DOI:
10.1039/D5EY00254K
(Paper)
EES Catal., 2026,
4, 134-145
Direct work function tuning via boron-acceptor substitution on an iron phthalocyanine ligand for a boosted oxygen reduction reaction in brine-seawater batteries
Received
23rd August 2025
, Accepted 29th August 2025
First published on 4th September 2025
Abstract
Highly conductive concentrated brine seawater can be reused as an electrolyte in aluminium–air seawater batteries used in on-board marine applications; however, the severe chloride corrosion in brine seawater often causes Pt-based oxygen reduction reaction (ORR) electrocatalysts at the cathode to degrade rapidly. Fe macrocyclic molecules, such as those in iron phthalocyanine (FePc), are reported to exhibit low affinity to chloride adsorption. On the other hand, the strongly bound O* and OOH* intermediates in the FeN4 active sites and the localized electron orbitals are well-known to restrict their ORR performance. In this study, by combining a room-temperature plasma-assisted material modification strategy with density functional theory (DFT) calculations and thermodynamic modelling, we successfully substituted boron as an acceptor on the FePc ligand to induce significant electron delocalization in the macrocyclic FePc structure, thereby reducing the ORR energy barrier of FePc. In an alkaline saline environment (0.1 M KOH + 1 M NaCl), B-FePc displays superior catalytic activity (0.932 V vs. RHE) at the half-wave potential with moderate stability, which surpassed the performance of a commercial 20 wt% Pt/Vulcan electrocatalyst and most of the recently reported electrocatalysts. When used as an air cathode catalyst in a brine seawater-based Al–air battery (1 M KOH + 1 M NaCl + seawater), B-FePc as a cathode catalyst exhibited a peak power density of 71.0 mW cm−2 and an exceptional stability following its discharging for 60 h at 20 mA cm−2 through a mechanical recharging process.
Broader context
Highly conductive concentrated brine seawater can be reused as an electrolyte in aluminium–air seawater batteries used in on-board marine applications; however, the severe chloride corrosion in brine seawater often causes degradation of the metal-based cathode catalysts rapidly. While single-atom FeN4 active sites in iron phthalocyanine (FePc) are reported to exhibit low affinity to chloride adsorption, the strongly bound O* and OOH* intermediates at FeN4 are well-known to restrict its ORR performance. In this study, we are the first to substitute boron as an acceptor on the FePc ligand to induce significant electron delocalization in the macrocyclic FePc structure via plasma engineering, thereby reducing the ORR energy barrier. In an alkaline saline environment, B-FePc displays superior catalytic activity (0.932 V vs. RHE) at the half-wave potential and high stability. When applied as an air cathode catalyst in a brine seawater-based Al–air battery, B-FePc as a cathode catalyst exhibited a peak power density of 71.0 mW cm−2 and an exceptional stability. Combined with our novel design of B-FePc as a cathode catalyst, the study realizes the feasibility of a sustainable brine-seawater battery for ships and global coastal regions.
|
Introduction
Recently, saline-concentrated brine seawater, considered a waste resource originating from the desalination systems of ships or desalination plants, has proved to be a promising conductive electrolyte that can be used in seawater-based batteries.1,2 Brine seawater, which contains a high chloride concentration, can improve the ionic conductivity of seawater-based electrolytes and Al oxidation through passive film breakdowns.3–6 However, Pt-based electrocatalysts often degrade because of their intrinsic intolerance to chloride adsorption and are thus unsuitable for use in brine seawater environments.7 Recent studies have highlighted the use of single iron atoms to serve as electrocatalysts for oxygen reduction reactions (ORRs) in seawater-based electrolytes, which will benefit from the low reaction energy barrier in the ORR and their low affinity to chloride adsorption.8,9 Macrocyclic metal complexes, such as iron phthalocyanine (FePc), would be promising single-atom iron-based components because of their outstanding catalytic properties, chemical stability, and electronic structure tunability. However, their highly symmetric geometry makes the electron distribution in FePc rather stable, limiting electron transport and overall ORR activity.10,11 Many studies have investigated the modulation of the FePc electronic structure through different approaches to optimise the catalytic performance of FePc further. One existing strategy involves the attachment of an Fe active centre to a conductive substrate made of carbon nanotubes, monolayer graphene, MXene, and highly conjugated polymers (metal–organic frameworks).12–18 All other approaches employ a coordination method to incorporate electron-withdrawing or electron-donating heteroatom dopants into central Fe atoms, which adds axial O-coordinated sites to break the electronic distribution symmetry of the central Fe single atoms or peripherally substituted phenylene carbon.16,19–22 Previous studies reported that the d-orbital level of the Fe atoms in the macrocyclic FePc could successfully lower the binding energy of the O* or OH* intermediates.17
A few studies have reported achieving advanced ORR catalytic activity by adding oxygen- or nitrogen-containing groups to the edges of macrocyclic molecules. However, previous studies have yet to report the effects of boron (B) substitution mainly owing to the technical challenges associated with the conventional thermal process used, which requires high-temperature pyrolysis at a temperature between 800 and 1100 °C to incorporate B dopants into phthalocyanine structures.23 Since the thermal decomposition temperature of FePc is in the range from 380 to 700 °C,16,24 applying B as an acceptor dopant in macrocyclic FePc without disrupting its ligand complex is hardly possible due to its low thermal stability.16,24 On the other hand, plasma engineering employs plasma-induced chemical reactions in place of conventional annealing processes with a unique surface modification approach that can directly modify macrocyclic structures at room temperature.25 Using plasma engineering, which can be considered a novel surface modification process, we successfully performed B-substitution for FePc macrocycle molecules without destroying the FePc ligand. The chemical and electronic structure analysis revealed that B-substitution for FePc macrocycles initiates charge transfer from the Fe centre to the surrounding ligand by working as an in-ligand acceptor, and it successfully modulates the FePc electronic structure for improved catalytic ORR activity. Additionally, by employing density functional theory (DFT) calculations and thermodynamics theory, we determined that boron as an acceptor preferentially replaces a specific carbon atom site right next to the FePc macrocycles and formed C–B–N bonding, which was confirmed by detailed characterization based on FTIR, XANES, EXAFS and XPS. DFT calculations revealed that the acceptor (B) substitution in FePc improved its ORR activity owing to (i) the less negative Gibbs free energy of *O and *OH intermediates by the increased FePc work function and (ii) improved electron transport facilitated by the delocalisation of the highest occupied molecular orbital (HOMO) level. The electrochemical performance of the B-substituted FePc (B-FePc) in 0.1 M KOH agreed well with the DFT calculation results, which showed a clear improvement in the half-wave potential of B-FePc (from 0.87 to 0.882 V vs. reversible hydrogen electrode (RHE)) and limiting the current density (from 5.54 to 5.90 mA cm−2) and kinetic current density (from 29.8 to 45.1 mA cm−2 at 0.85 V vs. RHE) with reference to those of pristine FePc. In alkaline seawater (0.1 M KOH + Seawater), B-FePc exhibited superior ORR catalytic activity and stability compared to commercial 20 wt% Pt/Vulcan. Initially and following 5000 durability test cycles, B-FePc demonstrated a kinetic current density that was approximately 8 times higher than that of 20 wt% Pt/Vulcan. When employed as the air cathode catalyst in an Al–air battery in brine seawater (1 M KOH + Seawater + 1 M NaCl), which had been discharging for 60 h at 20 mA cm−2, the B-FePc cathode catalyst still performed with a peak power density of 71.0 mW cm−2 and outstanding stability.
Experimental procedures
Preparation of B-FePc via plasma engineering
The B-FePc electrocatalysts used in the study were modified by plasma engineering, with a similar experimental setup as reported in previous studies.26 First, 40 mmol boric acid was dissolved in 50 mL of ethanol for 2 h under magnetic stirring to obtain a boric acid solution. Then, 40 mmol of boric acid contained in 25 mL of the prepared boric acid solution was mixed with 75 ml of the solution obtained by dispersing 100 mg of FePc in N-methyl-2-pyrrolidone, to obtain the solution precursors required for plasma engineering. The generated plasma was then discharged into the mixed solution through a pair of 2-mm graphite electrodes using a bipolar pulse power supply (MPP-HV02, Kurita Seisakusho, Kyoto, Japan). The plasma was kept at a voltage of 1.6 kV, a frequency of 25 kHz, and a pulse width of 1.0 μs for 10 min. The discharged precursor solution was then filtered using a 55 mm polytetrafluoroethylene filter and washed with deionised water until the washed solution became transparent. The powder samples were dried in an oven for 10 h at 80 °C.
Analysis of the structural and chemical composition of B-FePc
The morphology and structure of plasma-engineered B-FePc electrocatalysts were analysed through X-ray diffraction (XRD; Rigaku, Ultima IV, Japan). The chemical composition of the electrocatalysts was characterised using X-ray photoelectron spectroscopy (XPS, JEOL, JPS-9010MC, Japan). The concentration of each metal in the synthesised catalysts was determined using inductively coupled plasma optical emission spectrometry (ICP-OES; PerkinElmer, Optima 8300, USA). Morphological and elemental concentration analyses were conducted using field emission scanning electron microscopy (TESCAN, MIRA3, Czech Republic), energy dispersive spectroscopy (EDS, TESCAN, MIRA3, Czech Republic), and transmission electron microscopy (Thermo Fisher Scientific, TALOS F200X, USA). Fourier transform infrared (FT-IR) transmittance spectra were recorded using a Nicolet iS50 FT-IR spectrometer (Thermo Fisher Scientific, USA). X-band electron paramagnetic resonance (EPR) spectra were recorded at room temperature using a Bruker EMX Plus instrument. Ultraviolet photoemission spectroscopy (UPS) was performed using an ultraviolet photoemission spectroscopy instrument (AXIS Supra, Kratos Analytical Ltd, United Kingdom).
Electrochemical analysis of B-FePc in alkaline and brine-seawater electrolytes
An electrochemical potentiostat (Biologic, VSP, Grenoble, France) was used to analyse the catalytic activities of the modified catalysts B-FePc, pristine FePc, and 20 wt% Pt/C (Fuel Cell Store, 20% Platinum on XC-72 carbon) in 0.1 M KOH and brine seawater (0.1 M KOH + 1 M NaCl) electrolyte. The catalyst ink used in the electrochemical analysis comprised a well-ground powder catalyst (2 mg) and conductive carbon (graphene nanoplatelets, 2 mg) in a mixed solution of distilled water (400 μL), ethanol (1584 μL), and Nafion 117 solution (16 μL), and ultrasonicated for 30 min. The well-dispersed catalyst ink (200 μg cm−2) was added dropwise on a well-polished 4 mm glassy carbon disk electrode to obtain the working electrode. For comparison purposes, the 20 wt% Pt/Vulcan XC 72-R electrocatalyst was prepared with 4 mg of the catalyst using the same ink composition. A platinum coil counter electrode, along with Hg/HgO (1 M NaOH) and Ag/AgCl (1 M KCl) reference electrodes, was used in the 0.1 M KOH and alkaline seawater electrolytes, respectively, to establish a standard three-electrode cell system towards determining the electrochemical performance of B-FePc. The catalytic activity of the ORR was determined using linear sweep voltammetry (LSV) performed under O2-saturated conditions at a scan rate of 5 mV s−1, a rotating speed of 1600 rpm, and a potential range between 0.4 and 1.0 V vs. RHE with 0.1 M KOH, 0.1 M KOH + 0.5 M NaCl (alkaline seawater), and 0.1 M KOH + 1 M NaCl (brine seawater) as the electrolytes. For the accelerated durability test, the cyclic voltammogram of B-FePc was measured from 0.6 to 1.0 V vs. RHE at a scan rate of 100 mV s−1 for 5000 cycles.
Preparation of the saline-concentrated brine-seawater-based Al–air battery
Al–air batteries were tested in both alkaline seawater (1 M KOH + seawater salt) and alkaline brine seawater (1 M KOH + 1 M NaCl + seawater salt) to mimic the natural and brine seawater conditions, respectively. The seawater salt was obtained from AF Perfect Water, Aquaforest, Brzesko, Poland. The batteries were assembled with the as-prepared catalysts loaded onto the gas diffusion layer electrode with a mass loading of 2 mg cm−2. Catalyst ink was prepared using 6 mg of the B-FePc electrocatalyst and 6 mg of conductive carbon in 6 mL of the mixed solution comprising 4752 μL of ethanol, 1200 μL of deionised water, and 48 μL of the Nafion®117 solution. The prepared catalyst was sprayed on a 6 cm2 gas diffusion layer. Polarisation curves of the Al–air seawater batteries were obtained at 20 mV s−1. A voltage retention test was conducted at 20 mA cm−2.
DFT calculations
The Vienna ab initio simulation package was used for DFT calculations.27,28 The projector augmented wave method and the general gradient approximation with the Perdew, Burke, and Ernzerhof exchange–correlation functional were employed for plane wave expansion.29–31 The Brillouin zone was sampled using Gamma-centered k-point grids of 1 × 1 × 1 for optimising cell parameters with a kinetic energy cut-off of 400 eV.32 The energy convergence criteria in the self-consistent field were set to 10−6 eV. All the geometric structures were fully relaxed, while the Hellman–Feynman forces were below 0.1 eV Å−1. The supercell dimensions for FePc were set at 22 Å × 22 Å × 22 Å to prevent interactions among the adjacent periodic FePc molecules caused by the vacuum.
Results and discussion
Insight into possible B-substitution in FePc
To verify the possibility of B-substitution into FePc, the bond enthalpies and vibration frequencies of FePc were first evaluated by the Gibbs free energy change equation. To simplify the calculations, the displacements of one B atom around the FePc ligand were considered with 4 possible structures (B1-FePc, B2-FePc, B3-FePc, and B4-FePc), as shown in Fig. 1(a). The Gibbs free energy change equation is calculated as follows:| |  | (1) |
where ΔEf is the total energy change derived from DFT calculations, ΔZPE is the zero-point energy change,  is the heat capacity contribution, T is the temperature, and ΔS is the change in entropy. The ΔZPE,
is the heat capacity contribution, T is the temperature, and ΔS is the change in entropy. The ΔZPE,  , and TΔS terms were expressed as:
, and TΔS terms were expressed as:| |  | (2) |
| | 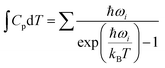 | (3) |
| |  | (4) |
where ℏ, ωi, and kB are the reduced Planck constant (ℏ = h/2π), the vibrational frequency eigenvalue, and the Boltzmann constant. All the atoms vibrated from their equilibrium positions, and the atomic positions in the FePc molecule model were obtained using the structural relaxation process. As shown in Fig. 1(b), all four possible structures differ greatly in the temperature required for boron substitution. According to thermodynamic predictions, out of four possible structures, the formation of B1-FePc is the only thermodynamically feasible structure, which includes a distinct B–C–N structure compared to other positions (B2, B3 and B4 consist of merely C–B–C chemical bonding). In addition, while traditional annealing processes often cause thermal decomposition of the FePc ligand during B-substitution, plasma engineering is expected to break some of the kinetic limitations via highly active species generated by the plasma-induced chemical reactions at room temperature and ensure the stability of the FePc ligand with B1-substitution.
 |
| | Fig. 1 (a) Atomic structure of FePc and four possible Boron doping sites, (b) calculated Gibbs free energy change (ΔG) by the formation of boron-doped FePc (four possible doping sites), (c) calculated adsorption free energy for the ORR on FePc (black) and boron-doped FePc (red, site 1) at a pH of 13 and a bias of 1.23 V. (d) Calculated orbital decomposed electron density of states of the Fe 3d orbitals in FePc and B1-FePc; (e) illustration of electron transfer from the 3d orbital of the central iron atom to the 2p orbital of the boron atom. | |
DFT calculation of the advanced ORR activity of B1-FePc
Adsorption energy is commonly used to determine the catalytic activity of a catalyst. The adsorption energies of the typical ORR intermediates in an alkaline medium following the four ORR catalytic reaction steps are calculated as shown in eqn (5)–(8).| | | * + O2(g) + H+ + e− → *OOH | (5) |
| | | *OOH + H+ + e− → *O + H2O(l) | (6) |
| | | *OH + H+ + e− → H2O(l) + * | (8) |
where O*, OOH*, and OH* are the typical reaction intermediates and * represents a catalytically active site. In previous studies, the desorption of O* or OH* intermediates was reported to be the rate-determining step of the ORR. Previous studies found that the incorporation of an electron-withdrawing group onto FePc macrocycle molecules delocalised the electron density of the central Fe atoms, thereby successfully lowering the binding energy of the O* or OH* intermediates.33 Gibbs free energies of the adsorbates were calculated using eqn (9).| | | ΔGads = ΔEads + ΔGpH + ΔGU − neΔV | (9) |
where ΔEads is the total energy change associated with adsorption and desorption, ΔGpH is −kBT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln10 × pH, which is the Gibbs free energy difference due to the change in proton concentration, ΔGU is −neU (U, n, and e are the applied potential, electron number, and transferred charge, respectively), and ΔV is the difference between the Fermi energy of the catalysts and hydrogen reduction level. The work functions of FePc (3.72 eV) and B–N/FePc (4.11 eV), which were calculated based on the UPS spectra shown in Fig. S1, were used to calculate the ORR (eqn (9)) required to prepare the energy diagram.
ln10 × pH, which is the Gibbs free energy difference due to the change in proton concentration, ΔGU is −neU (U, n, and e are the applied potential, electron number, and transferred charge, respectively), and ΔV is the difference between the Fermi energy of the catalysts and hydrogen reduction level. The work functions of FePc (3.72 eV) and B–N/FePc (4.11 eV), which were calculated based on the UPS spectra shown in Fig. S1, were used to calculate the ORR (eqn (9)) required to prepare the energy diagram.
Our calculations shown in Fig. 1(c) demonstrate that the largest ORR energy barrier of pristine FePc is 5.85 eV at the desorption of OH* intermediates under alkaline conditions (pH = 13) with the bias U = 1.23 V. In the energy diagram, B1-FePc shows two energy barriers: 0.37 and 1.95 eV for *OH formation and desorption, respectively. To improve the ORR activity, strong OH* binding has to be prevented.34 Thus, the surface reaction rate of oxygen reduction in FePc was improved by B-substitution because of the reduction in *OH desorption energy.
To gain an enhanced understanding of the mechanism that led to an increase in the work function of FePc via B substitution, we compared the electronic structures of FePc and B1-FePc, as shown in Fig. 1. The Fe atom in B1-FePc in Fig. 1(d) has fewer occupations of spin-down states of dz2 compared to the Fe atom in the pristine FePc. The reason for this can be understood as the doped boron atom accepts an electron from the 3d orbital of the iron atom. As a result, the magnetic moment of the Fe atom is increased from 2.0μB to 3.0μB, while the magnetic moment of the B atom (Fig. S2) results in 0μB, by reducing the down-spin electron occupation and increasing the work function. According to the above findings and the isosurfaces of the electron at the HOMO level in Fig. 1(e), the HOMO level is occupied together by the 2p orbital of the C atoms and the 3d orbital of the Fe atom in the B1-FePc model, while the HOMO level is occupied merely by the central Fe atom in the FePc model. Therefore, we can reason that the B1-FePc system has a lower energy barrier of electron transport from carbon to Fe during the ORR compared to that of pristine FePc. As shown in Fig. 1(e), B1-FePc has higher delocalization of electrons at the HOMO level compared to FePc, which agrees well with the findings of a previous study that reported the enhancement of the electron transport efficiency facilitated through a conjugate of the FePc molecules and a strong delocalisation effect on the central FePc moieties via adjacent polymer FePc molecules.17
Structural analyses of B-FePc
After confirming the preferred B substitution into FePc as the B1-FePc structure by thermodynamic calculations, the morphology and structural properties of the as-synthesized catalysts, referred to as B-FePc hereafter, were analysed in detail. The SEM images of pristine FePc and B-FePc are shown in Fig. 2(a) and Fig. S3. While pristine FePc exhibited a spherical agglomerated shape, the morphology of B-FePc was needle-like. The morphological differences between FePc and B-FePc can be due to the possible exfoliation of the agglomerated FePc following plasma engineering.35 According to SEM-EDS results, the boron content in B-FePc was approximately 1.87 at%, confirming successful B doping into FePc (Fig. S4 and Table S1). Moreover, the high-resolution transmission electron spectroscopy images showed that B-FePc had a stacked planar morphology without any undesired metallic particles resulting from the decomposition/agglomeration of atomic Fe, a common problem associated with other often reported thermal-based processes (Fig. 1(b) and Fig. S5). According to the selected area electron diffraction patterns, high-angle annular dark-field imaging results, and EDS images illustrated in Fig. 1(c)–(h), the B-substitution of the FePc ligand has not changed the original structure of FePc following plasma engineering; thus, the electronic structure modification effect of B on the FePc macrocycles towards the ORR can be further clarified. Also, from Fig. S3, HR-TEM, HAADF and EDS images of pristine FePc show a similar morphology to B-FePc, which supports that plasma engineering preserves the original structure of the pristine FePc.
 |
| | Fig. 2 (a) Field emission scanning electron microscopy image of B-FePc, (b) high-resolution transmission electron microscopy image of B-FePc, (c) selected area electron diffraction pattern of B-FePc, and (d) high-angle annular dark-field image of B-FePc, (e)–(h) EDS mapping images of B, C, N, and Fe in B-FePc. | |
In the XRD patterns of B-FePc (Fig. S6), identical diffraction peaks corresponding to pristine FePc were observed at 7.1°, 15.5°, and 24.9°. These peaks signify the presence of FePc in its a-form and the stacking of FePc along the a-direction, indicating that the crystalline structures of FePc had not changed following the substitution of boron atoms.36 According to ICP-OES results, the Fe contents in pristine FePc and B-FePc were 9 and 8.9 wt%, respectively, which indicates that the Fe contents remained almost identical following B-substitution (Table S2). In addition, UPS and EPR analyses were performed to analyse the changes in the electronic structure of FePc generated by B-substitution, and the electronic structure of B-FePc was compared with that of pristine FePc. As shown in Fig. S7, the cut-off energies in the UPS spectra of FePc and B-FePc are 17.48 and 17.09 eV, respectively, which indicate that the work function has increased from 3.72 to 4.11 eV following B-substitution. Furthermore, the maximum energies of the valence band (Evbm) of FePc and B-FePc are 2.65 and 2.35 eV, respectively, indicating that the HOMO in B-FePc is closer to the Fermi level than that of pristine FePc. Thus, B-substitution within the macrocycle molecules withdraws the valence electrons at the HOMO of FePc, while boron serves as an acceptor dopant for the FeN4 moiety.37 An increase in the work function by B-substitution could reduce the potential energy drop in charge transfer between the central Fe atoms and oxygen, facilitating the desorption of OH*, which is determined as the rate-determining step of the ORR in FePc38 in DFT calculations. The EPR analysis results shown in Fig. S8 also supported the strong electron delocalisation of B-FePc, with a g-value of 2.075.39
Identification of the types and states of chemical bonding in B-FePc
The major types of chemical bonding of B-FePc were first identified by FT-IR analysis. In Fig. 3(a), both pristine FePc and B-FePc showed typical C–H bending peaks (attributed to the out-of-plane deformation arising from the phthalocyanine skeleton) at 1086, 1119, and 1165 cm−1, while the characteristic peaks of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration appeared at 1288 and 1332 cm−1.40 Unlike pristine FePc, B-FePc shows an extra peak at 1389 cm−1, which can be attributed to in-plane B–N stretching, generally detected from sp2-hybridised hexagonal boron nitride.41 In addition, the peaks of pristine and B-FePc indicate that the original structure of FePc was maintained even after the B-substitution of the ligand. Based on the 4 possible structures of B substitution, only B1-FePc demonstrates this distinct bonding of B–N, where the other 3 structures (B2-FePc, B3-FePc and B4-FePc) would have presented other types of B–C bonding instead.
C stretching vibration appeared at 1288 and 1332 cm−1.40 Unlike pristine FePc, B-FePc shows an extra peak at 1389 cm−1, which can be attributed to in-plane B–N stretching, generally detected from sp2-hybridised hexagonal boron nitride.41 In addition, the peaks of pristine and B-FePc indicate that the original structure of FePc was maintained even after the B-substitution of the ligand. Based on the 4 possible structures of B substitution, only B1-FePc demonstrates this distinct bonding of B–N, where the other 3 structures (B2-FePc, B3-FePc and B4-FePc) would have presented other types of B–C bonding instead.
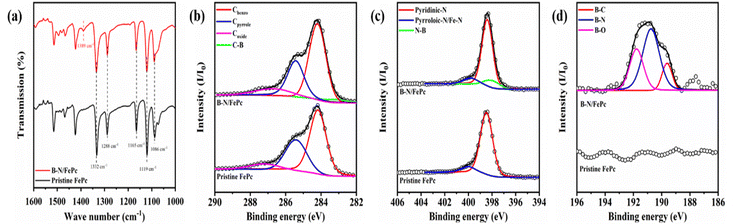 |
| | Fig. 3 (a) Fourier transform infrared transmittance spectra, and (b)–(d) X-ray photoelectron spectra of C1s, N1s, and B1s. | |
The chemical bonding structures were further investigated by various X-ray based analyses. In Fig. 3(b) and (c), the narrow C 1s and N 1s spectra exhibit a clear shift following the B-substitution of the FePc ligand. The C 1s spectra indicate the C-benzo emissions resulting from the 24 carbon atoms in the benzo groups and C-pyrrole emissions originating from the 8 carbon atoms bonded to the pyrrole nitrogen atoms, recorded at 284.2 and 285.4 eV, respectively.42 However, the binding energy of carbon in B-FePc moved towards a more negative value owing to the presence of C–B bonds (283.0 eV).43 The N 1s spectra of both pristine FePc and B-FePc exhibited trends similar to those of the C 1s spectra, revealing two significant bonds: pyridinic N, indicative of bonding with the outer carbon present at 398.8 eV in the Fe coordination structure, and pyrrolic N/Fe–N, representing the bonding with the inner carbon of the Fe coordination structure at 400.4 eV.44 Similar to the C 1s spectra of B-FePc, the N 1s spectra of B-FePc showed a clear shift towards more negative binding energies owing to forming the B–N bond (398.0 eV).45 Furthermore, the B 1s spectra in Fig. 3(d) of B-FePc can be deconvoluted into B–C, B–N, and B–O bonds at 189.6, 190.8 and 191.8 eV, respectively. This bonding state of B-FePc indicated that B–N and B–C bonds have been incorporated successfully into the hexagonal carbon structure,33,34,46,47 which agrees with the proposed B-substitution position as B1-FePc from theoretical calculation since only the B1-FePc structure consisted of a distinct C–B–N bonding simultaneously. In addition, as shown in Fig. S9, the Fe3+ ratio slightly increases from 43% in pristine FePc to 49% in B-FePc, further confirming that the electron delocalisation had originated from the central Fe atoms. From Fig. S9, increasing the boric acid concentration from 10 mmol to 20 mmol does not induce further changes in the chemical structure, as observed in the Fe 2p spectra and Fe2+/Fe3+ ratio (Fig. S9(a) and (b)), or in the electronic structure, as shown by the EPR analysis (Fig. S9(c)). This is because 10 mmol of boric acid already provides a boron content over 50 times higher than the maximum soluble amount of the chemical in FePc (568.38 g mol−1). Therefore, only 10 mmol of boric acid was applied to modify the FePc structure in this study.
Electrochemical evaluation of pristine and B-FePc in an alkaline electrolyte
Electrochemical analyses of as-synthesized catalysts under 0.1 M KOH electrolyte were conducted to verify the statement of advanced ORR catalytic activity of B-FePc based on theoretical calculations. Fig. 4(a) shows the LSV curves of the FePc and B-FePc electrocatalysts obtained under alkaline conditions. The LSV curves revealed a distinct improvement in the catalytic activity towards the ORR. With reference to pristine FePc, B-FePc exhibited an increase in the half-wave potential from 0.87 to 0.882 V vs. RHE and limiting current density from 5.54 to 5.90 mA cm−2 and a decrease in the Tafel slope from 39 to 37 mV dec−1, which proves the effect of B-FePc on advanced ORR activity (Fig. 4(b)). Moreover, the kinetic current density of B-FePc calculated at 0.85 V vs. RHE is over 45 mA cm−2, which exceeds the kinetic current density of FePc and 20 wt% Pt/Vulcan by more than 50% and 150%, respectively (Fig. 4(c)). Additionally, the mass activity per Fe content unit increased by approximately 60% following B-substitution (Fig. 4(c)). The calculated charge transfer resistances of FePc and B-FePc were 28 and 21 Ω, respectively (Fig. S10 and Table S3), indicating that the B-substitution in FePc macrocycles led to disorder and tortuosity within the carbon domains, improving the charge transfer properties and reaction kinetics throughout the electrocatalytic process. Based on the theoretical calculation results and electrochemical performance, we confirmed that the B-FePc macrocycles considerably improved the ORR activity in an alkaline environment. Combining the theoretical calculation and experimental characterization, the conceptual schematic of B-FePc is shown in Fig. 4(d). Since we have confirmed that the only feasible B-substitution site was B1-FePc, the advanced ORR activity of B-FePc can be explained as follows: the B-substitution at the B1 position serves as an acceptor dopant for the central Fe atoms, which can delocalize its HOMO electrons. This delocalization of the HOMO electrons of the central Fe atoms causes an adsorption energy change between the oxygen intermediates and the central Fe atoms of FePc, resulting in enhanced ORR catalytic activity, as illustrated in Fig. 4(e).
 |
| | Fig. 4 (a) Linear sweep voltammetry (LSV) profiles of 20 wt% Pt/Vulcan, Pristine FePc and B-FePc in 0.1 M KOH, (b) Tafel slope of each sample derived from (a), (c) calculated kinetic current density and mass activity at 0.85 V vs. RHE from LSV profiles, (d) the atomic structures and the calculated isosurfaces of HOMO levels in FePc and B1-FePc, and the isovalue of the isosurface was set to be 0.003 e Å−3, (e) schematic of the work function changes of B-FePc. | |
Electrochemical evaluation of B-FePc in a saline-concentrated brine seawater electrolyte
In general, abundant soluble ions in an electrolyte can accelerate charge transfer to enhance electrocatalytic ORR. However, the state-of-the-art Pt-based catalysts often suffer from chloride adsorption on their surfaces, which greatly hinders the charge transfer and reaction kinetics.48,49 Therefore, we firstly evaluated the electrochemical performance of B-FePc as an ORR electrocatalyst in alkaline saline (0.1 M KOH + 0.5 M NaCl) and in brine saline (0.1 M KOH + 1 M NaCl) electrolytes to identify its performance against chloride adsorption at normal to high chloride concentrations. As shown in Fig. 5(a), B-FePc exhibits superior catalytic activity in electrolytes with high chloride concentrations than in commercial 20 wt% Pt/Vulcan. As shown in Fig. 5(b), B-FePc exhibits exceptional ORR catalytic activity (0.917 V vs. RHE) at the half-wave potential, surpassing the corresponding performance of the commercial 20 wt% Pt/Vulcan electrocatalyst and most of the reported electrocatalysts under alkaline saline conditions.5,50–56 In Fig. 5(c), B-FePc also displays superior catalytic activity (0.932 V vs. RHE) at the half-wave potential under alkaline brine conditions with an elevated chloride concentration (0.1 M KOH + 1 M NaCl). Compared to alkaline saline, B-FePc under brine saline conditions shows a half-wave potential shifted towards a more positive value, while the reaction current density decreased at a higher chloride concentration. This behaviour would have resulted from the influence of chloride ions on the reaction current density occurring through ion adsorption at the catalysts.50 The Tafel slope of 20 wt% Pt/Vulcan increased from 84.6 to 88.8 mV dec−1, showing a decrease of reaction kinetics. In contrast, the Tafel slope of B-FePc only changes slightly from 35.8 to 36.6 mV dec−1 with an increase in chloride concentration (Fig. 5(d)), indicating that the influence of chloride on the kinetics in B-FePc is lower than that of the Pt catalyst. According to the accelerated stress test results shown in Fig. 5(e), B-FePc showed moderate stability with a mere decrease of 11 mV in the half-wave potential after 5000 cycles. In contrast, the half-wave potential of 20 wt% Pt/Vulcan degraded by 29 mV after undergoing an identical durability test, mainly caused by the chloride adsorption on the Pt catalysts. As shown in Fig. S11, B-FePc exhibits a kinetic current density approximately eight times higher than that of 20 wt% Pt/Vulcan both initially and after the durability test. The results show that B-FePc not only presents a higher catalytic activity and reaction kinetics, but also better stability at high chloride concentrations than 20 wt% Pt/Vulcan. Finally, we compared the electrochemical surface area (ECSA) and catalytic activity of pristine FePc and B-FePc in alkaline brine (Fig. S12). Fig. S12(a) and (b) show that both pristine FePc and B-FePc exhibit an almost rectangular shape in the potential range of 1.01 V to 1.13 V vs. RHE. As shown in Fig. S12(c), the calculated ECSA from the CV profiles is 5.0 mF cm−2 for pristine FePc and 4.2 mF cm−2 for B-FePc. The slight decrease in ECSA for B-FePc might be attributed to a minor Fe loss after plasma engineering, as confirmed by ICP results, where a slightly lower Fe content was observed in B-FePc. Despite the reduced active site density, B-FePc demonstrates superior catalytic activity in alkaline brine compared to pristine FePc, suggesting the enhanced intrinsic activity induced by B-substitution into FePc (Fig. S12(d)).
 |
| | Fig. 5 (a) Linear sweep voltammetry (LSV) profiles of 20 wt% Pt/C and B-FePc in 0.1 M KOH + 0.5 M NaCl, (b) catalytic activity of recently reported ORR electrocatalysts in alkaline seawater, (c) LSV profiles of 20 wt% Pt/C and B-FePc in 0.1 M KOH + 1 M NaCl, (d) Tafel slopes of 20 wt% Pt/Vulcan and B-FePc calculated from (a, filled) and (c, unfilled) and (e) accelerated stress test results of B-FePc and 20 wt% Pt/Vulcan in 0.1 M KOH + 1 M NaCl after they have undergone 5000 test cycles. | |
Practical application of B-FePc in Al–air brine seawater batteries
Instead of alkaline saline and brine saline electrolytes used in the electrochemical evaluation, the performance of B-FePc as a cathode catalyst in a practical Al–air seawater battery was evaluated under representative electrolytes used in practical seawater and brine seawater batteries, namely alkaline seawater (1 M KOH + seawater salt) and alkaline brine (1 M KOH + 1 M NaCl + seawater salt), respectively. From Fig. 6(a), the discharge performance of B-FePc in a brine seawater Al–air battery exhibits a peak power density of 71.0 mW cm−2, which is approximately 20% higher than that of an alkaline seawater Al–air battery (60.4 mW cm−2). The result implies that although chloride ions can potentially impede and diminish catalytic ORR, the improved ionic conductivity of an Al–air battery in a brine seawater electrolyte can lead to superior air cathode discharge performance. Moreover, the high saline concentration of the brine seawater electrolyte ensures high conductivity for OH−, thereby augmenting the oxidation reaction at the anodic surface.57 Thus, the anodic potential was lower in the brine–seawater electrolyte than in the seawater electrolyte, in particular, at a high-current range, as shown in Fig. 6(b). Furthermore, B-FePc presents highly stable discharging at 20 mA cm−2, while the cell voltage was mostly affected by the Al loss at the anode that occurred during the discharge process (Fig. 6(c)). As shown in Fig. 6(d), B-FePc exhibits outstanding stability as an air cathode catalyst after 60 h of its discharge at 20 mA cm−2 with the mechanical recharging of the Al anodes in brine seawater Al–air batteries. In summary, as depicted in Fig. 6(e), the development of B-FePc to serve as an advanced cathode catalyst in Al–air seawater batteries exhibits not only high ORR activity but also good stability even under a saline-concentrated brine seawater environment, which could make a significant breakthrough in the applications of brine seawater-based electrochemical devices.
 |
| | Fig. 6 (a) Discharge profiles of B-FePc in alkaline seawater (1 M KOH + seawater salt) and alkaline brine seawater (1 M KOH + 1 M NaCl + seawater salt), (b) discharge profiles of the cell components of an Al–air battery in alkaline seawater and alkaline brine seawater (solid: cathode; dash: Al anode), (c) cell durability analysis of each Al–air battery cell component in alkaline brine seawater, (d) overall durability analysis of the Al–air battery placed in alkaline brine for over 50 h, and (e) schematic of the brine seawater based Al–air battery. | |
Conclusions
A high-performance and stable B-FePc cathode catalyst was developed in this study for Al–air batteries operating under brine seawater environments. Our unique catalyst design involved the plasma-engineered surface modification as well as DFT and thermodynamic calculations to validate B-substitution onto FePc macrocyclic molecules (B-FePc) towards facilitating advanced ORR catalytic activities. According to DFT calculations, the increased work function arising from the introduction of acceptor B-doping resulted in the simultaneous less negative Gibbs free energy of the OOH* intermediate and the reduced electron occupation of Fe atoms in FePc, thereby lowering the energy barrier of the electron hopping from carbon atoms to FeN4 active sites. The reduced electron occupation of the Fe atoms was verified using the theoretical electronic structure and magnetic moment value from DFT calculations, and experimental EPR signals. The electrochemical performance agrees well with DFT calculation results, where B-FePc showed an improvement in its half-wave potential (0.87 to 0.882 V vs. RHE), limiting current density (5.54 to 5.90 mA cm−2), and kinetic current density (29.8 to 45.1 mA cm−2) with reference to pristine FePc. The electrochemical performance of B-FePc in a saline electrolyte demonstrated impressive ORR catalytic activity and stability compared with commercial 20 wt% Pt/Vulcan. B-FePc displayed a kinetic current density that was higher than that of 20 wt% Pt/Vulcan by approximately 8 times. When applied as an air cathode catalyst in an Al–air battery in a brine seawater electrolyte, the B-FePc catalyst demonstrated a peak power density of 71.0 mW cm−2 and superior stability after 60 h of discharge at 20 mA cm−2 with mechanical recharging. Thus, this study provides insights into the feasible modification of FePc into B-FePc using direct work function control for advanced and stable cathode catalysts in brine seawater-based batteries. The B-substitution into FePc offers a highly cost-effective and sustainable solution for recycling abundant waste-concentrated brine for ships and many rapidly growing desalination industries across the global coastal regions.
Conflicts of interest
There are no conflicts to declare.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and/or its supplementary information (SI). See DOI: https://doi.org/10.1039/d5ey00254k.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (RS-2024-00343361) and the 2024 Post-Doc. Development Program of Pusan National University. Y. Xu and H. Choi wish to thank financial supports from Suzhou Science and Technology Development Planning Programme (SYC2022101), the Suzhou Industrial Park High Quality Innovation Platform of Functional Molecular Materials and Devices (YZCXPT2023105), and Xi’an Jiaotong-Liverpool University for the financial support extended by the Research Development Fund (RDF-23-01-089) and the Advanced Materials Research Center (AMRC).
References
- J. Yu, B.-Q. Li, C.-X. Zhao and Q. Zhang, Seawater electrolyte-based metal–air batteries: from strategies to applications, Energy Environ. Sci., 2020, 13, 3253–3268 RSC.
- S. M. Hwang, J.-S. Park, Y. Kim, W. Go, J. Han, Y. Kim and Y. Kim, Rechargeable Seawater Batteries—From Concept to Applications, Adv. Mater., 2019, 31, 1804936 CrossRef.
- S. Wu, Q. Zhang, J. Ma, D. Sun, Y. Tang and H. Wang, Interfacial design of Al electrode for efficient aluminum-air batteries: issues and advances, Mater. Today Energy, 2020, 18, 100499 CrossRef CAS.
- S. A. Raza, M. R. A. Karim, T. Shehbaz, A. A. Taimoor, R. Ali and M. I. Khan, Effect of pH and Concentration on Electrochemical Corrosion Behavior of Aluminum Al-7075 T6 Alloy in NaCl Aqueous Environment, J. Electrochem. Sci. Technol., 2022, 13, 213–226 CrossRef CAS.
- J. Yu, C.-X. Zhao, J.-N. Liu, B.-Q. Li, C. Tang and Q. Zhang, Seawater-based electrolyte for zinc–air batteries, Green Chem. Eng., 2020, 1, 117–123 CrossRef.
- P. M. Natishan and W. E. O’Grady, Chloride Ion Interactions with Oxide-Covered Aluminum Leading to Pitting Corrosion: A Review, J. Electrochem. Soc., 2014, 161, C421 CrossRef CAS.
- T. J. Schmidt, U. A. Paulus, H. A. Gasteiger and R. J. Behm, The oxygen reduction reaction on a Pt/carbon fuel cell catalyst in the presence of chloride anions, J. Electroanal. Chem., 2001, 508, 41–47 CrossRef CAS.
- Y. Zhan, Z.-B. Ding, F. He, X. Lv, W.-F. Wu, B. Lei, Y. Liu and X. Yan, Active site switching of Fe-N-C as a chloride-poisoning resistant catalyst for efficient oxygen reduction in seawater-based electrolyte, Chem. Eng. J., 2022, 443, 136456 CrossRef CAS.
- R. Meng, C. Zhang, Z. Lu, X. Xie, Y. Liu, Q. Tang, H. Li, D. Kong, C.-N. Geng, Y. Jiao, Z. Fan, Q. He, Y. Guo, G. Ling and Q.-H. Yang, An Oxygenophilic Atomic Dispersed Fe–N–C Catalyst for Lean-Oxygen Seawater Batteries, Adv. Energy Mater., 2021, 11, 2100683 CrossRef CAS.
- H. Zhang, Z. Zhang, Z. Zhang, Y. Li, Y. Hou, P. Liu, B. Xu, H. Zhang, Y. Liu and J. Guo, Highly dispersed ultrasmall iron phthalocyanine molecule clusters confined by mesopore-rich N-doped hollow carbon nanospheres for efficient oxygen reduction reaction and Zn-air battery, Chem. Eng. J., 2023, 469, 143996 CrossRef CAS.
- Y. Sun, P. Cai, D. Yang and X. Yao, Single-site catalysis in heterogeneous electro-Fenton reaction for wastewater remediation, Chem. Catal., 2022, 2, 679–692 CAS.
- W. Zhang, C. Shu, J. Zhan, S. Zhang, L.-H. Zhang and F. Yu, Deep Electronic State Regulation through Unidirectional Cascade Electron Transfer Induced by Dual Junction Boosting Electrocatalysis Performance, Adv. Sci., 2023, 10, 2304063 CrossRef CAS.
- Y. Lee, J. H. Ahn, H. Jang, J. Lee, S. Yoon, D.-G. Lee, M. G. Kim, J. H. Lee and H.-K. Song, Very strong interaction between FeN 4 and titanium carbide for durable 4-electron oxygen reduction reaction suppressing catalyst deactivation by peroxide, J. Mater. Chem. A, 2022, 10, 24041–24050 RSC.
- N. Helsel and P. Choudhury, Regulating electronic descriptors for the enhanced ORR activity of FePc-functionalized graphene via substrate doping and/or ligand exchange: a theoretical study, J. Phys. Chem. C, 2022, 126, 4458–4471 CrossRef CAS.
- L. Li, X. Tang, S. Huang, C. Lu, D. Lützenkirchen-Hecht, K. Yuan, X. Zhuang and Y. Chen, Longitudinally Grafting of Graphene with Iron Phthalocyanine-based Porous Organic Polymer to Boost Oxygen Electroreduction, Angew. Chem., Int. Ed., 2023, 62, e202301642 CrossRef CAS PubMed.
- K. Chen, K. Liu, P. An, H. Li, Y. Lin, J. Hu, C. Jia, J. Fu, H. Li and H. Liu, Iron phthalocyanine with coordination induced electronic localization to boost oxygen reduction reaction, Nat. Commun., 2020, 11, 4173 CrossRef CAS PubMed.
- A. Kumar, G. Yasin, M. Tabish, D. K. Das, S. Ajmal, A. K. Nadda, G. Zhang, T. Maiyalagan, A. Saad and R. K. Gupta, A catalyst-free preparation of conjugated poly iron-phthalocyanine and its superior oxygen reduction reaction activity, Chem. Eng. J., 2022, 445, 136784 CrossRef CAS.
- Y. Wang, K. Wu, J. Kröger and R. Berndt, Structures of phthalocyanine molecules on surfaces studied by STM, AIP Adv., 2012, 2, 041402 CrossRef CAS.
- G. Wang, X. Feng, R. Ren, Y. Wang, J. Meng and J. Jia, Theoretical Study on ORR/OER Bifunctional Catalytic Activity of Axial Functionalized Iron Polyphthalocyanine, Molecules, 2023, 29, 210 CrossRef PubMed.
- M. Urbani, M.-E. Ragoussi, M. K. Nazeeruddin and T. Torres, Phthalocyanines for dye-sensitized solar cells, Coord. Chem. Rev., 2019, 381, 1–64 CrossRef CAS.
- K. M. Zhao, S. Liu, Y. Y. Li, X. Wei, G. Ye, W. Zhu, Y. Su, J. Wang, H. Liu and Z. He, Insight into the mechanism of axial ligands regulating the catalytic activity of Fe–N4 sites for oxygen reduction reaction, Adv. Energy Mater., 2022, 12, 2103588 CrossRef CAS.
- S. Li, L. Xia, J. Li, Z. Chen, W. Zhang, J. Zhu, R. Yu, F. Liu, S. Lee and Y. Zhao, Tuning structural and electronic configuration of FeN4 via external S for enhanced oxygen reduction reaction, Energy Environ. Mater., 2024, 7, e12560 CrossRef CAS.
- E. Proietti, F. Jaouen, M. Lefevre, N. Larouche, J. Tian, H. Herranz and J. Dodelet, Iron-based cathode catalyst with enhanced power density in polymer electrolyte membrane fuel cells, Nat. Commun., 2011, 2, 1427 Search PubMed.
- M. P. Vinod, T. Kr Das, A. J. Chandwadkar, K. Vijayamohanan and J. G. Chandwadkar, Catalytic and electrocatalytic properties of intrazeolitically prepared iron phthalocyanine, Mater. Chem. Phys., 1999, 58, 37–43 CrossRef CAS.
- L. Qin, N. Takeuchi, K. Takahashi, J. Kang, K. H. Kim and O. L. Li, N2/Ar plasma-induced surface sulfonation on graphene nanoplatelets for catalytic hydrolysis of cellulose to glucose, Appl. Surf. Sci., 2021, 545, 149051 CrossRef CAS.
- O. L. Li, N. Phan, J. Kim, H. Choi, D. H. Lee, Y. Yang, W. Yao, Y.-R. Cho and S. G. Lee, Insights on boosting oxygen evolution reaction performance via boron incorporation into nitrogen-doped carbon electrocatalysts, Appl. Surf. Sci., 2020, 528, 146979 CrossRef CAS.
- M. Orio, D. A. Pantazis and F. Neese, Density functional theory, Photosynth. Res., 2009, 102, 443–453 CrossRef CAS.
- C. J. Bartel, Review of computational approaches to predict the thermodynamic stability of inorganic solids, J. Mater. Sci., 2022, 57, 10475–10498 CrossRef CAS.
- J. Hafner, Ab-initio simulations of materials using VASP: Density-functional theory and beyond, J. Comput. Chem., 2008, 29, 2044–2078 CrossRef CAS.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B, 1976, 13, 5188 CrossRef.
- S. Yuan, J. Peng, Y. Zhang, D. J. Zheng, S. Bagi, T. Wang, Y. Roman-Leshkov and Y. Shao-Horn, Tuning the Catalytic Activity of Fe-Phthalocyanine-Based Catalysts for the Oxygen Reduction Reaction by Ligand Functionalization, ACS Catal., 2022, 12, 7278–7287 CrossRef CAS.
- X. Tan, H. Li, W. Zhang, K. Jiang, S. Zhai, W. Zhang, N. Chen, H. Li and Z. Li, Square-pyramidal Fe-N4 with defect-modulated O-coordination: Two-tier electronic structure fine-tuning for enhanced oxygen reduction, Chem. Catal., 2022, 2, 816–835 CAS.
- M. Evangelisti, J. Bartolome, L. J. Dejongh and G. Filoti, Magnetic properties of α-iron(II) phthalocyanine, Phys. Rev. B:Condens. Matter Mater. Phys., 2002, 66, 144410 CrossRef.
- T. Liu, F. Zhang, L. Ruan, J. Tong, G. Qin and X. Zhang, Facile synthesis and characterization of crystalline iron phthalocyanine, Mater. Lett., 2019, 237, 319–322 CrossRef CAS.
- G. Shao, Work Function and Electron Affinity of Semiconductors: Doping Effect and Complication due to Fermi Level Pinning, Energy Environ. Mater., 2021, 4, 273–276 CrossRef CAS.
- J. H. Zagal and M. T. M. Koper, Reactivity Descriptors for the Activity of Molecular MN4 Catalysts for the Oxygen Reduction Reaction, Angew. Chem., Int. Ed., 2016, 55, 14510–14521 CrossRef CAS.
- A. F. Paterson, A. Savva, S. Wustoni, L. Tsetseris, B. D. Paulsen, H. Faber, A. H. Emwas, X. Chen, G. Nikiforidis, T. C. Hidalgo, M. Moser, L. P. Maria, J. Rivnay, I. McCulloch, T. D. Anthopoulos and S. Inal, Water stable molecular n-doping produces organic electrochemical transistors with high transconductance and record stability, Nat. Commun., 2020, 11, 3004 CrossRef CAS.
- S. Yin, Y. Chen, Q. Hu, M. Li, Y. Ding, Y. Shao, J. Di, J. Xia and H. Li, In-situ preparation of iron(II) phthalocyanine modified bismuth oxybromide with enhanced visible-light photocatalytic activity and mechanism insight, Colloids Surf., A, 2019, 575, 336–345 CrossRef CAS.
- S. Jager, K. Bewilogua and C. P. Klages, Infrared spectroscopic investigations on h-BN and mixed h/c-BN thin films, Thin Solid Films, 1994, 245, 50–54 CrossRef.
- O. Snezhkova, F. Bischoff, Y. He, A. Wiengarten, S. Chaudhary, N. Johansson, K. Schulte, J. Knudsen, J. V. Barth, K. Seufert, W. Auwarter and J. Schnadt, Iron phthalocyanine on Cu(111): Coverage-dependent assembly and symmetry breaking, temperature-induced homocoupling, and modification of the adsorbate-surface interaction by annealing, J. Chem. Phys., 2016, 144, 094702 CrossRef PubMed.
- Y. Kang, Z. Chu, D. Zhang, G. Li, Z. Jiang, H. Cheng and X. Li, incorporate boron and nitrogen into graphene to make BCN hybrid nanosheets with enhanced microwave absorbing properties, Carbon, 2013, 61, 200–208 CrossRef CAS.
- D. Deng, X. Chen, L. Yu, X. Wu, Q. Liu, Y. Liu, H. Yang, H. Tian, Y. Hu, P. Du, R. Si, J. Wang, X. Cui, H. Li, J. Xiao, T. Xu, J. Deng, F. Yang, P. N. Duchesne, P. Zhang, J. Zhou, L. Sun, J. Li, X. Pang and X. Bao, A single iron site confined in a graphene matrix for the catalytic oxidation of benzene at room temperature, Sci. Adv., 2015, 1(11) DOI:10.1126/sciadv.1500462.
- L. Wang, C. Wang, Z. Zhang, J. Wu, R. Ding and B. Lv, Thermal induced BCN nanosheets evolution and its usage as metal-free catalyst in ethylbenzene dehydrogenation, Appl. Surf. Sci., 2017, 422, 574–581 CrossRef CAS.
- Q. Liu, C. Chen, M. Du, Y. Wu, C. Ren, K. Ding, M. Song and C. Huang, Porous Hexagonal Boron Nitride Sheets: Effect of Hydroxyl and Secondary Amino Groups on Photocatalytic Hydrogen Evolution, ACS Appl. Nano Mater., 2018, 1, 4566–4575 CrossRef CAS.
- X. Xu, T. Yuan, Y. Zhou, Y. Li, J. Lu, X. Tian, D. Wang and J. Wang, Facile synthesis of boron and nitrogen-doped graphene as efficient electrocatalyst for the oxygen reduction reaction in alkaline media, Int. J. Hydrogen Energy, 2014, 39, 16043–16052 CrossRef CAS.
- J. H. Zagal and M. T. M. Koper, Reactivity Descriptors for the Activity of Molecular MN4 Catalysts for the Oxygen Reduction Reaction, Angew. Chem., Int. Ed., 2016, 55, 14510–14521 CrossRef CAS.
- A. F. Paterson, A. Savva, S. Wustoni, L. Tsetseris, B. D. Paulsen, H. Faber, A. H. Emwas, X. Chen, G. Nikiforidis, T. C. Hidalgo, M. Moser, L. P. Maria, J. Rivnay, I. McCulloch, T. D. Anthopoulos and S. Inal, Water stable molecular n-doping produces organic electrochemical transistors with high transconductance and record stability, Nat. Commun., 2020, 11, 3004 CrossRef CAS.
- N. Helsel and P. Choudhury, Regulating Electronic Descriptors for the Enhanced ORR Activity of FePc-Functionalized Graphene via Substrate Doping and/or Ligand Exchanged: A Theoretical Study, J. Phys. Chem. C, 2022, 126, 4458–4471 Search PubMed.
- G. Shao, Work Function and Electron Affinity of Semiconductors: Doping Effect and Complication due to Fermi Level Pinning, Energy Environ. Mater., 2021, 4, 273–276 CrossRef CAS.
- T. J. Schmidt, U. A. Paulus, H. A. Gasteiger and R. J. Behm, The oxygen reduction reaction on a Pt/carbon fuel cell catalyst in the presence of chloride anions, J. Electroanal. Chem., 2001, 508, 41–47 Search PubMed.
- Q. He, J. Li, Y. Qiao, S. Zhan and F. Zhou, Investigation of two-electron ORR pathway of non-metallic carbon-based catalysts with P-C bond structure in Cl−-bearing electrolytes, Appl. Catal., B, 2023, 339, 123087 CrossRef CAS.
- S. Wu, X. Liu, H. Mao, T. Cui, B. Li, G. Zhou and L. Wang, Realizing high-efficient oxygen reduction reaction in alkaline seawater by tailoring defect-rich hierarchical heterogeneous assemblies, Appl. Catal., B, 2023, 330, 122634 Search PubMed.
- S. Wu, X. Liu, H. Mao, J. Zhu, G. Zhou, J. Chi, Z. Wu and L. Wang, Unraveling the Tandem Effect of Nitrogen Configuration Promoting Oxygen Reduction Reaction in Alkaline Seawater, Adv. Energy Mater., 2024, 2400183 Search PubMed.
- X. Liu, H. Mao, G. Liu, Q. Yu, S. Wu, B. Li, G. Zhou, Z. Li and L. Wang, Metal doping and Hetero-engineering of Cu-doped CoFe/Co embedded in N-doped carbon for improving trifunctional electrocatalytic activity in alkaline seawater, Chem. Eng. J., 2023, 451, 138699 CrossRef CAS.
- H. Mao, X. Liu, S. Wu, G. Sun, G. Zhou, J. Chi and L. Wang, Built-In Electric Fields and Interfacial Electron Modulation: Enhancement of Oxygen Reduction Reaction in Alkaline Seawater, Adv. Energy Mater., 2023, 13, 2302251 CrossRef CAS.
Footnote |
| † S. Kim and Y. Xu equally contributed to this work. |
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *c and
Oi Lun
Li
*c and
Oi Lun
Li

 is the heat capacity contribution, T is the temperature, and ΔS is the change in entropy. The ΔZPE,
is the heat capacity contribution, T is the temperature, and ΔS is the change in entropy. The ΔZPE,  , and TΔS terms were expressed as:
, and TΔS terms were expressed as:







