 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCharging dynamics of angstrom-scale pores of MXene electrodes with ionic-liquid electrolytes
Ming Chen ,
Sebastian Ostoja-Petkowski and
Alexei A. Kornyshev*
,
Sebastian Ostoja-Petkowski and
Alexei A. Kornyshev*
Department of Chemistry, Faculty of Natural Sciences, Imperial College, Molecular Sciences Research Hub, White City Campus, Wood Lane, London, W12 0BZ, UK. E-mail: a.kornyshev@imperial.ac.uk
First published on 17th January 2026
Abstract
Transition-metal carbides, MXenes, with angstrom-narrow slit pores are promising electrodes for high-power energy storage, particularly interesting when used with non-volatile ionic-liquid electrolytes. Yet pore charging of such slits remains challenging. Here, we investigate the charging dynamics of ultrathin MXenes immersed in ionic liquid using constant-potential molecular dynamics simulations. Contrary to the prevailing view that the charging process is governed predominantly by pore size, our results uncover a voltage-regulated shift of kinetic control: at low polarization, the charging time is limited mainly by geometric confinement, whereas at high polarization, the relaxation becomes dictated by the applied voltage itself. By introducing the time-resolved charging parameter, we reveal that charging is inherently collective and dynamic, rather than a simple monotonic ion accumulation. The concomitant non-monotonic change in in-pore conductivity further substantiates this picture, reflecting a sequence of voltage-driven structural transitions—from ionic crowding, to field-induced disorder, and finally to a highly packed ionic layer under strong electric fields. Our study unravels the fine details within the picture of angstrom-scale MXene charging dynamics, crucial for understanding the performance of MXene-based supercapacitors.
1. Introduction
Electric energy storage (EES) is playing a crucial role in the global transition from fossil-fuel-based energy systems toward electricity-driven renewable infrastructures.1–3 Among EES technologies, supercapacitors are valued for their non-faradaic, potential-driven charge storage, delivering high-power, ultra-fast charging, and long cycle durability for high-power applications.1–5 Their broader deployment, however, is impeded by the intrinsically limited energy density.6 Enhancing their energy storage without compromising the power density would enable their more widespread application.7–9To address this, sustained research efforts have been devoted to enhancing the energy density of supercapacitors through increasing the electrode/electrolyte interface via finer porosity, widening the operating voltage, and improving charge accommodation.8,9 Ionic liquids (ILs), with their broad electrochemical stability window (about 3 V or more) and excellent thermal stability, have therefore attracted significant interest as promising electrolytes.8 Parallel to electrolyte development, rational engineering of electrode architecture remains essential.6,10 A well-established principle is that high capacitance per electrode volume or weight and energy efficiency are achieved when pore walls are nanoscale-thin and the pore dimensions closely match the size of (de)solvated ions.11–14 Conventional porous carbon electrodes, however, comprise irregular, highly tortuous pore networks with broad size distributions, complicating deterministic pore tuning at the angstrom-to-nanometer scale.8 The advent of two-dimensional (2D) layered materials has opened new opportunities in this regard. MXenes (Mn+1XnTx), a family of transition-metal carbides and nitrides first reported by Naguib et al.,15 combine metallic conductivity, rich surface terminations, and exceptional ion-storage capability.16–18 Critically, their lamellar architecture enables precise control over interlayer spacing, allowing the creation of ultranarrow, angstrom-scale slit pores that can be engineered to match the dimensions of electrolyte ions. Ti3C2Tx, for example, can deliver volumetric capacitances approaching 1500 F cm−3.10 Coupling MXenes with ILs, therefore, represents a particularly promising strategy for achieving high-energy-density supercapacitors.19,20
Despite growing progress in MXene-based supercapacitors, energy storage and ion-charging dynamics in angstrom-narrow pores remain insufficiently resolved. In nanopores built from graphene/carbon electrodes or conductive metal–organic frameworks (MOFs), the charging process has been extensively investigated, yielding a detailed picture of how ionic liquids respond when confined.7,8,11,21–25 The confinement has been shown to induce superionic regimes and quasi-two-dimensional ordering.22 Strikingly, further confinement does not necessarily slow transport, but may enhance ion mobility, which can even exceed bulk values with quasi-two-dimensional ionic arrangements.21 A recent simulation by Mo et al.11 revealed a non-monotonic relationship between pore size and charging rate: some sub-nanometer pores actually charge faster than wider pores. This unexpected acceleration of charging—rather than the conventional slowing down—is attributed to a pore-size-induced structural re-arrangements of ions.11 However, these physical pictures arise from slit or pore models without chemical complexity on the pore walls.
By contrast, MXene slit pores introduce additional complexity through heterogeneous surface terminations (–O, –F, –OH, etc.) and intrinsic electrode polarizability.15,26 Given the sensitivity of in-pore ionic structure to both confinement and interfacial character, structural transitions in ion distributions inside the pores might be significantly altered by the structure and properties of MXene/IL interfaces. Although Xu et al.27 provided valuable insights into MXene-based supercapacitors and documented structural responses during charge–discharge cycles, their simulations relied on the constant-charge method, which constrains the electrode charges and therefore cannot faithfully represent potential-controlled charging processes. The constant-potential method (CPM) addresses this limitation by allowing electrode charges to fluctuate self-consistently in response to the electrolyte under fixed electrode potentials.28 While Sampaio et al.19 have applied the CPM to MXene systems, the emphasis of their work was on equilibrium properties, including energy density and capacitance. In contrast, the collective transport phenomena expected under strong confinement have not been fully resolved. Consequently, a dedicated investigation is required to clarify whether and how such structure-driven charging acceleration manifests under realistic MXene confinement.
Herein, to elucidate the energy-storage performance, especially the charging mechanism of a supercapacitor based on MXene electrodes and IL electrolytes, we conduct constant-potential MD simulations for Ti3C2O2 electrodes with pure 1-ethyl-3-methylimidazolium tetrafluoroborate ([Emim][BF4]) electrolyte. Different from earlier pore-size-dependent charging views, we show that confined 2D MXene nano-slits charge through a complex voltage-controlled rebalance in time, rather than a single static mode established after a potential step. Specifically, a voltage jump first drives rapid counter-ion enrichment, followed by faster co-ion clearance, before the final settlement of ions inside the pore, subject to the value of the set electrode potential.29 This unequal pacing of ion entry and ion exit determines the time dependence of the transient charging current. While the standard Transition Line Model (TLM) cannot give a closed analytical form for the transient current, when the equilibrium value of capacitance depends on electrode potential (which, in most cases, is the case), TLM can still, to some extent, recover the main relaxation trends and time constants when the fitted curves are supplied with realistic, voltage-resolved capacitance as input. In this paper, we investigate the effect of voltage and confinement on the charging dynamics of angstrom-scale MXene pores, thereby advancing our understanding of ion-guided energy storage in MXene-based supercapacitors.
2. Method
2.1. Molecular dynamics simulation
As illustrated in Fig. 1, the MD simulation system consists of two identical and symmetric conductive MXenes, Ti3C2O2, immersed in [Emim][BF4] electrolyte. Four different MXene electrodes, with interlayer spacing varying from 0.7 nm to 1.2 nm, were investigated. Since operando X-ray diffraction measurements indicate that the interlayer distance of MXenes varies only slightly (within approximately 0.2 Å) during the charging process, and reversibly returns during the subsequent discharge, the MXene layers can be reasonably approximated as rigid within the considered potential window.30 The Lennard–Jones force-field parameters for MXene atoms are derived from Xu et al.27 For the electrolyte, coarse-grained force fields are adopted for [Emim][BF4],31 which have been proven to correctly reproduce the conductivity and solvation structure while dramatically conserving the computational resources.31–34 Both the positive and negative electrodes exhibit symmetric structures, with a length of 6.0 nm. For both configurations, the simulation cells were chosen to be large enough to reproduce the bulk state in the central region of the electrolyte reservoir connected with the two electrodes, and periodic boundary conditions were applied in all directions. Specific system parameters are given in Fig. 1. | ||
| Fig. 1 Schematics of the molecular dynamics simulation setup. (a and b) Front view (a) and top view (b) of the MD system. The pore size, d, is defined by the gap between the surfaces of two inner oxygen sheets, ranging from 0.7 nm to 1.2 nm, and the slit length is 6.00 nm along the Z-direction. The coarse-grained model was adopted for IL [Emim][BF4]. The gray, black, and red spheres represent Ti, C, and O atoms, respectively, and the remaining colored spheres represent [Emim][BF4]. (c) Structure of Ti, C, and O atoms. (d) Schematic representations of the coarse-grained models of the 1-ethyl-3-methylimidazolium tetrafluoroborate ([Emim][BF4]). | ||
2.2. Constant-potential implementation
Simulations were conducted using the customized MD software GROMACS.35,36 In contrast to conventional constant-charge models (CCMs),27 where fixed and uniformly distributed atomic charges are prescribed on electrode atoms, the CPM23,28,36,37 was employed to allow fluctuations of the charges on electrode atoms to ensure an adequate description of the electrode polarization effects in the presence of electrolytes. Cyclic voltammetry measurements indicate that MXene-based supercapacitors operating in aprotic ionic-liquid electrolytes are dominated by electric double-layer charging within the electrochemical stability window (approximately 3–4 V), with negligible pseudocapacitive contributions.37–39 Accordingly, the present simulations focus on electric double-layer capacitance (EDLC) behavior. The V-rescale thermostat was utilized to couple the temperature to 350 K in the NVT ensemble.40 The cutoff radii for short-range electrostatics and van der Waals interactions were set at 1.2 nm, while long-range electrostatic interactions were computed using the particle-mesh Ewald (PME) algorithm.41 The PME k-space grid spacing was set to 0.1 nm. The system was first annealed at 500 K for 5 ns to eliminate initial structural bias, followed by a 5 ns cooling stage to 350 K. The temperature was then held at 350 K for 10 ns to reach a bulk-equilibrated configuration under null applied potential. To determine the microstructure and capacitance, simulations were performed under applied cell voltages (the potential difference between the two electrodes) ranging from 0 to 4 V for 10 ns, ensuring equilibrium was reached, followed by 10 ns production runs for analysis. To investigate the charging dynamics, three independent runs were performed to certify the accuracy of the simulation results. Note that the employment of the CPM should lead to similar results to the modification of interionic interactions in the pore by the superionic effect.292.3. Transmission-line model
As illustrated in Fig. 2a, the charging process generally involves the electrostatic attraction of counter ions (anions for positively polarized electrodes, and cations for negatively polarized electrodes) to the electrode and the exclusion of the co-ions (cations for positively polarized electrodes, and anions for negatively polarized electrodes), forming an electrical double-layer structure.42–44 Simultaneously, the electrolyte ions encounter resistance upon permeating the nanopores. The corresponding equivalent circuit diagram is presented in Fig. 2b, which follows the common transmission-line model (TLM), in which the z-axis denotes the direction of the MXene pore (z = 0 denoting the entrance of the pore, and z = L referring to the end of the pore). Along the axial direction, parameters R and C stand for the ion diffusion resistance and capacitance per unit length, respectively. Considering a micro-element dz, the resistance is Rdz, and the relative interface capacitance is Cdz. It is worth noting that L is half the length of the slit for open systems, whereas it equals the slit length for closed systems.23,45 | ||
| Fig. 2 Schematic of the transmission-line model. (a) Schematic of the charging process within a typical nanopore. (b) Corresponding equivalent circuit in the transmission-line model. R and C are the resistance and the capacitance ‘per unit length’ inside the electrodes, respectively. | ||
Owing to the high electronic conductivity of Mxenes,18,46 as compared to the ionic conductivity inside the pore (0.3–5 S m−1),23 the MXene electrode can be reasonably treated as equipotential during charging, which is a key assumption in TLM theory. For simplicity, we set the electrode potential to 0, and consider Ψ = Ψ(z, t) as the potential drop between the electrode and the middle of the ionic film inside the pore. The ionic current is defined as i = i (z, t). Applying Ohm's law of resistance and the response of the capacitor, one gets:42,43
| Ψ − (Ψ + dΨ) = iRdz | (1) |
 | (2) |
Combining these two equations leads to the master equation of the TLM:
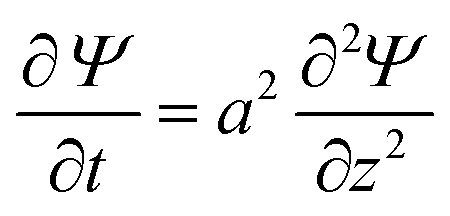 | (3) |
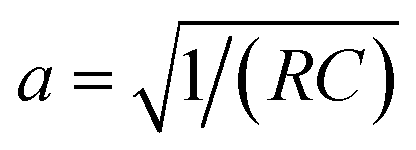 .
.
In this scenario, a constant potential difference is applied between the positive and negative electrodes of the supercapacitor, representing a conventional charging methodology commonly employed in industrial applications. The temporal evolution of the total charge (Q) on a given electrode comes out as a solution of eqn (3) (for more details see ref. 45) as follows:
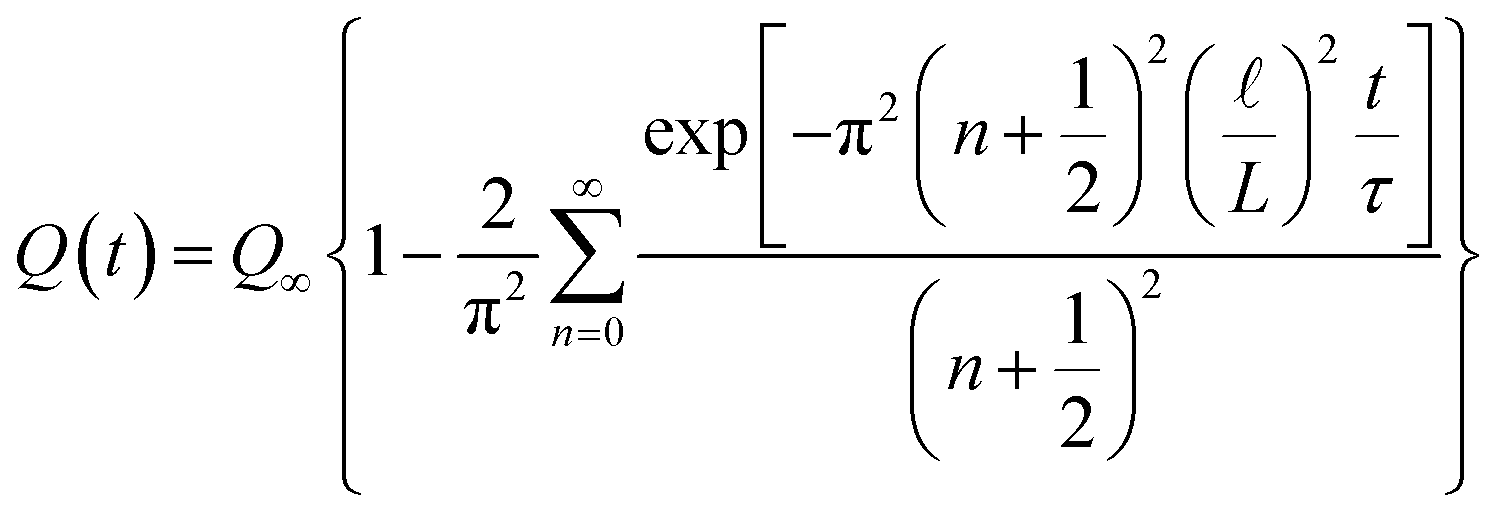 | (4) |
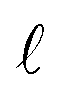 is the pore volume divided by its surface area (for a slit pore it is just half of the pore width) and L is half the length of the pore in an open slit system (with entrances on both sides of the pore); τ denotes the intrinsic relaxation time:21,23
is the pore volume divided by its surface area (for a slit pore it is just half of the pore width) and L is half the length of the pore in an open slit system (with entrances on both sides of the pore); τ denotes the intrinsic relaxation time:21,23
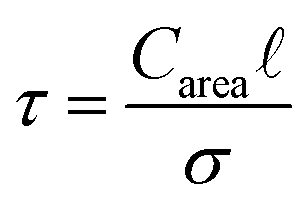 | (5) |
 .45
.45
3. Results and discussion
3.1. Equilibrium charge and ion distribution inside MXene nano-sheets
To uncover how angstrom-nanoscale confinement affects ionic behaviour, we first concentrate on the equilibrium ion arrangement inside MXene slit pores, as shown in Fig. 3. The ion number-density profiles along the pore-normal (x direction) reveal a continuous structural rearrangement dictated by pore width (Fig. 3a–d). The electrode potential drop, ΔΨ, is defined as the potential between the electrode and the bulk of the electrolyte reservoir relative to the potential of zero charge (PZC), that is: ΔΨ = (Ψelectrode − Ψbulk) − (Ψelectrode − Ψbulk)|PZC.47 At the smallest pore sizes of 0.7 and 0.8 nm, under negative polarization (ΔΨ = −1 V), cations and anions condense in a single, highly mixed layer centered near the pore midplane, indicating that the available free volume is only sufficient for single-plane packing (Fig. 3a and b). As the pore width increases to 1.0 nm (Fig. 3c), a more conventional stratified structure begins to emerge. The cation density develops a primary maximum at approximately 0.43 nm from the MXene surface, driven by stronger coulombic interactions between the positively charged ions and the polarized MXene wall. A pronounced shoulder appears at ∼0.33 nm from each of the pore walls, which progressively enhances with increasing electrode polarization. In contrast to cations, anions remain preferentially located near the center of the pore and are gradually excluded from the slit as the negative polarization strengthens. As shown in Fig. 3d, in the wider 1.2 nm pore, cation and anion populations become spatially decoupled, adopting distinct planes across the slit. The cation density in this regime simplifies to a single dominant peak, markedly different from the dual-peak cation structure observed at 1.0 nm. This shift reflects the fact that confinement continues to impose orientational constraints even when volumetric exclusion becomes less severe, altering how ions pack and screen the surface charge. | ||
| Fig. 3 Ion structure within the pore under negative polarization (−1 V). Columns in this figure correspond to the data obtained for different pore widths: 0.7, 0.8, 1.0, and 1.2 nm. First row (a–d): layer-resolved ion number-density distributions. Second row (e–h): corresponding axial (through-pore) ion number-density distributions. Third row (i–l): corresponding 2D in-pore number-density maps from a top-down view. In all panels, the solid red and dashed blue lines denote cations and anions, respectively. | ||
To further validate these structural trends, we examine the ionic distribution in the in-pore direction (z-axis, Fig. 3e–h). A motif characterized by “–cation–anion–cation–” alternations is observed at the PZC, where ions organize into lock-in stripes with lattice-like interlocking in the slit. Under the negatively polarized electrode, at 0.7 nm (Fig. 3e), both species still exhibit noticeable static ion-wave modulations, reflecting strong inter-ionic correlations under extreme confinement. However, the lateral ion distributions become much more homogeneous and almost featureless at 0.8 nm (Fig. 3f), suggesting minimal frustration in-plane and transiently weakened ion–wall coupling. When the pore expands to 1.0 nm (Fig. 3g), ion-wave patterns reappear for both species as layered structures begin to nucleate, yet still partially overlap. At 1.2 nm (Fig. 3h), a clear contrast emerges: anions populate nearly uniformly along the pore, while cations maintain pronounced stripe-like layering motifs near the MXene wall, confirming that interfacial accumulation of counter-ions with quasi-periodic lateral distribution becomes the defining structural pattern in wider pores.
Notably, a pronounced accumulation of ions is observed near the MXene pore inlet, characteristic of an entrance effect arising from the ‘competition’ between strong electrostatic attraction toward the charged surface and confinement-induced steric and entropic constraints that hinder ion penetration into the slit.7,48–50 This effect is further amplified in narrower channels, where reduced free volume and overlapping EDLs increase the barrier for ion entry, causing the pore entrance to act as a bottleneck that transiently concentrates ions at the pore entrance, before they get squeezed into the pore.48–50
The XZ-plane 2D number-density maps (Fig. 3i–l) visualize these trends more clearly. Collectively, these results demonstrate that the pore width fundamentally regulates ion packing symmetry and spatial delocalization, providing a microscopic basis for the pore size-dependent ion distribution behaviour observed in MXene-based supercapacitors.
To understand the voltage-driven ion redistribution in MXene nano-slits, we quantified the equilibrium charge-balancing mechanism. We evaluated the total change in ion population inside each MXene pore by resolving the relative contributions of the net co-ion expulsion and counter-ion insertion, as compared to their equal presence at the PZC, expressed as Δρ = ρ(Δψ) − ρ(0V). As shown in Fig. 4, a quite expected general trend is observed across all slit widths: with increasing cell voltage from 0 to 2 and 4 V, counter-ions are progressively enriched within the pore, generating positive Δρ values; meanwhile, co-ions are gradually depleted, producing negative Δρ values.
 | ||
| Fig. 4 Voltage-induced changes in ion population inside MXene pores. (a–d) Change in the equilibrium pore ion population, Δρ = ρ(Δψ) − ρ(0V), in pores with widths of 0.7 nm (a), 0.8 nm (b), 1.0 nm (c), and 1.2 nm (d). The negative values indicate a decrease in the number of ions inside the pore. | ||
However, the magnitude of co-ion exclusion is not uniform but strongly dependent on slit width. In the most confined pores, 0.7 and 0.8 nm, co-ion expulsion is strongest, indicating that additional counter-ion insertion is volumetrically compensated by simultaneous co-ion removal due to severe steric constraints. As the pore expands to 1.0 nm and 1.2 nm, the symmetry of the exchange relaxes, suggesting that the slit can support partial interfacial ion accumulation without enforcing strict one-for-one ion displacement. This trend demonstrates that confinement fundamentally regulates not only the spatial packing of ions but also the equilibrium ion population inside MXene pores.
We subsequently evaluated the capacitance of the MXene slit pores. As summarized in Table 1, the single-electrode specific integral capacitance of the supercapacitor can be estimated via 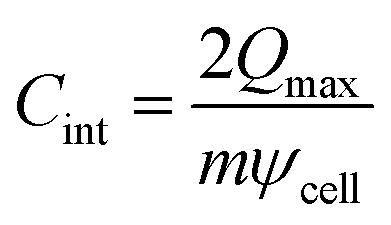 , where Qmax represents the maximum charge accumulated on the electrode, m is the electrode mass, and ψcell is the applied voltage between the positive and negative electrode surfaces. The comparison across pores of 0.7, 0.8, 1.0, and 1.2 nm shows that for slits up to 1.0 nm, the capacitance changes only marginally between 2 and 4 V, staying close to 20 F g−1, indicating pore-width-independent behaviour under strong confinement. When the pore expands beyond 1.0 nm, additional charge can be accommodated inside the pore, allowing the capacitance to increase by approximately 40%, reaching 28.42 F g−1 at 2 V. This increase stems from a wider MXene slit, which enables interfacial counter-ion accumulation without enforcing equivalent co-ion expulsion (Fig. 4). The resulting capacitance values are in quantitative agreement with those reported by Sampaio et al.,19 for the Ti3C2F2 system (15–30 F g−1), and with similar values obtained for Ti3C2(OH)2 systems.20
, where Qmax represents the maximum charge accumulated on the electrode, m is the electrode mass, and ψcell is the applied voltage between the positive and negative electrode surfaces. The comparison across pores of 0.7, 0.8, 1.0, and 1.2 nm shows that for slits up to 1.0 nm, the capacitance changes only marginally between 2 and 4 V, staying close to 20 F g−1, indicating pore-width-independent behaviour under strong confinement. When the pore expands beyond 1.0 nm, additional charge can be accommodated inside the pore, allowing the capacitance to increase by approximately 40%, reaching 28.42 F g−1 at 2 V. This increase stems from a wider MXene slit, which enables interfacial counter-ion accumulation without enforcing equivalent co-ion expulsion (Fig. 4). The resulting capacitance values are in quantitative agreement with those reported by Sampaio et al.,19 for the Ti3C2F2 system (15–30 F g−1), and with similar values obtained for Ti3C2(OH)2 systems.20
| 0.7 nm | 0.8 nm | 1.0 nm | 1.2 nm | |
|---|---|---|---|---|
| 2 V | 19.83 F g−1 | 19.29 F g−1 | 21.24 F g−1 | 28.42 F g−1 |
| 4 V | 20.61 F g−1 | 19.88 F g−1 | 20.92 F g−1 | 25.31 F g−1 |
3.2. Charge accumulation in porous MXene electrodes
To quantify how the applied potential drives ion insertion and removal in the Ti3C2O2 pore, the charging parameter, X, originally introduced by Forse et al.51 was calculated via:
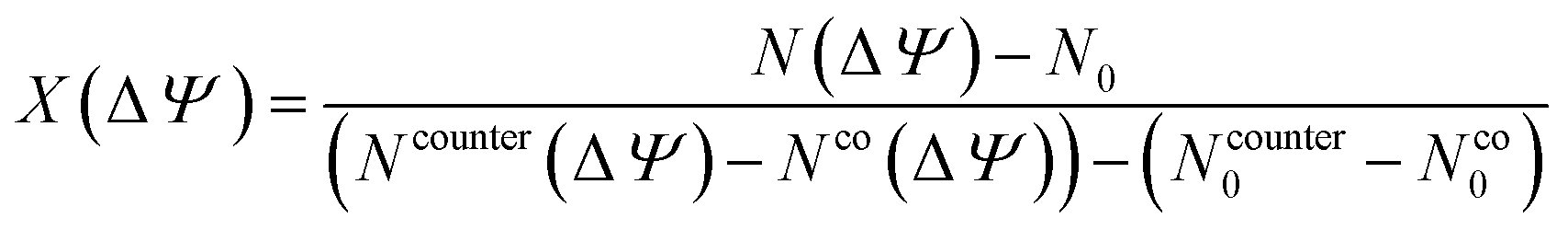 | (6) |
The calculated X values for all pore sizes and electrode potentials are summarized in Table 2. In the narrowest pores (0.7 and 0.8 nm), X remains negative at −2 V (−0.43 and −0.59, respectively) and −1 V (−0.53 and −0.73, respectively), indicating that the charging process in these strongly confined slits is dominated by co-ion (anion) exclusion coupled to limited counter-ion insertion. At 1.0 nm, the sign of X changes, reaching 0.18 at −2 V, showing that the pore can transiently accommodate positive charge without enforcing symmetric ion removal. When the slit width further increases to 1.2 nm, X becomes negative again (−0.15 at −2 V and −0.48 at −1 V), indicating that although wider pores favor interfacial counter-ion (cation) accumulation, enhanced polarization can strengthen co-ion exclusion and reduce the net ionic population available for screening. On the positive electrode, X remains close to zero for the 0.7 nm pores at 2 V (0.03), but it stabilizes around 0.2 in 1.0 and 1.2 nm slits. The positive values of the charging parameter in larger pores are in line with the experimental observations for Ti3C2F2–[Emim][BF4]-based supercapacitors,19 indicating that surface charging can now proceed through steady counter-ion buildup with less need for co-ion withdrawal.
| 0.7 nm | 0.8 nm | 1.0 nm | 1.2 nm | ||
|---|---|---|---|---|---|
| Negative electrode | −2 V | −0.43 | −0.59 | 0.18 | −0.15 |
| −1 V | −0.53 | −0.73 | −0.06 | −0.48 | |
| Positive electrode | 1 V | 0.22 | −0.18 | 0.21 | 0.19 |
| 2 V | 0.03 | −0.35 | 0.21 | 0.16 | |
These equilibrium charging parameters describe how interface charge is balanced by ion addition or removal, but they do not answer a key dynamic question: how do ions actually move in and out of the MXene slit as the electrode begins to charge? To probe this, we tracked the time-dependent in-pore charging parameter X(ΔΨ, t) for a 1.0 nm MXene electrode, as shown in Fig. 5a. After applying a potential, the pore does not charge through a single static ionic configuration. Instead, X(ΔΨ, t) evolves continuously over several nanoseconds, highlighting the kinetic competition inherent to confined nano-slits.
 | ||
| Fig. 5 Charging mechanism parameter X in a 1.0 nm MXene pore. (a) Time evolution of the voltage-dependent charging parameter X(ΔΨ, t) after a potential step, illustrating the relaxation toward equilibrium. The color scale denotes X(ΔΨ, t) resolved over an applied voltage ΔΨ, after time t. (b) Equilibrium values of X at different applied voltages, showing a clear asymmetry in charging response with applied voltage. | ||
Under weak polarization (e.g., |ΔΨ| ≤ 1 V), X(t) reveals a striking two-speed dynamic competition. A prominent transient feature appears in both positive and negative polarizations: counter-ions rapidly migrate toward the pore walls due to strong electrostatic attraction. This sudden ion influx can briefly oversaturate the interfacial region within the first ∼100 ps, pushing X to exceed the ideal permselective adsorption limit, often rising above 1 and occasionally reaching ∼2. This corresponds to a short-lived overcharging state rather than a stable ion-selection effect. Immediately afterward, the system begins a rebalancing process: steric confinement and electrostatic repulsion drive co-ions out of the pore more aggressively than counter-ions can be continually inserted. This slower, confinement-restricted process produces sharp negative excursions of X(t), sometimes dropping below −1, indicating that the pore experiences the initial counter-ion overshot followed by confinement-forced rebalancing, before relaxing toward ion exchange at longer times. This overshoot behaviour is also observed for weak polarization at the negative electrode, confirming that it is a general kinetic response of sub-nanometer confinement, not a polarity-specific anomaly. The overshoot is therefore a direct signature of this dynamic mismatch between the fast insertion process and the slower expulsion process, reflecting a short-lived overcharging state followed by confinement-forced rebalancing.
Crucially, such complex transient dynamics are significantly suppressed within higher polarizations (|ΔΨ| > 1 V). In this regime, counter-ions are inserted more steadily and co-ion exclusion becomes less abrupt, giving X(t) a calm stabilization toward the final value without strong oscillation. Note that a similar picture was predicted in a phenomenological theory of charging of nanoscale pores.29 Increasing the pore size from 1.0 to 1.2 nm preserves the qualitative charging mechanism (Fig. S1–S4), while substantially reducing the overshoot amplitude and accelerating the relaxation of X(t) toward equilibrium (within ∼3 ns).
The final equilibrium X values were extracted across the full voltage region, as shown in Fig. 5b. At equilibrium, distinct asymmetrical behaviour (related to the shapes and volumes of cations and anions) emerges between the electrodes. On the positive electrode, the 1.0 nm MXene slit pores favor ion exchange, but counter-ion (anion – a smaller ion) insertion remains slightly stronger than co-ion expulsion. On the negative electrode, however, the mechanism diverges for moderate polarization steps, with X becoming negative (∼−0.43), showing that anion exclusion is more dominant than cation insertion under sterically constrained negative interfacial charging. Importantly, at more negative polarizations, X begins to increase, gradually approaching zero and tending toward positive values, indicating that the charging mechanism shifts from anion expulsion to counter-ion/co-ion exchange.
Overall, charging in angstrom-scale MXene slit pores is not dictated by a simple ion arrangement after a voltage step. Instead, it proceeds through a collective mode in which ions migrate in and out of the slit through a time-dependent rebalance between counter-ion insertion and co-ion exclusion. This cooperative mechanism—revealed here for MXene–IL pores for the first time—highlights a complex yet physically coherent dynamic picture of in-pore charging.
3.3. Charging dynamics
We now turn to the charging process by tracking the time evolution of the accumulated charge on the MXene electrodes under different applied voltages (Fig. 6). Earlier research has made major progress in rationalizing the charging dynamics of ILs confined in sub-nanometer slit pores.11,19,21,24,29 Even in electrolytic solutions, not speaking about ILs, when the pore width approaches the electric double-layer thickness, ions tend to pack in very close proximity, leading to strong ion–ion coupling and correlated motion that cannot be described well by classical models that assume dilute or weakly interacting ions.21 Mesoscale models, particularly the TLM, have been shown to capture surface charge relaxation on polarizable quasi two-dimensional pores to some extent.21,23,24 This motivates us to fit the charging curves using TLM expressions, rather than a semi-empirical bi-exponential form,19 following the schematics outlined in the Method section (Fig. 2). It should be noted that the standard TLM solution works strictly under the assumption that pore capacitance does not change with voltage.45 Even though this is a known limitation, we keep eqn (4) and (5) as a reasonable approximation, using the measured capacitance at each applied potential as an effective input to the TLM. | ||
| Fig. 6 Charging process at the nanoscale. (a–d) Evolution of the total charge (Q) on layered Ti3C2O2 electrodes of supercapacitors with [Emim][BF4], dependent on the pore size of 0.7 nm (a), 0.8 nm (b), 1.0 nm (c), and 1.2 nm (d). e is the elementary charge. The dashed lines represent the fitting of MD data by the transmission-line model. (e and f) Intrinsic relaxation time (e) and equivalent ionic conductivity (f) of [Emim][BF4] in the MXene pores at different cell voltages. | ||
Fig. 6 presents the time evolution of the charge stored in our studied 0.7–1.2 nm slit pores of a MXene electrode, for 2 and 4 V ‘cell voltages’. Despite the aforementioned approximation, the TLM reproduces the overall relaxation trend surprisingly well and captures the characteristic timescale of the charging process, confirming that the TLM can, to a reasonable extent (Fig. 6a–d), reflect the dominant features of pore charging dynamics in confined ionic liquids.
Not unexpectedly, however, the narrowest 0.7 nm slit shows the largest fitting error. Here, the simulated curve relaxes faster at short times and flattens more slowly at long times than the TLM curve. A likely reason for this is that charge relaxation in this extreme confinement is affected more strongly by the voltage-dependence of capacitance, which cannot be fully described by a simple linear TLM approximation. More generally, at such extreme confinement, separation of resistive motion and capacitive charging, intrinsic to the TLM, must be taken with a pinch of salt.
Beyond tracking total charge, we then extracted the intrinsic relaxation time for the charging process based on the TLM (Fig. 6e). Under a 2 V potential step, the relaxation time displays a non-monotonic dependence on slit width, decreasing from 0.12 ns at 0.7 nm to 0.065 ns at 0.8 nm, but rising again as the pore widens to 1.2 nm. This behaviour agrees with earlier observations in graphene slit pores,11 reflecting a crossover in the dominant charging mechanism: at 0.7 nm, charging dynamics are governed by strong nanoconfinement, where single-layer ion packing severely restricts ion transport; meanwhile, increasing the pore width to 0.8 nm induces an ion-packing transition at a half-integer multiple of the effective ionic radius, thereby facilitating counter-ion recruitment and accelerating charge relaxation.11 This trend is further corroborated by the in-pore ionic conduction (Fig. 6f), where the in-pore ionic conductivity rises sharply from the one in the smallest slit to the one in 0.8 nm wide slit, increasing by nearly a factor of two, while remaining almost unchanged for further pore widening. In contrast, stepping the voltage window to 4 V fundamentally alters the charging response, giving nearly monotonic decay without observable oscillatory inversion, accompanied by a steady increase in in-pore ionic conductivity (Fig. 6f). This monotonic relaxation trend aligns with conventional views of pore charging, governed by progressive co-ion depletion and interfacial counter-ion insertion under strong voltage bias.21,52,53 The contrasting responses of each charging process indicate that charging kinetics in sub-nanopores are determined not only by confinement-regulated ion structure, but also by the applied voltage.
3.4. Voltage-affected in-pore ionic conductivity
To conduct a more detailed examination of the charging process, we investigated the voltage-affected, in-pore ionic conductivity (σz), calculated based on the Green–Kubo equation , where Jz(t) is the component of electric current along the z-direction.
, where Jz(t) is the component of electric current along the z-direction.
As seen in Fig. 7a, σz exhibits a non-monotonic dependence on electrode potential. On the positive electrode side, two distinct regimes emerge. At moderate electrode polarizations (in the interval between 0 V and 0.75 V), the conductivity initially decreases. This is attributed to ionic enrichment, which just leads to stronger crowding due to an increase in overall ion density (Fig. 7b and c), leading to a decrease in free ions (Fig. S5). The latter emerges from an influx of counter-ions with an insufficient ‘exodus’ of co-ions, and that crowding seems to restrict individual ion mobility within the pore. However, when the potential is further increased (e.g., ΔΨ > 0.75 V), the conductivity exhibits an anomalous increase. This may be caused by a disbalance in the population of counter-ions and co-ions (Fig. 7b), which creates a field-induced ion-pair decoupling (analogous to the Wien effect54) and a more disordered arrangement of ions that likely facilitates the self-diffusion and migration of ions.21 An interesting and opposite trend is observed on the negative electrode side. In the interval between 0 V and −0.75 V, the conductivity increases. This is primarily a result of ion depletion, as co-ion exclusion significantly outweighs counter-ion insertion, leading to a net reduction in ionic concentration and a less constrained (Fig. 7c), more disordered ionic arrangement. However, at more negative polarizations (below −1 V), the conductivity decreases again. This final decline is linked to the enrichment of the pore with counter ions (cations) and the formation of a highly packed ionic layer (not yet Wigner crystal-like, although the latter might form at further, even more negative electrode polarizations21).
 | ||
| Fig. 7 Mechanism of voltage-affected conductivity along the z-direction for a 1.0 nm slit pore. (a) Voltage-affected conductivity along the z-direction. The results of the calculation using the Green–Kubo equation. The accompanying snapshots (side and top views) illustrate the corresponding ion arrangements at negatively or positively polarized electrodes. Ti3C2O2 MXene sheets are shown in gray, cations in red, and anions in blue. (b and c) Voltage-affected cation and anion number densities (b) and total ion number density (c) in the pore. | ||
4. Conclusion
We have elucidated the voltage-driven charging mechanism of ILs confined within angstrom-narrow slit pores of a Ti3C2O2 MXene with various pore widths. Rather than being dictated solely by pore width, as reported in earlier work, we reveal that under low polarization, pore charging is governed primarily by the space available inside the slit, whereas at stronger polarizations, the ion response becomes increasingly defined by voltage rather than by pore width alone. Importantly, unlike previously reported equilibrium-only charging parameters, we provide an explicit description of how the charging parameter X(ΔΨ, t) evolves with charging time and applied polarization, uncovering a characteristic non-monotonic sequence that includes:(i) initial overshoot (transient overcharging with counter-ions), and
(ii) subsequent expulsion of co-ions, with
(iii) relaxation toward a final equilibrium value of the stored charge.
The non-monotonic in-pore conductivity further corroborates this mechanism, directly reflecting voltage-affected structural changes from ionic crowding to structural disorder and then to quasi-lattice states at high fields.
These findings complement existing understanding by offering new insight into the charging dynamics of MXene pores with ionic liquids. It should be noted that surface terminations of MXenes are expected to play an important role in charging behavior by modifying ion–surface interactions and local polarization, which may in turn influence both the magnitude and dynamics of charge storage.20 A systematic investigation of surface termination effects warrants further investigation.
Author contributions
M. C., S. O. P., and A. A. K. contributed to the design of the research. M. C. and S. O. P. performed calculations. A. A. K. supervised the work. All authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no competing interests.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: additional analyses of the charging mechanisms in MXene pores with different pore sizes, including ion number differences, voltage-dependent charging parameters, and the voltage-dependent proportion of free ions inside the pore. See DOI: https://doi.org/10.1039/d5fd00151j.
Acknowledgements
The authors acknowledge the support of the Leverhulme Trust under Grant No. RPG-2022-142 and Innovate UK – EXANST grant #10133246.References
- P. Simon and Y. Gogotsi, Perspectives for electrochemical capacitors and related devices, Nat. Mater., 2020, 19, 1151 CrossRef CAS PubMed.
- P. Simon, Y. Gogotsi and B. Dunn, Where Do Batteries End and Supercapacitors Begin?, Science, 2014, 343, 1210 CrossRef CAS PubMed.
- S. Fleischmann, Y. Zhang, X. Wang, P. T. Cummings, J. Wu, P. Simon, Y. Gogotsi, V. Presser and V. Augustyn, Continuous transition from double-layer to Faradaic charge storage in confined electrolytes, Nat. Energy, 2022, 7, 222 CrossRef CAS.
- X. Liu, D. Lyu, C. Merlet, M. J. A. Leesmith, X. Hua, Z. Xu, C. P. Grey and A. C. Forse, Structural disorder determines capacitance in nanoporous carbons, Science, 2024, 384, 321 CrossRef CAS PubMed.
- H. Shao, Y.-C. Wu, Z. Lin, P.-L. Taberna and P. Simon, Nanoporous carbon for electrochemical capacitive energy storage, Chem. Soc. Rev., 2020, 49, 3005 RSC.
- M. Salanne, B. Rotenberg, K. Naoi, K. Kaneko, P. L. Taberna, C. P. Grey, B. Dunn and P. Simon, Efficient storage mechanisms for building better supercapacitors, Nat. Energy, 2016, 1, 16070 CrossRef CAS.
- K. Breitsprecher, C. Holm and S. Kondrat, Charge Me Slowly, I Am in a Hurry: Optimizing Charge–Discharge Cycles in Nanoporous Supercapacitors, ACS Nano, 2018, 12, 9733 CrossRef CAS PubMed.
- S. Kondrat, G. Feng, F. Bresme, M. Urbakh and A. A. Kornyshev, Theory and Simulations of Ionic Liquids in Nanoconfinement, Chem. Rev., 2023, 123, 6668 CrossRef CAS PubMed.
- M. V. Fedorov and A. A. Kornyshev, Ionic liquids at electrified interfaces, Chem. Rev., 2014, 114, 2978 CrossRef CAS PubMed.
- M. R. Lukatskaya, S. Kota, Z. Lin, M.-Q. Zhao, N. Shpigel, M. D. Levi, J. Halim, P.-L. Taberna, M. W. Barsoum, P. Simon and Y. Gogotsi, Ultra-high-rate pseudocapacitive energy storage in two-dimensional transition metal carbides, Nat. Energy, 2017, 2, 17105 CrossRef CAS.
- T. Mo, S. Bi, Y. Zhang, V. Presser, X. Wang, Y. Gogotsi and G. Feng, Ion Structure Transition Enhances Charging Dynamics in Subnanometer Pores, ACS Nano, 2020, 14, 2395 CrossRef CAS PubMed.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P. L. Taberna, Anomalous increase in carbon capacitance at pore sizes less than 1 nanometer, Science, 2006, 313, 1760 CrossRef CAS PubMed.
- E. Raymundo-Piñero, K. Kierzek, J. Machnikowski and F. Béguin, Relationship between the nanoporous texture of activated carbons and their capacitance properties in different electrolytes, Carbon, 2006, 44, 2498 CrossRef.
- C. Largeot, C. Portet, J. Chmiola, P.-L. Taberna, Y. Gogotsi and P. Simon, Relation between the Ion Size and Pore Size for an Electric Double-Layer Capacitor, J. Am. Chem. Soc., 2008, 130, 2730 CrossRef CAS PubMed.
- M. Naguib, O. Mashtalir, J. Carle, V. Presser, J. Lu, L. Hultman, Y. Gogotsi and M. W. Barsoum, Two-Dimensional Transition Metal Carbides, ACS Nano, 2012, 6, 1322 CrossRef CAS PubMed.
- Q. Hu, D. Sun, Q. Wu, H. Wang, L. Wang, B. Liu, A. Zhou and J. He, MXene: a new family of promising hydrogen storage medium, J. Phys. Chem. A, 2013, 117, 14253 CrossRef CAS PubMed.
- M. Ghidiu, M. R. Lukatskaya, M.-Q. Zhao, Y. Gogotsi and M. W. Barsoum, Conductive two-dimensional titanium carbide ‘clay’ with high volumetric capacitance, Nature, 2014, 516, 78 CrossRef CAS PubMed.
- E. Pomerantseva, F. Bonaccorso, X. Feng, Y. Cui and Y. Gogotsi, Energy storage: The future enabled by nanomaterials, Science, 2019, 366, eaan8285 CrossRef CAS PubMed.
- A. M. Sampaio, S. Bi, M. Salanne and L. J. A. Siqueira, Molecular dynamics simulations of ionic liquids confined into MXenes, Energy Storage Mater., 2024, 70, 103502 CrossRef.
- X. Sun, Y. Li, Y. Wang, Z. Liu, K. Dong and S. Zhang, Effect of Interlayer Spaces and Interfacial Structures on High-Performance MXene/Ionic Liquid Supercapacitors: A Molecular Dynamics Simulation, Langmuir, 2024, 40, 2220 CrossRef CAS PubMed.
- S. Kondrat, P. Wu, R. Qiao and A. A. Kornyshev, Accelerating charging dynamics in subnanometre pores, Nat. Mater., 2014, 13, 387 CrossRef CAS PubMed.
- S. Kondrat and A. Kornyshev, Superionic state in double-layer capacitors with nanoporous electrodes, J. Phys.:Condens. Matter, 2011, 23, 022201 CrossRef CAS PubMed.
- S. Bi, H. Banda, M. Chen, L. Niu, M. Chen, T. Wu, J. Wang, R. Wang, J. Feng, T. Chen, M. Dincă, A. A. Kornyshev and G. Feng, Molecular understanding of charge storage and charging dynamics in supercapacitors with MOF electrodes and ionic liquid electrolytes, Nat. Mater., 2020, 19, 552 CrossRef CAS PubMed.
- M. Chen, T. Wu, L. Niu, T. Ye, W. Dai, L. Zeng, A. A. Kornyshev, Z. Wang, Z. Liu and G. Feng, Organic Solvent Boosts Charge Storage and Charging Dynamics of Conductive MOF Supercapacitors, Adv. Mater., 2024, 36, 2403202 CrossRef CAS PubMed.
- T. Mo, Z. Wang, L. Zeng, M. Chen, A. A. Kornyshev, M. Zhang, Y. Zhao and G. Feng, Energy Storage Mechanism in Supercapacitors with Porous Graphdiynes: Effects of Pore Topology and Electrode Metallicity, Adv. Mater., 2023, 35, 2301118 CrossRef CAS PubMed.
- J. Xu, X. Hu, X. Wang, X. Wang, Y. Ju, S. Ge, X. Lu, J. Ding, N. Yuan and Y. Gogotsi, Low-Temperature pseudocapacitive energy storage in Ti3C2T MXene, Energy Storage Mater., 2020, 33, 382 CrossRef.
