DOI:
10.1039/D5GC04080A
(Paper)
Green Chem., 2026,
28, 295-308
A green and efficient strategy for leaching critical metals from spent LiNixCoyMnzO2 cathodes: modulating the dielectric SiO2 contact-electro-catalytic activity
Received
5th August 2025
, Accepted 10th November 2025
First published on 13th November 2025
Abstract
Owing to the global transition toward carbon neutrality, lithium-ion batteries (LIBs) are experiencing unprecedented demand growth as key energy storage components. A contact-electro-catalysis (CEC) method has been utilized in recycling spent LIBs for some time due to the process economy, efficiency and environmental sustainability. However, systematic investigations into the effects of the microstructure and surface chemical environment of dielectric materials on their catalytic activity in the CEC process remain scarce. Here, we demonstrated that the metal leaching efficiency simultaneously depended on the size and porous structure of SiO2 microspheres. Appropriate size and narrow distribution could dominantly diminish the scattering intensity of ultrasonic waves to gather energy and then facilitate the formation of cavitation bubbles to generate abundant electrons and radicals. Similarly, the porous structure also promoted the generation of electrons and radicals by enhancing aqueous-phase contact interfaces. Moreover, we manipulated the surface chemical environment of porous SiO2 microspheres by incorporating sodium alginate (SA) and polyethyleneimine (PEI). Hydroxyl, carboxyl and amino groups and –COO−/–NH3+-microstructures not only accelerated electron transfer from water molecules to SiO2 microspheres, inducing hydroxyl radicals, but also boosted electron transfer from SiO2 microspheres to O2, generating superoxide radicals. Consequently, under the experimental conditions established in this study, approximately 100% leaching efficiency of metals in the spent ternary cathode was achieved in 8 h at 80 °C due to the synergistic effect of hydroxyl and superoxide radicals. We anticipate this innovative strategy will significantly expand the applicability of CEC in LIB recycling, thereby addressing the growing global demand for sustainable LIBs.
Green foundation
1. Our study presents an innovative finding that the metal leaching efficiency in contact-electro-catalysis processes is simultaneously governed by the microstructure and surface chemical environment of SiO2 microspheres.
2. Our work developed a cost-effective green process for achieving perfect (approximately 100%) leaching efficiencies of Li, Co, Mn, and Ni in spent LiNixCoyMnzO2 cathodes by carefully optimizing the size, pore structure, and surface chemistry of SiO2 microspheres.
3. This research makes significant contributions to both fundamental understanding of catalytic mechanisms and technological advancements in applying contact-electro-catalysis for greener LIB recycling processes.
|
1. Introduction
Aligned with the 2015 Paris Agreement, numerous nations have intensified efforts to attain carbon neutrality.1 Substantial mitigation of carbon emission rates represents a critical imperative for achieving the goal. However, the extensive use of traditional fossil fuels in transportation modes, such as cars, ships and airplanes, leads to serious carbon emissions, which critically impedes the successful achievement of carbon neutrality goals.2,3 Against this pressing backdrop, accelerating the exploitation of new clean energies and the development of advanced energy storage devices has become urgent.4,5 Currently, lithium-ion batteries (LIBs) have emerged as transformative energy storage devices, facilitating the transition from fossil fuel dependence to renewable energy integration across global energy industries.6,7 Moreover, a green transportation revolution is initiated by many governments through utilizing electric vehicles (EVs) to replace fossil fuel vehicles.3,5,8,9 Consequently, the electric vehicle (EV) industry rapidly expands, leading to a continuously surging demand for rechargeable LIBs. As we know, the capacity and durability of many commercial LIBs primarily depend on the cathode materials. Several dominant cathode materials are commonly employed in commercial LIBs, such as ferrous lithium phosphate (LiFePO4, abbreviated as LFP), ternary materials (LiNixCoyMnzO2, abbreviated as NCM), lithium nickel oxide (LiNiO2, abbreviated as LNO), lithium manganese oxide (LiMn2O4, abbreviated as LMO), and lithium cobalt oxide (LiCoO2, abbreviated as LCO).5,10 In contrast to other cathode materials, high energy density, excellent safety performance and versatile applications of NCM materials make them a preferred choice for EVs.11
To date, ternary LIBs have dominated a majority of the global EV battery market share, and the demand for them will continue growing. However, the corresponding demand for critical raw materials such as lithium (Li), manganese (Mn), cobalt (Co), and nickel (Ni) has already exceeded global supply capacities, threatening the sustainable manufacture of LIBs.12,13 Moreover, a significant number of LIBs in electric vehicles may reach their end-of-life after approximately 5 to 8 years, and 8 million tons of LIBs are expected to be discarded by 2040.14–16 Spent LIBs contain heavy metals and noxious organic substances, which may harm the ecological environment and human health if not properly treated.17–19 Nevertheless, spent LIBs should be treated not as waste but as recoverable sources of critical materials. For instance, spent NCM LIBs have abundant valuable metals such as Mn, Co, Ni and Li, and their contents are much higher than those in primary minerals.11,20,21 Therefore, recycling spent NCM LIBs is very crucial for sustainable resource utilization and environmental conservation.
At present, numerous efforts have been made to develop the effective recycling process of spent NCM LIBs, primarily involving direct regeneration, pyrometallurgy and hydrometallurgy.5,22 During the direct regeneration process, satisfactory electrochemical performance can be regained by directly repairing the crystalline structural defects of spent NCM materials. However, the performance of regenerated NCM materials is significantly impacted by their purities, posing a substantial challenge for industrial applications.3,19,23–26 The pyrometallurgy process exhibits advantages such as simplicity, high productivity and short processing time, but its high-temperature requirements can result in issues such as environmental pollution and huge energy consumption,3,5,21,27 which contradicts the principles of green chemistry. The hydrometallurgical process is considered the mainstream process for recycling spent NCM materials, due to its high recovery rate for critical metals, low energy consumption, and good processing stability.28 However, inorganic acids are commonly used as leaching agents in hydrometallurgical processes.21,29 Highly corrosive inorganic acids can produce harmful gases, posing risks to the environment and human health, which is also inconsistent with the principles of green chemistry. Consequently, organic acids such as malic acid,30 citric acid31 and succinic acid32 have emerged as promising alternatives. Howbeit, to enhance the leaching efficiency of critical metals, additional reducing agents are typically required in conjunction with organic acids such as glucose and hydrogen peroxide (H2O2).26 As a result, the recycling costs increase and economic returns decrease.
To advance spent LIB recycling technologies from laboratory research to practical industrial applications, it is crucial to address the green chemistry challenges inherent in the traditional process.33 Recently, a CEC method has been utilized to recycle spent LCO cathode materials, featuring low energy consumption and the use of nontoxic reagents.34 Specifically, in the citric acid-leaching process, reactive oxygen species are generated through contact electrification between liquid and dielectric SiO2, and they can effectively promote the leaching of critical metals, replacing traditional reducing agents. Moreover, SiO2 catalysts exhibited excellent recyclability, which adheres to the green chemistry principle of reagent reuse in the recycling process. Although CEC holds great application potential due to its high efficiency and green nature, significant challenges remain in catalyst development, performance enhancement, and application scalability.35 Among these, the development of high-performance catalysts is particularly critical, where physicochemical properties such as wettability and electronegativity can be tailored through the rational design of catalyst structures and surface functional groups to reduce the solid–liquid electron transfer barrier and improve charge excitation. Inspiringly, the electrification ability of SiO2 as the catalyst may be enhanced by adjusting the microstructure and surface chemical environment to further optimize the leaching efficiency of critical metals. Sodium alginate (SA) possesses plentiful hydroxyl (–OH) and carboxyl (–COOH) functional groups, which exhibit both significant hydrophilicity and excellent electron-donating abilities.36,37 If SA is employed to decorate SiO2, the enhanced hydrophilicity of SA@SiO2 can promote their interfacial contact with water molecules, facilitating electron transfer and subsequent hydroxyl radical generation. Concurrently, the electron-donating ability of carboxylate (COO−) groups may accelerate electron transfer from SA@SiO2 microspheres to O2, thereby generating superoxide radicals. Correspondingly, the generation of the oxygen species H2O2 will be significantly enhanced through the coupling and reactions of the above-mentioned radicals. Moreover, hydroxyl and carboxy groups can act as versatile reaction sites for chemical modification.38 Polyethyleneimine (PEI) contains numerous hydrophilic amine groups, and its protonated amine groups and carboxylate (COO−) groups of SA can form zwitterionic microstructures, which can serve as an electronic porter.39,40 Thus, it is speculated that anchoring SA and PEI molecular chains onto the surface of SiO2 could accelerate electron transfer in the CEC process, resulting in the generation of more reactive oxygen species.
Herein, SiO2 nanoparticles and porous microspheres were utilized to leach Li, Ni, Co and Mn from spent NCM cathode materials via the CEC technology. The effects of SiO2 microstructures on the leaching efficiency were systematically investigated by adjusting the size of nanoparticles and porous microspheres. Interestingly, the porous structure of SiO2 microspheres endowed them with stronger catalytic capability than SiO2 nanoparticles of the same size. Subsequently, SA and PEI were anchored onto the surface of optimal porous SiO2 microspheres by ionic crosslinking and hydrogen bonding. As expected, the leaching efficiency of critical metals was significantly enhanced with the introduction of SA and PEI chains. Furthermore, the surface chemical environment was further precisely modulated by controlling the PEI content and molecular weight, and the underlying mechanism of promoting metal leaching efficiency was systematically investigated. Overall, this work introduces a new optimization strategy for CEC technology by controllably tuning the microstructure and surface chemical environment of SiO2.
2. Experimental section
2.1. Materials
SiO2 particles (99.9%, Shanghai MCC Advanced Materials Co., Ltd), porous SiO2 microspheres (99.9%, Shanghai Baiyu Fluid Equipment Co., Ltd), citric acid monohydrate (C6H8O7·H2O, 99%, Aladdin), sodium alginate ((C6H7NaO6)n, Sinopharm Chemical Reagent Co., Ltd), CaCl2 (≥96%, Sinopharm Chemical Reagent Co., Ltd), polyethyleneimine (50%, Macklin), and black mass from discarded LiNixCoyMnzO2 cathodes (it is sourced from a battery-recycling company in Guizhou, China, where it underwent pretreatment steps of mechanical separation and thermal processing) were used in our work.
2.2. Fabrication of SA@SiO2 and PEI–SA@SiO2 microspheres
As shown in Fig. S1, sodium alginate was dissolved in deionized water at 80 °C to obtain a homogeneous aqueous solution (1.5 wt%). Subsequently, porous SiO2 microspheres were immersed in an SA solution to achieve complete impregnation through capillary action. The SA-saturated SiO2 microspheres were uniformly dispersed in a 0.25 mol L−1 CaCl2 solution under ultrasonication (200 W, 40 kHz). The following ionic crosslinking reaction proceeded for 40 min, enabling the formation of stable SA networks on the microsphere surfaces. The resulting SA@SiO2 microspheres were collected by centrifugation (4000 rpm, 5 min), purified with deionized water, and further functionalized via immersion in a polyethyleneimine solution at 60 °C for 1.5 h. After centrifugal purification, the PEI–SA@SiO2 microspheres were washed thoroughly with deionized water and finally dried in a vacuum oven at 80 °C for 3 h.
2.3. Metal leaching using the CEC method
Under ultrasonication (600 W, 40 kHz), 0.12 g of porous SiO2 microspheres, SA@SiO2 or PEI–SA@SiO2 functional microspheres were uniformly dispersed in 0.2 mol L−1 aqueous citric acid solution. Subsequently, 800 mg spent LiNixCoyMn2O2 powders were introduced into the system at a solid-to-liquid ratio of 20 g L−1, and the ultrasound-assisted leaching reaction was conducted at a constant temperature of 80 °C. Upon reaction completion, the mixed solution was centrifuged at 4000 rpm for 20 min, and the supernatant was collected for quantitative analysis of metal ion concentrations via inductively coupled plasma optical emission spectrometry (ICP-OES, detection limits: 0.1–10 ppb). The instrument was calibrated by the internal standard method. This involved adding a known concentration of an element not present in the samples (the internal standard), which shares similar physicochemical properties with the analyte, to both the calibration standards and the samples. A calibration curve was then constructed using the measured signal ratio of the analyte to the internal standard. The metal leaching efficiency (η, %) was then calculated according to eqn (1). Error bars on the resulting data represent ±1 standard deviation from the mean.| | 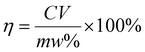 | (1) |
where C (mg L−1) denotes the concentration of Li+, Co2+, Mn2+ or Ni2+, V (L) is the leachate volume (40 mL), m (g) represents the mass of LiNixCoyMn2O2 powder (800 mg), and w% is the mass fraction of metals in the degraded LiNixCoyMn2O2 powder.
2.4. Characterization
The diameter distribution of SiO2 particles and porous SiO2 microspheres was estimated using a laser particle size analyzer (Bettersize 2600, Fangxu Technology (Shanghai) Co., Ltd, China). The BET surface area of SiO2 particles and porous SiO2 microspheres was tested using a surface area and porosity analyzer (NOVO-1000e, Quantachrome Instruments, America). The active species were identified by electron spin resonance spectroscopy (ESR, A300 spectrometer, Bruker, America) during the leaching reaction. Specifically, 0.1 mM trapping agents of 5,5-dimethyl-1-pyrroline N-oxide (DMPO) and 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) were added to the reaction system, which selectively captured hydroxyl radicals (˙OH), superoxide radicals (˙O2−) and unpaired electrons, respectively. The morphology of porous SiO2 microspheres before and after modification was observed by field-emission scanning electron microscopy (SEM, Quanta 250 FEG, FEI, America) and transmission electron microscopy (TEM, Tecnai G2 F20, FEI, America). The surface chemical composition of porous SiO2 microspheres before and after modification was investigated by energy-dispersive X-ray analysis (EDX, JSM-7500F, JEOL, Japan), attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FTIR, NEXUS570, Nicolet, America), and X-ray photoelectron spectroscopy (XPS, PHI-1600, PerkinElmer Co., America) with Mg-Kα as the radiation source. The zeta potential of porous SiO2 microspheres before and after modification was measured using a zeta potential analyzer (SurPASS, Anton Parr, Austria) at 25 °C ± 1 °C. The composition of the trace residual solids, obtained by centrifugation after the leaching process, was analyzed using a thermogravimetric analyzer (TGA, Q50, TA Instruments Waters, America).
3. Results and discussion
3.1. Effects of SiO2 microstructures on the metal extraction by the CEC technology
In the contact-electro-catalysis (CEC) process, numerous cavitation bubbles must be induced during the propagation of ultrasonic waves to achieve the continuous contact and separation of liquids and solids, which can constantly produce free radicals through the electron transfer in contact electrification. In theory, the size and morphology of solid particles inevitably affect the propagation of acoustic waves in liquid, which, in turn, influences the generation of cavitation bubbles due to the corresponding energy change.41–43 Therefore, the effects of SiO2 microstructures on the metal leaching efficiency were systematically investigated in Fig. 1–3. The wavelength and frequency of ultrasonic waves were 37 μm and 40 kHz, respectively. When the size of SiO2 particles ranged from 1 μm to 10 μm, ultrasound Rayleigh scattering was primarily induced.44 It could cause ultrasonic wave energy to be widely distributed, lowering the local pressure and reducing the formation of cavitation bubbles. The larger the particle size, the greater the scattering intensity. Surprisingly, along with the increase in the size of SiO2 particles from 1 μm to 10 μm, the leaching efficiency of metals was significantly elevated (Fig. 1a). Specifically, the leaching efficiencies of Li, Co, Mn and Ni were up to 90.2%, 91.4%, 90.1% and 91.6%, respectively, in the presence of 10 μm SiO2 particles. To understand the underlying mechanisms of the above-mentioned phenomena, the size distribution of SiO2 particles was measured, which is presented in Fig. 1b–f. Obviously, SiO2 particles with a larger size possessed a narrower distribution, which could counteract the negative effects from increasing size and dominantly diminish the scattering intensity to gather more energy and facilitate the formation of cavitation bubbles. Accordingly, abundant hydroxyl radicals and electrons were generated at the interface of deionized water and SiO2 with the decrease in size distribution, which promoted the extraction of Li, Co, Mn and Ni metals from spent NCM cathode materials. When the size of SiO2 particles increased to 20 μm, which is relatively close to the ultrasonic wave of 37 μm, strong ultrasound geometric scattering was mainly induced, leading to the uneven distribution of sound velocity and largely suppressing the accumulation of energy. This was not conducive to the generation of cavitation bubbles, hydroxyl radicals and electrons. Consequently, the leaching efficiencies of Li, Co, Mn and Ni metals were sharply reduced to 81.6%, 84.0%, 84.0% and 85.4% with the further increase in the size of SiO2 particles to 20 μm. Subsequently, the leaching efficiency of metals was further optimized by adjusting the dosage of 10 μm SiO2 particles and reaction time. As shown in Fig. 1g, the leaching efficiency initially increased and then decreased as the dosage grew and reached the highest value at 120 mg. This is because excessive SiO2 particles enhanced the scattering of ultrasound waves, leading to a reduced efficiency.45 Similarly, the leaching efficiency was initially elevated and then lowered with the increase in reaction time (Fig. 1h), which was also reported in the previous literature.34 In the end, higher leaching efficiencies of Li (93.6%), Co (93.6%), Mn (94.8%) and Ni (95.3%) metals were achieved at the reaction time of 12 h.
 |
| | Fig. 1 Leaching efficiency of metals using the CEC technology (effects of the SiO2 particle size (a), dosage (g) and leaching time (h) on the leaching efficiency of Li, Co, Mn and Ni from the spent NCM batteries at 90 °C) and diameter distribution of SiO2 particles ((b) 1 μm; (c) 2 μm; (d) 5 μm; (e) 10 μm; and (f) 20 μm). | |
 |
| | Fig. 2 Leaching efficiency of metals using the CEC technology (effects of the porous SiO2 microsphere temperature (a) and leaching time at 80 °C (b) on the leaching efficiency of Li, Co, Mn and Ni from the spent NCM batteries) and ESR spectra of the DMPO-trapped radical adducts for hydroxyl radicals (c) and the TEMPO-trapped radical adducts for electrons (d). | |
 |
| | Fig. 3 Leaching efficiency of metals using the CEC technology ((a) effects of the porous SiO2 microsphere size on the leaching efficiency of Li, Co, Mn and Ni from the spent NCM batteries at 80 °C), diameter distribution of porous SiO2 microspheres ((b) 2 μm; (c) 5 μm; (d) 10 μm; and (e) 15 μm), BET surface area of porous SiO2 microspheres (f), and photographs of the filtrate before and after leaching (g). | |
Besides the size of SiO2 particles, the influences of their porous structures on the metal leaching efficiency were explored in detail by adjusting the temperature and time in Fig. 2a and b. Interestingly, when 10 μm SiO2 particles were replaced by porous SiO2 microspheres, higher leaching efficiencies of Li (93.8%), Co (93.6%), Mn (95.3%) and Ni (95.9%) metals could be attained at a lower temperature (80 °C) and a much shorter time of 8 h. The reasons were mainly attributed to the following two aspects. On the one hand, porous SiO2 microspheres had a much narrower size distribution than that of SiO2 particles, which further curtailed the ultrasound Rayleigh scattering and accelerated the formation of cavitation bubbles. On the other hand, the surface area of porous SiO2 microspheres (203.06 m2 g−1) was approximately 117 times greater than that of SiO2 particles (1.74 m2 g−1), and thus more solid–liquid contact electrification was triggered during the collapse of cavitation bubbles, which generated copious free radicals to catalyze the leaching of metals.46 Significantly, the phenomenon of reduced leaching efficiency due to excessively long reaction time reoccurred. To be specific, the leaching efficiencies were diminished to 91.9% (Li), 91.1% (Co), 92.3% (Mn) and 92.5% (Ni) when the reaction time was extended to 12 h. Therefore, the effects of reaction time on hydroxyl radicals and electrons were qualitatively assessed using spin-trapping reagents of 5,5-dimethyl-1-pyrroline N-oxide (DMPO) and 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) in Fig. 2c and d. As depicted in Fig. 2c, the representative quadruplet peaks of DMPO–OH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:1) were observed after introducing the hydroxyl radical trapping agent DMPO under identical reaction conditions, confirming the generation of hydroxyl radicals during the CEC catalytic process. However, the peak intensity gradually weakened after 40 min, indicating that the generation of hydroxyl radicals was suppressed with the increasing reaction time. Additionally, TEMPO was utilized to evaluate the electrons in the CEC catalytic process. In Fig. 2d, three peaks with an intensity ratio of 1:1:1 were observed, but they quickly disappeared with the increasing reaction time and did not reappear, demonstrating that the electrons were created and adequately participated in the leaching reaction of metals. In other words, extended reaction time reduced the leaching efficiency of metals primarily by suppressing hydroxyl radicals.
:2:2:1) were observed after introducing the hydroxyl radical trapping agent DMPO under identical reaction conditions, confirming the generation of hydroxyl radicals during the CEC catalytic process. However, the peak intensity gradually weakened after 40 min, indicating that the generation of hydroxyl radicals was suppressed with the increasing reaction time. Additionally, TEMPO was utilized to evaluate the electrons in the CEC catalytic process. In Fig. 2d, three peaks with an intensity ratio of 1:1:1 were observed, but they quickly disappeared with the increasing reaction time and did not reappear, demonstrating that the electrons were created and adequately participated in the leaching reaction of metals. In other words, extended reaction time reduced the leaching efficiency of metals primarily by suppressing hydroxyl radicals.
Moreover, the above-mentioned results and analyses illustrated that the metal leaching efficiency simultaneously depended on the size and porous structure of SiO2 microspheres. Therefore, the effects of porous SiO2 microspheres of different sizes on the leaching efficiency of Li, Co, Mn and Ni metals were further investigated, as shown in Fig. 3a. The leaching efficiency gradually increased to the highest value with the increase in size to 10 μm and then sharply declined with the further increase in size. To elaborate the synergistic effects of sizes and porous structures, the size distribution of porous SiO2 microspheres was tested, which is displayed in Fig. 3b–e, and their N2 adsorption isotherms were measured, which are shown in Fig. S2. The corresponding BET surface areas, derived from the isotherms, are summarized in Fig. 3f. Obviously, the size distribution first narrowed and then widened before and after the increase in size to 10 μm, respectively (Fig. 3b–e), and the surface area first slightly decreased and then dramatically increased (Fig. 3f). As a result, in contrast to 2 μm, 5 μm, 15 μm porous SiO2 microspheres, 10 μm porous SiO2 microspheres exhibited the narrowest size distribution and relatively high surface area, indicating that it possessed very strong catalytic capability. This could be further confirmed by the filtrate color before and after leaching, as presented in Fig. 3g. The filtrate exhibited a pronounced dark gray color before the leaching process, which changed significantly afterward. Notably, when 10 μm microspheres were used, the resulting filtrate after leaching showed a distinct color difference compared to those obtained with 2 μm, 5 μm, and 15 μm porous SiO2 microspheres. The black color faded markedly, yielding a bright green solution. Furthermore, these results are in line with those of SiO2 particles. However, the leaching efficiency of metals in the presence of porous SiO2 microspheres was evidently higher than that in the presence of SiO2 particles with the same size, suggesting that the porous structure endowed SiO2 with stronger catalytic capability.
3.2. Construction of the surface chemical environment of porous SiO2 microspheres
To further improve the leaching efficiency of metals, SA was initially anchored onto the porous SiO2 microspheres using calcium ions as the crosslinking agent.47 Subsequently, PEI was utilized to functionalize the resulting SA@SiO2 microspheres. Field-emission scanning electron microscopy (FE-SEM) and transmission electron microscopy (TEM) were conducted to investigate the microstructural evolution of porous SiO2 microspheres before and after modification (Fig. 4a and b). Comparative FE-SEM analysis revealed significant morphological differences between the modified and unmodified microspheres (Fig. 4a). Conspicuous deposits appeared on the surface of SA@SiO2 and PEI–SA@SiO2 microspheres, and the deposition density exhibited a positive correlation with the increase in PEI content. Furthermore, TEM analysis further demonstrated substantial morphological transformations following microsphere functionalization (Fig. 4b). Specifically, pristine porous SiO2 microspheres featured smooth, well-defined edges. However, SA@SiO2 and PEI–SA@SiO2 microspheres exhibited distinct edge irregularities, which were gradually intensified with the increase in PEI content. Afterwards, EDX mappings of PEI(0.6%)–SA@SiO2 microspheres were performed to further understand the observed microstructural evolution. As shown in Fig. 4c, silicon, oxygen, carbon, calcium and nitrogen elements were present on the surface of PEI(0.6%)–SA@SiO2 microspheres, confirming that SA and PEI molecules might be successfully anchored onto porous SiO2 microspheres.
 |
| | Fig. 4 SEM (a) and TEM (b) images of the porous SiO2 microspheres before and after modification, and EDX mappings (c) of the PEI(0.6%)–SA@SiO2 microspheres. | |
Accordingly, a comprehensive evaluation of surface chemical composition and properties was implemented for porous SiO2 microspheres and their functionalized counterparts (SA@SiO2 and PEI–SA@SiO2) through FTIR spectroscopy, XPS and zeta potential analysis. Fig. 5a shows the FTIR spectra of porous SiO2, SA@SiO2 and PEI–SA@SiO2 microspheres. SA@SiO2 displayed symmetric and asymmetric stretching vibration peaks of the –COO− group at 1410 and 1610 cm−1 and asymmetric stretching vibration peak of the –OH group at 3359 cm−1, respectively, demonstrating the successful anchoring of SA.38,48–50 Following the incorporation of PEI into SA@SiO2, obvious asymmetric and symmetric stretching vibration peaks of –CH2 were observed at 2830 and 2947 cm−1, and the peak at 3359 cm−1 became broad and strong due to the partially overlapping N–H stretching vibrations and O–H stretching vibrations.37,49,51 Moreover, N–H bending vibration peaks in primary and secondary amine groups appeared at 1604 and 1469 cm−1.48,52,53 Notably, the increasing PEI loading induced a systematic shift of the characteristic peak from 1604 cm−1 to 1589 cm−1, accompanied by the emergence of a new peak at 1557 cm−1. This was ascribed to the protonation of –NH2 groups, which enabled the ionic cross-linking between PEI and SA@SiO2. The XPS spectra of SA@SiO2 and PEI–SA@SiO2 microspheres are presented in Fig. 5b. In addition to the peaks assigned to Si 2p, Si 2s, and O 1s, the peaks of SA@SiO2 at 284.6 and 347.8 eV corresponded to C 1s and Ca 2p.54–56 The peak of PEI–SA@SiO2 at 401.0 eV corresponded to N 1s.57 Moreover, the surface zeta potential evolution of porous SiO2 microspheres before and after modification was assessed, as shown in Fig. 5c. Under neutral conditions (pH = 7), porous SiO2 microspheres possessed negligible surface charge. In contrast, SA@SiO2 and PEI–SA@SiO2 exhibited very strong negative and positive charges, respectively. These agreed well with the above-mentioned FTIR results, further confirming the successful incorporation of SA and PEI into porous SiO2 microspheres. SA was first successfully anchored on SiO2 microspheres with the aid of porous microstructure and calcium ions, and then PEI was grafted onto SA@SiO2 microspheres through hydrogen bonding and ionic cross-linking between –COO− and –NH3+ groups (Fig. 5d). Significantly, PEI–SA@SiO2 acquired stronger positive charges at pH = 3, showing that more protonated amine groups were formed under acidic conditions (pH < 5). Correspondingly, plentiful hydrophilic zwitterionic microstructures consisting of protonated amine groups and carboxylate (COO−) groups were created, which might markedly enhance the metal leaching efficiency through effectively promoting electron transfer and significantly facilitating radical generation.
 |
| | Fig. 5 (a) FTIR spectra, (b) XPS spectra and (c) surface zeta potential of the porous SiO2 microspheres before and after modification, and schematic (d) of the PEI–SA@SiO2 microsphere formation. | |
3.3. Metal extraction and mechanism investigations of functionalized porous SiO2 microspheres in the CEC-leaching process
To confirm the above-mentioned speculation, the effects of the surface chemical environment of porous SiO2 microspheres on the metal leaching efficiency were systematically evaluated, as shown in Fig. 6a–c. While the leaching efficiencies of Li, Co, Mn and Ni were only slightly improved by the immobilization of SA on porous SiO2 microspheres, they were substantially boosted following the subsequent incorporation of PEI (Fig. 6a). Furthermore, the PEI–SA microstructures were optimized by adjusting the PEI content to further enhance the metal leaching efficiency. As expected, the leaching efficiency of Li, Co, Mn and Ni arrived at nearly 100% along with the increase in PEI content from 0.1 wt% to 0.6 wt% (Fig. 6b). The nearly superimposable thermogravimetric curves of original PEI(0.6%)–SA@SiO2 microspheres and purified solid residues (containing PEI(0.6%)–SA@SiO2 microspheres) across the 40–500 °C range further substantiated this finding (Fig. S3). However, the leaching efficiency of metals sharply decreased at higher PEI contents. It is surmised that excess amino groups in the PEI–SA microstructures might adsorb hydroxyl radicals via hydrogen bonding and partially quench them through electron transfer,58,59 leading to a decrease in the metal leaching efficiency. It is worth noting that, besides the PEI content, the availability of amino groups also depends on another key factor: PEI molecular weight. Consequently, at the optimal PEI content, the influence of molecular weight on the metal leaching efficiency was systematically estimated in Fig. 6c. Similarly, the leaching efficiencies of Li, Co, Mn, and Ni also exhibited a non-monotonic dependence on PEI molecular weight, initially increasing before decreasing beyond an optimal value. This further demonstrated that an overabundance of amino groups in the PEI–SA microstructures adversely affected the metal leaching efficiency. Nevertheless, the leaching efficiency of all metals still remained over 80% at the highest PEI content (1.0 wt%) and molecular weight (70000 Da).
 |
| | Fig. 6 Leaching efficiency of metals at 80 °C by manipulating the surface chemical environment of the porous SiO2 microspheres (SA and PEI (a), PEI contents (b), and PEI molecular weights (c)), ESR spectra of the TEMPO-trapped radical adducts for electrons (d) and the DMPO-trapped radical adducts for hydroxyl (e) and superoxide radicals (f), and proposed mechanism of the metal leaching by the PEI–SA@SiO2 microspheres (g). | |
It is well established that high-valent metal ions in the LIB cathode are inert and, notably, resistant to dissolution without any reducing agent. Therefore, reducing agents play a critical role in facilitating effective metal leaching. H2O2 serves as the predominant reducing agent in the organic acid-leaching processes of traditional hydrometallurgy. Of note, the CEC leaching process features the in situ generation of H2O2via reducing free radicals and active electrons in the contact electrification between liquid and dielectric materials, as described in eqn (2)–(6).34,60 Correspondingly, reducing free radicals and active electrons were assessed in the presence of PEI(0.6%)–SA@SiO2 microspheres to reveal the governing mechanism of the surface chemical environment of porous SiO2 microspheres in the CEC-leaching process. Although the three characteristic peaks of TEMPO-e−1 gradually weakened with the increase in reaction time, they persisted and remained detectable even at 50 minutes (Fig. 6d). This phenomenon distinctly differed from the above-mentioned observations in the presence of porous SiO2 microspheres, where complete disappearance occurred (Fig. 2d). Therefore, as predicted, the incorporation of PEI and SA significantly enhanced the generation of electrons through improving the interfacial contact with water molecules. Electrons function as crucial active species in the leaching process. They can not only directly participate in the reduction of high-valent metal ions but also induce the formation hydroxyl radicals and superoxide radicals. Water molecules lost electrons to form water radical cations (H2O+) (eqn (2)), which subsequently reacted with water to generate hydroxyl radicals (˙OH) (eqn (3)). Simultaneously, PEI(0.6%)–SA@SiO2 microspheres captured electrons from water molecules to form an excited state (PEI(0.6%)–SA@SiO2*). The captured electrons were then rapidly transferred to oxygen, thereby generating superoxide radicals (˙O2−) (eqn (4)). Ultimately, abundant H2O2 molecules with strong reducing activity are generated through the hydroxyl radical coupling and the reaction of superoxide radicals with excited-state microspheres and H+, according to eqn (5) and (6). However, the characteristic peaks of DMPO–OH (1:2:2:1) were very weak (Fig. 6e), demonstrating that amino groups in the PEI–SA microstructures indeed consumed some hydroxyl radicals via hydrogen bonding and electron transfer. Moreover, strong characteristic peaks corresponding to DMPO–OOH were clearly observed with the increasing reaction time (Fig. 6f), indicating the generation of numerous superoxide radicals. Hence, the perfect (approximately 100%) metal leaching efficiency was attributable to the fact that the combination of hydroxyl and superoxide radicals remarkably promoted the formation of plentiful H2O2 molecules. Moreover, the strong activation of superoxide radicals might enable over 80% metal leaching efficiency from PEI–SA@SiO2 microspheres prepared at elevated PEI content and molecular weight. According to the above-mentioned results and analyses, the introduction of SA and PEI enhanced the interfacial contact between SiO2 microspheres and water molecules by improving surface hydrophilicity, thereby accelerating electron transfer from water molecules to SiO2 microspheres and inducing hydroxyl radical (˙OH) formation (Fig. 6g). Although the abundant amino groups in PEI depleted some hydroxyl radicals, once electrons were transferred from water molecules to the SiO2 microsphere surface, the SA–PEI zwitterionic microstructure inevitably promoted rapid electron migration across the microsphere surface. As a result, the electronegativity and electron-donating capacity of –COO− groups were substantially enhanced, which facilitated efficient electron transfer from SiO2 microspheres to O2, resulting in superoxide radical (˙O2−) generation.61 Consequently, the synergistic effect of hydroxyl and superoxide radicals induced copious H2O2 molecules and achieved exceptional metal leaching efficiencies at a lower PEI content and molecular weight. Moreover, at a higher PEI content and molecular weight, where superoxide radicals became the dominant active species, the system still maintained relatively high metal leaching efficiencies (>80%) through the generated H2O2 molecules, according to eqn (6):
| | 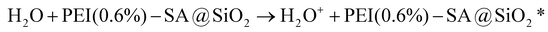 | (2) |
| | | H2O + H2O+ → ˙OH + H3O+ | (3) |
| | 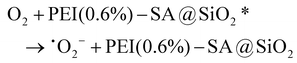 | (4) |
| | | PEI(0.6%)–SA@SiO2* + ˙O2− + 2H+ → H2O2 + SiO2 | (6) |
3.4. Green and cost-effective recycling of PEI–SA@SiO2 microspheres during leaching
Besides the green preparation of the catalyst and environmentally friendly metal leaching processes, the sustainable recycling of the catalyst should be evaluated, aligning with the green chemistry principle of promoting reagent reuse. Accordingly, we systematically assessed the recycling of PEI(0.6%)–SA@SiO2 microspheres over five leaching cycles. As depicted in Fig. 7a, the mixture after leaching was subjected to centrifugation at 4000 rpm for 20 min to separate various metal ions and solid residues. Subsequently, the obtained residue (containing PEI(0.6%)–SA@SiO2 microspheres) was washed with deionized water, vacuum-dried at 60 °C for 8 h, and then directly reused in the next leaching cycle. After five consecutive cycles, the chemical composition of the solid residues was analyzed using the FTIR spectra. The resulting spectrum was compared with that of the fresh PEI(0.6%)–SA@SiO2 microspheres to assess structural changes, as presented in Fig. 7b. The presence of characteristic peaks for –COO− groups (from SA) and –NH2 groups (from PEI) in the FTIR spectrum of the solid residues indicated that the hydrophilic zwitterionic microstructures on the SiO2 microsphere surface remained largely intact. Furthermore, the morphology of PEI(0.6%)–SA@SiO2 microspheres before and after five leaching cycles was explored, as shown in Fig. S4. The recycled microspheres retained the spherical morphology of the original ones, demonstrating that the structure was not significantly destroyed even after leaching five cycles. More importantly, the leaching efficiency of metal ions showed no dramatic reduction over five cycles (Fig. 7c), demonstrating the excellent reusability of the PEI(0.6%)–SA@SiO2 microspheres and their potential for cost reduction. To further confirm the economic feasibility of this process, a preliminary evaluation was conducted based on 1 kg of spent LiNixCoyMnzO2 powder. A detailed description of the cost calculation method is presented in the SI. The total cost is US$19.27 for 1 kg of spent LiNixCoyMnzO2 powder, which is very outstanding compared to conventional acid leaching processes and other reported CEC-assisted acid leaching approaches in the spent LIB recycling, as summarized in Table 1. Therefore, the developed process for recycling spent NCM batteries is both economical and eco-friendly, with promising prospects for large-scale industrial implementation.
 |
| | Fig. 7 Recycling flow chart of the PEI(0.6%)–SA@SiO2 microspheres in the CEC leaching process (a), their FTIR spectra before and after five leaching cycles (b), and leaching efficiency over five consecutive cycles (c). | |
Table 1 Cost assessment of metal leaching techniques in NCM LIB recycling
| Cathode type |
Technique |
S/L ratio (g L−1) |
Conditions |
Cost ($ per kg) |
Ref. |
| NCM |
Acid leaching + CEC |
10 |
80 mg SiO2, 20 mol citric acid, 70 °C, 360 min |
23.13 |
34
|
| NCM |
Acid leaching + CEC |
20 |
80 mg SiO2, 10 mol malic acid, 80 °C, 360 min |
28.14 |
60
|
| NCM |
Acid leaching |
20 |
25 mol citric acid, 75 mol H2O2, 90 °C, 60 min |
30.79 |
62
|
| NCM |
Acid leaching |
20 |
75 mol Citric acid, 80 °C, 120 min |
73.43 |
63
|
| NCM |
Acid leaching |
20 |
50 mol citric acid, 250 mol H2O2, 95 °C, 120 min |
70.02 |
64
|
| NCM111 |
Acid leaching |
30 |
67 mol citric acid, 67 mol H2O2, 80 °C, 90 min |
70.20 |
65
|
| NCM |
Acid leaching + CEC |
20 |
120 mg SiO2, 10 mol citric acid, 80 °C, 480 min |
19.27 |
This work |
4. Conclusion
We proposed an innovative and highly effective approach to boost the catalytic activity of dielectric SiO2 in the CEC-leaching process by engineering the microstructure and surface chemical environment. The 10 μm SiO2 microspheres exhibited superior metal leaching efficiencies owing to their uniform size distribution and porous architecture, which facilitated efficient electron and radical generation by minimizing ultrasonic wave scattering and maximizing aqueous-phase contact interfaces. Subsequently, the incorporation of SA and PEI not only accelerated electron transfer from water molecules to SiO2 microspheres but also promoted rapid electron migration across the microsphere surface and enhanced the electron-donating capacity of –COO− groups. Consequently, abundant superoxide radicals were generated by electron transfer from SiO2 microspheres to O2, and the perfect (approximately 100%) metal leaching efficiency of Li, Co, Mn and Ni in the spent NCM cathode was achieved in 8 h at 80 °C due to the synergistic effect of hydroxyl and superoxide radicals. In future studies, we will pursue advanced engineering of dielectric materials’ microstructure and surface chemistry to enable rapid and efficient metal leaching. This will involve systematic replacement of the current SiO2–SA–PEI system with alternative high-performance dielectric materials and tailored functional monomers to simultaneously enhance surface charge density, interfacial electron transfer efficiency and radical diffusion. Overall, this study makes significant contributions to both fundamental understanding of catalytic mechanisms and technological advancements in applying contact-electro-catalysis for LIB recycling.
Author contributions
Man Yang: investigation. Shuhao Qin: funding acquisition, supervision, methodology. Youchang Yang: methodology, supervision. Mingmi Wu: investigation. Ting Lei: investigation. Yufei Liu: data curation. Huiju Shao: funding acquisition, conceptualization, investigation, writing-original draft, review & editing.
Conflicts of interest
There are no conflicts to declare.
Data availability
Data will be made available on request.
Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d5gc04080a.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22368017), the Natural Science Foundation of Guizhou Province (ZD[2025]002), the Science and Technology Planning Project of Guizhou Province (2023[035], 2023[001], [2024]042, and [2024]002-1), and the Guizhou Science and Platform Talents (QKHPTRC-CXTD [2023] 012).
References
- X. Zhang and M. Y. Zhu, Green Chem., 2024, 26, 7656–7717 RSC
 .
.
- I. Bošković and A. Radivojević, J. Build. Eng., 2023, 66, 105908 CrossRef .
- X. L. Li, X. G. Li, Q. Gao, X. D. Shi, W. H. Gao, S. Yan, Z. Y. Wang, X. N. Zhu and X. Z. Qin, Sep. Purif. Technol., 2025, 358, 130251 CrossRef CAS .
- G. Palomo-Vélez, G. Perlaviciute, N. Contzen and L. Steg, Environ. Res. Commun., 2021, 3, 115004 CrossRef .
- L. Y. Yu, X. B. Liu, S. S. Feng, S. Z. Jia, Y. Zhang, J. X. Zhu, W. W. Tang, J. K. Wang and J. B. Gong, Chem. Eng. J., 2023, 476, 146733 CrossRef CAS .
- S. Kim, J. Kim and J. Kim, Chem. Eng. J., 2025, 504, 158699 CrossRef CAS .
- F. M. N. U. Khan, M. G. Rasul, A. S. M. Sayem and N. K. Mandal, J. Energy Storage, 2023, 71, 108033 CrossRef .
- W. H. Yu, Y. Guo, Z. Shang, Y. C. Zhang and S. M. Xu, eTransportation, 2022, 11, 100155 CrossRef .
- A. J. Niri, G. A. Poelzer, S. E. Zhang, J. Rosenkranz, M. Pettersson and Y. Ghorbani, Renewable Sustainable Energy Rev., 2024, 191, 114176 CrossRef .
- B. M. Bresolin, A. Zanoletti and E. Bontempi, Sep. Purif. Technol., 2025, 363, 131918 CrossRef CAS .
- Z. G. Cun, P. Xing, C. Y. Wang, H. Q. Li, S. Y. Ma, Z. H. Sun, Q. Wang and X. Guan, Resour., Conserv. Recycl., 2024, 202, 107390 CrossRef CAS .
- Q. Peng, X. Q. Zhu, J. Li, Q. Liao, Y. M. Lai, L. Zhang, Q. Fu and X. Zhu, Waste Manage., 2021, 134, 100–109 CrossRef CAS .
- G. Ko, S. Park, W. Kim, S. Kim, J. Bae and K. Kwon, J. Energy Chem., 2025, 103, 723–734 CrossRef CAS .
- X. Lai, Q. W. Chen, X. P. Tang, Y. Q. Zhou, F. R. Gao, Y. Guo, R. Bhagat and Y. J. Zheng, eTransportation, 2022, 12, 100169 CrossRef .
- G. H. Zhu, Q. Yang, X. Y. Guo, D. W. Yu, A. M. Mitrašinović, Q. H. Tian, H. Feng and K. Zhang, J. Environ. Chem. Eng., 2025, 13, 115173 CrossRef CAS .
- Q. Zhang, N. Q. Sun, W. G. Lv, Y. Wang and Z. Sun, Chem. Eng. J., 2025, 503, 158383 CrossRef CAS .
- G. W. Zhang, X. Yuan, Y. Q. He, H. F. Wang, W. N. Xie and T. Zhang, Waste Manage., 2020, 115, 113–120 CrossRef CAS PubMed .
- Y. S. Liao, S. S. Gong, G. G. Wang, T. Wu, X. L. Meng, Q. Huang, Y. F. Su, F. Wu and R. M. Kelly, J. Environ. Chem. Eng., 2022, 10, 108627 CrossRef CAS .
- C. L. Liu, J. X. Yu, J. Hu, J. B. Xu, A. Yu, T. T. Liu, Z. B. Wang, X. B. Luo, C. J. Deng, F. Luo, J. W. He and G. S. Zeng, J. Environ. Chem. Eng., 2024, 12, 112586 CrossRef CAS .
- W. J. Yu, Y. Li, B. C. Deng, J. Wang, G. Q. Mao, H. Tong, H. B. He and X. Y. Guo, J. Environ. Chem. Eng., 2024, 12, 114171 CrossRef CAS .
- X. H. He, Y. P. Wen, X. Y. Wang, Y. R. Cui, L. B. Li and H. Z. Ma, Waste Manage., 2023, 157, 8–16 CrossRef CAS .
- P. C. Zhao, Y. Li, X. Y. Wang, X. M. Liu, P. Gao, P. Guo, X. X. Wang, C. F. Wu and B. X. Shen, Sep. Purif. Technol., 2025, 357, 129988 CrossRef CAS .
- Y. X. Fan, Y. L. Kong, P. X. Jiang, G. H. Zhang, J. L. Cong, X. Y. Shi, Y. K. Liu, P. Zhang, R. Y. Zhang and Y. H. Huang, Chem. Eng. J., 2023, 463, 142278 CrossRef CAS .
- R. H. Zhang, Z. F. Meng, X. T. Ma, M. Y. Chen, B. Chen, Y. D. Zheng, Z. Y. Yao, P. Vanaphuti, S. Bong, Z. Z. Yang and Y. Wang, Nano Energy, 2020, 78, 105214 CrossRef CAS .
- Y. D. Zheng, R. H. Zhang, P. Vanaphuti, J. Z. Fu, Z. Z. Yang and Y. Wang, ACS Sustainable Chem. Eng., 2021, 9, 6087–6096 CrossRef CAS .
- E. Fan, L. Li, Z. P. Wang, J. Lin, Y. X. Huang, Y. Yao, R. J. Chen and F. Wu, Chem. Rev., 2020, 120, 7020–7063 CrossRef CAS .
- S. Z. Li, X. Wu, Y. Z. Jiang, T. Zhou, Y. Zhao and X. P. Chen, Waste Manage., 2021, 136, 18–27 CrossRef CAS .
- T. Nshizirungu, M. Rana, Y. T. Jo and J. H. Park, J. Hazard. Mater., 2021, 414, 125575 CrossRef CAS .
- J. Li, Y. Tian, B. Yang, B. Q. Xu, J. X. Hu and S. J. Yao, Sep. Purif. Technol., 2025, 354, 129035 CrossRef CAS .
- L. Li, J. B. Dunn, X. X. Zhang, L. Gaines, R. J. Chen, F. Wu and K. Amine, J. Power Sources, 2013, 233, 180–189 CrossRef CAS .
- W. F. Gao, C. M. Liu, H. B. Cao, X. H. Zheng, X. Lin, H. J. Wang, Y. Zhang and Z. Sun, Waste Manage., 2018, 75, 477–485 CrossRef CAS PubMed .
- L. Li, W. J. Qu, X. X. Zhang, J. Lu, R. J. Chen and F. Wu, J. Power Sources, 2015, 282, 544–551 CrossRef CAS .
- Y. K. Li, W. G. Lv, H. L. Huang, W. Y. Yan, X. K. Li, P. G. Ning, H. B. Cao and Z. Sun, Green Chem., 2021, 23, 6139 RSC .
- H. F. Li, A. Berbille, X. Zhao, Z. M. Wang, W. Tang and Z. L. Wang, Nat. Energy, 2023, 8, 1137–1144 CrossRef CAS .
- X. N. Li and W. S. Tong, J. Mater. Chem. A, 2024, 12, 19783 RSC .
- X. L. Su, X. Yang, H. Long, Y. H. Li, W. Y. Sun, T. L. Mo, H. X. Lyu, A. Cavaco-Paulo, H. B. Wang and J. Su, Int. J. Biol. Macromol., 2024, 279, 134929 CrossRef CAS .
- H. Y. Shen, L. Wang, F. M. Zhang, J. J. Luo, K. Han, Z. Zhang, C. Zhang, H. Yuan, Z. L. Wang and Y. K. Pang, Nano Energy, 2025, 136, 110677 CrossRef CAS .
- Q. Xin, Q. L. Wang, K. W. Luo, Z. W. Lei, E. M. Hu, J. W. Lv, H. Q. Wang, F. Liang, F. Hu and H. Q. Wang, Desalination, 2024, 585, 117758 CrossRef CAS .
- Y. Yu, H. T. Liu, P. Wang, X. W. Kong, H. C. Jin, X. M. Chen, J. M. Chen and D. Z. Chen, J. Hazard. Mater., 2025, 481, 136527 CrossRef CAS PubMed .
- W. Gao, S. H. Qin, K. Li, M. M. Wu, M. Yang and H. J. Shao, Sep. Purif. Technol., 2025, 359, 130857 CrossRef CAS .
- K. Kitao, M. Tani, M. Yamane, S. Inui, M. Yamada and T. Norisuye, Colloids Surf., A, 2024, 690, 133807 CrossRef CAS .
- M. I. Khan, S. X. Wang, E. Ullah, M. Sajjad, L. B. Zhang and L. K. Fu, J. Mol. Liq., 2024, 410, 125545 CrossRef CAS .
- C. Canciani, E. Colleoni, V. P. Sarvothaman, P. Guida and W. L. Roberts, Environ. Adv., 2024, 17, 100570 CrossRef CAS .
- F. J. Twagirayezu, Phys. Lett. A, 2021, 400, 127323 CrossRef CAS .
- İ. Deveci and B. Mercimek, Ultrason. Sonochem., 2019, 51, 197–205 CrossRef PubMed .
- S. Chakraborty, G. Deka and H. Dutta, Food Phys., 2025, 2, 100040 CrossRef .
- L. Peng, Q. Y. Long, C. Z. Liang, L. C. Fang and Y. F. Xu, Biochem. Eng. J., 2025, 219, 109731 CrossRef CAS .
- L. H. Fan, Y. Q. Lu, L. Y. Yang, F. F. Huang and X. K. Ouyang, J. Colloid Interface Sci., 2019, 554, 48–58 CrossRef CAS PubMed .
- Z. X. Huang, W. Y. Li, S. Y. Xu, X. P. Xu and M. R. Ou, Int. J. Biol. Macromol., 2024, 279, 135004 CrossRef CAS .
- S. H. Zhang, X. Y. Yuan, M. T. Li, K. Y. Gong, C. Y. Zhou, X. P. Gao, M. Y. Li and F. Q. Fan, Int. J. Biol. Macromol., 2025, 290, 138918 CrossRef CAS PubMed .
- Y. Hui, R. K. Liu, J. W. Lan, L. Z. M. Li, Z. H. Xiao, A. R. Xu and X. F. Wei, Int. J. Biol. Macromol., 2024, 272, 132842 CrossRef CAS .
- Y. L. Feng, H. Wang, J. H. Xu, X. S. Du, X. Cheng, Z. L. Du and H. B. Wang, J. Hazard. Mater., 2021, 416, 125777 CrossRef CAS PubMed .
- Q. X. Yuan, Y. H. Pan, L. B. Yang, X. B. Yin, Y. Z. Wei and X. P. Wang, Chem. Eng. J., 2025, 505, 159595 CrossRef CAS .
- X. T. Lin, Z. K. Wang, X. X. Jiang, T. X. Ning, Y. Jiang and A. X. Lu, Ceram.
Int., 2023, 49, 38499–38508 CrossRef CAS .
- P. T. Liu, K. Freiberg, D. C. Grinter and V. Sivakov, Results Phys., 2024, 61, 107733 CrossRef .
- K. Xing, S. Cao, Y. S. Song, M. Y. Chen, Z. W. Gu, Q. Y. Li, X. X. Han, B. S. Zou and J. L. Zhao, Appl. Surf. Sci., 2024, 673, 160887 CrossRef CAS .
- F. F. Zhang, H. J. Shao, Y. F. Diao, K. Li, W. Gao and S. H. Qin, J. Membr. Sci., 2024, 690, 122210 CrossRef CAS .
- D. D. Miller and S. S. C. Chuang, J. Phys. Chem. C, 2016, 120, 1147–1162 CrossRef CAS .
- Y. Wen, J. S. Huang, H. Liu, J. Li, T. Y. Liu, A. S. Belousov, S. Yang and H. Li, Chem. Eng. J., 2025, 515, 163596 CrossRef CAS .
- H. F. Li, Y. F. Chen, Z. W. Cui, Y. S. Su, Z. Q. Liu, X. L. Dong, Z. L. Wang and W. Tang, J. Hazard. Mater., 2025, 484, 136769 CrossRef CAS PubMed .
- L. J. Li, J. L. Zhao, X. D. Zhao, Z. M. Zhou and G. H. Jing, Sep. Purif. Technol., 2025, 354, 129005 CrossRef CAS .
- L. P. He, S. Y. Sun, Y. Y. Mu, X. F. Song and J. G. Yu, ACS Sustainable Chem. Eng., 2017, 5, 714–721 CrossRef CAS .
- X. H. Zhang, H. B. Cao, Y. B. Xie, P. G. Ning, H. J. An, H. X. You and F. Nawaz, Sep. Purif. Technol., 2015, 150, 186–195 CrossRef CAS .
- X. P. Chen, B. L. Fan, L. P. Xu, T. Zhou and J. R. Kong, J. Cleaner Prod., 2016, 112, 3562–3570 CrossRef CAS .
- F. Meng, Q. C. Liu, R. Kim, J. X. Wang, G. Liu and A. Ghahreman, Hydrometallurgy, 2020, 191, 105160 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab,
Youchang
Yang
b,
Mingmi
Wu
a,
Ting
Lei
b,
Yufei
Liu
a and
Huiju
Shao
*ab,
Youchang
Yang
b,
Mingmi
Wu
a,
Ting
Lei
b,
Yufei
Liu
a and
Huiju
Shao









