymers such as poly(lactic-co-glycolic acid) (PLGA), polycaprolactone (PCL), poly(ethylene glycol) diacrylate (PEGDA), and gelatin methacrylate (GelMA) have also been explored for similar applications; however, these are either partially bio-derived (e.g., PLGA, GelMA)34,35 or entirely synthetic (e.g., PCL, PEGDA),36,37 and often show limitations such as higher stiffness, slow or uncontrolled degradation, solvent-dependent processing, or poor surface interaction. In contrast, PGS is synthesized from naturally occurring, FDA-approved monomers: glycerol and sebacic acid through a solvent-free process, offering a sustainable and clean route to elastomer formation. The physicochemical and mechanical characteristics of PGS are tunable,38–40 very much similar to those of PDMS. PGS shows a glass transition temperature Tg ∼ −40 °C, which is higher than that of PDMS: −123 °C, yet it remains flexible at room temperature. The mechanical properties of PGS, assessed by varying the crosslinking time, shows a tunable modulus of 0.1–1.4 MPa, comparable to that of PDMS: 0.3–3 MPa.41 Because of these favorable characteristics, PGS has been used for many biomedical applications, including hard and soft tissue engineering, controlled drug delivery, and tissue adhesives.42–47 In addition, PGS has been explored for vascular graft applications, where porous structures generated by electrospinning, extrusion, or porogen-leaching have been designed to enhance tissue integration and endothelialization.48 Beyond such porous designs, we show here that PGS can also be used as a platform for making microchannels suitable for a variety of process applications. Although microchannels made of PGS have been previously reported using techniques such as soft lithography49 and laser ablation,50 these approaches generally require expensive equipment, molds, and complex processing steps. In contrast, the present work demonstrates a simple, mold-free, and cost-effective etching-based approach for fabricating microchannels. The use of an easily available etchant further enhances the practicality and scalability of the process.
In particular, we have synthesized PGS by systematically varying the crosslinking time to tune its mechanical, thermal and surface properties. We have developed a controlled etching method to generate microchannels of different types on its surface and have optimized it by systematically varying the associated parameters. Our method has led to the generation of various microchannel geometries: Y-shaped, spiral, Y-shaped channels with curved extensions, and Y-shaped channels featuring rectangular grooves on the channel surface.
2. Materials and methods
Materials
Sebacic acid (98%, molecular weight 202.25) was purchased from Ottokemi. Glycerol anhydrous (≥98%) was purchased from Merck Life Science Private Limited. Sodium hydroxide pellets, tri-sodium citrate dihydrate, tris-hydrochloride AR, bovine albumin and chloroform were purchased from Loba Chemie Pvt Ltd. and trichloro (1H,1H,2H,2H-perfluorooctyl) silane was purchased from Sigma-Aldrich. The last two chemicals were used for the fluorocarbon coating of the glass plates. Citric acid monohydrate was procured from RANKEM. Citric acid monohydrate and tri-sodium citrate dihydrate were used for the preparation of citrate buffer solution, while tris-hydrochloride was used for preparing Tris buffer solutions. Millipore water was used in all cases, wherever required.
Synthesis of the poly(glycerol sebacate) (PGS) blocks
PGS was synthesized as reported in the literature.32 Equimolar amounts of sebacic acid and glycerol anhydrous were taken in a conical flask and heated at 130 °C with continuous stirring under N2 for 24 h. After 24 hours the slightly yellowish, viscous prepolymer obtained was poured on a fluorocarbon coated glass mold and kept in the oven at 120 °C for crosslinking (Fig. 1a) (Scheme 1). The prepolymer was crosslinked for different crosslinking times, tcr = 12–48 hours, and the samples were named accordingly, PGStcr. For tcr < 12 hours, the films were insufficiently cured, liquid like, and could not be used as freestanding films. In contrast, for tcr > 48 hours, the films turned overly stiff and brittle with negligible flexibility. Therefore, the tcr was chosen between 12 and 48 hours to achieve a balance between mechanical integrity for handling and the flexibility necessary for integration into microfluidic channel fabrication. The PGS films were colorless and transparent (Fig. 1a) when pre-polymerized under inert conditions, however, they turned brownish (Fig. S1a) in the absence of an inert atmosphere because of oxidation.51 To confirm successful synthesis of PGS, Fourier-transform infrared spectroscopy (FTIR) was conducted on the synthesized PGS and its precursors (Fig. 1b). For sebacic acid, the signal at 1694 cm−1 corresponds to the carboxylic group and in glycerol the strong peak at 3305 cm−1 corresponds to the O–H bond. As PGS is formed, the signal at 1694 cm−1 becomes more intense and gets shifted to 1734 cm−1, which belongs to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of the ester group. The O–H signal appears reduced, suggesting consumption of hydroxyl groups during polymerization. The data matches with that reported in the literature showing successful synthesis of PGS.32,52 FTIR spectra of PGS films crosslinked for different time intervals, PGS12–48hours, did not show any significant changes in peak positions (Fig. S1b), indicating that the overall chemical structure remains largely unaltered during the crosslinking process. However, a slight decrease in the intensity of the OH stretching band was observed for PGS24–48hours compared to PGS12hours, suggesting consumption of hydroxyl groups during crosslinking.
O of the ester group. The O–H signal appears reduced, suggesting consumption of hydroxyl groups during polymerization. The data matches with that reported in the literature showing successful synthesis of PGS.32,52 FTIR spectra of PGS films crosslinked for different time intervals, PGS12–48hours, did not show any significant changes in peak positions (Fig. S1b), indicating that the overall chemical structure remains largely unaltered during the crosslinking process. However, a slight decrease in the intensity of the OH stretching band was observed for PGS24–48hours compared to PGS12hours, suggesting consumption of hydroxyl groups during crosslinking.
 |
| | Fig. 1 (a) The schematic depicts the process of synthesizing the PGS material. Equimolar amounts of sebacic acid and glycerol anhydrous were heated at 130 °C for 24 hours with continuous stirring under N2. After 24 hours, the resulting viscous prepolymer was poured on a fluorocarbon coated glass housing and crosslinked for 12–48 hours at 120 °C. The crosslinked films were colourless and optically transparent. To check their optical transparency, IITK was printed on an A4 sheet, the PGS films were placed over it and a photograph was taken. (b) The FTIR spectra of PGS confirm its successful synthesis from the precursors. Sebacic acid exhibits a sharp CO signal at 1694 cm−1 which corresponds to the carboxylic group. Glycerol shows a broad O–H signal at 3305 cm−1 and a C–O signal at 1032 cm−1. In PGS, the appearance of a strong signal at 1734 cm−1 corresponding to CO of the ester group validates the formation of the ester bond. | |
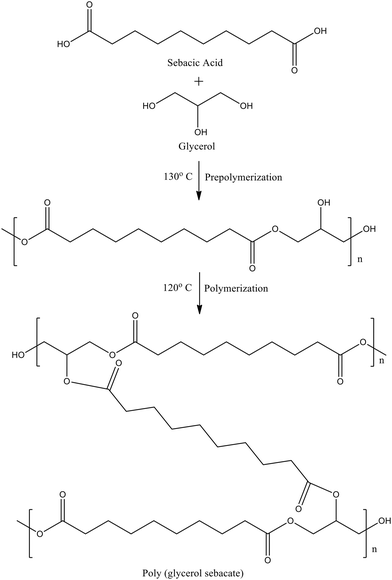 |
| | Scheme 1 The reaction scheme illustrates the synthesis of PGS, where the first stage, prepolymerization, involves the formation of ester bonds between glycerol and sebacic acid to yield a viscous prepolymer, followed by curing, in which further crosslinking occurs to produce the final elastomeric PGS network. | |
Estimation of mechanical properties of PGS for application suitability
Microfluidic devices are generally employed across wide-ranging applications: from soft and flexible platforms such as wearable sensors and tissue scaffolds to rigid systems used in high-pressure analytical setups.53–55 To assess the potential applicability of our fabricated channels in such diverse environments, the mechanical properties of PGS were required to be evaluated; it was done by carrying out a tensile test on PGS12–48hours samples (details of the test instrument and sample size are presented in SOM).
Estimation of thermal properties of PGS
In order to understand the temperature range through which a microfluidic device made on the PGS platform can be used, the glass transition temperature, Tg, was obtained. The PGS12–48hours samples were subjected to differential scanning calorimetry (DSC), which confirmed (Fig. S2) that the elastomers were semi-crystalline as reported earlier.56,57 The Tg of these samples was found to vary from −37.38 to −39.51 °C for tcr = 12–48 hours, suggesting negligible dependence of Tg on tcr (Table S1). Since most microfluidic devices are operated at room temperature, PGS was considered to be a suitable material for making microfluidic channels.