- K. Xu, Z. Lin, C. Merlet, P. L. Taberna, L. Miao, J. Jiang and P. Simon, Tracking Ionic Rearrangements and Interpreting Dynamic Volumetric Changes in Two-Dimensional Metal Carbide Supercapacitors: A Molecular Dynamics Simulation Study, ChemSusChem, 2018, 11, 1892 CrossRef CAS PubMed.
- L. Zeng, J. Peng, J. Zhang, X. Tan, X. Ji, S. Li and G. Feng, Molecular dynamics simulations of electrochemical interfaces, J. Chem. Phys., 2023, 159, 091001 CrossRef CAS PubMed.
- S. Kondrat and A. Kornyshev, Charging Dynamics and Optimization of Nanoporous Supercapacitors, J. Phys. Chem. C, 2013, 117, 12399 CrossRef CAS.
- G. Li, N. Boulanger, B. Gurzęda, S. Bi, C. Hennig and A. V. Talyzin, Operando X-Ray Diffraction Study of MXene Electrode Structure in Supercapacitors with Alkali Metal Electrolytes, Small Sci., 2025, 5, e202500367 CrossRef CAS PubMed.
- C. Merlet, M. Salanne and B. Rotenberg, New Coarse-Grained Models of Imidazolium Ionic Liquids for Bulk and Interfacial Molecular Simulations, J. Phys. Chem. C, 2012, 116, 7687 CrossRef CAS.
- C. Merlet, M. Salanne, B. Rotenberg and P. A. Madden, Influence of solvation on the structural and capacitive properties of electrical double layer capacitors, Electrochim. Acta, 2013, 101, 262 CrossRef CAS.
- C. Merlet, C. Pean, B. Rotenberg, P. A. Madden, B. Daffos, P. L. Taberna, P. Simon and M. Salanne, Highly confined ions store charge more efficiently in supercapacitors, Nat. Commun., 2013, 4, 2701 CrossRef CAS PubMed.
- C. Pean, B. Rotenberg, P. Simon and M. Salanne, Multi-scale modelling of supercapacitors: From molecular simulations to a transmission line model, J. Power Sources, 2016, 326, 680 CrossRef CAS.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation, J. Chem. Theory Comput., 2008, 4, 435 CrossRef CAS PubMed.
- L. Zeng, T. Wu, T. Ye, T. Mo, R. Qiao and G. Feng, Modeling galvanostatic charge–discharge of nanoporous supercapacitors, Nat. Comput. Sci., 2021, 1, 725 CrossRef PubMed.
- Z. Liang, C. Zhao, W. Zhao, Y. Zhang, P. Srimuk, V. Presser and G. Feng, Molecular Understanding of Charge Storage in MoS2 Supercapacitors with Ionic Liquids, Energy Environ. Mater., 2021, 4, 631 CrossRef CAS.
- Q. Fan, R. Zhao, M. Yi, P. Qi, C. Chai, H. Ying and J. Hao, Ti3C2-MXene composite films functionalized with polypyrrole and ionic liquid-based microemulsion particles for supercapacitor applications, Chem.–Eng. J., 2022, 428, 131107 CrossRef CAS.
- K. Liang, R. A. Matsumoto, W. Zhao, N. C. Osti, I. Popov, B. P. Thapaliya, S. Fleischmann, S. Misra, K. Prenger, M. Tyagi, E. Mamontov, V. Augustyn, R. R. Unocic, A. P. Sokolov, S. Dai, P. T. Cummings and M. Naguib, Engineering the Interlayer Spacing by Pre-Intercalation for High Performance Supercapacitor MXene Electrodes in Room Temperature Ionic Liquid, Adv. Funct. Mater., 2021, 31, 2104007 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, Canonical sampling through velocity rescaling, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- T. R. Gingrich and M. Wilson, On the Ewald summation of Gaussian charges for the simulation of metallic surfaces, Chem. Phys. Lett., 2010, 500, 178 CrossRef CAS.
- R. de Levie, On porous electrodes in electrolyte solutions: I. Capacitance effects, Electrochim. Acta, 1963, 8, 751 CrossRef.
- R. de Levie, On porous electrodes in electrolyte solutions—IV, Electrochim. Acta, 1964, 9, 1231 CrossRef CAS.
- Y. Lin, C. Lian, M. U. Berrueta, H. Liu and R. van Roij, Microscopic Model for Cyclic Voltammetry of Porous Electrodes, Phys. Rev. Lett., 2022, 128, 206001 CrossRef CAS PubMed.
- A. A. Kornyshev, R. M. Twidale and A. B. Kolomeisky, Current-Generating Double-Layer Shoe with a Porous Sole: Ion Transport Matters, J. Phys. Chem. C, 2017, 121, 7584 CrossRef CAS.
- A. VahidMohammadi, J. Rosen and Y. Gogotsi, The world of two-dimensional carbides and nitrides (MXenes), Science, 2021, 372, eabf1581 CrossRef CAS PubMed.
- S. Bi, R. Wang, S. Liu, J. Yan, B. Mao, A. A. Kornyshev and G. Feng, Minimizing the electrosorption of water from humid ionic liquids on electrodes, Nat. Commun., 2018, 9, 5222 CrossRef PubMed.
- T. Mo, L. Zeng, Z. Wang, S. Kondrat and G. Feng, Symmetrizing cathode-anode response to speed up charging of nanoporous supercapacitors, Green Energy Environ., 2022, 7, 95 CrossRef CAS.
- K. Breitsprecher, M. Janssen, P. Srimuk, B. L. Mehdi, V. Presser, C. Holm and S. Kondrat, How to speed up ion transport in nanopores, Nat. Commun., 2020, 11, 6085 CrossRef CAS PubMed.
- T. Mo, J. Peng, W. Dai, M. Chen, V. Presser and G. Feng, Horn-like Pore Entrance Boosts Charging Dynamics and Charge Storage of Nanoporous Supercapacitors, ACS Nano, 2023, 17, 14974 CrossRef CAS PubMed.
- A. C. Forse, C. Merlet, J. M. Griffin and C. P. Grey, New Perspectives on the Charging Mechanisms of Supercapacitors, J. Am. Chem. Soc., 2016, 138, 5731 CrossRef CAS PubMed.
- C. Pean, C. Merlet, B. Rotenberg, P. A. Madden, P. L. Taberna, B. Daffos, M. Salanne and P. Simon, On the dynamics of charging in nanoporous carbon-based supercapacitors, ACS Nano, 2014, 8, 1576 CrossRef CAS PubMed.
- R. Tivony, S. Safran, P. Pincus, G. Silbert and J. Klein, Charging dynamics of an individual nanopore, Nat. Commun., 2018, 9, 4203 CrossRef PubMed.
- P. Robin, N. Kavokine and L. Bocquet, Modeling of emergent memory and voltage spiking in ionic transport through angstrom-scale slits, Science, 2021, 373, 687 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |