
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sustainable conversion of Penaeopsis serrata waste into phosphorylated chitosan for agricultural drought mitigation
Fatima
El Amerany
 *a,
Oumaima
Ait Ali
b and
Mohammed
Rhazi
b
*a,
Oumaima
Ait Ali
b and
Mohammed
Rhazi
b
aLaboratory of Natural Resources and Environment, Polydisciplinary Faculty of Taza, Department of Biology, Sidi Mohamed Ben Abdellah University, PO Box 1223, Taza, 35000, Morocco. E-mail: el.amerany.fatima@gmail.com; fatima.elamerany1@usmba.ac.ma
bInterdisciplinary Laboratory in Bio-Resources, Environment and Materials, Higher Normal School, Department of Biology, Cadi Ayyad University, PO Box 575, Marrakech, 40000, Morocco
First published on 17th November 2025
Abstract
The disposal of marine waste and the intensifying effects of drought on tomato productivity are major environmental and agricultural challenges. This study valorizes shrimp shell waste by synthesizing a non-water-soluble phosphorylated chitosan (PCh) with a high degree of phosphoric substitution and evaluates its potential to enhance tomato growth under drought in a controlled greenhouse experiment. The work also addresses the limited understanding of how physicochemical properties of marine waste-derived polymers influence plant responses to drought stress. The extraction method yielded pure chitin representing about one-third of the shrimp shells. The resulting chitosan (Ch) showed strong fat- and water-binding capacities, a low degree of acetylation, and low molecular weight, suggesting its potential to improve soil moisture retention. Among the synthesized PChs, PCh1 exhibited the lowest crystallinity and highest degree of phosphorylation, reflecting enhanced structural modification and functionality. Application of PCh1 to drought-stressed tomato plants significantly increased shoot length (by up to 155%), root length (by 17%), photosynthetic pigment content (by 79–153%), and relative water content (by 12%) compared to drought-stressed controls, while reducing electrolyte leakage and oxidative stress. These findings indicate that PCh1, with a high degree of phosphoric substitution and more amorphous regions, has a stronger ability to interact with water and soil particles and enhance nutrient availability, thereby improving plant resilience and productivity under drought. This study supports circular economy strategies and highlights the potential of PCh1 as a sustainable soil conditioner. Future research should evaluate its long-term field performance under diverse crops, soils, and irrigation conditions.
Sustainability spotlightThis study advances circular economy principles by converting seafood waste into value-added agricultural material, phosphorylated chitosan (PCh). The approach reduces environmental burden, enhances soil health, and provides a sustainable pathway for resource recovery. Furthermore, this biodegradable polymer (PCh) supports sustainable agriculture by improving plant growth and resilience to water stress. The findings highlight PCh as an environmentally friendly alternative to synthetic agrochemicals, contributing to reduced chemical inputs and improved food security. |
1. Introduction
Tomato (Solanum lycopersicum L.) is one of the most important vegetables in the world, cultivated for its nutritional and economic importance. However, its production has increasingly been constrained by drought stress, a major abiotic factor limiting yield and fruit quality. Conventional approaches to mitigate drought stress include breeding drought-resistant varieties and developing new cultivars.1,2 Although the use of breeding methods has successfully increased the quality and quantity of tomatoes as well as the ability of plants to recover quickly from injury, the use of these methods is limited due to the complexity of drought-related traits. Consequently, there is growing interest in sustainable, low-cost alternatives that can enhance tomato resilience under water-limited conditions.Among these alternatives, bio-based compounds such as biofertilizers and biopolymers have attracted considerable attention. Chitosan (Ch), a biodegradable and non-toxic polysaccharide derived from chitin,3 is widely used as a biostimulant, an elicitor, and a source of nitrogen.4 Ch can improve plant growth, enhance tolerance to environmental stresses, and reduce dependence on agrochemicals,5–8 particularly when applied in combination with phosphorus,9 an essential mineral involved in plant energy metabolism, root development, nutrients assimilation, and osmotic adjustment under water stress conditions.10,11
Previous studies from Asia and North Africa have demonstrated that Ch derivatives containing phosphate groups positioned between polymer chains, such as Ch–tripolyphosphate nanoparticles, are more effective in enhancing plant growth and improving drought stress tolerance in tomato,12Catharanthus roseus,13 and soybean.14 This enhanced efficiency is attributed to increased surface reactivity and improved physicochemical characteristics compared with unmodified Ch. However, no studies have yet investigated Ch derivatives in which phosphate groups are covalently bound to the polymer backbone. This highlights a critical gap in understanding how direct phosphorylation influences both the structural properties of Ch and its biological activity in plants. Combining both in a single formulation may synergistically improve phosphorus availability and its slow release, while enhancing plant resilience to water deficit by integrating the nutrient and biostimulant properties of each component.
Recent advances in chemical modification have led to the development of phosphorylated Ch (PCh).15 This new product is produced when one (or more) unit (s) of Ch is (are) interacted with phosphate molecules. This modification of Ch, to form PCh, affords properties for specific end use in some fields, mainly biomedical and biotechnological fields.16 However, PCh has never been applied to plants or soil, highlighting an important knowledge gap and its unexplored potential as a sustainable agricultural material. Despite the promising performance of this product, its synthesis has certain drawbacks, particularly the use of N,N-dimethylformamide (DMF), a volatile solvent known for its hepatotoxicity,17 and methanesulfonic acid, a corrosive and irritant compound that can cause skin and eye damage.18 Both chemicals pose environmental and health hazards, highlighting the need for greener synthesis approaches to broaden its practical applicability. Developing eco-friendly Ch derivatives with different functions is therefore crucial to reduce chemical fertilizer dependence and improve crop resilience under climate-induced water scarcity.
Marine biomass provides a valuable but underutilized source of chitin. Every year, approximately 8 million tonnes of marine waste, specifically shells of shrimp, lobster, and crab, are discarded in the sea or dumped in landfill.19 The disposal of this waste is costly; however, it can be valorized to avoid environmental pollution. For example, shrimp shells can be used as a source of calcium carbonate, carotenoids, proteins, amino acids, and chitin. Several shrimp species such as Litopenaeus vannamei, Fenneropenaeus chinensis, Metapenaeus monoceros, and Parapenaeus longirostris20–23 have been exploited for chitin and Ch production. In contrast, others, such as Penaeopsis serrata, remain unexplored, and it is not yet known how much chitin is present in them. Valorizing this waste not only reduces environmental burden but also offers opportunities for sustainable agricultural applications.
In this study, we synthesized PCh from P. serrata shells using dimethylacetamide (DMAc), non-toxic solvent, under different reaction conditions. The properties of the resulting products were compared with those obtained by using DMF. The objectives of this work were (i) to assess the chitin potential of P. serrata shells for PCh production, (ii) to develop a greener synthesis route for PCh, and (iii) to evaluate the effects of PCh on tomato growth and drought tolerance. To our knowledge, this is the first study to synthesize PCh using DMAc as a greener solvent and to evaluate its bioactivity in plants under drought stress. By pursuing these goals, we aim to demonstrate the potential of PCh as a sustainable material that both valorizes marine waste and enhances crop resilience under water-limited conditions.
2. Materials and methods
2.1. Biological material
The raw material used in this study for the production of chitin, Ch, and PCh was the shells of shrimps (P. serrata), collected from the Atlantic Ocean, Casablanca, Morocco. Species identification was conducted based on morphological characteristics (color, rostrum shape, and body segmentation) and compared with diagnostic descriptions and photographs reported by Poupin and Corbari.24P. serrata shells were washed, dried at 50 °C for 24 h, and finely ground using a Retsch RM 200 mill to obtain powder with a particle size of less than 0.5 mm.2.2. Ash content quantification
The ash content reflects the level of inorganic compounds in a biological sample. The ash content of a raw material (shrimp shells) was determined by heating dried shells (5 g) at 600 °C for 14 h and weighing the obtained product after cooling in a desiccator containing silica gel.25 Three replicates were performed for this analysis. The ash content was calculated using the following formula (A.1): | (A.1) |
2.3. Chitin isolation and chitosan (Ch) preparation
Shells were treated with chemical reagents in order to dissolve calcium carbonate and to solubilize proteins, and therefore to extract chitin. Chitin was isolated through two main steps: demineralization and deproteinization. For the 1st step, 1 g shells were mixed with 0.25 M HCl solution at room temperature for 3 h, and the ratio was 40 mL g−1.26 The 2nd step was performed with an alkaline treatment. The demineralized shells were treated with 1 M NaOH at 70 °C for 24 h and the ratio was 20 mL g−1.26 The conversion of chitin to Ch was achieved by using 60% (w/v) NaOH solution. The reaction was carried out at room temperature for 24 h, followed by heating at 110 °C for 10 h, using a solid-to-liquid ratio of 15 mL g−1.27 Demineralization, deproteinization, and deacetylation steps were not repeated. After each step, the obtained solid was washed with distilled water until the pH became neutral, then dried, and weighed to calculate the yield.2.4. Carotenoid contents
To evaluate the quality of chitin and Ch, the levels of pigments, especially carotenoids, were measured. The levels of carotenoids in shells, demineralized materials, chitin, and Ch samples were determined according to the method described by Su et al.28 using three independent samples. Samples were extracted several times with 80% acetone until they became colorless. The extracts were centrifuged at 3000×g for 1 min and DO of the supernatant was measured at 478 nm. The total carotenoids were determined as astaxanthin, using the equation of Simpson and Haard (A.2):29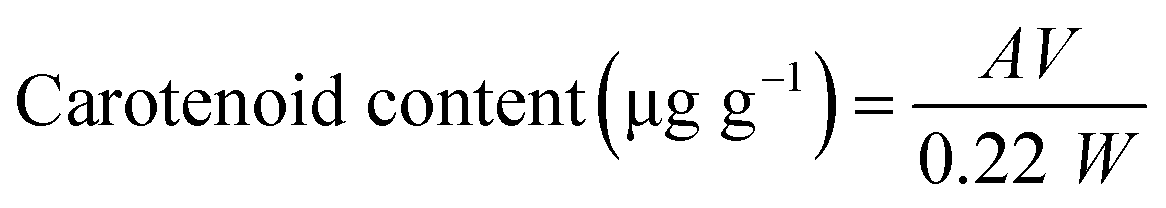 | (A.2) |
2.5. Levels of free proteins and proteins attached to chitin
Free proteins were quantified by analyzing the liquids separated from the residue after the deproteinization process as well as those obtained after the washing step. The levels of proteins in all extracts were determined by Bradford's method.30 The absorbance of the resulting solution was determined at 595 nm. Bovine serum albumin (BSA) was used as standard.The levels of protein still attached to chitin were measured according to Lyon Jr et al.31 method, with some modification. Chitin samples were mixed with 5 mL of phosphate-buffered saline (PBS-T, containing Tween® 20, pH 7.4), with a mild agitation. After 2 h, the mixture was centrifuged at 1750×g for 5 min to separate supernatant, containing proteins, from chitin backbone. The extraction procedure was then repeated twice using 2.5 and 2 mL of buffer, respectively, to recover residual proteins. The protein content in the combined supernatants was determined as previously described, using PBS-T as a blank and BSA as the standard.
2.6. Water and fat binding capacities of Ch
Water binding capacity (WBC) of Ch was measured according to the method of Wang and Kinsella,32 with slight modification, using three independent samples. In a centrifuge tube, 150 mg of Ch was mixed with 3 mL of distilled water to ensure the dispersion of the sample. Then, the mixture was left at room temperature for 30 min and shaken for 5 s every 10 minutes. Afterward, the mixture was centrifuged at 2900×g for 25 min. The supernatant was slightly removed and the tubes were weighted. These steps were also followed to measure the fat binding capacity (FBC), but the water was replaced by soya oil.WBC and FBC were calculated using the following formulas33 ((A.3) and (A.4)):
 | (A.3) |
 | (A.4) |
2.7. Phosphorylation of chitosan
The preparation of PCh was done according to the method of Subhapradha et al.,34 with some modifications in order to:-Assess the effect of other solvent DMAc, in comparison to DMF.
-See if a complete solubilization of Ch in a solvent is required before adding a ortho-phosphoric acid.
Thus, the 4 methods were performed as follows:
Method 1 for PCh1: Ch powder was mixed with DMAc for 1 min. Then, urea and ortho-phosphoric acid were added to Ch solution. The reaction was carried out at 165 °C for 1 h.
Method 2 for PCh2: Ch powder was dissolved in DMAc for 45 min. Then, urea and ortho-phosphoric acid were added to Ch solution. The reaction was carried out at 165 °C for 1 h.
Method 3 for PCh3: Ch powder was mixed with DMF for 1 min. Then, urea and ortho-phosphoric acid were added to Ch solution. The reaction was carried out at 159 °C for 1 h.
Method 4 for PCh4: Ch powder was dissolved in DMF for 45 min. Then, urea and ortho-phosphoric acid were added to Ch solution. The reaction was carried out at 159 °C for 1 h.
At the end of each reaction, the obtained residue was washed with distilled water.
2.8. Solubility of Ch and PCh
Ch and PCh were dissolved in three solvents (distilled water, 1% acetic acid, and 0.5% acetic acid) to test their solubility.33 In a tube, 10 mg of a sample was added to 10 mL of the solution and left under stirring for 3 h. After centrifugation (1750×g for 10 min), the undissolved residue was collected, dried in an oven at 5 °C, and weighed.The solubility of polymers was calculated according to the following eqn (A.5):
 | (A.5) |
2.9. Physicochemical characterization
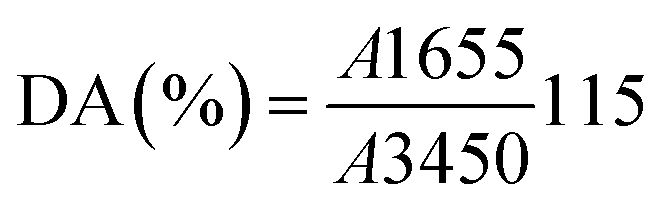 | (A.6) |
In this procedure, 250 mg of Ch was dissolved in 0.1 M HCl under continuous stirring at room temperature until complete dissolution was achieved. Subsequently, three drops of methyl orange indicator were added to the Ch solution, which was then titrated with 0.1 M NaOH solution. The titration was considered complete when the solution color changed from pink to yellow, and the volume of NaOH consumed was recorded.
The DA was calculated according to the following equation:37
 | (A.7) |
| [η] = KMva | (A.8) |
 | (A.9) |
For PCh1, the intensity of the amorphous domain was higher than that of crystalline domain; therefore, eqn (A.10) was used:
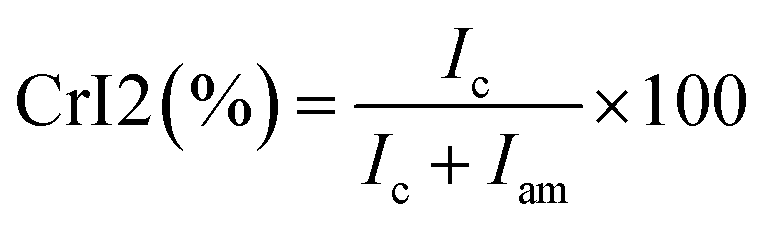 | (A.10) |
The crystallite size was calculated using the following equation:42
 | (A.11) |
Also, the minerals that existed in materials were identified and quantified using the energy dispersive X-ray combined with SEM. The analysis was performed at a magnification of ×5000, with an accelerating voltage of 10 kV, a takeoff angle of 33.3°, a live time of 30 s, and an amplifier time of 7.68 µs. The system resolution was 125.9 eV. Phosphorus (P) and nitrogen (N) contents were obtained from the EDX spectra provided by SEM laboratory at the Centre d'Analyse et de Caractérisation (CAC), Marrakesh, Morocco. Three samples were analyzed as replicates. The degree of phosphoric substitution (DS) of PCh was calculated according to eqn (A.12):43
 | (A.12) |
2.10. Plant materials, treatments, and analysis
PCh1 solution (3 mg mL−1) was prepared by dissolving PCh in 0.15% acetic acid (AA) and the pH of the solution was neutralized to 5.6 by adding NaOH.Seeds (S. Lycopersicum cv. Campbell33) were sterilized and germinated at 28 °C for 9 d. Then, the germinated seeds were transferred to trays containing peat and irrigated once a day for a period of 3 weeks. Plants were grown under greenhouse conditions (225 µmol m−2 s−1 light intensity, 16 h photoperiod, 65% relative humidity, and temperature range of 25 to 28 °C). Afterward, seedlings were transferred to pots of 1.1 kg, and the substrate used was composed of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 peat:agricultural soil.
:3 peat:agricultural soil.
Pots were weighed and irrigated daily with water to maintain 50% of the field capacity (FC),44 which was determined gravimetrically. Briefly, the dry peat–agricultural soil mixture was weighed (P1), then saturated with water and allowed to drain freely. Once drainage ceased, the pot was weighed again (P2). The difference between P2 and P1 represented the water content corresponding to 100% FC. During the experiment, pots were maintained at 50% FC by weighing them daily and replenishing the water lost through evaporation and transpiration with water to reach the target weight.
After 3 weeks, PCh1 solution and water, containing 0.15% AA, were applied near the root system, each 5 days for a period of 35 days. After the 5th application of treatments, the plant received no water for 11 days to impose drought stress. Thus, the design of the experiment was completely randomized with four treatments. For each treatment, 6 plants were used as independent biological replicates (Table 1).
| Treatments | Acronyms |
|---|---|
| 0.15% acetic acid | Control |
| 3 mg mL−1 of PCh1 | PCh1 |
| 0.15% acetic acid + drought for 11 days | DS |
| 3 mg mL−1 of PCh1 + drought for 11 days | PCh1 + DS |
At the end of the experiment, different analyses, such as leaf number (LN), stem length (SL), root length (RL), and plant fresh and dry weight (PFW and PDW), were carried out to highlight the effect of PCh1 under drought stress. The length of the aerial and underground parts was measured by using a graduated ruler. In addition, seedlings were weighed to measure their fresh weight. Then, they were placed in an oven (80 °C) for 48 h to determine their dry weight.
In addition, the levels of leaves pigments such as chlorophylls (chla and chlb) and carotenoids were measured according to Arnon method.45 Fresh tomato leaves were homogenized with liquid nitrogen and then with 80% acetone. Afterwards, the absorbance of the collected supernatant was measured at 663 nm, 645 nm, and 470 nm, using a UV/visible spectrophotometer (Specord 210 Plus, Analytic Jena, Lena, Germany). Pigment levels were calculated using Arnon eqn (A.13)–(A.15):
 | (A.13) |
 | (A.14) |
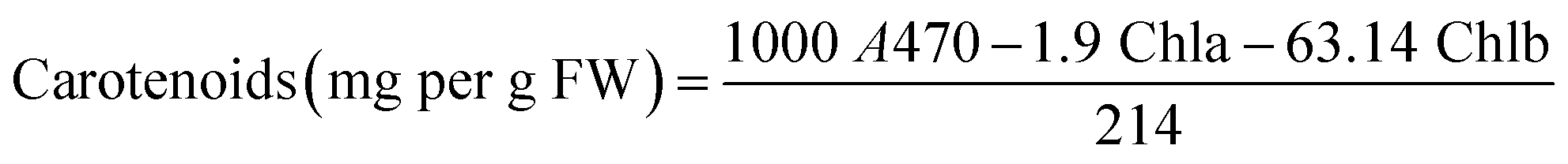 | (A.15) |
Protein contents in leaves were determined according to Bradford's method30 after homogenizing leaves with 0.02 M potassium phosphate buffer, pH 7.6.
For measuring relative water content (RWC), leaves were weighed, just after harvesting to determine their fresh weights (W1). Afterward, they were stored in distilled water for 24 h, drained with filter paper, and weighed (W2). Finally, they were dried in an oven to determine their dry weight (W3). RWC was measured using the equation of Azam-Ali and Squire.46
 | (A.16) |
The effect of PCh1 on membrane's permeability of tomato leaves was evaluated by measuring the electrolyte concentration. Fresh leaves were harvested, rinsed with distilled water, and gently dried with paper towels. In tube, leaf disks (≈20 disks of 1 cm2) were prepared and immersed in 25 mL deionized water. The tubes were shaken for 24 h at 100 rpm and 20 °C, and the initial electrical conductivity (ECi) was then measured. Afterwards, the tubes were incubated for 1 h at 80 °C and placed again on the shaker for 24 h at 100 rpm and 20 °C. The final electrical conductivity (ECf) was then determined. Electrolyte leakage was calculated using the following formula:47
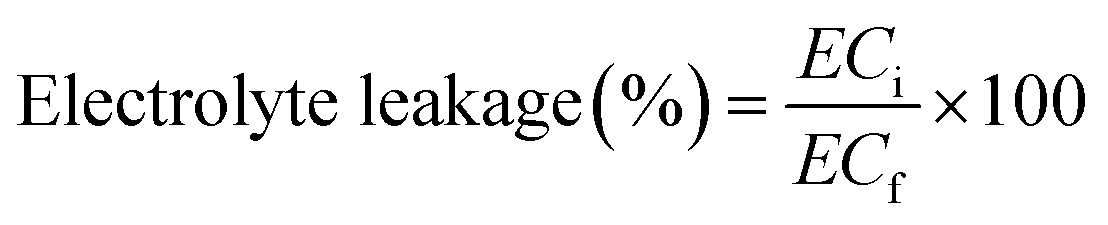 | (A.17) |
Hydrogen peroxide (H2O2) accumulation was assessed using two complementary approaches: histochemical detection48 and biochemical quantification.49
For the histochemical detection, leaflets from the third fully expanded leaves (from the top) of stressed (SD) and stressed + treated (SD + Ch1) plants were collected. The samples were gently rinsed with distilled water to remove dust or soil residues and carefully blotted dry with soft paper. The leaflets were then immediately immersed in a 1 mg mL−1 solution of 3,3′-diaminobenzidine (DAB) and incubated overnight under light. After incubation, the leaflets were transferred to tubes containing absolute ethanol and placed in a boiling water bath until complete chlorophyll removal. Once cooled, the decolorized leaflets were preserved in glycerol for subsequent visual examination of H2O2 deposition.
For the biochemical quantification, fresh leaves from all four treatments were homogenized in 5 mL of 0.1% (w/v) trichloroacetic acid (TCA) and centrifuged at 5000×g for 10 min. Then, 1 mL of the supernatant was mixed with 1 mL of 10 mM potassium phosphate buffer (pH 7.0) and 2 mL of 1 M potassium iodide (KI). The reaction mixture was incubated in darkness for 1 h, and the absorbance was measured at 390 nm. The H2O2 concentration was quantified using three independent biological replicates and a standard curve generated with known H2O2 concentrations.
2.11. Statistical analysis
All data were analyzed using Costat software, which provides reliable tools for the analysis of variance (ANOVA) and Duncan's test (p < 0.05). In addition, a t-test (p < 0.05) was performed to evaluate significant differences between two measurements. Principal Component Analysis (PCA) and Heatmaps were generated to analyze growth (PFW, PDW, SL, LN, and RL) and biochemical parameters (chla, chlb, carotenoids, proteins, RWC, and electrolyte leakage) using MetaboAnalyst. Prior to PCA, the data were normalized by calculating log2 fold changes and then applying Pareto scaling to ensure variance stabilization and comparability across variables.3. Results and discussion
3.1. Color of the produced products
Fig. 1 shows that the shrimp shells of Penaeopsis serrata have a pink-orange color, while chitin and Ch have white flakes, which means that carotenoids were partially or completely removed after demineralization, deproteinization and deacetylation steps. Rasweefali et al.50 reported that removing shrimp minerals can increase the accessibility of alkali treatments to proteins and therefore facilitate removal of pigments, such as β-carotene, attached to proteins and chitin. In addition, all PChs were characterized by a dark brown color (Fig. 1). This color change is attributed to non-enzymatic browning reactions, mainly involving the covalent interaction between the amino groups of urea and the carbonyl groups of Ch monomers under heating conditions, leading to the formation of Maillard-type condensation products.51 The progression of the Maillard reaction is not only promoted by elevated temperature but also facilitated when the molecular weight of Ch is lower.52 Similar browning behavior has also been reported in previous studies during the phosphorylation of Ch.16,53 | ||
| Fig. 1 Pictures showing the color of shells, chitin, Ch, and PChs. | ||
3.2. Ash content and the yield of chitin
The ash content was measured to estimate the level of inorganic compounds in shell samples. The shells of Penaeopsis serrata consisted of 28 ± 0.10% ash content and the major mineral elements were nitrogen (N, 4.48%) and calcium (Ca, 4.23%) (Fig. 2 and Table 2). The mineral contents of Penaeopsis serrata were higher than those of Litopenaeus vannamei (8.57%), and Macrobrachium rosenbergii (13.31%), however these levels were lower in comparison to those of Solenocera hextii (32.16%), Fenneropenaeus chinensis (33%), and Pandalus borealis (43.61%).21,54–56 This variation has happened because the mineral content was depended on the geographical location and growth stage of shrimps. | ||
| Fig. 2 Mineral composition of shrimp shells. 3 biological replicates were used. | ||
| Samples | Ash (%) | Yield (%) |
|---|---|---|
| a ND, not determined. | ||
| Shrimp shells | 28 ± 0.10 | ND |
| Chitin | ND | 27.77 ± 4.7 |
In addition, the yield of chitin was 27.77 ± 4.7% (Table 2). The yield of the obtained chitin was higher than those extracted from Parapenaeus longirostris, Penaeus semisulcatus, and Metapenaeus affinis using similar extraction conditions,25,26 and it was similar to those isolated from Antarctic krill (Euphausia superba) under different conditions (demineralization using 1.7 M HCl for 6 h at room temperature and deproteinization using 2.5 M NaOH for 1 h at 75 °C).57 These results indicate that chitin yield depends on the species, and may also be influenced by the age and geographic origin of the species.
3.3. Evaluation of chitin and Ch quality
To assess the efficiency of the applied methods for extracting chitin and preparing Ch, the levels of carotenoids, proteins, and minerals were measured.Fig. 3A and B show that the carotenoid contents were around 7.27 µg kg−1. These contents were very low, in comparison to those of other species, such as Exopalaemon carinicauda (10.84 × 103 µg kg−1), Penaeus monodon (48.9 to 51 ×103 µg kg−1), and Penaeus indicus (43.9 ×103 µg kg−1).27,28,58,59 In addition, the carotenoids contents were reduced during demineralization, deproteinization, and deacetylation steps by about 31%, 94%, and 98% respectively, in comparison to the control (Fig. 3A), indicating that applying a diluted organic acid and concentrated base, as well as high temperature, may remove carotenoids attached to proteins and chitin. These results are in accordance with the data published by Rasweefali et al.50,56 who found that raising the temperature to 100 °C or applying alkaline treatment to shrimp shells removes pigments.50,56 Even though the produced chitin and Ch has 0.00154 ppm and 0.00097 ppm, respectively, of carotenoids (Fig. 3B), the blanching step is not required because 1 ppm carotenoids is considered acceptable to avoid the changes in the chemical structure of chitin and Ch.
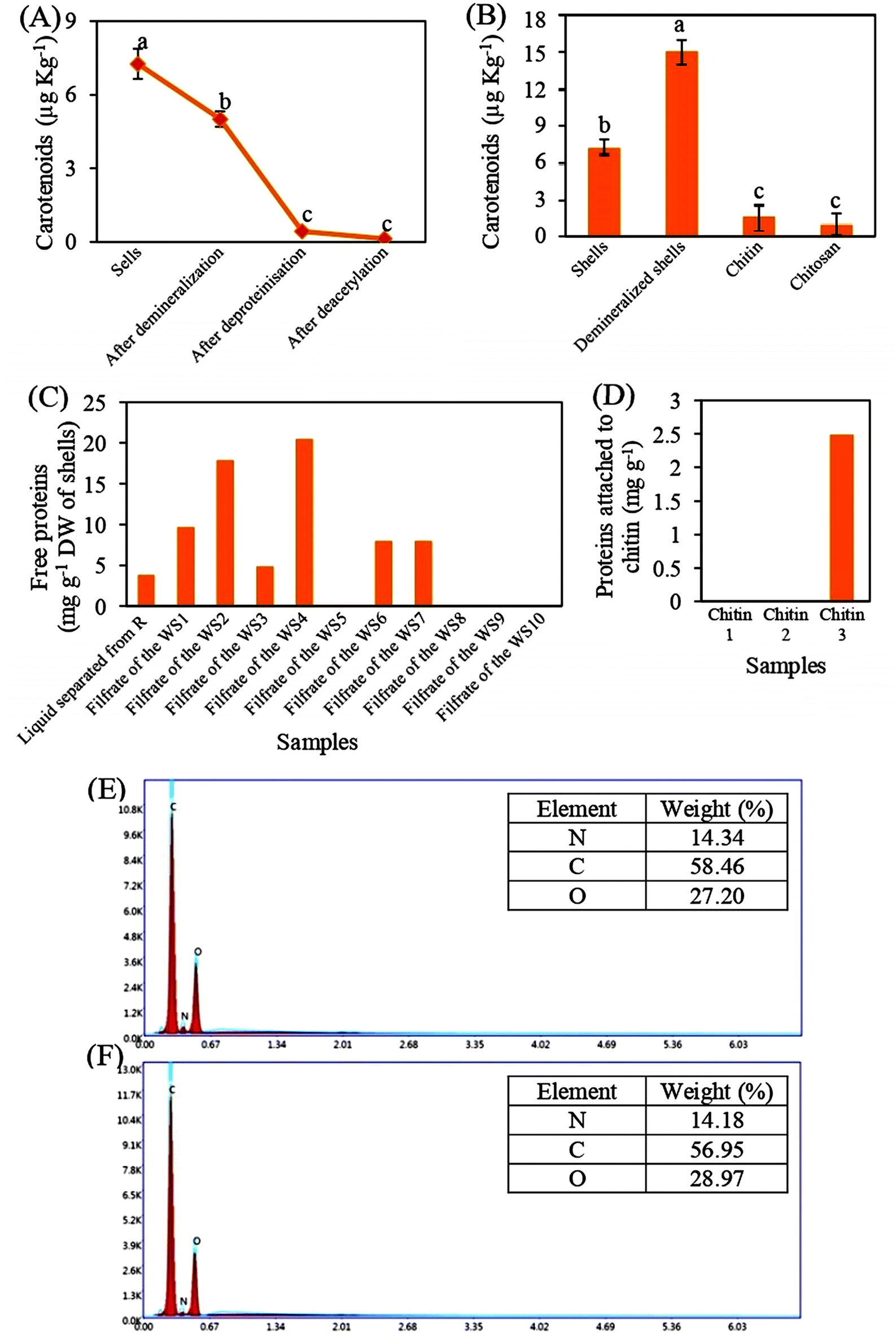 | ||
| Fig. 3 Measurement of purity of chitin and Ch. Carotenoids levels in shells (before and after demineralization, deproteinization, and deacetylation steps), demineralized shells, chitin, and Ch (A and B). Levels of proteins not attached (C) and attached (D) to chitin after the deproteinization process. Mineral composition in demineralized shells (E) and chitin (F).R, residues; WS1, first washing step. 3 biological replicates were used. | ||
Moreover, Fig. 3C shows that the total amount of proteins extracted by 1 M NaOH was equal to 3.7 mg g−1 and the large amount of proteins (68.4 mg g−1) was removed after the application of 7 washing steps. Some studies reported that the deproteinization step of shells is considered efficient when 1 M NaOH solution is used or the reaction time is taken more than 24 h.60 In addition, the analysis of three samples of chitin shows that a small quantity of proteins, which was equal to 2.48 mg g−1, was found only in one chitin sample (Fig. 3D). This could be due to the presence of a stable complex between chitin and proteins which limiting their isolation.61 To our knowledge, there have been no studies that conduct an isolation of chitin with 0% proteins. All chemical and enzymatic methods allow the production of chitin that contains about 4% to 10% of residual protein content.60 For many food and industrial application, the presence of less than 5% protein impurities in chitin is considered acceptable, as these residues can be removed in subsequent processing steps.62 This applies to our case as well, since the obtained chitin will be used as a raw material for Ch production, where the remaining proteins will be eliminated during the deacetylation step using a concentrated NaOH.
Regarding mineral analysis, Fig. 3E shows that the demineralization step successfully removed all the attached minerals (P, K, Mg, Ca, B, and Na) from the chitin skeletons. Also, the deproteinization step was not exerted a negative effect on the chitin structure, particularly on the –NH2 group because the level of nitrogen was not altered during the application of a concentrated base (Fig. 3F). Similar data was obtained on other species.26
3.4. Determination of fat and water binding capacity, acetylation degree, and molecular weight of Ch
FBC and WBC were measured to see if Ch can be used as a binder material in a wide range of applications. Both of them are considered as the functional properties of polymer that varies with the preparation method and depends on its characteristics.63 The data showed that FBC and WBC values were found to be equal to 444.44% and 397.78%, respectively (Table 3).| Analysis | Chitosan |
|---|---|
| FBC (%) | 444.44 ± 67.62 |
| WBC (%) | 397.78 ± 77.81 |
| DA obtained by FTIR (%) | 4.38 ± 0.43 |
| DA obtained by acid–base titration (%) | 3.78 ± 0.62 |
| M v (kDa) | 73.72 ± 19.24 |
Although these parameters represent different binding mechanisms (hydrophobic and hydrophilic, respectively), their values were not statistically different according to t-test analysis (p > 0.05). Similar results have been obtained by Cho et al.64 who found that FBC values of commercial Ch were ranging from 314 to 535%. However, WBC values were reduced in comparison to those found in the literature.65 Water molecules can easily penetrate in Ch with a high degree of deacetylation (DDA) because the removal of acetyl groups can increase the binding sites for OH groups and NH2 groups. A study found that chitin treated with 45% NaOH at 100 °C for 3 h has a high WBC, in comparison to those treated for 6 h, which indicates that the time of deacetylation can also influence WBC values.50
Deacetylation of chitin is generally considered successful when the DA of the final product is less than 50%.38 Also, it is not so easy, especially when a high DDA is required. In this study, the DA of Ch, measured by FTIR and acid–base titration, was equal to 4.38% and 3.78, respectively (Table 3). Statistical analysis (t-test) revealed no significant difference between the two values, indicating that the produced Ch contained a very low proportion of acetyl groups. These data are in agreement with the results obtained by Arrouze et al.66 In addition, the obtained Ch from shrimp shells of Penaeopsis serrata had a low molecular weight (Mv, 73.72 KDa). The reduction in the Mv of Ch might happen because the deacetylation step takes a long time under certain conditions (high temperature and concentrated basic solution).67
3.5. Solubility of Ch and PCh
Ch solubility depends on different factors such as its Mv, its DA, its degree of crystallinity, and the pH and temperature of a solution used for solubilization.68 All the previous works agree that the acidic solvents are the most appropriate solution to dissolve molecules containing amino groups including Ch69 and the most used solvent was 1% acetic acid.70 Thus, in order to determine which solvent will tend to make our Ch and its derivative, PCh, soluble, water and two doses of acetic acid (0.5% and 1%) were used for solubility test. Table 4 shows that the obtained Ch was soluble in acetic acids (1% and 0.5%) and swelled when distilled water was used. Ch was soluble in a diluted acetic acid because of the protonation of its amino groups, and it was insoluble in water due to the presence of the acetyl groups.71| Samples | Solubility (%) | Degree of P substitution (%) | ||
|---|---|---|---|---|
| Distilled water | 1% acetic acid | 0.5% acetic acid | ||
| a Solubility varies between 42 and 53%. b Solubility varies between 29 and 41%. | ||||
| Ch | Swelling | Soluble | Soluble | 0 |
| PCh1 | Insoluble | Moderately solublea | Partially solubleb | 4.88 |
| PCh2 | Insoluble | Moderately solublea | Partially solubleb | 0.82 |
| PCh3 | Insoluble | Moderately solublea | Partially solubleb | 1.31 |
| PCh4 | Insoluble | Moderately solublea | Partially solubleb | 1.07 |
All the produced PChs were insoluble in distilled water, and exhibited partial and moderate solubility in 0.5% and 1% acetic acid, respectively (Table 4). These results are inconsistent with those found in the literature,34 which generally describe PCh as a water-soluble polymer, with solubility value reaching up to 78%. This discrepancy can be attributed to several factors, such as the intrinsic physico-chemical characteristics of Ch, the use of solvents (DMAc and DMF) during phosphorylation, which may promote intra- and intermolecular crosslinking, and the possible formation of ammonium phosphate salt that linked between amino (–NH2) and phosphate (–PO–(OH)2) group, which reduces the number of free amino sites available for protonation. Consequently, the reduced protonation capacity limits solubility in acidic media.
This interpretation is supported by the findings of Tachaboonyakiat et al.,43 who demonstrated that PCh solubility is inversely proportional to both DDA of Ch and its degree of substitution (DS) with phosphate groups. In other words, PCh becomes less soluble when the DDA of Ch is high or when DS exceeds 0.3%. A higher DDA indicates a greater number of –NH2 groups, which can be protonated and subsequently form intra or intermolecular linkages with negatively charged phosphate atoms, thereby decreasing solubility. Similarly, a higher DS increases the density of phosphate groups capable of forming ionic crosslinks with ammonium groups, further reducing solubility.
Based on our data, the DDA of the obtained Ch ranged between 95.62% and 96.22%, suggesting that the reduced solubility of the resulting PChs may be related to the high DDA of the precursor Ch and potentially to an elevated DS. To confirm this hypothesis, the determination of the DS of the produced PChs is needed.
3.6. DS of PCh and the analysis of the surface of materials
The degree of P substitution of PCh was determined by energy-dispersive X-ray spectroscopy (EDX) coupled with SEM, as shown in Table 4. In our experiment, the degree of P substitution of all PCh samples was more than 0.82%, which proves that the insolubility of PCh in water and acetic acid solutions was due to the formation of a complexe between amino (–NH2) group and phosphate (–PO–(OH)2) group. In addition, our data showed that the P substitution degree was induced when ortho-phosphoric acid was added to Ch before being completely dissolved in DMAc (PCh1) or DMF (PCh3). The highest P substitution degree (4.88%) was recorded in PCh1 and this value was also higher than those reported by Tachaboonyakiat et al.43 These results indicate the potential of PCh1 to enhance phosphorus availability upon application; therefore, it is essential to further evaluate its actual performance on plant growth and drought tolerance.To get more information about the morphology of materials as well as the changes occurred during different physio-chemical processes, the surfaces of demineralized shells, chitin, Ch, and PCh1 produced by using DMAc (Method 1), were analyzed by SEM. Fig. 4 shows that demineralized shells contained at least 7 layers (marked by arrows) and they were colonized by bacteria (marked by a red circle), especially cocci and bacilli, which means that using 0.25 N HCl for 3 h could not totally destroy or remove the microorganisms. In addition, the surface of the layer marked by the red arrow (Fig. 4A) was characterized by small holes and this feature was typical to chitin surface as shown in Fig. 4B. Layers marked by yellow arrows might be proteins or pigments because they were no longer visible in chitin's surface (Fig. 4A and B). In addition, both groups of bacteria disappeared after the deproteinization process (Fig. 4A and B), and this might be due to the application of 1 M NaOH at 70 °C.72Fig. 4C shows that the surface of chitin was slightly changed after the deacetylation process. Holes in Ch surface became wider, in comparison with that of chitin (Fig. 4B and C) and this is caused by the elimination of acetyl groups and swelling of chitin at 110 °C.73 The structure of PCh1 is so different from that of chitin and Ch (Fig. 4D). There is a change from an homogenous and semi-smooth fibrous structure to a non-homogeneous structure, which may indicate that Ch has undergone a chemical modification through the incorporation of phosphate.
 | ||
| Fig. 4 SEM images represent the surface of demineralized shells (A), chitin (B), Ch (C), and PCh1 (D). Arrows indicate demineralized shell layers. The area marked by a circle shows the presence of bacteria. Bars represent 10 µm. | ||
3.7. Identification of the functional groups of materials using FTIR spectroscopy
The most important functional groups in chitin, Ch, and PCh1 were identified by FTIR spectroscopy (Fig. 5). Regarding the chitin spectrum, a broad peak detected between 3775 and 3000 cm−1 was assigned to O–H and N–H groups.74 In addition, the peak at 2884.97 cm−1 corresponded to C–H stretching vibrations of aliphatic hydrocarbons (alkane groups). Characteristic amide bands were also observed. The peaks at 1656.31 (amide I, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), 1627.75, and 1562.32 cm−1 (amide II, N–H bending coupled with C–N stretching) (Fig. 5), reflected the presence N-acetyl groups.34,75,76 The absence of absorption peaks at 859.40 cm−1 (Fig. 5), which are characteristic of the stretching and bending vibrations of aragonite, confirms that the treatments have removed all CaCO3 present in the shells.77
O stretching), 1627.75, and 1562.32 cm−1 (amide II, N–H bending coupled with C–N stretching) (Fig. 5), reflected the presence N-acetyl groups.34,75,76 The absence of absorption peaks at 859.40 cm−1 (Fig. 5), which are characteristic of the stretching and bending vibrations of aragonite, confirms that the treatments have removed all CaCO3 present in the shells.77
 | ||
| Fig. 5 FTIR spectra of chitin, Ch and PCh1, and their absorbance bands. | ||
Treating chitin with basic solvent, as well as high temperature, leads to changes in its chemical structure; therefore, some peaks that were typical for chitin (i.e., 1627.75 and 1562.32 cm−1) were shifted and new peaks appeared (i.e., 1594.87 cm−1) (Fig. 5). The peak observed around 1594.87 cm−1 was attributed to N–H bending of free NH2 groups.78 These changes indicate successful removal of acetyl groups and increased amine availability.
Regarding PCh1 spectrum, the peak observed at 1632 cm−1 was assigned to the amide I (CO stretching),79 while new absorption bands (Fig. 5) at 1085.20 and 558 cm−1 represented P–OH group.80,81 The characteristic NH2 band around 1594.87 cm−1 became less distinct, likely due to overlap or modification caused by the interaction of amino groups with phosphate. These features confirm the successful phosphorylation of Ch.
3.8. Crystallinity
The analysis of the X-ray diffraction (XRD) patterns and crystallinity indices (CrI) of chitin, Ch, and PCh1 provides valuable insights into the structural changes that occur upon modification of these polymers. The XRD patterns of chitin shows characteristic peaks at 9.35° and 19.40°, along with lower-intensity peaks at 12.77° and 23.35° (Fig. 6), which are typical for the α-crystalline structure of chitin. These observations are consistent with previous reports by Hao et al.82 and Huang et al.83 | ||
| Fig. 6 X-ray diffraction (XRD) patterns of chitin, Ch, and PCh1. | ||
For Ch, the main diffraction peaks at 9.35° and 19.40° shifted to 10.42° and 20.26°, respectively, accompanied by a reduction in their intensity (Fig. 6). This agrees with the findings of Zhang et al.,84 who reported that the intensity of the characteristic α-chitin peaks decreases during deacetylation. Moreover, the appearance of a new diffraction peak at 29.88° (Fig. 6), as also reported in other studies on Ch derived from pen shells,85 suggests a partial structural reorganization rather than an overall increase in the crystallinity of the Ch sample.
Upon phosphorylation, the XRD pattern of Ch (now PCh1) exhibited a distinct crystalline state compared to that of Ch (Fig. 6). The peak at 20° disappeared after phosphorylation, likely due to the disruption of hydrogen bonding involving the –NH2 and –OH groups of Ch.86 This disappearance was accompanied by the emergence of new peaks at 11.60° and 16.73°, corresponding to amorphous regions,87,88 while those at 18.33°, 23.78°, and 29.13° were attributed to crystalline regions,85 confirming the reduction in crystallinity after phosphorylation. Additionally, small peaks at 33.73°, 37.90, and 45.17 were also observed in PCh1, similar to those reported for PCh derived from the cuttlebone of Sepia pharaonis.88 These reflections may be associated with phosphate-related crystalline domains or residual mineral components incorporated during the phosphorylation process.
The CrI values (Table 5) obtained using the two different eqn (A.9) and (A.10) for chitin and Ch were not significantly different according to t-test analysis, confirming the suitability of eqn (A.10) for determining the CrI of PCh1, which exhibits high-intensity peaks in the amorphous regions. Table 5 shows also that CrI values of chitin and Ch were ranged from 94% to 95% and from 73% to 79%, respectively. The higher CrI of chitin reflects its well-ordered crystalline structure, while the lower CrI of Ch results from the reduced DA,89 which disrupts the crystalline order. The introduction of phosphate groups into Ch further reduced its CrI to approximately 17–42%, as also evidenced by the XRD diffractogram. Similar findings have attributed this decrease to the weakening of hydrogen bonds between the –NH2 and –OH groups.86
| Parameters | Samples | ||
|---|---|---|---|
| Chitin | Ch | PCh1 | |
| a ND, not determined; * or **, indicate no significant difference between values in the same column, while Δ denote no significant difference between values in the same row, according to t-test (p < 0.05). | |||
| CrI1 at ≈20° | 94.50 ± 6.90* | 73.39 ± 6.26** | ND |
| CrI2 at ≈20° | 95.05 ± 5.99* | 79.12 ± 4.12** | ND |
| CrI2 at ≈18° | ND | ND | 17.01 ± 1.66 |
| CrI2 at ≈23° | ND | ND | 41.84 ± 5.59 |
| Crystallite size (nm) at ≈ 20° | 11.52 ± 3.21Δ | 11.46 ± 2.12Δ | ND |
| Crystallite size (nm) ≈ 18° | ND | ND | 10.59 ± 1.15 |
| Crystallite size (nm) ≈ 23° | ND | ND | 77.07 ± 15.73 |
To the best of our knowledge, this is the first report analyzing the crystal size of PCh1. The data (Table 5) show that PCh1 contains crystallites of varying sizes. The crystals detected at 18° were very small (≈11 nm) and comparable to those observed in chitin and Ch, whereas those detected at 23° were slightly larger (≈77 nm). These results indicate that the chemical modification of Ch through phosphorylation alters the crystalline domain size, resulting in a more heterogeneous crystalline structure. This structural reorganization may also contribute to the reduced solubility of PCh1, as the formation of larger crystalline domains can hinder solvent penetration and limit polymer chain mobility.
3.9. Effects of water deficit and/or PCh1 on tomato growth
Drought stress (DS) is the devastating abiotic stress that limits crop productivity, particularly in semi-arid to arid land. The present study investigated the protective role of PCh1 against drought stress. As shown in Fig. 7, DS significantly reduced plant growth compared with the control (p < 0.05); whereas PC1 application, alone or in combination with DS, markedly improved growth performance. Similar findings were reported by Bakhoum et al.,90,91 who demonstrated that climate-induced stress significantly affected the physiological performance of crops. | ||
| Fig. 7 Treated and non-treated plants. | ||
To determine the main factors responsible for treatment effectiveness, a Principal Component Analysis (PCA) plot and Hierarchical Clustering Heatmaps were created. PCA plot explained about 87.4% of the total variation (Fig. 8A), with PC1 and PC2 accounting for 50.2% and 37.2%, respectively. Most importantly, a clear distinction of treatments was observed, hence Control, DS, PCh1, and PCh1 + DS treatments were placed in the middle, left, right, and the top, respectively, indicating distinct plant responses. The heatmap showed that all measured parameters are grouped into 4 clusters: 1st cluster represents chlorophyll a level, PFW, carotenoid levels, and chlorophyll b levels, 2nd cluster represents SL, LN, and PDW, 3rd cluster represents EL, and 4th cluster represents protein levels, RL, and RWC (Fig. 8B).
 | ||
| Fig. 8 Score plot of principal component analysis (A) and hierarchical clustering heatmaps (B) showing PCh1 effects on tomato growth exposed to drought stress (DS). 3 replicates were used for each treatment (Control, DS, PCh1, and PCh1 + DS). PFW, plant fresh weight; SL, stem length; LN, leaves number; PDW, plant dry weight; EL, electrolyte leakage; RL, root length; RWC, relative water content. | ||
Under DS, most growth and biochemical parameters declined significantly (p < 0.05), except EL, which increased by 47% compared to the control. This increase is attributed to oxidative membrane injury caused by the accumulation of reactive oxygen species (ROS) such as H2O2, hydroxyl radicals, and superoxide anions.92,93 In contrast, PCh1 + DS-treated plants exhibited significantly lower EL (p < 0.05) during the withholding of water (Fig. 8B), indicating reduced oxidative damage and better membrane integrity, likely due to the activation of antioxidant defense pathways by Ch derivatives.94,95
To verify this hypothesis, H2O2 accumulation in the leaflets of SD and PCh1 + DS plants was analyzed using DAB staining. As shown in Fig. 9A, leaflets from both treatments displayed brown coloration, confirming the presence of H2O2; however, the staining was markedly more intense in DS leaves, particularly along the veins and margins, indicating higher oxidative stress. In contrast, PCh1 + DS leaflets exhibited noticeably weaker staining, suggesting that PCh1 application reduced H2O2 accumulation under drought conditions.
 | ||
| Fig. 9 Effect of treatments on H2O2 production. (A) H2O2 deposition in leaves (DS on the left, PCh1 + DS on the right). (B) Quantification of H2O2 levels in treated plants, based on three biologically independent replicates. | ||
Biochemical quantification supported these observations (Fig. 9B). H2O2 content did not differ significantly (p < 0.05) among the Control, PCh1, and PCh1 + DS treatments, whereas DS plants showed a significant 30% increase in H2O2 levels relative to the control (p < 0.05). These results demonstrate that PCh1 mitigates drought-induced oxidative stress by limiting H2O2 accumulation in plant tissues. These findings are in agreement with whose of Dawook et al.96 who reported that applying Ch or related bio-stimulants reduced H2O2 accumulation under drought stress through enhanced antioxidant enzyme activity, including catalase (CAT), superoxide dismutase (SOD), and peroxidase (POX).
In addition, PCh1 treatment significantly enhanced growth metrics like SL even under drought conditions. The SL of DS plants significantly decreased by 32% compared with the control (p < 0.05), whereas PCh1 and PCh1 + DS increased SL by 66% and 75%, respectively (Fig. 8B). These findings are consistent with the report of Bañon et al.,97 who demonstrated that drought stress inhibits plant growth by reducing cell division, cell size, and cell elongation. The promotion in SL under PCh1 application may be associated with improved cytokinin activity, as well as enhanced water and nutrient uptake. Previous studies demonstrated that Ch can stimulate the biosynthesis of phytohormones, specifically cytokinins, which promote cell division and elongation,10 while phosphate contributes to improved nutrient assimilation and root development.98
Moreover, PCh1 application significantly increased the levels of Chla (by 39%), Chlb (by 32%), carotenoids (by 24%), and protein (by 139%), as well as RL (by 38%) and RWC (by 10%), compared with the control (p < 0.05) (Fig. 8B). The combined treatment (PCh1 + DS) elicited similar physiological responses, particularly enhancing photosynthetic pigment biosynthesis, which may contribute to improved photosynthetic efficiency under stress (Fig. 8B). Similar stimulative effects of Ch and its derivatives on drought-stressed plants were reported by Dawood et al.96 in Vicia faba and by Sadak et al.99 in other crops. It is plausible that Ch application under drought conditions promoted protein turnover, leading to the release of amino acids (i.e., glycine) that serve as precursors for pigment biosynthesis. This interpretation aligns with the findings of Abdel-Razik et al.100 (2024), who reported that Ch combined with amino acids enhanced plant growth and biochemical responses, including increased Chla and Chlb synthesis, under stress conditions.
To gain deeper insights into the mechanisms underlying PCh1-mediated drought tolerance and growth enhancement, particularly regarding amino acid release and pigment formation, comprehensive metabolite profiling should be conducted. Furthermore, analyzing the activity of key antioxidant enzymes such as SOD, CAT, and POX would help elucidate the pathways through which PCh1 mitigates oxidative damage under drought conditions.
4. Conclusions
This study demonstrates that solvent choice and treatment duration significantly influence the chemical and structural properties of PCh. Treatment of Ch with ortho-phosphoric acid in the presence of DMAc produced a derivative with a high degree of phosphoric substitution (4.88%) and markedly reduced crystallinity (17–42%). These modifications enhanced the polymer's amorphous and flexible structure, improving its water-retention capacity and nutrient-binding potential, which contributed to enhanced tomato growth and reduced oxidative stress under drought. The findings highlight the potential of PCh, particularly under optimized synthesis conditions, as a sustainable material to improve agricultural productivity and resilience in arid environments.Future research should evaluate the long-term stability and biodegradability of PCh under field conditions, assess its environmental safety, and explore scalable synthesis and application methods. This work provides a foundation for developing biopolymer-based soil conditioners that support circular economy goals and contribute to climate-resilient agriculture.
Author contributions
Conceptualization, methodology, formal analysis, and validation: [Fatima El Amerany]; investigation and writing – original draft: [Fatima El Amerany and Oumaima Ait Ali]; resources and writing – review & editing: [Fatima El Amerany and Mohammed Rhazi]. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this study are available within the article (tables and figures).Acknowledgements
Authors wish to thank «Le Centre d'Analyse et de Caractérisation (CAC) de Marrakech – UCA» and «La Cité de l'Innovation de Fès – USMBA» for supporting this study.References
- S. S. Solankey, R. K. Singh, D. K. Baranwal, D. K. Singh and I. J. Veg, Scienece, 2015, 21, 496–515, DOI:10.1080/19315260.2014.902414.
- K. E. Chiwina, G. Bhattarai, H. Xiong, N. K. Joshi, R. W. Dickson, T. M. Phiri, I. Alatawi, Y. Chen, Z. Stansell, K.-S. Ling and A. Shi, Agronomy, 2024, 14, 380, DOI:10.3390/agronomy14020380.
- R. Román-Doval, S. P. Torres-Arellanes, A. Y. Tenorio-Barajas, A. Gómez-Sánchez and A. A. Valencia-Lazcano, Polymers, 2023, 15, 2867, DOI:10.3390/polym15132867.
- M. Stasińska-Jakubas and B. Hawrylak-Nowak, Molecules, 2022, 27, 2801, DOI:10.3390/molecules27092801.
- F. Rassaei, Environ. Prog. Sustainable Energy, 2024, 43, e14301, DOI:10.1002/ep.14301.
- F. El Amerany, A. Meddich, S. Wahbi, A. Porzel, M. Taourirte, M. Rhazi and B. Hause, Int. J. Mol. Sci., 2020, 21, 535, DOI:10.3390/ijms21020535.
- F. El Amerany, O. Ait Ali and M. Rhazi, Euro-Mediterr. J. Environ. Integr., 2025, 1–16, DOI:10.1007/s41207-024-00719-5.
- F. El Amerany, M. Naimi and M. Rhazi, J. Biomech., 2025, 406, 244–254, DOI:10.1016/j.jbiotec.2025.07.022.
- F. El Amerany, M. Rhazi, S. Wahbi, M. Taourirte and A. Meddich, Sci. Hortic., 2020, 261, 109015, DOI:10.1016/j.scienta.2019.109015.
- F. Khan, A. B. Siddique, S. Shabala, M. Zhou and C. Zhao, Plants, 2023, 12, 2861, DOI:10.3390/plants12152861.
- A. Tariq, K. Pan, O. A. Olatunji, C. Graciano, Z. Li, F. Sun, X. Sun, D. Song, W. Chen, A. Zhang, X. Wu, L. Zhang, D. Mingrui, Q. Xiong and C. Liu, Front. Plant Sci., 2017, 8, 1561, DOI:10.3389/fpls.2017.01561.
- N. G. Mohamed and M. A. Abdel-Hakeem, Plant Gene, 2023, 34, 100406, DOI:10.1016/j.plgene.2023.100406.
- E. F. Ali, A. M. El-Shehawi, O. H. M. Ibrahim, E. Y. Abdul-Hafeez, M. M. Moussa and F. A. S. Hassan, Plant Physiol. Biochem., 2021, 161, 166–175, DOI:10.1016/j.plaphy.2021.02.008.
- N. J. Methela, A. Pande, M. S. Islam, W. Rahim, A. Hussain, D. S. Lee, B.-G. Mun, N. P. M. J. Raj, S.-J. Kim, Y. Kim and B.-W. Yun, BMC Plant Biol., 2023, 23, 639, DOI:10.1186/s12870-023-04640-x.
- M. O. Ekeoma, B. C. Oleleme and S. Sabar, J. Chem. Soc. Niger., 2024, 49, 116–128, DOI:10.46602/jcsn.v49i1.955.
- U. Anushree, P. Punj, Vasumathi and S. Bharati, Glycoconjugate J., 2023, 40, 19–31, DOI:10.1007/s10719-022-10093-5.
- H. Liu, M. J. Li, X. N. Zhang, S. Wang, L. X. Li, F. F. Guo and T. Zeng, Ecotoxicol. Environ. Saf., 2022, 238, 113609, DOI:10.1016/j.ecoenv.2022.113609.
- K. Binnemans and P. T. Jones, Green Chem., 2024, 26, 8583–8614, 10.1039/D4GC02031F.
- N. Yan and X. Chen, Nature, 2015, 524, 155–157, DOI:10.1038/524155a.
- T. S. Trung, L. H. Tram, N. Van Tan, N. Van Hoa, N. C. Minh, P. T. Loc and W. F. Stevens, Carbohydr. Res., 2020, 489, 107913, DOI:10.1016/j.carres.2020.107913.
- X. Hu, Z. Tian, X. Li, S. Wang, H. Pei, H. Sun and Z. Zhang, ACS Omega, 2020, 5, 19227–19235, DOI:10.1021/acsomega.0c02705.
- F. El Amerany, A. Meddich, S. Wahbi, M. Taourirte and M. Rhazi, J. Crop Health, 2023, 75, 1485–1495, DOI:10.1007/s10343-023-00837-0.
- A. Ewais, R. A. Saber, A. Abdel Ghany, A. Sharaf and M. Sitohy, SN Appl. Sci., 2023, 5, 365, DOI:10.1007/s42452-023-05602-6.
- J. Poupin and L. Corbari, Zootaxa, 2016, 4190, 1–107, DOI:10.11646/zootaxa.4190.1.1.
- E. M. Dahmane, M. Taourirte, N. Eladlani and M. Rhazi, Int. J. Polym. Anal. Charact., 2014, 19, 342–351, DOI:10.1080/1023666X.2014.902577.
- F. A. Al Sagheer, M. A. Al-Sughayer, S. Muslim and M. Z. Elsabee, Carbohydr. Polym., 2009, 77, 410–419, DOI:10.1016/j.carbpol.2009.01.032.
- N. M. Sachindra, N. Bhaskar and N. S. Mahendrakar, Waste Manag., 2006, 26, 1092–1098, DOI:10.1016/j.wasman.2005.07.002.
- F. Su, B. Huang, J. Liu and J. Crus, Biol, 2018, 38, 523–530, DOI:10.1093/jcbiol/ruy049.
- B. K. Simpson and N. F. Haard, J. Appl. Biochem., 1985, 7, 212–222 CAS.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254, DOI:10.1016/0003-2697(76)90527-3.
- D. R. Lyon, B. R. Smith, N. Abidi and J. L. Shamshina, Molecules, 2022, 27, 3983, DOI:10.3390/molecules27133983.
- J. C. Wang and J. E. Kinsella, J. Food Sci., 1976, 41, 286–292, DOI:10.1111/j.1365-2621.1976.tb00602.x.
- S. Kumari, S. H. Kumar Annamareddy, S. Abanti and P. Kumar Rath, Int. J. Biol. Macromol., 2017, 104, 1697–1705, DOI:10.1016/j.ijbiomac.2017.04.119.
- N. Subhapradha, P. Ramasamy, S. Sudharsan, P. Seedevi, M. Moovendhan, A. Srinivasan, V. Shanmugam and A. Shanmugam, Bioact. Carbohydr. Diet. Fibre, 2013, 1, 148–155, DOI:10.1016/j.bcdf.2013.03.001.
- J. Brugnerotto, J. Lizardi, F. M. Goycoolea, W. Argüelles-Monal, J. Desbrières and M. Rinaudo, Polymer, 2001, 42, 3569–3580, DOI:10.1016/S0032-3861(00)00713-8.
- A. Baxter, M. Dillon, K. D. Anthony Taylor and G. A. F. Roberts, Int. J. Biol. Macromol., 1992, 14, 166–169, DOI:10.1016/S0141-8130(05)80007-8.
- J. Dutta, Heliyon, 2022, 8, e09924, DOI:10.1016/j.heliyon.2022.e09924.
- M. Rinaudo, M. Milas and P. Le Dung, Int. J. Biol. Macromol., 1993, 15, 281–285, DOI:10.1016/0141-8130(93)90027-J.
- G. A. F. Roberts and J. G. Domszy, Int. J. Biol. Macromol., 1982, 4, 374–377, DOI:10.1016/0141-8130(82)90074-5.
- J. Brugnerotto, J. Desbrières, G. Roberts and M. Rinaudo, Polymer, 2001, 42, 09921–09927, DOI:10.1016/S0032-3861(01)00557-2.
- J. Kumirska, M. Czerwicka, Z. Kaczyński, A. Bychowska, K. Brzozowski, J. Thöming and P. Stepnowski, Mar. Drugs, 2010, 8, 1567–1636, DOI:10.3390/md8051567.
- G. Kitagawa, T. Mizuta, M. Akamatsu and S. Ifuku, High-yield chitin extraction and nanochitin production from cricket legs, Carbohydr. Polym. Technol. Appl., 2025, 100816, DOI:10.1016/j.carpta.2025.100816.
- W. Tachaboonyakiat, N. Netswasdi, V. Srakaew and M. Opaprakasit, Polym. J., 2010, 42, 148–156, DOI:10.1038/pj.2009.317.
- A. Torrecillas, C. Guillaume, J. J. Alarcón and M. C. Ruiz-Sánchez, Plant Sci., 1995, 105, 169–176, DOI:10.1016/0168-9452(94)04048-6.
- D. I. Arnon, Plant Physiol., 1949, 24, 1–15, DOI:10.1104/pp.24.1.1.
- S. N. Azam-Ali and G. R. Squire, Principles of Tropical Agronomy, Cambridge University Press, United Kingdom, 2002 Search PubMed.
- G. Szalai, T. Janda, E. Páldi and Z. Szigeti, J. Plant Physiol., 1996, 148, 378–383, DOI:10.1016/S0176-1617(96)80269-0.
- H. G. Gowtham, B. Singh, M. Murali, N. Shilpa, M. Prasad, M. Aiyaz, K. N. Amruthesh and S. R. Niranjana, Microbiol. Res., 2020, 234, 126422, DOI:10.1016/j.micres.2020.126422.
- V. Velikova, I. Yordanov and A. J. P. S. Edreva, Plant Sci., 2000, 151, 59–66, DOI:10.1016/S0168-9452(99)00197-1.
- M. K. Rasweefali, S. Sabu, K. V. Sunooj, A. Sasidharan and K. A. M. Xavier, Carbohydr. Polym. Technol. Appl., 2021, 2, 100032, DOI:10.1016/j.carpta.2020.100032.
- P. Sacco, M. Cok, F. Scognamiglio, C. Pizzolitto, F. Vecchies, A. Marfoglia, E. Marsich and I. Donati, Molecules, 2020, 25, 1534, DOI:10.3390/molecules25071534.
- Y. Xie, J. Ding, Y. Li, P. Wei, S. Liu and R. Yang, Foods, 2024, 13, 3572, DOI:10.3390/foods13223572.
- R. F. B. de Souza, F. C. B. de Souza, A. Thorpe, D. Mantovani, K. C. Popat and A. M. Moraes, Int. J. Biol. Macromol., 2020, 143, 619–632, DOI:10.1016/j.ijbiomac.2019.12.004.
- Z. Liu, Q. Liu, D. Zhang, S. Wei, Q. Sun, Q. Xia, W. Shi, H. Ji and S. Liu, Foods, 2021, 10, 2603, DOI:10.3390/foods10112603.
- J. Pohling, D. Dave, Y. Liu, W. Murphy and S. Trenholm, Green Chem., 2022, 24, 1141–1151, 10.1039/D1GC03140F.
- M. K. Rasweefali, S. Sabu, K. S. Muhammed Azad, M. K. Raseel Rahman, K. V. Sunooj, A. Sasidharan and K. K. Anoop, Adv. Biomarker Sci. Technol., 2022, 4, 12–27, DOI:10.1016/j.abst.2022.03.001.
- A. Renny, S. Nyi Mekar, H. Eli, A. Yuli, N. Aliya, L. Jutti and S. Sri Adi, J. Appl. Pharm. Sci., 2020, 10, 140–149, DOI:10.7324/JAPS.2020.101218.
- T. Paul, S. K. Halder, A. Das, K. Ghosh, A. Mandal, P. Payra, P. Barman, P. K. Das Mohapatra, B. R. Pati and K. C. Mondal, 3 Biotech., 2015, 5, 483–493, DOI:10.1007/s13205-014-0245-6.
- S. S. Pattanaik, P. B. Sawant, K. A. M. Xavier, K. Dube, P. P. Srivastava, V. Dhanabalan and N. K. Chadha, Aquaculture, 2020, 515, 734594, DOI:10.1016/j.aquaculture.2019.734594.
- I. Younes and M. Rinaudo, Mar. Drugs, 2015, 13, 1133–1174, DOI:10.3390/md13031133.
- M. S. Benhabiles, N. Abdi, N. Drouiche, H. Lounici, A. Pauss, M. F. A. Goosen and N. Mameri, Food Hydrocolloids, 2013, 32, 28–34, DOI:10.1016/j.foodhyd.2012.11.035.
- J. Pohling, V. V. Ramakrishnan, A. Hossain, S. Trenholm and D. Dave, Mar. Drugs, 2024, 22, 445, DOI:10.3390/md22100445.
- H. K. No, K. S. Lee and S. P. Meyers, J. Food Sci., 2000, 65, 1134–1137, DOI:10.1111/j.1365-2621.2000.tb10252.x.
- Y. I. Cho, H. K. No and S. P. Meyers, J. Agric. Food Chem., 1998, 46, 3839–3843, DOI:10.1021/jf971047f.
- A. El-araby, L. El Ghadraoui and F. Errachidi, Molecules, 2022, 27, 8285, DOI:10.3390/molecules27238285.
- F. Arrouze, M. Essalhi, M. Rhazi, J. Desbrières and A. Tolaimate, J. Mater. Environ. Sci., 2017, 8, 2251–2258 CAS.
- G. Yang, E. Lam and A. Moores, ACS Sustain. Chem. Eng., 2023, 11, 7765–7774, DOI:10.1021/acssuschemeng.3c00367.
- I. Aranaz, A. R. Alcántara, M. C. Civera, C. Arias, B. Elorza, A. Heras Caballero and N. Acosta, Polymers, 2021, 13, 3256, DOI:10.3390/polym13193256.
- G. Romanazzi, F. M. Gabler, D. Margosan, B. E. Mackey and J. L. Smilanick, Phytopathology, 2009, 99, 1028–1036, DOI:10.1094/PHYTO-99-9-1028.
- A. R. N. Pontillo, S. Koutsoukos, T. Welton and A. Detsi, Mater. Adv., 2021, 2, 3954–3964, 10.1039/D0MA01008A.
- C. K. S. Pillai, W. Paul and C. P. Sharma, Prog. Polym. Sci., 2009, 34, 641–678, DOI:10.1016/j.progpolymsci.2009.04.001.
- C. E. Starliper, B. J. Watten, D. D. Iwanowicz, P. A. Green, N. L. Bassett and C. R. Adams, J. Adv. Res., 2015, 6, 501–509, DOI:10.1016/j.jare.2015.02.005.
- J. Zhang, W.-R. Xu and Y.-C. Zhang, RSC Adv., 2022, 12, 22631–22638, 10.1039/D2RA03417D.
- T. K. Varun, S. Senani, N. Jayapal, J. Chikkerur, S. Roy, V. B. Tekulapally, M. Gautam and N. Kumar, Vet. World, 2017, 10, 170–175, DOI:10.14202/vetworld.2017.170-175.
- S. Abirami and D. Nagarajan, Extraction of Chitin from Shrimp Shell Wastes by Using Bacillus Licheniformis and Lactobacillus Plantarum, Int. J. Recent Res. Asp., 2018, 307–315 Search PubMed.
- A. Pawlak and M. Mucha, Thermochim. Acta, 2003, 396, 153–166, DOI:10.1016/S0040-6031(02)00523-3.
- D. Chakrabarty and S. Mahapatra, J. Mater. Chem., 1999, 9, 2953–2957, 10.1039/a905407c.
- R. Valentin, B. Bonelli, E. Garrone, F. Di Renzo and F. Quignard, Biomacromolecules, 2007, 8, 3646–3650, DOI:10.1021/bm070391a.
- Z. Chen, Y. Yu, Q. Zhang, Z. Chen, T. Chen and J. Jiang, Polym. Adv. Technol., 2019, 30, 1933–1942, DOI:10.1002/pat.4625.
- C. O. de Lima, A. L. M. de Oliveira, L. Chantelle, E. C. Silva Filho, M. Jaber and M. G. Fonseca, Colloids Surf., B, 2021, 198, 111471, DOI:10.1016/j.colsurfb.2020.111471.
- M. E. Zarif, S. A. Yehia-Alexe, B. Bita, I. Negut, C. Locovei and A. Groza, Polymers, 2022, 14, 5241, DOI:10.3390/polym14235241.
- G. Hao, Y. Hu, L. Shi, J. Chen, A. Cui, W. Weng and K. Osako, Sci. Rep., 2021, 11, 1646, DOI:10.1038/s41598-021-81318-0.
- W.-C. Huang, D. Zhao, C. Xue and X. Mao, Mar. Life Sci. Technol., 2022, 4, 384–388, DOI:10.1007/s42995-022-00129-y.
- Y. Zhang, C. Xue, Y. Xue, R. Gao and X. Zhang, Carbohydr. Res., 2005, 340, 1914–1917, DOI:10.1016/j.carres.2005.05.005.
- B. P. Sudatta, V. Sugumar, R. Varma and P. Nigariga, Int. J. Biol. Macromol., 2020, 163, 423–430, DOI:10.1016/j.ijbiomac.2020.06.291.
- S. Subramaniam, K. Y. Foo, E. M. Yusof, A. H. Jawad, L. D. Wilson and S. Sabar, Int. J. Biol. Macromol., 2021, 193, 1716–1726, DOI:10.1016/j.ijbiomac.2021.11.009.
- N. A. H. Rosli, K. S. Loh, W. Y. Wong, T. K. Lee and A. Ahmad, Membranes, 2021, 11, 675, DOI:10.3390/membranes11090675.
- M. Subramanian, Y. Manogaran and P. Ramasamy, Cureus, 2024, 16, DOI:10.7759/cureus.69951.
- A. C. Valbuena, Adv. Chitin Sci., 2012, 14, 98–103 Search PubMed.
- G. S. Bakhoum, M. M. Tawfik, M. O. Kabesh and M. S. Sadak, Vegetos, 2025, 1–12, DOI:10.1007/s42535-025-01198-x.
- G. S. Bakhoum, M. M. Tawfik, M. O. Kabesh and M. S. Sadak, Biocatal. Agric. Biotechnol., 2023, 51, 102794, DOI:10.1016/j.bcab.2023.102794.
- V. Alexieva, I. Sergiev, S. Mapelli and E. Karanov, Plant, Cell Environ., 2001, 24, 1337–1344, DOI:10.1046/j.1365-3040.2001.00778.x.
- G. S. Bakhoum, M. S. Sadak and M. Thabet, J. Soil Sci. Plant Nutr., 2023, 23, 6612–6631, DOI:10.1007/s42729-023-01514-x.
- S. Ackah, Y. Bi, S. Xue, S. Yakubu, Y. Han, Y. Zong, R. A. Atuna and D. Prusky, Front. Plant Sci., 2022, 13, 959762, DOI:10.3389/fpls.2022.959762.
- R. S. Hanafy and M. S. Sadak, J. Soil Sci. Plant Nutr., 2023, 23, 2433–2450, DOI:10.1007/s42729-023-01197-4.
- M. G. Dawood, M. El-Awadi and M. S. Sadak, J. Soil Sci. Plant Nutr., 2024, 24, 5696–5709, DOI:10.1007/s42729-024-01934-3.
- S. Bañon, J. Ochoa, J. A. Franco, J. J. Alarcón and M. J. Sánchez-Blanco, Environ. Exp. Bot., 2006, 56, 36–43, DOI:10.1016/j.envexpbot.2004.12.004.
- F. Chen, Q. Li, Y. Su, Y. Lei and C. Zhang, Molecules, 2023, 28, 2053, DOI:10.3390/molecules28052053.
- M. S. Sadak, M. M. Tawfik and G. S. Bakhoum in, Nanomaterial-Plant Interactions Role of Chitosan and Chitosan-Based Nanomaterials in Plant Sciences, ed. S. Kumar and S. V. Madihally, Academic Press, 2022, pp. 475–501, DOI:10.1016/B978-0-323-85391-0.00013-7.
- T. A. Abdel-Razik, B. A. Bakry and M. S. Sadak, Egypt. Pharm. J., 2024, 23, 630–642, DOI:10.4103/epj.epj_23_24.
| This journal is © The Royal Society of Chemistry 2026 |