Estimation of surface energy of PGS
In the context of microfluidic applications, surface energy is an important factor, which determines the range of materials that can be handled in the channel, ease of fluid flow through it, possibility of adsorption of a species on the channel wall and so on. It is therefore natural to see a variety of surface chemical modification routes that have been successfully developed for PDMS to meet the needs of a large range of applications. In the same spirit, we wanted to know the surface energy of PGS. It is worth noting also that although PGS has been explored extensively as a material of bio-medical applications and tissue engineering, its surface energy has not been estimated to sufficient detail. For example, PGS has been inconsistently reported as both hydrophilic32,58 and hydrophobic59,60 in the literature. It is not known also if PGS undergoes surface reconstruction in the presence of a polar liquid. So, it was important to estimate the surface energy of PGS. The surface energy of the PGS films was estimated by the contact angle method. 3 μl drops of water (γpL = 51.0 mN m−1, γdL = 21.8 mN m−1) and hexadecane (γpL = 0.0 mN m−1, γdL = 27.47 mN m−1) were placed over PGS films and advancing contact angles were recorded using a CCD camera. The initial contact angle of water on PGS films crosslinked for 12–48 hours was found to vary from 106–95°. Using these initial contact angle values, the surface energy of PGS films was calculated by using the Owens–Wendt equation (eqn (S1)).61
Results and discussion
Elastic modulus of PGS layers
Fig. 2 shows the results from tensile tests for different PGS samples. Fig. 2a shows a PGS12hours sample at three different stages: before test, under tension, and after failure. The image reveals that the sample fractured at the center rather than near the grips, indicating uniform stress distribution, and that it returned to its original shape after failure, demonstrating elastic behavior. In Fig. 2b we show the tensile stress–strain data for PGS12–48hours samples, where stress is denoted by σ and strain by ε. It is worth noting that the ε goes as high as 235 ± 29% and extensibility increases with the decrease in tcr, much beyond the linear elastic limit. The ultimate tensile strength (UTS), at which the sample finally ruptures, increases (Fig. 2b) with the increase in tcr possibly because of the increase in cross-linking density. The sample nevertheless behaves elastically, as evident from Fig. 2b. Therefore, the neo-Hookean model was employed to relate the true stress data, σ′ = σλ, to the extension ratio, λ = 1 + ε.62 The plot in Fig. 2c shows that σ′ varies linearly with 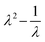 , with the slope defining the elastic constant, E, of the material. The E for different PGS samples was found to vary from 0.13 to 1.4 MPa respectively, a range in which silicone is also used as a microfluidic platform (Table S1). In order to obtain a quantitative relation between E and tcr, in Fig. 2d, Young's modulus E was plotted against tcr to show that E increases super-linearly with tcr: E ∼ t3/2cr. This observation can be rationalized by considering essentially the kinetics of the crosslinking process. It has been shown earlier15 that during the pre-polymerization stage, the primary hydroxyl groups (–OH) of glycerol react with carboxyl groups (–COOH) of sebacic acid leaving behind one secondary –OH group which later on, during the crosslinking process, reacts with a –COOH group to form the crosslinked network. So, crosslinking at the second stage is essentially driven by 1/3rd the number of moles of glycerol in the reacting solution. This consideration implies that the rate of crosslinking reaction varies as C1/3g, where Cg = ng/V is the molar concentration of glycerol per unit volume:
, with the slope defining the elastic constant, E, of the material. The E for different PGS samples was found to vary from 0.13 to 1.4 MPa respectively, a range in which silicone is also used as a microfluidic platform (Table S1). In order to obtain a quantitative relation between E and tcr, in Fig. 2d, Young's modulus E was plotted against tcr to show that E increases super-linearly with tcr: E ∼ t3/2cr. This observation can be rationalized by considering essentially the kinetics of the crosslinking process. It has been shown earlier15 that during the pre-polymerization stage, the primary hydroxyl groups (–OH) of glycerol react with carboxyl groups (–COOH) of sebacic acid leaving behind one secondary –OH group which later on, during the crosslinking process, reacts with a –COOH group to form the crosslinked network. So, crosslinking at the second stage is essentially driven by 1/3rd the number of moles of glycerol in the reacting solution. This consideration implies that the rate of crosslinking reaction varies as C1/3g, where Cg = ng/V is the molar concentration of glycerol per unit volume:| | 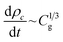 | (1) |
where ρc = nc/V defines the crosslinking density in the network. Note that during crosslinking, each glycerol molecule leads to one crosslinked joint, implying essentially that nc = ng. Substitution of this expression in eqn (1) and integration yields ρc ∼ t3/2cr. Since E scales linearly with the crosslinking density, E is indeed expected to scale as E ∼ t3/2cr. The intercept in this plot shows that it is only at tcr > 11 hours that a finite E is attained. The mechanical properties of PGS demonstrate that it can serve as a substrate with a wide range of tunable elastic moduli, making it suitable for fabricating microchannels.
 |
| | Fig. 2 The figure illustrates the tensile test results of PGS12–48hours. (a) Representative images of a PGS12hours film during tensile testing show the elastic nature of the material. (b) The tensile stress–strain response of PGS shows that the material can sustain strain up to 235 ± 29%, with extensibility decreasing as the tcr increases. This behavior extends well beyond the linear elastic regime. (c) To account for the large deformations, the data is fitted using the neo-Hookean model, where true stress, σ′, is plotted against λ2 − 1/λ (λ is the extension ratio) with the slope yielding the elastic modulus, E, of the material (linear fits: R2 = 0.99–1). (d) The plot of E versus t3/2cr suggests a super linear relation. | |
Surface energy
In Fig. 3a, we show sessile drops of DI water and hexadecane dispensed on a PGS24hours surface. The images represent the quasi-equilibrium state of the drops at a short time scale of ∼5 min. Similar observations are also made for different other surfaces, all of which depict the hydrophobic character of the PGS surface. To estimate the surface energy γs, the contact angle values of water and hexadecane on these surfaces were used. The table in Fig. 3 shows that the dispersive component of surface energy, γds, was nearly identical for all surfaces and was very similar to that of PDMS: 21 mJ m−2, whereas the polar component, γps, increased with the increase in tcr (Fig. 3b). In fact, this increase was super-linear: γps ∼ t3/2cr.
 |
| | Fig. 3 The figure shows the wetting behaviour of PGS films, investigated by the contact angle method. (a) The advancing contact angle images of water and hexadecane on the PGS24hours surface show the non-polar nature of the polymer. (b) The γs of the PGS increased with increasing tcr, as shown in the table, primarily due to an increase in the γps, while the γds remained largely unchanged. | |
At a longer time scale, tobs, the drops were found to spread with the decrease in contact angle (Fig. 4a). The rate of spreading was slow enough that it appeared to happen quasi-statically. To examine this effect systematically, the PGS films were placed inside a closed chamber with controlled humidity to minimize evaporation. PGS films, of all different extent of crosslinking, showed that the advancing contact angle, θa, of the water drop decreased with the spreading of the drop (Fig. 4b); however, the spreading stopped within 30 min following which the spreading diameter, dc, was nearly constant; the contact angle, θa, however continued to decrease till reaching an equilibrium value within ∼50 min. The spreading data for all surfaces were pulled together to show that dc increased with tobs as dc ∼ t1/16.5obs (Fig. 4c). It is worth noting that the exponent, 1/16.5, was significantly smaller than 1/10 as predicted for the spreading of a viscous liquid on a perfectly wettable rigid substrate: Tanner's 1/10th law.63 This result implied that, in contrast to such a substrate, the PGS surface was hydrophobic initially and turned partially wettable over time in response to water in contact with it. This behaviour can be attributed to reorientation or segmental mobility of molecules at the vicinity of the surface, in response to a contacting material, in this case, a polar liquid like water. This reorientation effect was expected to be less pronounced with PGS films crosslinked to a larger extent, e.g. PGS48hours (ref. 64 and 65) because of the constraints imposed by the crosslinking points. For the PGS12hours films, higher chain mobility and the presence of more unreacted polar groups such as hydroxyl or carboxyl functionalities are expected to result in more pronounced migration or orientation of hydrophilic segments towards the surface upon exposure to water, facilitating stronger interactions with water molecules and thus reducing the contact angle to a larger extent.66 This effect was reversible, implying that a PGS surface turned hydrophilic via wetting could be turned back to hydrophobic by drying in air; and this cycle could be repeated.
 |
| | Fig. 4 (a) The sequence of images captures the quasi-static spreading of a water drop dispensed on a typical PGS24hours surface. (b) The advancing contact angle, θa, of the drop decreases with the consequent increase in the spreading diameter, dc, on the substrate over time, tobs. Here open and filled symbols represent respectively dcvs. tobs and θavs. tobs. (c) dc attains a constant value within 30 min beyond which it remains unaltered. Within this time, dc varied with tobs as dc ∼ 1/t1/16.5obs. | |
Swelling of PGS
To examine if the observed decrease in θa results from absorption of water, the PGS12hours and PGS48hours samples (three 1.0 cm × 1.0 cm × 0.12 cm samples of each of these films) were weighed and immersed inside pools of three different liquids: water, hexadecane and hexane respectively. At fixed time intervals, the films were removed from the respective liquid pool and their surface was dried by blowing N2 gas; the weight of the films was measured, and the films were re-immersed in the liquid. Analysis of swelling data (eqn (S2) and Fig. S3) shows that for a polar molecule like water, swelling was negligibly small within 1 hour and was @4% v/v over ∼50 hours; the samples swelled insignificantly in hexadecane too (1% v/v over 50 hours). It was conjectured that hexadecane, with large molecular size, could diffuse only to a limited extent into the polymer network. So, hexane, a smaller non-polar solvent, was subsequently used to better evaluate the polymer's swelling behavior in a non-polar environment. PGS showed negligible swelling in hexane as well (5% v/v by the above period). These results suggest that the decrease in θa on PGS films in the experiment of Fig. 4 was not because of the swelling of the crosslinked network of the solid. It is worth noting also that the least crosslinked PGS, PGS12hours, showed a higher initial θa compared to more crosslinked surfaces. This behavior was consistent with previous studies on hydrophilic gels that report high initial θa due to the preferential surface alignment of hydrophobic chain segments.67 For PGS, reduced crosslinking likely allows greater segmental mobility, facilitating the migration of hydrophobic moieties to the surface, thereby increasing initial hydrophobicity. In contrast, increased crosslinking constrains this mobility, resulting in reduced surface reconstruction and a lower initial θa. The observed transition of the PGS from hydrophobic to hydrophilic in the presence of polar liquids indicates also that for applications requiring hydrophilic surfaces, PGS does not require a surface treatment process except being in contact with water for @30 min. Secondly, the PGS surface is expected to be compatible with both polar and non-polar organic liquids to qualify as a material of choice for microfluidic applications.
Effect of pH on surface wettability of PGS
In addition to the above three liquids, buffer solutions of different pH were also used to examine the sensitivity of the PGS material towards pH. For this purpose, drops (3 μL) of tris HCl buffer of different pH: 3.5, 5.5, 7.4, 9.5 and 11.5 were dispensed on PGS12hours samples and θa was measured. θa decreased with time for all cases, however the extent of spreading varied with pH (Fig. S4a). Maximum spreading was observed at neutral pH (7.4), while at basic pH (9.5 and 11.5), polymer swelling at the droplet site made contact angle measurement unreliable (Fig. S4b). The increased swelling observed with the basic pH buffer is attributed to the deprotonation of carboxylic acid groups in PGS, resulting in the formation of negatively charged –COO− groups that promote water uptake.68 However, no swelling was observed in the PGS24hours and PGS48hours samples when buffer solution droplets of pH 9.5 were placed on their surfaces and monitored over time (Fig. S4c). These results show that the PGS is sensitive to basic conditions, whereas it remains stable in acidic and neutral environments.
Fabrication of microchannels by etching
PGS is known to undergo surface degradation in neutral buffer solutions, in the presence of enzymes and under alkaline conditions.32,69,70 Based on this concept, we have developed a controlled degradation or etching method using aqueous NaOH as the etchant to create microchannels in PGS blocks. NaOH, being a strong base, hydrolyzes the ester bonds of PGS71,72 so that water-soluble hydrolysis products, primarily glycerol and sodium sebacate, are released into the solution, leading to surface erosion. To spatially confine the location for etching, we utilized a transparent polyester sheet as a mask, on which we created desired patterns in the form of through holes by laser cutting, and this sheet was then placed in intimate contact with the PGS film (12–24 hours crosslinked); 1 M aqueous NaOH (referred to as aqNaOH) solution was then poured on this sheet (Fig. 5a). The base come into contact with the PGS film at the location of the holes, leading to the spatially controlled etching. After 3–4 hours the sheet was removed from the PGS film, and the resultant channel was washed thoroughly with DI water and was sonicated to remove any trace of left-over base on the etched surface and was then dried at room temperature. The etched microchannels were analyzed using an optical profilometer for measuring the channel depth. Fig. 5b and c shows a typical Y-shaped channel, its side view and an optical micrograph of the channel surface. The image shows that the etching yielded a nearly flat surface. The width of the channel closely matched that of the mask suggesting that etching happed normal to the mask surface. Thus, channels of precise depth, with a flat and smooth surface, could be formed by this method. To demonstrate the generality of the fabrication process, polyester sheets were laser-cut to form different other channel geometries: a Y-shaped channel with a curved extension and a spiral channel (Fig. 5d). Using these sheets as masks, etching of the PGS surface, as in Fig. 5a, led to corresponding different micro-channel shapes (Fig. 5d).
 |
| | Fig. 5 (a) The schematic diagram shows the process of making microchannels in PGS by etching, using aqueous NaOH solution as an etchant. Desired patterns were made on a transparent polyester sheet, which was then placed over the PGS film, and a 1 M aqueous NaOH solution was poured over it. After 3–4 hours, the sheet was removed, and the channel was thoroughly washed with DI water and dried at room temperature. (b and c) The images show the top and side views of a typical “Y” channel. The width of the channel closely matches with that of the mask used to make it. (d) The images demonstrate fabrication of various planar channel geometries, including Y-shaped channels with a curved tail and spiral channels. (e) The sequence of images shows the magnified view of the channels etched for different times with 1.0 M aqNaOH. (f) The plot shows the channel height hchannel data from different sets of experiments (Cetch = 0.1–1 M, tetch = 2–8 hours) scaled linearly with the quantity tetch·Cetch. | |
Variation of channel depth with concentration of etchant and time of etching
To understand the effect of etching time, tetch, and etchant concentration, Cetch, on the channel depth, hchannel, these parameters were systematically varied: tetch from 2–8 hours and Cetch from 0.1 to 1.0 M. The sequence of image in Fig. 5e shows the channels which were fabricated by subjecting a layer of PGS12hours to different etching times. Here hchannel was found to increase with tetch. In fact, hchannel increased linearly with both Cetch and tetch, as shown in the plot of Fig. 5f. Here, hchannel data from all different experiments, plotted against the quantity, Cetch·tetch, were found to fall on a straight line, implying that longer exposure of PGS to the etchant resulted in dissolution of more bonds of the polymer; similarly, aqNaOH of increasing concentration also resulted in a larger extent of dissolution of bonds. It was noted that aqNaOH with concentration less than 0.1 M didn't result in etching of the PGS surface but caused its swelling; therefore, Cetch < 0.1 M was not used in these experiments. While etching leads to precise channel height, it doesn't appear to attain a sharp corner. Analysis of the side view of images in Fig. 5(e) shows that the radius of curvature of the corner, ρ, increases with hchannel. Nevertheless, ρ remains significantly smaller than the width of the channel. Importantly, for processes occurring inside a microchannel, a blunt corner prevents the occurrence of a dead zone thereby enhancing the process efficiency.73
Etching in multiple stages
We also explored making complex microchannels by carrying out etching in multiple stages (Fig. 6). For example, a PGS block was first etched to fabricate a Y-shaped channel. In the second stage a strip of transparent polyester sheet was laser cut to form parallel rectangular openings of width 200 μm. The width of the strip was identical to that of the channel width, so that it could be placed and adhered on the smooth surface of the channel. 1 M aqNaOH solution was then poured over the sheet and was left to etch the exposed surface of PGS for 6 hours. Following this step, the polyester sheet was removed, and the channel was thoroughly washed with DI water and dried at room temperature to form a Y-channel with a grooved surface. The resulting channels were approximately 80 μm wide, with grooves in between measuring ∼480 μm in width and 85 μm in height. It is worth noting that the mask used for making these channels had an opening of 200 μm, the channels that got generated, however, were of width ∼80 μm. The reduced width could possibly be due to dewetting of the etching liquid on the mask surface.
 |
| | Fig. 6 The diagram illustrates the fabrication of a channel via etching in multiple steps. In particular, rectangular grooves of width: ∼480 μm and height: ∼85 μm were created within a prefabricated Y-shaped channel; a polyester sheet, having width similar to that of the channel and laser patterned with small rectangular openings (200 μm), was brought into intimate contact with the PGS block. The etchant was then dispensed over the polyester sheet and was left for 6 hours, allowing etching of the exposed regions, which resulted in the rectangular grooves on the surface of the Y-shaped channel. | |
Does etching lead to chemical modification of PGS?
To know if etching induces any chemical changes in the PGS, these surfaces were subjected to FTIR and Raman spectroscopy. In Fig. 7a and b we present the FTIR and Raman spectra of the etched channel surface and that of pristine PGS. The FTIR spectra of the two surfaces completely overlap suggesting that etching of the PGS network didn't introduce any chemical change in the material; the Raman spectra (details of the test instrument presented in SOM) for both surfaces show similar peaks with small alteration in intensity suggesting some alteration in the crosslinked structure of the molecules at the etched surface. These results suggest furthermore that the zone of reaction was limited at the vicinity of the surface only. To assess if the etched surface retained its wetting behavior as the pristine PGS surface, after etching, the advancing water contact angle, θa, was measured on the etched surface. The θa within the first 5 minutes was as high as 124 ± 8°, indicating a hydrophobic surface. The θa however decreased with time very much similar to the pristine PGS surface, except with one difference: here the θa decreased more rapidly with time than that on the pristine PGS (Fig. 7c). This observation can be attributed to the increased surface roughness after etching, as confirmed by profilometer images (Fig. 7d).
 |
| | Fig. 7 (a and b) The plots show respectively the FTIR and Raman spectra of the etched channel surface and that of the pristine PGS film. In both cases the spectra of the two surfaces nearly overlap. (c) A drop of water was dispensed on the etched surface to examine its wettability. The inset shows the droplet image demonstrating how the θa was measured. The plot shows variation of θa over time on both pristine and etched PGS surfaces. (d) The image shows a typical surface profile of an etched channel captured using an optical profilometer. | |
Flow experiments through microchannels built on the PGS platform
Mixing of two liquids
To examine if the microchannels fabricated above were suitable for common microfluidic operations, these were used for examining mixing of two differently colored aqueous solutions. For this purpose, the PGS block containing a typical “Y” shaped microchannel was bonded to a plasma-oxidized glass slide, and two small holes were created at the inlet sides. MilliQ water was dyed using food colors: ANUJA bright green and ANUJA orange red respectively and the liquid samples were pumped into the channel through its two inlets. The flow rate of the liquid streams was maintained at 200 μL min−1 which, for the channel used (width = 2 mm, height, hchannel = 215 μm), corresponds to Reynolds number, Re = 3, confirming laminar flow. In this regime, mixing between the two liquid streams was expected to occur solely via diffusion. In corroboration with that, Fig. 8a shows that the liquid streams were distinctly parallel to each other with a diffusion mixing zone, the width of which increases axially. Microscopy images (Fig. 8a) taken at different time intervals, tobs, confirm the qualitative observation that insignificant mixing occurs between the two streams. A similar experiment was done also for a channel with a curved tail and a spiral channel. While the extent of mixing was not characterized in these channels, the successful passage of two-colored liquids through the complex channel geometries confirms their functionality.
 |
| | Fig. 8 (a) Sequence of images demonstrates flow experiments that could be carried out in different channels. In particular two differently colored aqueous media were pumped into two different inlets of the channel. The flow being laminar, the liquid streams were found to remain parallel to each other. (b–d) In a different set of experiments, buffer solutions of different pH were incubated in PGS wells for different times and the UV-vis spectra of these liquid samples collected at time intervals: 2, 4, and 24 hours were obtained. | |
Leaching of solute from the channel wall
We examined also the possibility of any contamination or leaching from the channel wall because of prolonged exposure to the liquid inside. For this purpose, we generated square shaped wells of 20 mm side and 2 mm height in PGS layers by etching with 1.0 M aqNaOH; we then incubated tris buffer solutions (pH 7.4 and 8.5) and citrate buffer solution (pH 3.5) inside these wells for different time intervals. The liquid was collected at regular time intervals and was subjected to UV-vis spectroscopy. Across the whole range of pH, the UV-vis spectra showed negligible change in absorbance from 2 to 4 hours of incubation, indicating minimal short-term leaching from the etched surface (Fig. 8b–d). However, a distinct increase in absorbance was observed after 24 hours of incubation, particularly under basic conditions: pH 8.5 (Fig. 8d). These results suggest that hydrolytic degradation of the ester linkage was more pronounced under alkaline conditions, leading to the release of soluble oligomers or degradation products into the buffer. In basic environments, e.g. pH 8.5, hydroxide ions can directly attack the electrophilic carbonyl carbon of the ester group, resulting in faster bond cleavage and release of degradation fragments. This process leads to surface erosion and leaching, which is reflected in the spectra of buffer solutions stored in PGS wells, where changes in the signal were observed at pH 8.5 after 24 hours. In contrast, under acidic conditions, e.g. pH 5.5, protonation of the carbonyl oxygen enhances the electrophilicity of the ester group, making it more susceptible to nucleophilic attack by water. However, this acid-catalyzed hydrolysis proceeds slowly, and such conditions showed comparatively weaker changes in the spectrum (Fig. 8b). Under physiological conditions (pH 7.4), the hydrolysis is minimally catalyzed, resulting in negligible degradation over short timeframes (Fig. 8c).74,75 The comparison of UV-vis spectra of buffer incubated in etched PGS wells and pristine PGS films revealed no significant differences, indicating that the etching does not enhance leaching from the material (Fig. S5). These findings suggest that the etched channels are expected to remain benign to aqueous media over neutral and acidic pH and also solvents but are susceptible to hydrolysis under alkaline conditions.
Micro-particle generation
As another demonstration, a T-shaped PGS channel (Fig. 9a) was employed to generate micro-particles. Aqueous Na-alginate solution (3% w/v), with a small amount of green food coloring for visualization, was introduced through the central inlet of the microchannel, while sunflower oil was supplied through the two side inlets of the channel. In the first set of experiments, the flow rate of aqueous Na-alginate solution, Qd, was kept constant at 33 μL min−1 while the flow rate of oil, Qc, was varied from 50 to 200 μL min−1. In the second set, Qc was maintained at 150 μL min−1 while the Qd was varied from 33 to 67 μL min−1. At the junction, the oil streams pinch the central alginate stream to form droplets (Fig. 9b) as the dispersed phase. The dispersion thus formed is directed through the outlet channel into a 20% w/v aqueous CaCl2 solution kept inside a Petri-dish. Upon contact, the droplets gel into Ca-alginate micro-particles, which settled at the bottom of the Petri-dish (optical micrograph as in Fig. 9c). The droplet size, ddroplet, from both sets of experiments were analyzed to yield the relevant scaling relations. In particular, ddroplet was found to scale as ∼Qd/Q1/2c (Fig. 9d). Qualitatively, the larger flow rate of the dispersed phase, Qd, led to a larger droplet size, whereas the enhanced shearing effect at higher velocity of the continuous phase, Qc, produced smaller droplets. These results match well with those obtained for PDMS based microfluidic channels76,77 commonly employed for generating emulsions and droplets, which are then used in applications such as particle and cell encapsulation.78,79
 |
| | Fig. 9 The figure shows the generation of micro-particles in the PGS microchannel. (a) The top view of the channel used for generation of micro-particles. (b) Na-alginate solution was introduced through the central inlet, while sunflower oil entered from the side inlets, forming discrete water-in-oil droplets at the junction. (c) The droplets were collected into an aqueous CaCl2 solution, where they gelled into Ca-alginate micro-particles that settled at the bottom; their optical micrographs are shown here. (d) The data from different sets of experiments were pulled together to show that ddroplet scales as ∼Qd/Q1/2c. | |
Hemolysis assay
After evaluating the compatibility of the channels with aqueous solutions of varying pH, hemocompatibility studies were performed to assess their behavior in a biologically relevant, non-aqueous environment such as blood. The hemocompatibility of pristine and etched PGS was evaluated according to the protocol reported in the literature.80 A fresh blood sample from a rat (without sacrificing it) in a sterile environment from the animal house at the Indian Institute of Technology Kanpur was obtained via retro orbital vein puncture and collected in a tube coated with anticoagulant ethylenediaminetetraacetic acid (EDTA) to prevent clotting. Red blood cells (RBCs) were isolated from the blood by centrifugation at 2000 rpm for 10 minutes and washed thrice with phosphate-buffered saline (PBS). Subsequently, 0.9 mL of the diluted RBC suspension was treated with 300 μL of Triton X-100, PBS, and pristine PGS and etched PGS. After 4 hours of incubation at room temperature, the samples were subjected to centrifugation at 2000 rpm for 10 minutes and the absorbance of the supernatant was measured at 540 nm using a UV Vis spectrophotometer. The quantitative hemolysis values, calculated using eqn (2), for both pristine PGS and etched PGS were less than 2%, indicating good hemocompatibility of the materials (Fig. 10A). The pristine PGS and etched PGS-treated samples appeared transparent, similar to the negative control: PBS; positive control: Triton X-100 turned red because of RBC lysis (Fig. 10B). These findings confirm that etching doesn't alter the hemocompatibility of the PGS.| |  | (2) |
Here, Asamples is the absorbance of the pristine PGS and etched PGS; Ap and An represent the absorbance of Triton X-100 and PBS respectively.
 |
| | Fig. 10 The figure shows the blood compatibility of pristine and etched PGS evaluated through hemolysis assay. (A) Hemolysis percentage for both the pristine and etched PGS was below 2%, indicating good hemocompatibility. (B) Representative images of precipitated RBCs treated with the positive control, negative control, pristine PGS and etched PGS. | |
Protein adsorption test
To evaluate the interaction of pristine and etched PGS surfaces with a typical protein, bovine serum albumin (BSA) was used as a model protein. Solutions of BSA in PBS (pH 7.4) at concentrations of 1.0 and 1.5 mg mL−1 were prepared, and polymer films (1 cm × 1 cm × 0.1 cm) of pristine PGS, etched PGS and PDMS (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 base-to-crosslinker ratio, cured at 80 °C for 3 hours) were incubated in these solutions for 2 to 4 hours. The optical density of the supernatant was measured using a UV-vis spectrophotometer and the corresponding protein concentrations were determined from a pre-established calibration curve. No measurable change in OD was observed after 2 hours of incubation; however, after 4 hours, OD was found to decrease to a limited extent. In Fig. 11a, we have presented the UV-vis spectra of the BSA solution before incubation, and after incubation with the pristine and etched PGS for 4 hours. The decrease in OD was observed to be somewhat larger for the solution incubated with the etched PGS than the one with pristine PGS. Unlike PGS samples, the OD of the 1.0 mg mL−1 BSA solution showed no change even after 4 hours of incubation with PDMS, whereas the 1.5 mg mL−1 BSA solution exhibited a decrease in OD under the same conditions. The adsorbed amount was calculated from the difference between the initial and final concentrations and is presented in Fig. 11b. Protein adsorption on the etched surface was consistently higher than on the pristine surface, which can be attributed to the increased surface roughness resulting from the etching process. Compared to PDMS (28 μg cm−2), the PGS exhibits substantially higher BSA adsorption, 110 and 197 μg cm−2 for pristine PGS and etched PGS respectively, which can be attributed to its higher hydrophobicity. Such higher protein adsorption can be advantageous in applications like tissue engineering or bio interface applications, as protein layers facilitate subsequent cell attachment and growth.81 To further confirm BSA adsorption on the substrate, contact angle and AFM analyses were performed after protein adsorption. The PGS surfaces exhibited lower contact angles, θa, (Fig. 11c) after BSA adsorption, with values of 62.5 ± 1.73° for pristine PGS and 56.5 ± 3.67° for etched PGS, indicating enhanced surface hydrophilicity due to the presence of protein molecules. In comparison, PDMS exhibited a relatively smaller change in θa (79.8 ± 6.24°) under identical conditions, as shown in Fig. 11c, suggesting limited surface modification. AFM images (Fig. 11d) also showed a noticeable change in surface texture and an increase in root mean square roughness, Rq = 1.23 nm (pristine) to 5.8 nm (after BSA adsorption) for PGS surfaces. ATR-FTIR spectra of the BSA-adsorbed PGS surfaces are presented in Fig. 11e which shows two additional peaks around 1260 cm−1 and 1630 cm−1, which can be attributed to the amide-III and amide-I vibrations of the adsorbed protein, respectively.82 These results collectively support successful BSA adsorption and further confirm the difference in surface behavior between PGS and PDMS.
:1 base-to-crosslinker ratio, cured at 80 °C for 3 hours) were incubated in these solutions for 2 to 4 hours. The optical density of the supernatant was measured using a UV-vis spectrophotometer and the corresponding protein concentrations were determined from a pre-established calibration curve. No measurable change in OD was observed after 2 hours of incubation; however, after 4 hours, OD was found to decrease to a limited extent. In Fig. 11a, we have presented the UV-vis spectra of the BSA solution before incubation, and after incubation with the pristine and etched PGS for 4 hours. The decrease in OD was observed to be somewhat larger for the solution incubated with the etched PGS than the one with pristine PGS. Unlike PGS samples, the OD of the 1.0 mg mL−1 BSA solution showed no change even after 4 hours of incubation with PDMS, whereas the 1.5 mg mL−1 BSA solution exhibited a decrease in OD under the same conditions. The adsorbed amount was calculated from the difference between the initial and final concentrations and is presented in Fig. 11b. Protein adsorption on the etched surface was consistently higher than on the pristine surface, which can be attributed to the increased surface roughness resulting from the etching process. Compared to PDMS (28 μg cm−2), the PGS exhibits substantially higher BSA adsorption, 110 and 197 μg cm−2 for pristine PGS and etched PGS respectively, which can be attributed to its higher hydrophobicity. Such higher protein adsorption can be advantageous in applications like tissue engineering or bio interface applications, as protein layers facilitate subsequent cell attachment and growth.81 To further confirm BSA adsorption on the substrate, contact angle and AFM analyses were performed after protein adsorption. The PGS surfaces exhibited lower contact angles, θa, (Fig. 11c) after BSA adsorption, with values of 62.5 ± 1.73° for pristine PGS and 56.5 ± 3.67° for etched PGS, indicating enhanced surface hydrophilicity due to the presence of protein molecules. In comparison, PDMS exhibited a relatively smaller change in θa (79.8 ± 6.24°) under identical conditions, as shown in Fig. 11c, suggesting limited surface modification. AFM images (Fig. 11d) also showed a noticeable change in surface texture and an increase in root mean square roughness, Rq = 1.23 nm (pristine) to 5.8 nm (after BSA adsorption) for PGS surfaces. ATR-FTIR spectra of the BSA-adsorbed PGS surfaces are presented in Fig. 11e which shows two additional peaks around 1260 cm−1 and 1630 cm−1, which can be attributed to the amide-III and amide-I vibrations of the adsorbed protein, respectively.82 These results collectively support successful BSA adsorption and further confirm the difference in surface behavior between PGS and PDMS.
 |
| | Fig. 11 The figure shows the protein adsorption results on pristine and etched PGS surfaces in comparison with PDMS, where BSA was used as a model protein. (a) The UV-vis spectra of the BSA solution before incubation, and after incubation with the pristine PGS and etched PGS for 4 hours. The absorbance peak at 280 nm, characteristic of aromatic amino acids such as tryptophan and tyrosine in proteins, was used to monitor the residual BSA concentration in solution. A more pronounced decrease in OD (absorbance) at 280 nm is observed for the etched PGS compared to the pristine PGS, where the change is relatively small, indicating higher protein uptake by the etched surface. (b) The bar chart shows the amount of protein adsorbed on pristine PGS, etched PGS and PDMS surfaces at initial BSA concentrations of 1.0 mg mL−1 and 1.5 mg mL−1, with an incubation time of 4 hours in both cases. Protein adsorption is consistently higher for the etched surface, possibly due to the increased surface roughness enhancing the available area for protein binding. (c and d) Contact angle and AFM analyses after BSA adsorption show reduced hydrophobicity and an increase in surface roughness, respectively, confirming protein adsorption on PGS as well as PDMS surfaces. (e) ATR-FTIR spectra of BSA-adsorbed PGS surfaces show new peaks at 1260 cm−1 and 1630 cm−1 corresponding to amide vibrations of BSA. | |
Potential application: membrane-based mass transfer in microchannels
To demonstrate the versatility of the fabricated PGS-based microchannels, a nylon membrane (0.45 μm pore size) was inserted between two parallel channels to form a dual-channel configuration (Fig. 12a). The two microchannels were bonded using PGS prepolymer and crosslinked at 120 °C to ensure sealing and structural integrity. An aqueous solution of methylene blue (MB) was introduced through the lower channel while deionized water was simultaneously flowed through the upper channel. The flow rate of both streams was maintained at 30 μL min−1. The outlet from the upper channel was analyzed using UV-vis spectroscopy to evaluate dye transport across the membrane. Aqueous solutions of MB of two different concentrations: 0.5 μM and 0.7 μM were used for which the dye concentration in the aqueous phase of the upper channel was 0.118 μM and 0.125 μM respectively. While detailed experiments are to be performed to estimate the dye permeability of the membrane, the results presented here highlight the potential applications of the PGS microchannel system involving controlled mass-transport, relevant for diffusion-based drug transport, dialysis modeling, or membrane characterization studies, complementing the material's known biocompatibility and biodegradability.
 |
| | Fig. 12 The schematic shows the dual-channel microfluidic configuration with an interposed nylon membrane for studying solute transport. (a) A Y-shaped microchannel is fabricated in a PGS block. A nylon membrane with 0.45 μm pores is placed over this channel, and a second PGS block containing a Y-shaped microchannel is bonded on top to complete the dual-channel configuration. (b) The actual image shows the assembled system, with the membrane sandwiched between the two PGS blocks. | |
3. Conclusion
In this report, we have demonstrated that PGS is a viable alternative to PDMS for making microfluidic devices. A detailed characterization of PGS was conducted, and its properties were compared with those of PDMS reported in the literature. The key outcomes of this comparison are summarized in Table 1. We established that microfluidic channels of various planar shapes can be etched into PGS using aqNaOH as an etchant. Flow tests confirmed the functionality of these channels. The UV-vis analysis of buffer solutions after incubation in etched PGS demonstrated excellent compatibility of PGS with physiological pH (7.4), showing minimal absorbance and suggesting negligible leaching or contamination. Higher absorbance observed at pH 8.5 indicates slight interaction due to the known degradation of PGS under basic conditions, which leads to material leaching at non-physiological pH levels. However, the leaching is solely attributed to the inherent degradation of PGS under basic conditions and is not influenced by etching. The successful demonstration of controlled dye transport across a membrane interface establishes the fabricated PGS microchannels as promising platforms for diffusion-based applications such as drug delivery, dialysis modeling, and bioanalytical studies. These findings highlight the versatility of PGS in microfluidic applications and provide a foundation for further exploration of its potential in lab-on-chip and biomedical devices.
Table 1 Summary of key property comparisons
| Property |
PGS |
PDMS |
| Young's modulus (MPa) |
0.13–1.4 MPa |
0.3–3.0 MPa |
| Surface energy (mJ m−2) |
25–28 mJ m−2 |
21 mJ m−2 |
| Swellability with polar liquids |
Negligible swelling in water |
Negligible swelling in water |
| Swellability with non-polar liquids |
Negligible swelling in hexadecane and hexane |
High swelling in hexane; also swells in hexadecane |
| Moldability/fabrication |
Moldability is moderate, less versatile compared to PDMS |
Excellent moldability, easy demolding |
| Biodegradability |
Biodegradable |
Non-biodegradable |
| Hemocompatibility |
Hemocompatible |
Low hemocompatibility |
| BSA adsorption |
110 μg cm−2 |
28 μg cm−2 |
Author contributions
Animangsu Ghatak conceived the idea, supervised and administered the project and wrote the manuscript; Yasmeena Ashraf curated and analyzed all data and wrote the original draft of the manuscript.
Conflicts of interest
There are no conflicts of interest to declare.
Data availability
All data of this study are included in the article and in the supplementary information (SI) in the form of figures, spectra, plots, and tables.
Supplementary information is available. See DOI: https://doi.org/10.1039/d5lc00851d.
Acknowledgements
This work was supported by Department of Science and Technology, Government of India in the form of grant: SR/FST/ETII-055/2013, Department of Biotechnology, Government of India in the form of grant: IC-12015(12)/3/2022-ICD-DBT and Department of Chemicals and Petrochemicals grant: PC-II-25014/3/2022-PC II-CPC (FTS: 3019123). YA and AG thank Dr. Ashiq Hussain Pandit for carrying out the hemolysis test.
References
- S. K. Tiwari, S. Bhat and K. K. Mahato, Design and fabrication of low-cost microfluidic channel for biomedical application, Sci. Rep., 2020, 10(1), 9215 CrossRef CAS PubMed.
- S. N. Bhatia and D. E. Ingber, Microfluidic organs-on-chips, Nat. Biotechnol., 2014, 32(8), 760–772 CrossRef CAS.
- D. C. Duffy, J. C. McDonald, O. J. Schueller and G. M. Whitesides, Rapid prototyping of microfluidic systems in poly (dimethylsiloxane), Anal. Chem., 1998, 70(23), 4974–4984 CrossRef CAS.
- P. F. Ng, K. I. Lee, M. Yang and B. Fei, Fabrication of 3D PDMS microchannels of adjustable cross-sections via versatile gel templates, Polymers, 2019, 11(1), 64 CrossRef PubMed.
- M. K. Verma, A. Majumder and A. Ghatak, Embedded template-assisted fabrication of complex microchannels in PDMS and design of a microfluidic adhesive, Langmuir, 2006, 22(24), 10291–10295 CrossRef CAS PubMed.
- A. Majumder, A. Ghatak and A. Sharma, Microfluidic adhesion induced by subsurface microstructures, Science, 2007, 318(5848), 258–261 CrossRef CAS PubMed.
- M. T. Guler, Fabricating plasma bonded microfluidic chips by CO2 laser machining of PDMS by the application of viscoelastic particle focusing and droplet generation, J. Manuf. Process., 2022, 73, 260–268 CrossRef.
- S. Y. Kim, H. B. Son and H. R. Lim, One-Step Fabrication of Microfluidic Channels in Polydimethylsiloxane:Influence of Laser Power on Channel Formation, Micromachines, 2025, 16(3), 282 CrossRef.
- W. T. Kahsai, U. H. Pham, J. S. Sankaran and S. M. Iqbal, Self-assembled synthesis and characterization of microchannels in polymeric membranes, J. Appl. Phys., 2012, 112(2), 024701 CrossRef.
- L. P. Gómez, P. Bollgruen, A. I. Egunov, D. Mager, F. Malloggi, J. G. Korvink and V. A. Luchnikov, Vapour processed self-rolled poly (dimethylsiloxane) microcapillaries form microfluidic devices with engineered inner surface, Lab Chip, 2013, 13(19), 3827–3831 RSC.
- A. A. Bhagat, S. S. Kuntaegowdanahalli and I. Papautsky, Continuous particle separation in spiral microchannels using dean flows and differential migration, Lab Chip, 2008, 8(11), 1906–1914 RSC.
- G. Guan, L. Wu, A. A. Bhagat, Z. Li, P. C. Chen, S. Chao, C. J. Ong and J. Han, Spiral microchannel with rectangular and trapezoidal cross-sections for size based particle separation, Sci. Rep., 2013, 3(1), 1475 CrossRef.
- M. K. Verma, S. R. Ganneboyina, V. Rakshith and A. Ghatak, Three-dimensional multihelical microfluidic mixers for rapid mixing of liquids, Langmuir, 2008, 24(5), 2248–2251 CrossRef CAS.
- T. T. Huang, D. G. Taylor, K. S. Lim, M. Sedlak, R. Bashir, N. S. Mosier and M. R. Ladisch, Surface-directed boundary flow in microfluidic channels, Langmuir, 2006, 22(14), 6429–6437 CrossRef CAS PubMed.
- T. Dahlberg, T. Stangner, H. Zhang, K. Wiklund, P. Lundberg, L. Edman and M. Andersson, 3D printed water-soluble scaffolds for rapid production of PDMS micro-fluidic flow chambers, Sci. Rep., 2018, 8, 1–10 CAS.
- A. D. Stroock, S. K. Dertinger, A. Ajdari, I. Mezic, H. A. Stone and G. M. Whitesides, Chaotic mixer for microchannels, Science, 2002, 295(5555), 647–651 CrossRef CAS.
- S. Sofela, S. Sahloul, C. Stubbs, A. Orozaliev, F. S. Refai, A. M. Esmaeel, H. Fahs, M. O. Abdelgawad, K. C. Gunsalus and Y. A. Song, Phenotyping of the thrashing forces exerted by partially immobilized C. elegans using elastomeric micropillar arrays, Lab Chip, 2019, 19(21), 3685–3696 RSC.
- J. Xiao, W. He, Z. Zhang, W. Zhang, Y. Cao, R. He and Y. Chen, PDMS micropillar-based microchip for efficient cancer cell capture, RSC Adv., 2015, 5(64), 52161–52166 RSC.
- H. Li, J. J. Whittenberg, H. Zhou, D. Ranganathan, A. V. Desai, J. Koziol, D. Zeng, P. J. Kenis and D. E. Reichert, Development of a microfluidic “click chip” incorporating an immobilized Cu (I) catalyst, RSC Adv., 2015, 5(8), 6142–6150 RSC.
- B. Huang, H. Wu, S. Kim, B. K. Kobilka and R. N. Zare, Phospholipid biotinylation of polydimethylsiloxane (PDMS) for protein immobilization, Lab Chip, 2006, 6(3), 369–373 RSC.
- C. Priest, P. J. Gruner, E. J. Szili, S. A. Al-Bataineh, J. W. Bradley, J. Ralston, D. A. Steele and R. D. Short, Microplasma patterning of bonded microchannels using high-precision “injected” electrodes, Lab Chip, 2011, 11(3), 541–544 RSC.
- M. Cernik, K. Poláková, L. Kubala, A. Vítečková Wünschová, A. Mac Gillavry Danylevska, M. Pešková and J. Vitecek, Luminal surface plasma treatment of closed cylindrical microchannels: A tool toward the creation of on-chip vascular endothelium, ACS Biomater. Sci. Eng., 2023, 9(5), 2755–2763 CrossRef CAS PubMed.
- A. I. Gomez-Varela, A. Viña, C. Bao-Varela, M. T. Flores-Arias, B. Carnero, M. Gonzalez-Peteiro, J. R. Gonzalez-Juanatey and E. Álvarez, Biocompatibility Testing of UV-Curable Polydimethylsiloxane for Human Umbilical Vein Endothelial Cell Culture on-a-Chip, ACS Omega, 2024, 9(28), 30281–30293 CrossRef CAS PubMed.
- H. J. Kim, D. Huh, G. Hamilton and D. E. Ingber, Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow, Lab Chip, 2012, 12(12), 2165–2174 RSC.
- M. J. Moorcroft, W. R. Meuleman, S. G. Latham, T. J. Nicholls, R. D. Egeland and E. M. Southern, In situ oligonucleotide synthesis on poly (dimethylsiloxane): a flexible substrate for microarray fabrication, Nucleic Acids Res., 2005, 33(8), e75 CrossRef PubMed.
- H. Li, M. Zhao, W. Liu, W. Chu and Y. Guo, Polydimethylsiloxane microfluidic chemiluminescence immunodevice with the signal amplification strategy for sensitive detection of human immunoglobin G, Talanta, 2016, 147, 430–436 CrossRef CAS PubMed.
- Y. J. Chuah, S. Kuddannaya, M. H. Lee, Y. Zhang and Y. Kang, The effects of poly (dimethylsiloxane) surface silanization on the mesenchymal stem cell fate, Biomater. Sci., 2015, 3(2), 383–390 RSC.
- M. Ebara, J. M. Hoffman, A. S. Hoffman and P. S. Stayton, Switchable surface traps for injectable beadbased chromatography in PDMS microfluidic channels, Lab Chip, 2006, 6(7), 843–848 RSC.
- A. L. Cordeiro, S. Zschoche, A. Janke, M. Nitschke and C. Werner, Functionalization of poly(dimethylsiloxane) surfaces with maleic anhydride copolymer films, Langmuir, 2009, 25(3), 1509–1517 CrossRef CAS.
- G. T. Roman, T. Hlaus, K. J. Bass, T. G. Seelhammer and C. T. Culbertson, Sol–gel modified poly(dimethylsiloxane) microfluidic devices with high electroosmotic mobilities and hydrophilic channel wall characteristics, Anal. Chem., 2005, 77(5), 1414–1422 CrossRef CAS.
- C. W. Li, Y. Zhu, J. Zhan, J. Ma, L. Gu, Y. Fang and C. Yi, Separation of polystyrene nanoparticles in polydimethylsiloxane microfluidic devices with a combined titania and sodium dodecyl sulfate inner coating, Microchim. Acta, 2017, 184(7), 2227–2239 CrossRef CAS.
- Y. Wang, G. A. Ameer, B. J. Sheppard and R. Langer, A tough biodegradable elastomer, Nat. Biotechnol., 2002, 20(6), 602–606 CrossRef CAS.
- J. L. Ifkovits, R. F. Padera and J. A. Burdick, Biodegradable and radically polymerized elastomers with enhanced processing capabilities, Biomed. Mater., 2008, 3(3), 034104 CrossRef.
- H. K. Makadia and S. J. Siegel, Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier, Polymers, 2011, 3(3), 1377–1397 CrossRef CAS PubMed.
- J. W. Nichol, S. T. Koshy, H. Bae, C. M. Hwang, S. Yamanlar and A. Khademhosseini, Cell-laden microengineered gelatin methacrylate hydrogels, Biomaterials, 2010, 31(21), 5536–5544 CrossRef CAS PubMed.
- M. A. Woodruff and D. W. Hutmacher, The return of a forgotten polymer—Polycaprolactone in the 21st century, Prog. Polym. Sci., 2010, 35(10), 1217–1256 CrossRef CAS.
- S. J. Bryant and K. S. Anseth, Hydrogel properties influence ECM production by chondrocytes photoencapsulated in poly (ethylene glycol) hydrogels, J. Biomed. Mater. Res., 2002, 59(1), 63–72 CrossRef CAS PubMed.
- J. M. Kemppainen and S. J. Hollister, Tailoring the mechanical properties of 3D-designed poly (glycerol sebacate) scaffolds for cartilage applications, J. Biomed. Mater. Res., Part A, 2010, 94(1), 9–18 CrossRef PubMed.
- S. L. Liang, W. D. Cook, G. A. Thouas and Q. Z. Chen, The mechanical characteristics and in vitro biocompatibility of poly (glycerol sebacate)-Bioglass® elastomeric composites, Biomaterials, 2010, 31(33), 8516–8529 CrossRef CAS PubMed.
- Q. Z. Chen, S. L. Liang, J. Wang and G. P. Simon, Manipulation of mechanical compliance of elastomeric PGS by incorporation of halloysite nanotubes for soft tissue engineering applications, J. Mech. Behav. Biomed. Mater., 2011, 4(8), 1805–1818 CrossRef CAS PubMed.
- S. Li, J. Zhang, J. He, W. Liu, Y. Wang, Z. Huang, H. Pang and Y. Chen, Functional PDMS elastomers: bulk composites, surface engineering, and precision fabrication, Adv. Sci., 2023, 10(34), 2304506 CrossRef CAS PubMed.
- R. Rai, M. Tallawi, A. Grigore and A. R. Boccaccini, Synthesis, properties and biomedical applications of poly(glycerol sebacate)(PGS): A review, Prog. Polym. Sci., 2012, 37(8), 1051–1078 CrossRef CAS.
- X. Zhao, Y. Wu, Y. Du, X. Chen, B. Lei, Y. Xue and P. X. Ma, A highly bioactive and biodegradable poly (glycerol sebacate)–silica glass hybrid elastomer with tailored mechanical properties for bone tissue regeneration, J. Mater. Chem. B, 2015, 3(16), 3222–3233 RSC.
- F. Yi and D. A. LaVan, Poly (glycerol sebacate) nanofiber scaffolds by core/shell electrospinning, Macromol. Biosci., 2008, 8(9), 803–806 CrossRef CAS PubMed.
- K. Mollazadeh-Moghaddam, H. Rezaei Nejad, A. Z. Chen, J. Ju, A. Tamayol, X. Liu, Y. S. Zhang, R. Oklu and A. Khademhosseini, Fracture-Resistant and Bioresorbable Drug-Eluting Poly (glycerol Sebacate) Coils, Adv. Ther., 2019, 2(3), 1800109 CrossRef.
- I. C. Becerril-Rodriguez and F. Claeyssens, Low methacrylated poly (glycerol sebacate) for soft tissue engineering, Polym. Chem., 2022, 13(23), 3513–3528 RSC.
- B. Louage, L. Tack, Y. Wang and B. G. De Geest, Poly (glycerol sebacate) nanoparticles for encapsulation of hydrophobic anti-cancer drugs, Polym. Chem., 2017, 8(34), 5033–5038 RSC.
- Z. Wang, M. Zhang, L. Liu, S. M. Mithieux and A. S. Weiss, Polyglycerol sebacate-based elastomeric materials for arterial regeneration, J. Biomed. Mater. Res., Part A, 2024, 112(4), 574–585 CrossRef CAS PubMed.
- N. Naik, D. Hanjaya-Putra, C. A. Haller, M. G. Allen and E. L. Chaikof, Rapid homogeneous endothelialization of high aspect ratio microvascular
networks, Biomed. Microdevices, 2015, 17(4), 83 CrossRef PubMed.
- A. Tevlek, B. Topuz, E. Akbay and H. M. Aydin, Surface channel patterned and endothelialized poly (glycerol sebacate) based elastomers, J. Biomater. Appl., 2022, 37(2), 287–302 CrossRef CAS PubMed.
- B. Godinho, R. Nogueira, N. Gama and A. Ferreira, Synthesis and Characterization of Poly (glycerol sebacate), Poly (glycerol succinate) and Poly (glycerol sebacate-co-succinate), J. Polym. Environ., 2024, 32(9), 4330–4347 CrossRef CAS.
- M. Gultekinoglu, Ş. Öztürk, B. Chen, M. Edirisinghe and K. Ulubayram, Preparation of poly (glycerol sebacate) fibers for tissue engineering applications, Eur. Polym. J., 2019, 121, 109297 CrossRef CAS.
- R. Takahashi, H. Miyazako, A. Tanaka, Y. Ueno and M. Yamaguchi, Tough, permeable and biocompatible microfluidic devices formed through the buckling delamination of soft hydrogel films, Lab Chip, 2021, 21(7), 1307–1317 RSC.
- Y. Wang, L. Shang, Y. Zhao and L. Sun, Microfluidic generation of multicomponent soft biomaterials, Engineering, 2022, 13, 128–143 CrossRef CAS.
- P. N. Nge, C. I. Rogers and A. T. Woolley, Advances in microfluidic materials, functions, integration, and applications, Chem. Rev., 2013, 113(4), 2550–2583 CrossRef CAS.
- X. Ding, Y. Chen, C. A. Chao, Y. L. Wu and Y. Wang, Control the mechanical properties and degradation of poly (glycerol sebacate) by substitution of the hydroxyl groups with palmitates, Macromol. Biosci., 2020, 20(9), 2000101 CrossRef CAS PubMed.
- B. Godinho, R. Nogueira, N. Gama and A. Ferreira, Synthesis and Characterization of Poly (glycerol sebacate), Poly (glycerol succinate) and Poly (glycerol sebacate-co-succinate), J. Polym. Environ., 2024, 32(9), 4330–4347 CrossRef CAS.
- A. K. Gaharwar, A. Patel, A. Dolatshahi-Pirouz, H. Zhang, K. Rangarajan, G. Iviglia, S. R. Shin, M. A. Hussain and A. Khademhosseini, Elastomeric nanocomposite scaffolds made from poly (glycerol sebacate) chemically crosslinked with carbon nanotubes, Biomater. Sci., 2015, 3(1), 46–58 RSC.
- Á. Conejero-García, H. R. Gimeno, Y. M. Sáez, G. Vilariño-Feltrer, I. Ortuño-Lizarán and A. Vallés-Lluch, Correlating synthesis parameters with physicochemical properties of poly (glycerol sebacate), Eur. Polym. J., 2017, 87, 406–419 CrossRef.
- R. Martín-Cabezuelo, G. Vilariño-Feltrer and A. Vallés-Lluch, Influence of pre-polymerisation atmosphere on the properties of pre-and poly (glycerol sebacate), Mater. Sci. Eng., C, 2021, 119, 111429 CrossRef.
- D. K. Owens and R. C. Wendt, Estimation of the surface free energy of polymers, J. Appl. Polym. Sci., 1969, 13(8), 1741–1747 CrossRef CAS.
-
L. G. Treloar, The physics of rubber elasticity, 1975 Search PubMed.
- L. H. Tanner, The spreading of silicone oil drops on horizontal surfaces, J. Phys. D: Appl. Phys., 1979, 12(9), 1473 CrossRef CAS.
- J. A. Kleingartner, H. Lee, M. F. Rubner, G. H. McKinley and R. E. Cohen, Exploring the kinetics of switchable polymer surfaces with dynamic tensiometry, Soft Matter, 2013, 9(26), 6080–6090 RSC.
- M. Inutsuka, H. Tanoue, N. L. Yamada, K. Ito and H. Yokoyama, Dynamic contact angle on a reconstructive polymer surface by segregation, RSC Adv., 2017, 7(28), 17202–17207 RSC.
- J. A. Crowe and J. Genzer, Creating responsive surfaces with tailored wettability switching kinetics and reconstruction reversibility, J. Am. Chem. Soc., 2005, 127(50), 17610–17611 CrossRef CAS.
- H. Lee, M. L. Alcaraz, M. F. Rubner and R. E. Cohen, Zwitter-wettability and antifogging coatings with frost-resisting capabilities, ACS Nano, 2013, 7(3), 2172–2185 CrossRef CAS.
- S. Yoon and B. Chen, Elastomeric and pH-responsive hydrogels based on direct crosslinking of the poly (glycerol sebacate) pre-polymer and gelatin, Polym. Chem., 2018, 9(27), 3727–3740 RSC.
- I. Pomerantseva, N. Krebs, A. Hart, C. M. Neville, A. Y. Huang and C. A. Sundback, Degradation behavior of poly (glycerol sebacate), J. Biomed. Mater. Res., Part A, 2009, 91(4), 1038–1047 CrossRef PubMed.
- Y. Wang, Y. M. Kim and R. Langer, In vivo degradation characteristics of poly (glycerol sebacate), J. Biomed. Mater. Res., Part A, 2003, 66(1), 192–197 CrossRef PubMed.
- I. Čorak, A. Tarbuk, D. Đorđević, K. Višić and L. Botteri, Sustainable alkaline hydrolysis of polyester fabric at low temperature, Materials, 2022, 15(4), 1530 CrossRef PubMed.
- D. G. Barrett, B. M. Lamb and M. N. Yousaf, Microfluidic etching and oxime-based tailoring of biodegradable polyketoesters, Langmuir, 2008, 24(17), 9861–9867 CrossRef CAS.
- X. Yang, O. Forouzan, J. M. Burns and S. S. Shevkoplyas, Traffic of leukocytes in microfluidic channels with rectangular and rounded cross-sections, Lab Chip, 2011, 11(19), 3231–3240 RSC.
- L. N. Woodard and M. A. Grunlan, Hydrolytic degradation and erosion of polyester biomaterials, ACS Macro Lett., 2018, 7, 976–982 CrossRef CAS.
- W. K. Lee and J. A. Gardella, Hydrolytic kinetics of biodegradable polyester monolayers, Langmuir, 2000, 16(7), 3401–3406 CrossRef CAS.
- V. Steijn, C. R. Kleijn and M. T. Kreutzer, Predictive model for the size of bubbles and droplets created in microfluidic T-junctions, Lab Chip, 2010, 10, 2513–2518 RSC.
- S. L. Anna and H. C. Mayer, Microscale tip streaming in a microfluidic flow focusing device, Phys. Fluids, 2006, 18, 121512 CrossRef.
- K. S. Huang, T. H. Lai and Y. C. Lin, Manipulating the generation of Ca-alginate microspheres using microfluidic channels as a carrier of gold nanoparticles, Lab Chip, 2006, 6(7), 954–957 RSC.
- M. D. Eqbal and V. Gundabala, Controlled fabrication of multi-core alginate microcapsules, J. Colloid Interface Sci., 2017, 507, 27–34 CrossRef CAS PubMed.
- X. Han, C. Saengow, L. Ju, W. Ren, R. H. Ewoldt and J. Irudayaraj, Exosome-coated oxygen nanobubble-laden hydrogel augments intracellular delivery of exosomes for enhanced wound healing, Nat. Commun., 2024, 15(1), 3435 CrossRef CAS PubMed.
- J. L. Ifkovits, J. J. Devlin, G. Eng, T. P. Martens, G. Vunjak-Novakovic and J. A. Burdick, Biodegradable fibrous scaffolds with tunable properties formed from photo-cross-linkable poly (glycerol sebacate), ACS Appl. Mater. Interfaces, 2009, 1(9), 1878–1886 CrossRef CAS.
-
H. Kaur, B. Rana, D. Tomar, S. Kaur and K. C. Jena, Fundamentals of ATR-FTIR spectroscopy and its role for probing in-situ molecular-level interactions, in Modern Techniques of Spectroscopy: Basics, Instrumentation, and Applications, Springer Singapore, Singapore, 2021, pp. 3–37 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab
*ab


 , with the slope defining the elastic constant, E, of the material. The E for different PGS samples was found to vary from 0.13 to 1.4 MPa respectively, a range in which silicone is also used as a microfluidic platform (Table S1). In order to obtain a quantitative relation between E and tcr, in Fig. 2d, Young's modulus E was plotted against tcr to show that E increases super-linearly with tcr: E ∼ t3/2cr. This observation can be rationalized by considering essentially the kinetics of the crosslinking process. It has been shown earlier15 that during the pre-polymerization stage, the primary hydroxyl groups (–OH) of glycerol react with carboxyl groups (–COOH) of sebacic acid leaving behind one secondary –OH group which later on, during the crosslinking process, reacts with a –COOH group to form the crosslinked network. So, crosslinking at the second stage is essentially driven by 1/3rd the number of moles of glycerol in the reacting solution. This consideration implies that the rate of crosslinking reaction varies as C1/3g, where Cg = ng/V is the molar concentration of glycerol per unit volume:
, with the slope defining the elastic constant, E, of the material. The E for different PGS samples was found to vary from 0.13 to 1.4 MPa respectively, a range in which silicone is also used as a microfluidic platform (Table S1). In order to obtain a quantitative relation between E and tcr, in Fig. 2d, Young's modulus E was plotted against tcr to show that E increases super-linearly with tcr: E ∼ t3/2cr. This observation can be rationalized by considering essentially the kinetics of the crosslinking process. It has been shown earlier15 that during the pre-polymerization stage, the primary hydroxyl groups (–OH) of glycerol react with carboxyl groups (–COOH) of sebacic acid leaving behind one secondary –OH group which later on, during the crosslinking process, reacts with a –COOH group to form the crosslinked network. So, crosslinking at the second stage is essentially driven by 1/3rd the number of moles of glycerol in the reacting solution. This consideration implies that the rate of crosslinking reaction varies as C1/3g, where Cg = ng/V is the molar concentration of glycerol per unit volume:











