
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Discontinuous coordination boosting ion transport in solid polymer electrolytes
Bochun
Liang†
 ab,
Xue-Yan
Huang†
c,
Shendong
Tan
a,
Tairan
Wang
b,
Chaoyuan
Ji
a,
Ting
Si
b,
Xi-Yao
Li
c,
Hao
Chen
ad,
Yaoshu
Xie
a,
Lu
Jiang
e,
Chen-Zi
Zhao
*c,
Jun
Fan
*b,
Tingzheng
Hou
*a and
Qiang
Zhang
cf
ab,
Xue-Yan
Huang†
c,
Shendong
Tan
a,
Tairan
Wang
b,
Chaoyuan
Ji
a,
Ting
Si
b,
Xi-Yao
Li
c,
Hao
Chen
ad,
Yaoshu
Xie
a,
Lu
Jiang
e,
Chen-Zi
Zhao
*c,
Jun
Fan
*b,
Tingzheng
Hou
*a and
Qiang
Zhang
cf
aInstitute of Materials Research, Tsinghua Shenzhen International Graduate School, Tsinghua University, Shenzhen, 518055, P. R. China. E-mail: tingzhenghou@sz.tsinghua.edu.cn
bDepartment of Materials Science and Engineering, City University of Hong Kong, Hong Kong, 999077, P. R. China. E-mail: junfan@cityu.edu.hk
cBeijing Key Laboratory of Complex Solid State Batteries & Tsinghua Center for Green Chemical Engineering Electrification, Department of Chemical Engineering, Tsinghua University, Beijing, 100084, P. R. China. E-mail: zcz@mail.tsinghua.edu.cn
dDepartment of Chemistry, University of Chinese Academy of Sciences, Beijing 100049, P. R. China
eDepartment of Materials Science and Engineering, City University of Hong Kong (Dongguan), Dongguan, 523808, P. R. China
fThe Innovation Center for Smart Solid State Batteries, Yibin 644002, P. R. China
First published on 27th January 2026
Abstract
Decoupling Li+ transport from polymer segmental dynamics is crucial for enhancing ionic conductivity (σ) and transference number (t+) in solid polymer electrolytes (SPEs). Herein, by studying four ether-based SPEs with varying oxygen density, we identify a transition from polymer relaxation-limited ion transport in poly(ethylene oxide) (PEO) to ion hopping-dominant transport in poly(tetrahydrofuran) (PTHF), poly(1,3-dioxolane) (PDOL), and poly(trioxymethylene) (PTOM). Molecular dynamics simulations and solid-state 7Li nuclear magnetic resonance reveal origins of the transition. In PTHF, weak solvation with lithium bond characteristics contributes to a less-shielded Li+ environment, while in PDOL and PTOM, the discontinuous coordination (DC) structure and multi-chain binding are pivotal. The presence of DC structures is experimentally confirmed by in situ attenuated total reflection Fourier transform infrared spectroscopy and supported by quantum chemistry calculations. As a result, PDOL and PTOM exhibit t+ values exceeding 0.5 and enhanced σ values of 4.3 × 10−3 and 8.5 × 10−3 S cm−1 at 373 K, respectively. The Li/SPEs/LiFePO4 cell with ex situ-prepared PDOL achieves a superior capacity retention of 90.8% after 50 cycles. This work underscores the significance of functional group spacing in tuning the transport mechanisms and demonstrates how the decoupling strategy can guide the bottom-up design of advanced SPEs.
Broader contextSolid polymer electrolytes (SPEs) are key candidates for enabling high-energy lithium metal batteries, yet their practical application is fundamentally restricted by the intrinsic coupling between ion transport and sluggish polymer segmental dynamics. This work presents a transformative molecular-level strategy to overcome this limitation by precisely tuning the functional group density along polymer backbones. Specifically, increasing the oxygen density in ether-based SPEs effectively decouples Li+ motion from segmental relaxation, leading to a 2–3 fold increase in both ionic conductivity and Li+ transference number compared with conventional poly(ethylene oxide) (PEO). The origin of the transition is identified as the formation of unique discontinuous coordination (DC) structures, corroborated by a combination of in situ spectroscopic analysis and theoretical calculations. Critically, the engineered SPE enables high-performance full Li|LiFePO4 cells that demonstrate a 5-fold increase in capacity retention after 50 cycles, confirming its device-level superiority. This performance is directly attributed to the concept of local functional group spacing control, providing a generalizable strategy applicable to energy storage materials where ion transport and interfacial stability are intertwined. Engineering coordination chemistry through polymer structure design is anticipated to serve as a pivotal approach to advance materials design for next-generation energy storage technologies. |
1. Introduction
Solid polymer electrolytes (SPEs) offer a promising solution for enabling the practical operation of high-energy-density lithium metal anodes, which are challenging to pair with routine organic liquid electrolytes due to safety concerns and dendrite formation.1–3 SPEs are cost-effective, easier to process, and exhibit superior interfacial compatibility,3–5 establishing them as a leading technical pathway for the commercialization of all solid-state batteries.6,7 Nevertheless, SPEs face significant limitations, particularly low ionic conductivity (σ) and low Li+ transference number (t+) at room temperature.8,9 Ether-based SPEs, such as poly(ethylene oxide) (PEO) and poly(tetrahydrofuran) (PTHF), have been extensively utilized as an ionic conductive matrix to facilitate the ionic conduction in SPEs.10–17 However, their room-temperature σ and t+ are limited to 10−4 S cm−1 and below 0.5, respectively.4,18 These limitations of ether-based SPEs largely arise from the strong dependence of Li+ transport on the segmental relaxation of the polymer chains coordinating Li+.4,18,19 Ether groups in PEO, with their strong coordinating capability, are effective in dissolving lithium salts and enhancing chain flexibility, thereby promoting Li+ transport via segmental motion of the polymer chains.20 However, the segmental dynamics are significantly hindered at or below room temperature due to polymer crystallization and the restricted relaxation of the polymer chains.21,22Decoupling Li+ movement from the overall polymer dynamics is one of the effective strategies to address these issues,4,18,19,23 which can be achieved through appropriate molecular design.9,24 The rational construction of packing-frustrated structures25–27 or liquid crystalline polymers28,29 can decouple the local sidechain relaxation from the main chain of polymer matrices. At the monomer level, modifying functional groups and polymer chemistry can reduce the coupling strength between Li+ and the polymer binding sites.16,30,31 Nevertheless, balancing the salt solubility and the coupling strength remains challenging.4 Alternatively, variations in oxygen spacing along the polymer backbone are expected to alter the Li+ solvation structure, which in turn can regulate Li+ dynamics. For instance, PTHF-based electrolytes exhibit higher Li+ transference numbers compared to PEO-based electrolytes due to looser ether oxygen coordination, resulting in weaker binding and easier decoupling.15,16,32 Recently, ether-based polymers with denser oxygen spacing, such as poly(1,3-dioxolane) (PDOL)33–36 and poly(trioxymethylene) (PTOM),37–39 have been reported to exhibit excellent transport properties. For instance, in situ synthesized PDOL electrolyte has demonstrated high ionic conductivity at room temperature (>10−3 S cm−1) and low interfacial resistances.33 Similarly, copolymers of PDOL and PTOM exhibit increasing ionic conductivity and Li+ transference number as the trioxymethylene fraction increases up to a critical point where crystallization occurs.37 These findings suggest the potential for effectively decoupling ion transport from segmental dynamics in ether-based SPEs by tuning oxygen spacing. However, uncovering the intrinsic design rules for this decoupling strategy requires a deeper understanding of the ion transport mechanisms, particularly the microscopic cation dynamics at the atomic level.34,40,41
Given the extremely small temporal and spatial scales involved in ion motion, it is challenging to reveal the ion transport mechanism when relying solely on experimental techniques.42 In this context, molecular dynamics (MD) simulations have become a valuable tool for investigating cation dynamics in SPEs.15,34,43–48 Recently, a theoretical framework has been proposed to quantify cation transport modes in MD trajectories of SPEs, including intrachain hopping, segmental relaxation, and interchain hopping.49–51 This approach, based on the dynamic bond-percolation model52,53 and the Rouse model,54 allows for the exploration of atomic-level mechanisms governing cation transport in SPEs, including the decoupling of Li+ transport from polymer relaxation. The advancements in computational and sampling techniques enable rigorous variable-controlled comparative studies and detailed mechanistic investigations, paving the way for the bottom-up design and optimization of ether-based SPEs.
In this contribution, we demonstrate that by controlling the oxygen density in ether-based polymers, PDOL and PTOM, which have a higher oxygen density along the polymer backbone, serve as superior ionic conductive phases for SPEs. Both PDOL and PTOM electrolytes exhibit higher ionic conductivity in the amorphous state and enhanced Li+ transference numbers. By quantifying the time scales of Li+ transport through intrachain hopping, polymer segmental relaxation, and interchain hopping, we reveal that Li+ transport in PEO is predominantly governed by polymer segmental dynamics, whereas in PTHF, PDOL, and PTOM, Li+ transport is primarily facilitated by ion hopping. Moreover, a unique solvation structure featuring discontinuous coordination sites are observed in the PDOL and PTOM electrolytes, which are validated by a combination of in situ attenuated total reflection Fourier transform infrared (ATR-FTIR) spectroscopy and quantum chemistry calculations. Further analysis of Li+ dynamics, energetics of Li+–polymer interaction, and solid-state nuclear magnetic resonance (ssNMR) characterization illustrates that these discontinuous coordination structures underlie the weaker correlation of PDOL and PTOM with Li+ and contribute to the enhanced Li+ transport.
2. Results and discussion
2.1. Effects of oxygen density on transport properties
The solvation structure of cations in liquid electrolytes is determined by the solvent type, salt selection, and concentration, all of which impact ionic transport properties.55–58 Similarly, modifying the Li+ solvation structure in SPEs offers a potential strategy for tuning Li+ transport modes. Unlike liquid electrolytes, where Li+ coordination can be adjusted by altering solvent concentration, in ether-based SPEs, the distribution of coordinating sites is fixed on the polymer backbone. Therefore, we hypothesize that modifying the spacing of these coordinating sites provides a direct means to influence Li+ transport.Therefore, we investigate the effect of oxygen density on the transport properties of ether-based SPEs using four linear polymer matrices, PTHF, PEO, PDOL, and PTOM, with a C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O ratio of 4:1, 2:1, 3:2, and 1:1, respectively (Fig. 1a). Each polymer was mixed with lithium bis(trifluoromethanesulfonyl)imide (LiTFSI), a widely used lithium salt in SPEs for lithium metal batteries due to its electron delocalization, and excellent thermal and electrochemical stability.59–62 A molar ratio of Li+ to ether oxygen (Oe) was set to 1:20 to form four SPE systems. Classical MD simulations were performed to obtain the key transport properties. Unless otherwise specified, these SPEs are referred to by their constituting polymers: PTHF, PEO, PDOL, and PTOM, respectively. Polymer chain lengths were selected according to a constant number of heavy atoms (or equivalently, a comparable molecular weight) criterion.
:O ratio of 4:1, 2:1, 3:2, and 1:1, respectively (Fig. 1a). Each polymer was mixed with lithium bis(trifluoromethanesulfonyl)imide (LiTFSI), a widely used lithium salt in SPEs for lithium metal batteries due to its electron delocalization, and excellent thermal and electrochemical stability.59–62 A molar ratio of Li+ to ether oxygen (Oe) was set to 1:20 to form four SPE systems. Classical MD simulations were performed to obtain the key transport properties. Unless otherwise specified, these SPEs are referred to by their constituting polymers: PTHF, PEO, PDOL, and PTOM, respectively. Polymer chain lengths were selected according to a constant number of heavy atoms (or equivalently, a comparable molecular weight) criterion.
 | ||
| Fig. 1 Effects of oxygen density on transport properties of ether-based polymer electrolytes. (a) Schematic illustration of ether-based polymers in SPEs systems, solvation structures, and the effect on Li+ transport in the contribution. The variation in oxygen density along the polymer backbone influences the Li+ solvation structure, tuning the Li+–Oe binding strength and results in optimal Li+ transport. Hydrogen atoms are omitted in the snapshot of the solvation structure for clarity. The calculated (b) ionic conductivity (σ), (c) Li+ transference number (t+) from MD simulations, and (d) Onsager transport coefficient analysis. The experimental measured (e) σ and (f) t+. (g) The measured and calculated Tg. | ||
Self-diffusion coefficients of Li+ and TFSI− were first determined by the mean-square displacement from MD simulations (Fig. S13). Across the studied temperature range, PTOM exhibits notably higher self-diffusion coefficients for Li+ and moderately higher coefficients for TFSI−, followed by PDOL. Although the difference between the Li+ self-diffusion coefficient of PTHF and those of PEO and PDOL is insignificant, PTHF presents a notably lower value for TFSI−. Clustering analysis (Fig. S14) reveals that the slow ion transport in PTHF stems from impaired salt dissociation and pronounced ion clustering, resulting from its sparse polar oxygen solvating sites.
Moreover, both ionic conductivity (σ) and lithium transference number (t+) were obtained by analyzing the MD trajectories based on the Onsager transport theory, which rigorously accounts for ion correlations in electrolytes.57,63Fig. 1b presents the calculated σ. A clear trend can be observed for the σ value: PTOM > PDOL > PEO > PTHF, indicating that oxygen density plays a significant role in governing the ionic conduction. However, a different pattern emerges for the calculated t+ (Fig. 1c). Relative to PEO, both increasing and decreasing the oxygen density lead to a pronounced enhancement in t+. Additionally, our evaluation shows that the concentration effect is minimal, with the trends of σ and t+ remaining consistent across varying concentrations (Fig. S15). This indicates that the differences in transport properties arise from variations in oxygen density. Onsager transport coefficient analysis was further performed to quantify the fractional contributions of different ion pair correlations to the total ionic conduction,63P(Lij), as presented in Fig. 1d. The results reveal a unique cation–anion correlation in PTHF, characterized by a positive value, indicative of the cation–anion clustering discussed above. This correlation negatively impacts the overall ionic conductivity due to the opposite charges carried by the cation and anion. In PEO, favorable salt dissociation leads to a pronounced self-anion–anion correlation, whereas the self-cation–cation correlation is weak, with a fraction even lower than that in PTHF. This highlights a strong coupling between Li+ transport and polymer dynamics in PEO. As the oxygen density increases in PDOL and PTOM, the fraction of self-cation–cation correlation rises, while that of self-anion–anion correlation decreases. Notably, the absolute magnitudes (Fig. S16) of both correlations increase, despite slightly aggravated ion clustering. This suggests that the enhanced ionic conductivity in PDOL and PTOM predominantly originates from accelerated Li+ diffusion. Collectively, these results suggest that distinct mechanisms govern the calculated σ and t+ in the four electrolytes, while the underlying physical and chemical origins merit further investigation.
Building on the amorphous-state simulation results, we conducted experimental measurements to evaluate the actual transport properties under concentrations and polymer molecular weights closely matched to those in the simulations. 1H nuclear magnetic resonance (NMR) spectra confirm the successful synthesis of these polymer matrices (Fig. S17). Fig. 1e presents the measured σ results. In the high-temperature range of 373–393 K, where differential scanning calorimetry (DSC) confirms suppressed crystallization (Fig. S18), σ decreases in the order PTOM > PDOL > PEO > PTHF, mirroring the MD trend and validating the predictions for amorphous-state conductivity. As the temperature decreases (363–333 K), σ drops more rapidly for PTOM and PDOL, altering the conductivity order to PEO > PTOM ≈ PDOL > PTHF. As the temperature further decreases to room temperature (323–298 K), a pronounced reduction in σ is observed for PEO, exhibiting the well-known non-Arrhenius temperature dependence,19 consistent with previous reports.4,60 The final conductivity ranking at room temperature is PTHF > PDOL > PTOM > PEO. The melting peak at around 50 °C (Fig. S18) attributes the drop in σ for PEO to its crystallization. Further wide-angle X-ray scattering (WAXS) measurements (Fig. S19) reveal that the addition of LiTFSI suppresses crystallization in PDOL and PTOM compared with the pristine polymer membranes. Among these electrolytes, PTHF exhibits the lowest activation energy, as its σ decreases more gradually with temperature, suggesting superior low-temperature performance. Furthermore, the measured t+ trend (Fig. 1f) also aligns with the MD prediction, where PEO exhibits the lowest value (0.23), while modifying the oxygen density, whether increasing or decreasing, significantly enhances t+, yielding values above 0.5. Overall, these results confirm that tuning oxygen density effectively optimizes the transport properties of SPEs, especially promoting Li+ transport. Additionally, as another important factor governing the ionic conductivity, the glass transition temperature (Tg) was obtained using DSC measurements and MD-simulated PVT relationships,64 as shown in Fig. 1g. The calculated Tg reproduces the trend of the measured values and agrees within approximately 30 K.64,65 This comparison rationalizes the observed room-temperature σ trend that PTHF, with the lowest Tg, exhibits the highest room-temperature σ, whereas PEO shows the opposite behavior.
2.2. Li+ transport modes
To reveal the influence of polymer chemistry and variations in Oe spacing on Li+ transport, the Li+–polymer interaction was first analyzed by extracting the Li+–Oe association lifetime (τs) (Fig. S20). Among these systems, PEO exhibits the largest τs, indicating the strong Li+–Oe association. In contrast, the weaker Li+–Oe association in other systems facilitates more frequent ion hopping, promoting more efficient Li+ transport. Additional ab initio molecular dynamics (AIMD) simulations were performed, which corroborate the Li+–Oe correlation observed using the classical molecular force field.The mechanistic differences in Li+ transport across these systems were investigated by quantifying the Li+ transport time scales in three distinct modes:49–51 intrachain hopping, polymer segmental relaxation, and interchain hopping (Fig. 2a). Specifically, τ1 represents the time required for Li+ to traverse a polymer chain via intrachain hopping. τ2 denotes the relaxation time of the polymer segment with Li+ coordination. The corresponding spatial scale in the absence of intrachain or interchain hopping is determined by the polymer radius of gyration (Fig. 2b), while τ3 corresponds to the waiting time for interchain hopping to occur, which can be treated as the renewal of the intrachain transport process and is crucial for long-range Li+ transport.51 Additionally, the Rouse time (τR) can be defined as the relaxation time of the whole polymer chain, which characterizes the overall polymer dynamics. Details of the approach can be found in Supplementary Note 5. The structural properties and calculated time scales for the PEO system were compared with previous studies under identical conditions using a polarizable force field,51 showing reasonable agreement (Table S5). Fig. 2c and d present the τR and τ2 values for the four investigated systems, respectively. The trends in τ2 and τR are similar, with PEO exhibiting the fastest polymer dynamics (lowest values), indicating that Li+ transport in PEO is predominantly facilitated via polymer relaxation. In contrast, τ1 and τ3 follow the opposite trend (Fig. 2e and f), where PEO shows the largest values, followed by PDOL, PTHF, and PTOM. Notably, for τ3, the difference between PEO and PTHF/PTOM exceeds an order of magnitude. Although PEO and PDOL exhibit similar amorphous σ, their underlying Li+ transport mechanisms differ significantly. Li+ transport in PEO is highly dependent on polymer segmental motion, resulting in poor room-temperature σ and low t+. The loosely spaced sites along the backbone of PTHF lead to weak Li+–Oe coupling, which enhances t+ but compromises salt dissociation. In contrast, PDOL and PTOM exhibit a more balanced Li+–Oe interaction, ensuring sufficient salt dissociation while also facilitating Li+ decoupling from the polymer chains for efficient ion transport. Overall, while Li+ transport in PEO is primarily governed by polymer relaxation, ion hopping plays a dominant role in PTHF, PDOL, and PTOM.
 | ||
| Fig. 2 Transport mode analysis of the electrolytes. Schematic illustrations of (a) the three transport modes and the corresponding time scales for Li+ transport, and (b) structure properties of polymer chains. The calculated (c) τR (Rouse relaxation time for whole polymer chains), (d) τ2 (Rouse relaxation time for the binding Oe), (e) τ1 (intrachain hopping), and (f) τ3 (interchain hopping). The statistic (g) persistence length and (h) radius of gyration. | ||
Given the pivotal role of polymer chains in determining ion transport mechanisms, we further investigated the structural variations and segmental dynamics within the simulated polymer systems. With the selected chain length (Fig. 1a) comprising comparable numbers of heavy atoms (C and O), the calculated contour lengths exhibit similar values across the four polymers (Fig. S21a). This indicates that the observed difference in transport modes arise from polymer chemistry rather than chain length. Furthermore, the persistence lengths and radius of gyration were calculated (Fig. 2g and h), indicating a chain flexibility trend of PEO > PDOL > PTOM > PTHF. The end-to-end distance (Fig. S21b) consistently reflects the order of chain flexibility, with PEO adopting more compact, coiled conformations that yield a smaller end-to-end distance, and simultaneously faster chain rearrangement. These structural trends are fully consistent with the relaxation behavior captured by the calculated τR and τ2.
Through the transport-mode analysis, we demonstrate that tuning the oxygen density along polymer backbones provides an effective strategy to decouple Li+ transport from polymer dynamics, thereby enhancing transport properties. To evaluate the generality of this approach, we performed additional MD simulations by replacing the TFSI− anion with other commonly used anions in SPEs, including FSI−, PF6−, and BF4− (Fig. S22 and S23). The enhancements in transport properties and the transition in Li+ transport modes induced by oxygen-density tuning are consistently observed across all anions studied. Additionally, unlike Li+ transport, anion transport is shown to be largely governed by salt dissociation and exhibits weak correlations with the polymer (Fig. S24).
2.3. Structural origins
The structural origin of these transitions in the ion transport modes is of particular interest, especially for PDOL and PTOM. They feature denser polar sites and higher molecular polarity (Table S6), where a stronger Li+–polymer interaction would be intuitively expected. To deepen the understanding of the differences in transition mode in these electrolytes, comprehensive analysis of the MD trajectories was conducted, which shows that in PTHF and PEO, when only one polymer chain is present in the Li+ solvation shell, Li+ typically coordinates with a continuous sequence of oxygen sites along the chain (Fig. 3a and Fig. S25), consistent with previous reports.32,40 However, in PDOL and PTOM, a discontinuous coordination (DC) structure emerges (Fig. 3b and Fig. S25, S26). Taking PTOM as an example (Fig. 3b), Li+ binds to nonadjacent oxygens along the polymer chain, leaving several uncoordinated oxygens between binding sites and forming an expanded solvation shell. AIMD simulations provide evidence at a high accuracy level of how the DC structure affects the Li+ solvation dynamics. As shown in Fig. 3c (PTOM) and Fig. S27 (PDOL), the evolution of coordinated and uncoordinated oxygens reveals frequent swapping between these two states, indicating significant competition in Li+ coordination. This dynamic instability disrupts the one-chain chelate structure and weakens the Li+–Oe binding. As a result, Li+ in PDOL and PTOM tends to coordinate with multiple polymer chains to eliminate the presence of uncoordinated Oe between coordinated Oe in its solvation shell (Fig. 3d and Fig. S25). In PEO, the one-chain chelate structure is the most stable and predominant. In PTHF, due to the loosely spaced Oe sites, certain degree of two-chain binding is observed. However, as discussed in the previous section, a significant number of TFSI− anions also participate in the Li+ solvation shell. As the oxygen density increases, multi-chain coordination becomes more prominent in PDOL and PTOM, indicating that multi-chain binding is thermodynamically more stable than the one-chain chelate structure, which facilitates interchain hopping of Li+. | ||
| Fig. 3 Structural origins of the transport mode transitions. Representative Li+ solvation structure in (a) PEO and (b) PTOM. Hydrogen atoms are omitted for clarity. The one-chain chelate of PEO displays continuous coordination, whereas that of PTOM exhibits discontinuous coordination. In PEO, the one-chain chelate is more stable than the multi-chain coordinate complex, while it is the opposite in PTOM. (c) The discontinuous coordination (DC) structure observed in the AIMD simulation of PTOM. The uncoordinated oxygen and the coordinated oxygen swap frequently. (d) The statistical binding polymer chain number of Li+. (e) Solid-state 7Li NMR spectra, (f) binding free energy of the representative Li+–polymer complex; the numbers represent the binding chain number of Li+, and (g) binding energy decomposition into contributions of permanent electrostatics (ELEC), Pauli repulsion (PAULI), dispersion (DISP), polarization (POL), and charge transfer (CT) for different systems. | ||
To demonstrate the enhanced interchain hopping transport assisted by the multi-chain binding, we compared the local Li+ dynamics in PEO, PDOL and PTOM by tracking the evolution of coordination and representative Li+ hopping events (Fig. S28). Li+ in PTOM engages in a dynamic three-chain coordination environment and completes interchain hopping within 0.2 ns, an order of magnitude faster than in PEO. This stark contrast highlights the critical role of multi-chain binding in accelerating Li+ transport via interchain hopping in systems like PTOM. Extended coordination trajectories further reveal system-specific transport characteristics as discussed in Fig. S29.
The structural features of Li+–polymer interaction were further examined using NMR spectroscopy and quantum chemistry calculations. Solid-state 7Li NMR spectroscopy was employed to probe the local environment of Li+ in the electrolytes (Fig. 3e). The lithium nuclei in PEO are the most shielded, indicating the strongest Li+–polymer interaction, while the shielding is the weakest in PTHF. This can be attributed to the looser oxygen coordination environment and the formation of lithium bonds, as evidenced by the fraction of Li+ with coordination number (CN) ≤4 (Fig. S30).66 The order of chemical shifts, from most shielded to least shielded, aligns with the order of Oe CN from high to low, as obtained from MD simulations (Fig. S31). This highlights the dominant role of Oe in determining the degree of Li+ shielding. Additionally, the full width at half maximum of the peaks is associated with the mobility of Li+. The order of peak width is 0.1259 (PTOM) < 0.1326 (PDOL) < 0.1330 (PTHF) < 0.4339 (PEO), which corroborates the order of the Li+ diffusion coefficient calculated from MD simulations (Fig. S13).
The Li+–polymer binding free energies were calculated based on representative Li+–polymer complexes sampled from MD simulations. For PTHF and PEO, we focused on one-chain chelate structures, which feature continuous coordination. For PDOL and PTOM, we randomly sampled both one-chain binding structures, exhibiting DC structures, and multichain binding structures, where coordination is typically more continuous, with fewer uncoordinated Oe atoms. The representative optimized structures and the calculated binding free energy (ΔG) results are shown in Fig. S32 and Fig. 3f, respectively. This trend in ΔG further supports our previous conclusions regarding the Li+ transport mechanisms in these electrolytes. Specifically, the DC structures in PDOL and PTOM result in smaller binding free energies (PDOL-1 and PTOM-1), making multichain binding thermodynamically favorable to minimize the number of uncoordinated Oe between coordinated ones. As a result, this leads to a stronger Li+–polymer interaction (more shielded) compared to PTHF. Additionally, energy decomposition analysis (EDA) was performed to assess the electronic structure origins of the Li+–polymer interaction. The binding energy was decomposed into contributions from permanent electrostatics (ELEC), Pauli repulsion (PAULI), dispersion (DISP), polarization (POL), and charge transfer (CT), as shown in Fig. 3g. In all structures, electrostatic interactions dominated the total binding energy, followed by polarization effects.
2.4. Identification of the discontinuous coordination structures
To validate the presence of DC structures in PDOL and PTOM, we conducted ATR-FTIR spectroscopy experiments for both pristine polymers and electrolytes (Fig. S33). The characteristic C–O–C stretching vibration modes in the range of 1170–1030 cm−1 were adopted to investigate the different Li+–OE coordination behavior for all four systems. For PTHF and PEO, a single coordinated C–O stretching peak or enveloped feature emerges upon salt addition, while more diverse changes were observed in PDOL and PTOM. Notably, pure PTOM exhibits a single C–O–C vibrational peak, which splits into multiple peaks upon salt addition, suggesting a more heterogeneous solvation environment and the presence of DC structures.To gain further insight into the ATR-FTIR spectra, quantum chemical calculations were performed to obtain the vibration modes of representative structures sampled from MD simulations. The calculated results were compared with the experimental spectra in Fig. 4a, b and Fig. S34. For PEO, the experimental peak near 1092 cm−1 broadens and slightly shifts to 1104 cm−1 upon the addition of salts (first row in Fig. 4a), consistent with shifted peaks from 1091 cm−1 to 1109 cm−1 predicted by the calculations (second and third row in Fig. 4a). A similar result was observed for PTHF (Fig. S34a), where the C–O peak at 1102 cm−1 shifts to 1060 cm−1, corresponding to the calculated shift from 1127 cm−1 to 1071 cm−1. For PTOM (Fig. 4b), the initial peak at 1088 cm−1 splits into four peaks upon salt addition, in excellent agreement with theoretical predictions. Specifically, the pristine PTOM chain exhibits a single C–O stretching mode at 1095 cm−1, whereas the Li+–PTOM complex displays multiple peaks at 1138, 1105, 1072, and 1034 cm−1. Notably, these peaks can be assigned to coordinated (red) and uncoordinated (orange) Oe atoms in the DC structures. In PDOL (Fig. S34b), although spectral complexity increases due to the asymmetrical ether groups in the monomer, we still identify shifted vibrational peaks corresponding to uncoordinated oxygens. Collectively, these results validate the presence of DC solvation structures in PTOM and PDOL, establishing a correlation between the theoretically predicted DC structures and experimental spectroscopy. Such structures weaken the Li+–polymer interaction, despite the higher polarity of the polymer backbone and increasing oxygen density, facilitating multi-chain binding and promoting faster Li+ hopping transport.
 | ||
| Fig. 4 The identification of the discontinuous coordination structure and its effect on Li+ transport behavior. The ATR-FTIR spectra of (a) PEO electrolyte and PEO polymer, as well as (b) PTOM electrolyte and PTOM polymer. The first row presents the experimental results, and the second and third rows display the theoretically calculated vibration modes and the corresponding structures. (c) Schematic illustration of the mechanism of the solvation structure affecting the Li+ transport in PEO and PTOM. | ||
Fig. 4c schematically illustrates how solvation structures with varying Oe spacings influence Li+ transport modes and properties. In PEO, Li+ transport is primarily governed by polymer segmental relaxation due to the tight binding between the polymer chain and Li+, which accounts for the experimentally observed non-Arrhenius behavior of ionic conductivity (σ drop at lower temperatures). While the tight binding promotes salt dissociation, it also restricts Li+ hopping transport, particularly at low temperatures when polymer dynamics slow down. In contrast, Li+ transport in PDOL and PTOM is dominated by ion hopping, which favors high room-temperature ionic conductivity and an elevated Li+ transference number. This enhanced transport results from the instability of their DC structures, which provide optimal Li+–polymer coupling driven by the competition between coordinated and uncoordinated Oe atoms. This competition facilitates the formation of multi-chain binding structures and promotes Li+ interchain hopping transport.
2.5. Electrochemical performance
To comprehensively assess the performance of these electrolytes, we performed electrochemical tests, beginning with the evaluation of electrochemical stability windows (ESWs) using linear sweep voltammetry (LSV) (Fig. 5a). The decomposition voltages, ranked from high to low, are 4.8 V, 4.4 V, 4.3 V, and 4.1 V for PDOL, PTHF, PTOM, and PEO, respectively. This trend is consistent with the calculated oxidation potentials of polymer–TFSI− complexes (Fig. 5b), where PDOL exhibits the highest oxidation stability and PEO exhibits the lowest oxidation stability. The optimized molecular geometries of the oxidated states further suggest potential proton transfer reactions in PEO and PTOM (Fig. S35), indicating electrochemical instability. These findings highlight PDOL's superior oxidative stability, making it compatible with high-voltage cathode materials. Critical current density (CCD) was measured using Li/SPEs/Li symmetric cells. PTHF exhibited the lowest CCD value due to severe interface polarization (Fig. S36a), whereas PTOM displayed “soft shorts”, as evidenced by a flat voltage profile lacking mass transfer (Fig. S36b).67,68 These results suggest that further optimization on their electrochemical interfacial stability is required for practical use in LMBs. The CCD values for PEO and PDOL are shown in Fig. 5c and d. Owing to its higher Li+ conductivity, PDOL demonstrated a higher CCD value (1.2 mA cm−2) compared to PEO (0.8 mA cm−2), with a significant reduction in voltage polarization. | ||
| Fig. 5 Electrochemical performance of the electrolytes. (a) The LSV curves. (b) The calculated oxidation potentials for polymer-TFSI− complexes. The CCD tests of Li/SPEs/Li symmetric cells for (c) PEO and (d) PDOL. The solid curve represents the voltage, and the dashed curve represents the current density. (e) The cycling performance of Li/PEO/LFP and Li/PDOL/LFP at 0.2C and 80 °C with a cathode mass loading of 5 mg cm−2. Filled circles represent the specific capacity, and open circles represent the coulombic efficiency. | ||
Full cells were assembled with LiFePO4 (LFP) cathodes and Li metal anodes to evaluate the practical cycling performance. The performance results for PEO and PDOL are shown in Fig. 5e. PDOL presents a 90.8% capacity retention after 50 cycles. This is notably superior to PEO, which retained only 17.7% of its capacity after 50 cycles. These results are consistent with previous reports of PDOL and PEO with ex situ preparation.69,70 In contrast, the Li/PTHF/LFP and Li/PTOM/LFP cells failed during initial cycling. The failure of the PTHF cell is attributed to its poor ionic conductivity and the resulting high internal resistance (Fig. S37). For PTOM, a “soft short” feature plays a key role. X-ray photoelectron spectroscopy (XPS) analysis of the solid–electrolyte interphase (SEI) composition reveals the underlying mechanism. Specifically, a notably higher Li+ fraction in the SEI of PTOM (Fig. S38) indicates Li-dendrite-induced soft shorting. Furthermore, C 1s XPS depth profiles (Fig. S39a) reveal a higher proportion of C–F species in PTOM, distinct from the LiF-dominated SEI observed in other systems (Fig. S39b). The difference is attributed to side-reactions of residual solvent during PTOM electrolyte preparation. Further work will therefore focus on optimizing the electrolyte preparation in order to fully realize PTOM's inherent transport advantages.
It is important to note that these results reflect the intrinsic electrochemical performance of the electrolyte. The primary aim of our work is to identify the structural factors that influence coordination chemistry and decoupling behavior, and to highlight the potential for further enhancement of ion transport performance through the rational design of functional group spacing and solvation structure regulation. Additional strategies, such as in situ polymerization,5,10,33,36,38 sidechain grafting,12,71,72 crosslinking,14,36 copolymerization,10,37,73 regulating chain-spacing,74,75 and composite polymer electrolytes,76,77 can be applied to further improve the full battery performance of SPEs.
3. Conclusions
A variable-controlled comparative study and an in-depth mechanistic investigation of the transport properties and mechanisms were conducted in four ether-based SPEs with varying oxygen densities along their backbones. Both experimental measurements and MD simulations confirm that modifying the oxygen density relative to PEO can enhance the Li+ transference number to values exceeding 0.5. In addition, PDOL and PTOM exhibit higher amorphous ionic conductivity, while PTHF demonstrates superior low-temperature ionic conductivity. The transport mode analysis reveals that in PEO, Li+ transport is primarily governed by polymer segmental relaxation, which leads to sluggish ion transport. Altering the oxygen density results in a transition in Li+ transport mode, from a predominant reliance on segmental relaxation to an increased contribution from ion hopping. In PTHF, this transition is driven by looser oxygen sites and weaker binding with lithium bond characteristics, though this also results in poor salt dissociation. In contrast, PDOL and PTOM, with denser oxygen sites, exhibit a DC structure and induced multi-chain binding, which are identified as the key factors underlying this transition. The presence of the discontinuous coordination structures is further validated through a combination of ATR-FTIR spectroscopy and quantum chemistry calculations, establishing a correlation between the microscopic DC structures and experimental characterization. Local Li+ dynamics analysis, 7Li solid-state NMR, and binding free energy results provide additional insights into how the DC structure enhances Li+ transport. Specifically, the instability of DC structures weakens the Li+–polymer interaction and drives the formation of multichain binding configuration, which further promotes Li+ interchain hopping transport. Electrochemical tests demonstrate that even with ex situ preparation, PDOL, which exhibits collectively superior Li+ conductivity, transference number, and electrochemical stability, achieves the best capacity retention of 90.8% after 50 cycles at 0.2C, significantly outperforming PEO. Overall, this work illustrates how rational molecular design can effectively decouple ion transport from polymer segmental dynamics, providing valuable insights for the bottom-up design of SPE materials.4. Methods
4.1. Materials
Poly(tetrahydrofuran) (PTHF, average Mn = 2900 g mol−1) and poly(trioxymethylene) (PTOM, average Mn = 3500 g mol−1) were purchased from Sigma-Aldrich. Poly(1,3-dioxolane) (PDOL, average Mn = 3000 g mol−1) was purchased from Shanghai DaoWin Co., Ltd. Poly(ethylene oxide) (PEO, average Mn = 8000 g mol−1) was purchased from Macklin. Acetonitrile (99.9% purity, extra dry with molecular sieves) and hexafluoroisopropanol (98% purity) were purchased from Innochem. The polyimide (PI, thickness = 25 µm) separator was purchased from Jiangxi Xiancai nanofiber Technology Co., Ltd. Lithium bis (trifluoro-methanesulfonyl) imide (LiTFSI, 99.9% purity) was purchased from Suzhou Duoduo Chemical Technology Co., Ltd.4.2. Preparation of solid-state polymer electrolytes
The polymer electrolytes were prepared by a solvent casting method. In particular, 1.00 g of PTHF and 0.199 g of LiTFSI (Li+:Oe =1:20) were dissolved in acetonitrile, and a homogeneous solution was formed by stirring for 4 h at 25 °C. The slurry was then cast onto the PI separator and dried in a vacuum oven at 60 °C for 12 h to remove the acetonitrile solvent. For the PEO polymer electrolyte, 1.00 g of PEO and 0.326 g of LiTFSI (Li+:Oe = 1:20) were dissolved in acetonitrile and a homogeneous solution was formed by stirring for 4 h at 25 °C. The forming and drying procedures were identical to those used for the PTHF polymer electrolyte. For the PDOL polymer electrolyte, 1.00 g of PDOL and 0.388 g of LiTFSI (Li+:Oe = 1:20) were dissolved in acetonitrile and a homogeneous solution was formed by stirring for 4 h at 60 °C. The forming and drying procedures were identical to those used for the PTHF polymer electrolyte. For the PTOM polymer electrolyte, hexafluoroisopropanol was used for dissolving due to the strong crystallinity of the pristine PTOM. 1.00 g of PTOM and 0.478 g of LiTFSI (Li+:Oe = 1:20) were dissolved in hexafluoroisopropanol and a homogeneous solution was formed by stirring for 4 h at 25 °C. The forming and drying procedures were identical to those used for the PTHF polymer electrolyte.
4.3. Classical molecular dynamics simulations
All classical molecular dynamics (MD) simulations were performed using the large-scale atomic/molecular parallel simulator (LAMMPS) package.78 The bonded and nonbonded interactions for polymers were described by the OPLS-AA force field,79,80 while parameters for Li+ and TFSI− were from ref. 81 and 82, respectively. The partial charges applied to Li+ and TFSI− were scaled to 0.7 to mitigate the overestimation of ion–ion interaction in nonpolarizable force fields.83 The geometric mean combining rule was used for mixing the parameters, and a cutoff distance of 10 Å was set for non-bonded interaction. The long-range coulombic interaction was computed by the particle–particle particle–mesh (PPPM) method with an accuracy of 10−4.84 The Nose-Hoover thermostat and barostat were used to control the temperature and pressure, respectively.854.4. Ab initio molecular dynamics
Ab initio molecular dynamics (AIMD) simulations were implemented using the QUICKSTEP module86 in the CP2K (v2022.2) package.87 The PBE exchange correlation functional88 was employed in conjunction with the DFT-D3(BJ) dispersion corrections.89 The molecularly optimized double-ζ-DZVP-MOLOPT-SR-GTH basis set90 was used with the Goedecker–Teter–Hutter pseudopotentials.91,92 A convergence test was performed for the PEO system (Fig. S3), indicating that a plane wave cut-off of 400 Ry is sufficient to obtain energies with an accuracy of 1 meV per atom compared to 800 Ry. Hence, the plane wave cut-off of 400 Ry with a reference grid cut-off of 50 Ry was used for all simulations. The orbital transformation (OT) method93 with a DIIS minimizer and a FULL KINETIC preconditioner was used to solve the Kohn–Sham equations.4.5. Quantum chemistry calculations
The binding free energy calculations were carried out using Gaussian 16.94 The geometry optimizations were performed at the B3LYP-D3(BJ)/6-311+g(d,p)95–97 level, while the energy calculations were performed at the B2PLYPD3/def2-TZVP98,99 level. The binding free energy ΔG was calculated using the following equation:| ΔG = Gcomplex − (GLi+ + Gpolymer) | (1) |
| G = H − TS | (2) |
The local polarity of the polymer was characterized using the molecular polarity index (MPI)103 and fraction of polar surface area, which were computed by the Multiwfn program.104 MPI is defined as:
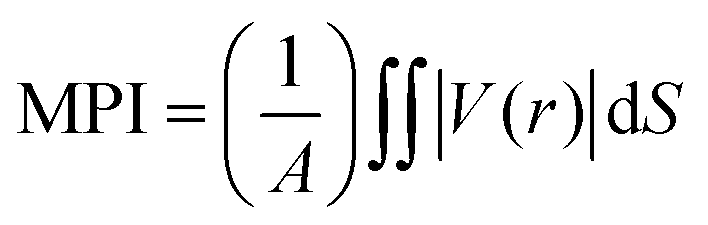 | (3) |
The energy decomposition analysis based on absolutely localized molecular orbitals (ALMO-EDA)105–107 was performed using Q-chem 6.2.2108 at the B3LYP-D3(BJ)/6-311+g(d,p) level using the same structure as used in free energy calculations. In the ALMO-EDA method, the binding energy of two fragments EB is decomposed into contributions of permanent electrostatics (ELEC), Pauli repulsion (PAULI), dispersion (DISP), polarization (POL), and charge transfer (CT):
| EB = ΔEELEC + ΔEPAULI + ΔEDISP + ΔEPOL + ΔECT | (4) |
The harmonic vibrational analysis for simulated infrared (IR) spectra was performed at the B3LYP-D3(BJ)/6-31++G(d) with an implicit polarizable continuum model (PCM). The dielectric constants were set to 3.0, 5.0, 2.8, and 4.8 for PTHF, PEO, PDOL, and PTOM, respectively.109–111 The oxidation potentials were calculated at the B3LYP-D3(BJ)/6-311G(d) level with an implicit polarizable continuum model (PCM); the oxidation potential (Eox) is calculated as follows:112
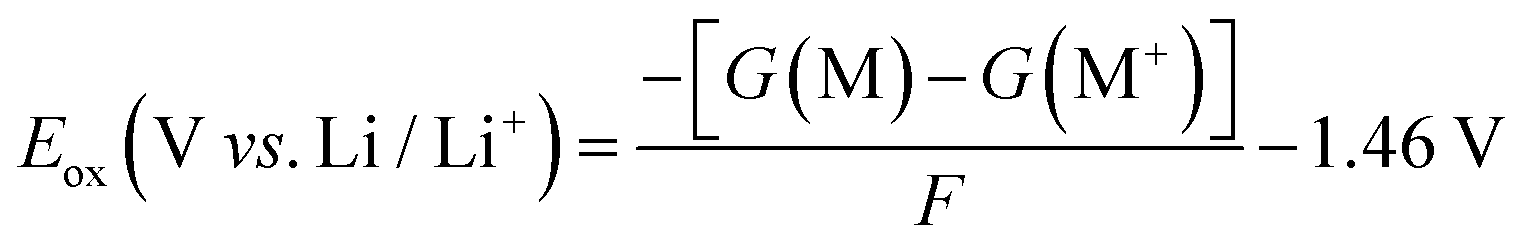 | (5) |
Author contributions
B. Liang, X. Huang, and T. Hou conceived the idea. T. Hou, J. Fan, C.-Z. Zhao, and Q. Zhang supervised the project. B. Liang, S. Tan, and C. Ji designed and implemented the MD simulations, B. Liang, T. Wang, T. Si, Y. Xie, and L. Jiang implemented the quantum chemistry calculations. X. Huang prepared the materials and cells, X.-Y. Huang, X.-Y. Li, and H. Chen performed the characterization and electrochemical tests. B. Liang, X.-Y. Huang, T. Hou, and C.-Z. Zhao wrote the manuscript. All authors commented on the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Data availability
The result data from all the simulations and experiments in this study are provided within the paper or in the supplementary information (SI) file. The SI file includes additional experimental details, simulation procedures, transport properties, glass transition temperature, coordination and clustering analysis, transport modes analysis, and additional results and discussions. See DOI: https://doi.org/10.1039/d5ee05901a.Additional data are available from the corresponding authors upon reasonable request. Source data are provided with this paper.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22409121, 22393900, 22409114, and 52394170), the Natural Science Foundation of Guangdong Province (2025A1515012161), the Special Funds for the Development of Strategic Emerging Industries in Shenzhen (XMHT20240108008), the Shenzhen Technical Plan Project (JCYJ20240813112111015), the Shenzhen Stable Support Program for Higher Education Institutions (WDZC20231126215806001), the Hong Kong Research Grant Council Collaborative Research Fund (C1002-21G and C1017-22G), the City University of Hong Kong Project (7006111), and the Beijing Municipal Natural Science Foundation (L247015).Notes and references
- J. B. Goodenough and Y. Kim, Chem. Mater., 2009, 22, 587–603 CrossRef.
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 Search PubMed.
- J. Janek and W. G. Zeier, Nat. Energy, 2023, 8, 230–240 CrossRef.
- Z. Song, F. Chen, M. Martinez-Ibanez, W. Feng, M. Forsyth, Z. Zhou, M. Armand and H. Zhang, Nat. Commun., 2023, 14, 4884 Search PubMed.
- P. Xu, Z.-Y. Shuang, C.-Z. Zhao, X. Li, L.-Z. Fan, A. Chen, H. Chen, E. Kuzmina, E. Karaseva, V. Kolosnitsyn, X. Zeng, P. Dong, Y. Zhang, M. Wang and Q. Zhang, Sci. China: Chem., 2023, 67, 67–86 CrossRef.
- J. T. Frith, M. J. Lacey and U. Ulissi, Nat. Commun., 2023, 14, 420 CrossRef CAS PubMed.
- A. Mauger, C. M. Julien, J. B. Goodenough and K. Zaghib, J. Electrochem. Soc., 2020, 167, 070507 Search PubMed.
- X. Lu, Y. Wang, X. Xu, B. Yan, T. Wu and L. Lu, Adv. Energy Mater., 2023, 13, 2301746 CrossRef CAS.
- N. Meng, F. Lian and G. Cui, Small, 2020, 17, 2005762 CrossRef PubMed.
- X. Y. Huang, C. Z. Zhao, W. J. Kong, N. Yao, Z. Y. Shuang, P. Xu, S. Sun, Y. Lu, W. Z. Huang, J. L. Li, L. Shen, X. Chen, J. Q. Huang, L. A. Archer and Q. Zhang, Nature, 2025, 646, 343–350 CrossRef CAS PubMed.
- R. Bouchet, S. Maria, R. Meziane, A. Aboulaich, L. Lienafa, J.-P. Bonnet, T. N. T. Phan, D. Bertin, D. Gigmes, D. Devaux, R. Denoyel and M. Armand, Nat. Mater., 2013, 12, 452–457 CrossRef CAS PubMed.
- S. Han, P. Wen, H. Wang, Y. Zhou, Y. Gu, L. Zhang, Y. Shao-Horn, X. Lin and M. Chen, Nat. Mater., 2023, 22, 1515–1522 Search PubMed.
- F. Pei, L. Wu, Y. Zhang, Y. Liao, Q. Kang, Y. Han, H. Zhang, Y. Shen, H. Xu, Z. Li and Y. Huang, Nat. Commun., 2024, 15, 351 CrossRef CAS PubMed.
- J. Chen, Y. Gao, L. Shi, W. Yu, Z. Sun, Y. Zhou, S. Liu, H. Mao, D. Zhang, T. Lu, Q. Chen, D. Yu and S. Ding, Nat. Commun., 2022, 13, 4868 CrossRef CAS PubMed.
- W. Jiang, D. Wang, W. Li and J. Zhang, Macromolecules, 2024, 57, 8682–8689 CAS.
- D. G. Mackanic, W. Michaels, M. Lee, D. Feng, J. Lopez, J. Qin, Y. Cui and Z. Bao, Adv. Energy Mater., 2018, 8, 1800703 Search PubMed.
- P. Ding, L. Wu, Z. Lin, C. Lou, M. Tang, X. Guo, H. Guo, Y. Wang and H. Yu, J. Am. Chem. Soc., 2023, 145, 1548–1556 CrossRef CAS PubMed.
- D. Bresser, S. Lyonnard, C. Iojoiu, L. Picard and S. Passerini, Mol. Syst. Des. Eng., 2019, 4, 779–792 Search PubMed.
- C. A. Angell, Electrochim. Acta, 2017, 250, 368–375 CrossRef CAS.
- Z. Xue, D. He and X. Xie, J. Mater. Chem. A, 2015, 3, 19218–19253 RSC.
- P. V. Wright, Br. Polym. J., 1975, 7, 319–327 CrossRef CAS.
- V. Bocharova and A. P. Sokolov, Macromolecules, 2020, 53, 4141–4157 Search PubMed.
- C. Austen Angell, Electrochim. Acta, 2019, 313, 205–210 CrossRef CAS.
- J. Wang, S. Li, Q. Zhao, C. Song and Z. Xue, Adv. Funct. Mater., 2020, 31, 2008208 CrossRef.
- A. L. Agapov and A. P. Sokolov, Macromolecules, 2011, 44, 4410–4414 Search PubMed.
- Y. Wang, A. L. Agapov, F. Fan, K. Hong, X. Yu, J. Mays and A. P. Sokolov, Phys. Rev. Lett., 2012, 108, 088303 CrossRef PubMed.
- Y. Wang, F. Fan, A. L. Agapov, T. Saito, J. Yang, X. Yu, K. Hong, J. Mays and A. P. Sokolov, Polymer, 2014, 55, 4067–4076 Search PubMed.
- C. T. Imrie, M. D. Ingram and G. S. McHattie, Adv. Mater., 1999, 11, 832–834 CrossRef CAS.
- Y. Wang, C. J. Zanelotti, X. Wang, R. Kerr, L. Jin, W. H. Kan, T. J. Dingemans, M. Forsyth and L. A. Madsen, Nat. Mater., 2021, 20, 1255–1263 CrossRef CAS PubMed.
- K. Kimura, J. Motomatsu and Y. Tominaga, J. Phys. Chem. C, 2016, 120, 12385–12391 CrossRef CAS.
- H. Xu, J. Yang, Y. Niu, X. Hou, Z. Sun, C. Jiang, Y. Xiao, C. He, S. Yang, B. Li and W. Chen, Angew. Chem., Int. Ed., 2024, 136, e202406637 CrossRef.
- F. S. Genier and I. D. Hosein, Macromolecules, 2021, 54, 8553–8562 CrossRef CAS.
- Q. Zhao, X. Liu, S. Stalin, K. Khan and L. A. Archer, Nat. Energy, 2019, 4, 365–373 CrossRef CAS.
- P. Kang, D. Chen, L. Wu, X. Yang and G. Sui, Chem. Eng. J., 2023, 455, 140931 CrossRef CAS.
- C. Z. Zhao, Q. Zhao, X. Liu, J. Zheng, S. Stalin, Q. Zhang and L. A. Archer, Adv. Mater., 2020, 32, e1905629 Search PubMed.
- S. Li, H. Hong, X. Yang, D. Li, Q. Xiong, D. Zhang, S. Wang, Z. Huang, H. Lv and C. Zhi, Adv. Mater., 2025, 37, e2504333 CrossRef PubMed.
- F. Liu, T. Li, Y. Yang, J. Yan, N. Li, J. Xue, H. Huo, J. Zhou and L. Li, Macromol. Rapid Commun., 2020, 41, e2000047 CrossRef PubMed.
- H. Wu, B. Tang, X. Du, J. Zhang, X. Yu, Y. Wang, J. Ma, Q. Zhou, J. Zhao, S. Dong, G. Xu, J. Zhang, H. Xu, G. Cui and L. Chen, Adv. Sci., 2020, 7, 2003370 Search PubMed.
- Z. Li, R. Yu, S. Weng, Q. Zhang, X. Wang and X. Guo, Nat. Commun., 2023, 14, 482 CrossRef PubMed.
- O. Borodin and G. D. Smith, Macromolecules, 2006, 39, 1620–1629 CrossRef CAS.
- P. Kang, L. Wu, D. Chen, Y. Su, Y. Zhu, J. Lan, X. Yang and G. Sui, J. Phys. Chem. B, 2022, 126, 4531–4542 CrossRef CAS PubMed.
- N. Yao, X. Chen, Z. H. Fu and Q. Zhang, Chem. Rev., 2022, 122, 10970–11021 CrossRef CAS PubMed.
- S. Mogurampelly, J. R. Keith and V. Ganesan, J. Am. Chem. Soc., 2017, 139, 9511–9514 CrossRef CAS PubMed.
- D. J. Brooks, B. V. Merinov, W. A. Goddard, B. Kozinsky and J. Mailoa, Macromolecules, 2018, 51, 8987–8995 CrossRef CAS.
- N. Molinari, J. P. Mailoa and B. Kozinsky, Chem. Mater., 2018, 30, 6298–6306 CrossRef CAS.
- L. J. Abbott and J. W. Lawson, Macromolecules, 2019, 52, 7456–7467 CrossRef CAS.
- C. Deng, M. A. Webb, P. Bennington, D. Sharon, P. F. Nealey, S. N. Patel and J. J. de Pablo, Macromolecules, 2021, 54, 2266–2276 CrossRef CAS.
- C. Deng, P. Bennington, R. J. Sánchez-Leija, S. N. Patel, P. F. Nealey and J. J. de Pablo, Macromolecules, 2023, 56, 8069–8079 CrossRef CAS PubMed.
- A. Maitra and A. Heuer, Phys. Rev. Lett., 2007, 98, 227802 CrossRef CAS PubMed.
- D. Diddens, A. Heuer and O. Borodin, Macromolecules, 2010, 43, 2028–2036 CrossRef CAS.
- D. Diddens and A. Heuer, J. Phys. Chem. B, 2014, 118, 1113–1125 CrossRef CAS PubMed.
- S. D. Druger, A. Nitzan and M. A. Ratner, J. Chem. Phys., 1983, 79, 3133–3142 CrossRef CAS.
- A. Nitzan and M. A. Ratner, J. Phys. Chem., 1994, 98, 1765–1775 CrossRef CAS.
- P. E. Rouse, J. Chem. Phys., 1953, 21, 1272–1280 Search PubMed.
- J. Self, K. D. Fong and K. A. Persson, ACS Energy Lett., 2019, 4, 2843–2849 CrossRef CAS.
- O. Borodin, G. A. Giffin, A. Moretti, J. B. Haskins, J. W. Lawson, W. A. Henderson and S. Passerini, J. Phys. Chem. C, 2018, 122, 20108–20121 CrossRef CAS.
- T. Hou, K. D. Fong, J. Wang and K. A. Persson, Chem. Sci., 2021, 12, 14740–14751 RSC.
- K. Yoshida, M. Nakamura, Y. Kazue, N. Tachikawa, S. Tsuzuki, S. Seki, K. Dokko and M. Watanabe, J. Am. Chem. Soc., 2011, 133, 13121–13129 Search PubMed.
- K. S. Ngai, S. Ramesh, K. Ramesh and J. C. Juan, Ionics, 2016, 22, 1259–1279 CrossRef CAS.
- C. D. Fang, Y. Huang, Y. F. Sun, P. F. Sun, K. Li, S. Y. Yao, M. Y. Zhang, W. H. Fang and J. J. Chen, Nat. Commun., 2024, 15, 6781 CrossRef CAS PubMed.
- X. Xie, P. Zhang, X. Li, Z. Wang, X. Qin, M. Shao, L. Zhang and W. Zhou, J. Am. Chem. Soc., 2024, 146, 5940–5951 CrossRef CAS PubMed.
- W. Liang, X. Zhou, B. Zhang, Z. Zhao, X. Song, K. Chen, L. Wang, Z. Ma and J. Liu, Angew. Chem., Int. Ed., 2024, 63, e202320149 CrossRef CAS PubMed.
- K. D. Fong, J. Self, B. D. McCloskey and K. A. Persson, Macromolecules, 2020, 53, 9503–9512 CrossRef CAS.
- J. Han, R. H. Gee and R. H. Boyd, Macromolecules, 2002, 27, 7781–7784 CrossRef.
- H. Gudla and C. Zhang, J. Phys. Chem. B, 2024, 128, 10537–10540 CrossRef CAS PubMed.
- N. Yao, X. Chen, S.-Y. Sun, Y.-C. Gao, L. Yu, Y.-B. Gao, W.-L. Li and Q. Zhang, Chem, 2025, 11, 102254 CAS.
- M. J. Counihan, K. S. Chavan, P. Barai, D. J. Powers, Y. Zhang, V. Srinivasan and S. Tepavcevic, Joule, 2024, 8, 64–90 CrossRef CAS.
- Q. Li, A. Chen, D. Wang, Z. Pei and C. Zhi, Joule, 2022, 6, 273–279 CrossRef.
- Z. Y. Wang, C. Z. Zhao, N. Yao, Y. Lu, Z. Q. Xue, X. Y. Huang, P. Xu, W. Z. Huang, Z. X. Wang, J. Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2025, 64, e202414524 CrossRef CAS PubMed.
- X. Yang, M. Jiang, X. Gao, D. Bao, Q. Sun, N. Holmes, H. Duan, S. Mukherjee, K. Adair, C. Zhao, J. Liang, W. Li, J. Li, Y. Liu, H. Huang, L. Zhang, S. Lu, Q. Lu, R. Li, C. V. Singh and X. Sun, Energy Environ. Sci., 2020, 13, 1318–1325 RSC.
- G. Zeng, S. Dai, X. Chen, L. Qiu, X. Kong, M. Huang and T. Wen, Macromolecules, 2024, 57, 1258–1265 CrossRef CAS.
- D. Rosenbach, A. Krimalowski, H. Erabhoina and M. Thelakkat, J. Mater. Chem. A, 2022, 10, 8932–8947 RSC.
- G. Ye, L. Zhu, Y. Ma, M. He, C. Zheng, K. Shen, X. Hong, Z. Xiao, Y. Jia, P. Gao and Q. Pang, J. Am. Chem. Soc., 2024, 146, 27668–27678 CrossRef CAS PubMed.
- C. Yang, Q. Wu, W. Xie, X. Zhang, A. Brozena, J. Zheng, M. N. Garaga, B. H. Ko, Y. Mao, S. He, Y. Gao, P. Wang, M. Tyagi, F. Jiao, R. Briber, P. Albertus, C. Wang, S. Greenbaum, Y. Y. Hu, A. Isogai, M. Winter, K. Xu, Y. Qi and L. Hu, Nature, 2021, 598, 590–596 CrossRef PubMed.
- Q. Shen, K. Li, Y. Song, R. Gao, Z. Li and Y. Wang, Adv. Funct. Mater., 2025, e21332 CrossRef.
- P. Shi, J. Ma, M. Liu, S. Guo, Y. Huang, S. Wang, L. Zhang, L. Chen, K. Yang, X. Liu, Y. Li, X. An, D. Zhang, X. Cheng, Q. Li, W. Lv, G. Zhong, Y. B. He and F. Kang, Nat. Nanotechnol., 2023, 18, 602–610 CrossRef CAS PubMed.
- K. Khan, M. B. Hanif, H. Xin, A. Hussain, H. G. Ali, B. Fu, Z. Fang, M. Motola, Z. Xu and M. Wu, Small, 2024, 20, e2305772 CrossRef PubMed.
- A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolintineanu, W. M. Brown, P. S. Crozier, P. J. int Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen, R. Shan, M. J. Stevens, J. Tranchida, C. Trott and S. J. Plimpton, Comput. Phys. Commun., 2022, 271, 108171 CrossRef CAS.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- G. A. Kaminski, R. A. Friesner, J. Tirado-Rives and W. L. Jorgensen, J. Phys. Chem. B, 2001, 105, 6474–6487 CrossRef CAS.
- K. P. Jensen and W. L. Jorgensen, J. Chem. Theory Comput., 2006, 2, 1499–1509 CrossRef CAS PubMed.
- J. N. Canongia Lopes and A. A. H. Pádua, J. Phys. Chem. B, 2004, 108, 16893–16898 CrossRef.
- I. Leontyev and A. Stuchebrukhov, Phys. Chem. Chem. Phys., 2011, 13, 2613–2626 Search PubMed.
- R. Hockney and J. Eastwood, Computer Simulation Using Particles, Adam Hilger, Bristol and New York, NY, USA, 1989, 120–165 Search PubMed.
- W. G. Hoover, Phys. Rev. A, 1985, 31, 1695 CrossRef PubMed.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS.
- T. D. Kuhne, M. Iannuzzi, M. Del Ben, V. V. Rybkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Schutt, F. Schiffmann, D. Golze, J. Wilhelm, S. Chulkov, M. H. Bani-Hashemian, V. Weber, U. Borstnik, M. Taillefumier, A. S. Jakobovits, A. Lazzaro, H. Pabst, T. Muller, R. Schade, M. Guidon, S. Andermatt, N. Holmberg, G. K. Schenter, A. Hehn, A. Bussy, F. Belleflamme, G. Tabacchi, A. Gloss, M. Lass, I. Bethune, C. J. Mundy, C. Plessl, M. Watkins, J. VandeVondele, M. Krack and J. Hutter, J. Chem. Phys., 2020, 152, 194103 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 Search PubMed.
- J. VandeVondele and J. Hutter, J. Chem. Phys., 2007, 127, 114105 CrossRef PubMed.
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 1703–1710 CrossRef CAS PubMed.
- C. Hartwigsen, S. Goedecker and J. Hutter, Phys. Rev. B:Condens. Matter Mater. Phys., 1998, 58, 3641–3662 CrossRef CAS.
- J. VandeVondele and J. Hutter, J. Chem. Phys., 2003, 118, 4365–4369 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Wallingford CT, 2016 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- L. Goerigk and S. Grimme, J. Chem. Theory Comput., 2011, 7, 291–309 CrossRef CAS PubMed.
- S. Grimme, J. Chem. Phys., 2006, 124, 034108 CrossRef PubMed.
- Y. P. Li, J. Gomes, S. M. Sharada, A. T. Bell and M. Head-Gordon, J. Phys. Chem. C, 2015, 119, 1840–1850 CrossRef CAS.
- S. Grimme, Chemistry, 2012, 18, 9955–9964 Search PubMed.
- G. Luchini, J. V. Alegre-Requena, I. Funes-Ardoiz and R. S. Paton, F1000Research, 2020, 9, 291 Search PubMed.
- Z. Liu, T. Lu and Q. Chen, Carbon, 2021, 171, 514–523 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 Search PubMed.
- P. R. Horn, Y. Mao and M. Head-Gordon, Phys. Chem. Chem. Phys., 2016, 18, 23067–23079 RSC.
- Y. Mao, M. Loipersberger, P. R. Horn, A. Das, O. Demerdash, D. S. Levine, S. Prasad Veccham, T. Head-Gordon and M. Head-Gordon, Annu. Rev. Phys. Chem., 2021, 72, 641–666 CrossRef CAS PubMed.
- P. R. Horn, Y. Mao and M. Head-Gordon, J. Chem. Phys., 2016, 144, 114107 CrossRef PubMed.
- E. Epifanovsky, A. T. B. Gilbert, X. Feng, J. Lee, Y. Mao, N. Mardirossian, P. Pokhilko, A. F. White, M. P. Coons, A. L. Dempwolff, Z. Gan, D. Hait, P. R. Horn, L. D. Jacobson, I. Kaliman, J. Kussmann, A. W. Lange, K. U. Lao, D. S. Levine, J. Liu, S. C. McKenzie, A. F. Morrison, K. D. Nanda, F. Plasser, D. R. Rehn, M. L. Vidal, Z. Q. You, Y. Zhu, B. Alam, B. J. Albrecht, A. Aldossary, E. Alguire, J. H. Andersen, V. Athavale, D. Barton, K. Begam, A. Behn, N. Bellonzi, Y. A. Bernard, E. J. Berquist, H. G. A. Burton, A. Carreras, K. Carter-Fenk, R. Chakraborty, A. D. Chien, K. D. Closser, V. Cofer-Shabica, S. Dasgupta, M. de Wergifosse, J. Deng, M. Diedenhofen, H. Do, S. Ehlert, P. T. Fang, S. Fatehi, Q. Feng, T. Friedhoff, J. Gayvert, Q. Ge, G. Gidofalvi, M. Goldey, J. Gomes, C. E. Gonzalez-Espinoza, S. Gulania, A. O. Gunina, M. W. D. Hanson-Heine, P. H. P. Harbach, A. Hauser, M. F. Herbst, M. Hernandez Vera, M. Hodecker, Z. C. Holden, S. Houck, X. Huang, K. Hui, B. C. Huynh, M. Ivanov, A. Jasz, H. Ji, H. Jiang, B. Kaduk, S. Kahler, K. Khistyaev, J. Kim, G. Kis, P. Klunzinger, Z. Koczor-Benda, J. H. Koh, D. Kosenkov, L. Koulias, T. Kowalczyk, C. M. Krauter, K. Kue, A. Kunitsa, T. Kus, I. Ladjanszki, A. Landau, K. V. Lawler, D. Lefrancois, S. Lehtola, R. R. Li, Y. P. Li, J. Liang, M. Liebenthal, H. H. Lin, Y. S. Lin, F. Liu, K. Y. Liu, M. Loipersberger, A. Luenser, A. Manjanath, P. Manohar, E. Mansoor, S. F. Manzer, S. P. Mao, A. V. Marenich, T. Markovich, S. Mason, S. A. Maurer, P. F. McLaughlin, M. Menger, J. M. Mewes, S. A. Mewes, P. Morgante, J. W. Mullinax, K. J. Oosterbaan, G. Paran, A. C. Paul, S. K. Paul, F. Pavosevic, Z. Pei, S. Prager, E. I. Proynov, A. Rak, E. Ramos-Cordoba, B. Rana, A. E. Rask, A. Rettig, R. M. Richard, F. Rob, E. Rossomme, T. Scheele, M. Scheurer, M. Schneider, N. Sergueev, S. M. Sharada, W. Skomorowski, D. W. Small, C. J. Stein, Y. C. Su, E. J. Sundstrom, Z. Tao, J. Thirman, G. J. Tornai, T. Tsuchimochi, N. M. Tubman, S. P. Veccham, O. Vydrov, J. Wenzel, J. Witte, A. Yamada, K. Yao, S. Yeganeh, S. R. Yost, A. Zech, I. Y. Zhang, X. Zhang, Y. Zhang, D. Zuev, A. Aspuru-Guzik, A. T. Bell, N. A. Besley, K. B. Bravaya, B. R. Brooks, D. Casanova, J. D. Chai, S. Coriani, C. J. Cramer, G. Cserey, A. E. DePrince, 3rd, R. A. DiStasio, Jr., A. Dreuw, B. D. Dunietz, T. R. Furlani, W. A. Goddard, 3rd, S. Hammes-Schiffer, T. Head-Gordon, W. J. Hehre, C. P. Hsu, T. C. Jagau, Y. Jung, A. Klamt, J. Kong, D. S. Lambrecht, W. Liang, N. J. Mayhall, C. W. McCurdy, J. B. Neaton, C. Ochsenfeld, J. A. Parkhill, R. Peverati, V. A. Rassolov, Y. Shao, L. V. Slipchenko, T. Stauch, R. P. Steele, J. E. Subotnik, A. J. W. Thom, A. Tkatchenko, D. G. Truhlar, T. Van Voorhis, T. A. Wesolowski, K. B. Whaley, H. L. Woodcock 3rd, P. M. Zimmerman, S. Faraji, P. M. W. Gill, M. Head-Gordon, J. M. Herbert and A. I. Krylov, J. Chem. Phys., 2021, 155, 084801 CrossRef CAS PubMed.
- A. Tsutsumi, K. Hikichi and M. Kaneko, Jpn. J. Appl. Phys., 1968, 7, 577 CrossRef CAS.
- M. Kumar and S. S. Sekhon, Eur. Polym. J., 2002, 38, 1297–1304 CrossRef CAS.
- C. H. Porter, J. H. L. Lawler and R. H. Boyd, Macromolecules, 2002, 3, 308–314 CrossRef.
- X. Jiao and X. Pan, J. Electroanal. Chem., 2021, 882, 114995 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2026 |