
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High-asymmetry bipolar membrane electrode assemblies generate a superconcentration of cations and hydroxide at a catalyst surface
Qiu-Cheng
Chen†
 a,
Wenjin
Zhu†
a,
Yiqing
Chen†
a,
Hongmin
An
ac,
Shuang
Yang
a,
Yong
Wang
a,
Yali
Ji
a,
Guangcan
Su
a,
Rui
Wang
a,
Jianan Erick
Huang
a,
Ji-Yoon
Song
a,
Jaerim
Kim
a,
Weiyan
Ni
a,
Charles
Musgrave
a,
Ke
Xie
*a and
Edward H.
Sargent
*ab
a,
Wenjin
Zhu†
a,
Yiqing
Chen†
a,
Hongmin
An
ac,
Shuang
Yang
a,
Yong
Wang
a,
Yali
Ji
a,
Guangcan
Su
a,
Rui
Wang
a,
Jianan Erick
Huang
a,
Ji-Yoon
Song
a,
Jaerim
Kim
a,
Weiyan
Ni
a,
Charles
Musgrave
a,
Ke
Xie
*a and
Edward H.
Sargent
*ab
aDepartment of Chemistry, Northwestern University, Evanston, IL, USA. E-mail: ted.sargent@northwestern.edu; ke-xie@northwestern.edu
bDepartment of Electrical and Computer Engineering, Northwestern University, Evanston, IL, USA
cDepartment of Chemical and Biomolecular Engineering, Korea Advanced Institute of Science and Technology, Daejeon, Republic of Korea
First published on 31st December 2025
Abstract
In electrochemical CO reduction reactions, a highly alkaline pH is typically desired to promote multicarbon liquid products and suppress hydrogen evolution, thus prioritizing pH ≥ 14 (e.g. 1 M KOH). However, bulk electrolytes with pH exceeding 14 are prone to produce corrosion of the catalyst and electrolyzer. Here we find that an engineered class of bipolar membrane assemblies (BPMEAs) achieves a superconcentration of local metal hydroxides, and generates a product slate consistent with a local electrolyte pH of 15. We report that, in a cathode:anion exchange layer (AEL):cation exchange layer (CEL):anode architecture, a high thickness ratio of CEL![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AEL generates a high local pH at the cathode, which was achieved by blocking the transport of hydroxide ions, generated on the cathode, over to the anode side. This enables production of C2+ liquids at a total faradaic efficiency of 93%, with an ethanol:ethylene productivity ratio of 70:1. Compared to anion-exchange membrane assemblies (AEMEAs) operating at the same 100 mA cm−2 current density for similar product selectivity, these BPMEA systems exhibit 28 hours of stable operation (compared to <30 minutes in AEMEAs), and a 12× lower rate of liquid product crossover, enabling us to report a liquid product concentration of 23 wt% on the cathode. Operando Raman spectroscopy shows that the optimal BPM enhances coverage, on the cathode catalyst, of surface-bound hydroxyl species, ∼5× higher than in AEM systems, thereby simultaneously maximizing the surface CO population. Mechanistic studies indicate that surface OH promotes hydroxylation of the CCH intermediate, steering the reaction pathway toward ethanol instead of ethylene, leading to strong preference towards liquid production.
:AEL generates a high local pH at the cathode, which was achieved by blocking the transport of hydroxide ions, generated on the cathode, over to the anode side. This enables production of C2+ liquids at a total faradaic efficiency of 93%, with an ethanol:ethylene productivity ratio of 70:1. Compared to anion-exchange membrane assemblies (AEMEAs) operating at the same 100 mA cm−2 current density for similar product selectivity, these BPMEA systems exhibit 28 hours of stable operation (compared to <30 minutes in AEMEAs), and a 12× lower rate of liquid product crossover, enabling us to report a liquid product concentration of 23 wt% on the cathode. Operando Raman spectroscopy shows that the optimal BPM enhances coverage, on the cathode catalyst, of surface-bound hydroxyl species, ∼5× higher than in AEM systems, thereby simultaneously maximizing the surface CO population. Mechanistic studies indicate that surface OH promotes hydroxylation of the CCH intermediate, steering the reaction pathway toward ethanol instead of ethylene, leading to strong preference towards liquid production.
Broader contextElectrochemical conversion of CO2-derived CO into liquid fuels like ethanol offers a promising route to decarbonize heavy industry and achieve carbon neutrality. However, existing systems face a critical trade-off: high alkalinity boosts liquid fuel production but accelerates corrosion, limiting device stability and practicality. This work addresses this challenge by designing an asymmetric bipolar membrane electrode assembly (BPMEA) that locally generates an ultra-high pH environment (pH ≈ 15) at the catalyst surface while maintaining benign bulk conditions. We demonstrate that this membrane architecture achieves 93% selectivity for liquid products, with a 70:1 ethanol-to-ethylene ratio, and operates stably for 28 hours (versus <30 minutes in conventional systems). Crucially, it reduces liquid product crossover by 12× and achieves unprecedented ethanol concentrations (23 wt%) on the cathode. By revealing how localized hydroxide superconcentration steers reaction pathways toward ethanol, this work provides a blueprint for stable, membrane-engineered electrolyzers. This advance bridges a key gap toward scalable production of carbon-neutral liquid fuels from CO2.
|
Introduction
The CO electroreduction reaction (CORR) is of interest in making multi-carbon products, and CO is generated efficiently from CO2 in solid-oxide electrolyzers.1–4 Compared with gaseous CO2RR products, liquid products enable convenient transport from the point of production to that of further processing or utilization.5 Since ethylene and ethanol share key intermediates in reaction mechanisms, significant efforts have focused on enhancing ethanol and liquid-product selectivity by modulating these competing pathways via catalyst engineering3,6–12 as well as through the design of membranes,13–17 ionomers,18,19 and electrolytes.20–29Alkalinity is known to facilitate C–C coupling and suppress the HER in favour of the CORR,1,30,31 and has been shown to enhance selectivity to acetate.1,32,33 Recent studies also suggested that the adsorbed OH7,34–36 is a key species to turn on the CO(2)-to-ethanol pathway, entailing that ethanol selectivity may also be increased by manipulating alkalinity.37,38 In these studies, the coverage of adsorbed OH and CO was tuned by catalyst modification and pulsed oxidation of a copper catalyst surface. Notably, the dependence of both ethanol production and the ethanol-to-ethylene ratio on bulk pH and surface OH/CO coverage has been proposed to be nonmonotonic, indicating a complex ethanol/ethylene production mechanism.34,35 In principle, increasing electrolyte alkalinity can directly regulate OH coverage. Indeed, we found that gradually increasing anolyte concentration steers the CORR toward ethanol production (Fig. S1). However, such a high alkalinity can destabilize oxygen evolution reaction (OER) catalysts39,40 and induce corrosion41,42 (Fig. S2).
In parallel to catalyst design, advancing CORR electrocatalysis relies increasingly on zero-gap membrane electrode assembly (MEA) electrolyzers to regulate reaction conditions.43,44 In these systems, the cathode and anode are disposed on either side of a polymeric ion exchange membrane in a zero-gap configuration, with gas supplied to the cathodic gas diffusion electrode (GDE), and electrolyte circulated on the anode (Fig. 1a).13,14,16,45–49 The choice of membrane will affect the pH and ions on the catalyst layers (Fig. 1b), affecting thereby the reaction pathway and thus the product distribution. The impact of membrane/assembly design on pH and ion egress has been studied and exploited in water electrolyzers.50–54
 | ||
| Fig. 1 MEA-CO electrolyzer configuration and membrane-dependent CORR performance. (a) Schematic illustrations of the interface structure in the MEA. (b) Transport of ionic species (M+ = Li+, Na+, K+, Cs+, etc.) in different membrane configurations: anion exchange membrane (AEM), cation exchange membrane (CEM), forward-bias bipolar membrane (f-BPM) and reverse-bias bipolar membrane (r-BPM). (c) Faradaic efficiency and liquid-product crossover of CORR liquid products in MEAs with different membranes or anolytes. (d) Key performance metrics (water migration rate, Dwater; FEs and concentration of ethanol and total liquid product; full cell voltages, Efull) for AEM, CEM, and f-BPM cells using 1 M KOH anolyte; (e) cell voltage and faradaic efficiencies for CO reduction in the optimized f-BPMEA system during a 28 hour operation. | ||
We focused herein on how membrane design impacts surface coverage of OH and CO when the composite membrane is rendered selective for the transport of specific ions,49,54–56 our goal to favor liquid production.
Results
CORR selectivity under different membrane configurations
We began by investigating CORR reactivity in a suite of candidate membrane configurations: AEM, CEM, forward-bias BPM and reverse-bias BPM; all in a zero-gap cell (Fig. 1a and Fig. S3) at 100 mA cm−2 current density. In 1 M KOH anolyte, the AEM cell (AEMEA) shows FEliquid of 60%, and the CEM and r-BPM cells (CEMEA and r-BPMEA) show 72% and 49%, respectively. In contrast, the f-BPM cell (f-BPMEA) achieves FEliquid of 93% and correspondingly low gas production (Fig. 1c and Fig. S4).Compared to the AEM, the cation exchange layer (CEL) in f-BPM serves as a blocker for OH− generated during the CORR (Fig. 1b, AEM vs. f-BPM), while transferring K+ from the anolyte, instead of H+ in r-BPM systems (Fig. 1b, r-BPM vs. f-BPM). The result is an increased KOH concentration on the cathode surface, known to promote the CORR to multi-carbon products.1,20,21,23 The product distribution is similar in an AEMEA in which a higher anolyte concentration, 10 M KOH, is employed; however, these systems suffer from instability (Fig. S2) and have a >12× higher liquid product crossover rate compared to f-BPMEA (Fig. 1c), the result of the electroosmotic drag effect in AEMEA. The f-BPMEA achieved a 2.5× higher liquid product concentration on the cathode compared to the AEMEA (23 wt% vs. 9 wt%, Fig. 1d, liquid wt%) and it operated at a similar full-cell voltage (Fig. 1d, Efull).
In a CEMEA, K+ migrates from the anolyte to cathode, with electroosmotic drag leading to the transport of water molecules (one study57 quantified 27 H2O per K+). This ion current inhibits the undesired transport of the liquid product to the anolyte, but the co-transport of H2O from the anode to cathode causes unacceptably high product dilution on the cathode. We observed that f-BPM hinders anode-to-cathode water crossover (Fig. 1d, Dwater, rate of water crossover), and, when one compares to the CEM, the cathode-side liquid product concentration is 6× higher (Fig. 1d, liquid wt%). The AEL of f-BPM provides cathode-to-anode OH− egress, dragging 4–7 H2O per OH−.57 This anion/water flow offsets the cation/water flow. We ran the f-BPMEA continuously for 28 hours, and found that it maintained high selectivity of ethanol and acetate over ethylene at 100 mA cm−2 (Fig. 1e). The corresponding energy efficiencies (EEs) for ethanol, and for total liquid products, were 22% and 40%, respectively. The concentrated ethanol output from the f-BPMEA enables a distillation cost that is less than half of that for the CEMEA (Note S5). While smaller cations (Li+, Na+) are anticipated to transport less water owing to their lower hydration numbers,58 the use of LiOH and NaOH as anolytes was found to shift product selectivity toward gas-phase products (Fig. S17).
We then studied the impact of the thickness ratio of the CEL to AEL on the CORR selectivity. In the AEMEA, n-propanol (31% FE) and ethylene (30% FE) are the main products, with 21% of FEethanol (Fig. 2a). We processed CELs of different thicknesses atop a prefabricated anion exchange layer (AEL) in order to produce BPMs, and placed these in the MEA in the forward bias configuration. Increasing the thickness of the CEL from 2 to 25 µm (0.04 to 0.5 CEL:AEL thickness ratio) leads to diminished FEHER, FEethylene and FEn-propanol, and progressively increasing selectivity toward ethanol and acetate (Fig. 2a). Further increasing the thickness ratio of CEL:AEL to 0.5 suppresses the FE of hydrogen and ethylene evolution reactions to below 1%. Interestingly, we then saw a rise in both the HER occurrence and ethylene production, and decreased ethanol and acetate selectivity, when we increased the CEL:AEL thickness ratio beyond 0.5 (Fig. 2a). Assuming that the CEL plays a role in keeping the OH− at the cathode side, a thicker CEL decreases the transport rate of OH− from the cathode surface to the anolyte, increasing the local pH of the catalyst layer.
 | ||
| Fig. 2 Characteristics of f-BPM for the CORR in MEA systems. (a) Faradaic efficiency profiles for CO reduction on the Cu catalyst using AEM, CEM, and f-BPM with varying CEL-to-AEL thickness ratios at −100 mA cm−2. (b) Potassium transport rate (DK+) in membranes with different CEL-to-AEL ratios; a ratio of 0 corresponds to the pristine AEM. (c) Schematic of species transport mechanisms in the MEA with an f-BPM. (d) COMSOL-simulated species concentration profiles from the cathode to anolyte for varying CEL-to-AEL thickness ratios. (e) Measured interfacial pH (cathode/membrane) for different CEL-to-AEL thickness ratios. (f) FEethanol and ethanol-to-ethylene selectivity as a function of DK+ and interfacial pH (pHCL/M) for tested membrane configurations. | ||
To explore the relationship between the membranes and the interfacial environment, we studied ion transport in the MEA. We track K+, instead of OH−, since the latter could originate from anolyte crossover or cathodic reactions. At the cathode side after the CORR, under 100 mA cm−2 galvanostatic electrolysis, we estimated the rate of K+ transport (DK+) from the anolyte to cathode by collecting the totality of liquids and gases exiting the cathode chamber and the gas outlet, and evaluated the composition using inductively coupled plasma (ICP) spectroscopy (Note S1). A rate of 0.5 µmol min−1 for DK+ was observed in the AEMEA system. When we introduced a CEL onto the AEM to form a f-BPM, we saw evidence of an increased DK+ value, with only 2 µm of CEL leading to 12× higher DK+ than the bare AEM (Fig. 2b). Thicker CEL@f-BPM further accelerates DK+, reaching 24 µmol min−1DK+ with a 1-to-1 CEL:AEL thickness ratio. Since (1) the ion diffusion constant (ci), electromigration (ϕ) and reaction rate are similar under identical applied current density and temperature in the AEL, and (2) CEL transport of OH− is much slower than of K+, the OH− generated during the COR began to accumulate between the cathode and the CEL, leading to K+ transport across the CEL from the anode to cathode through ion-correlations (μi) that achieve electroneutrality on the cathode surface.59 When we changed the anolyte from 1 M KOH to neutral 0.5 M K2SO4, we saw similar DK+ and CORR product distribution, from which we surmise that the cathode surface environment is independent of the anolyte (Fig. S5).
We propose therefore this picture of the f-BPM system: the majority of OH− generated from the cathodic reaction accumulates between the AEL and CEL, forming superconcentrated KOH at the interface. KOH diffuses through the AEL and causes the high alkalinity of the cathodic local environment (Fig. 2c).16,60 COMSOL simulation of the ion distribution within f-BPMs vs. thickness ratios also supports the higher CEL:AEL ratio leading to higher pH and [K+] near the cathode catalyst layer (Fig. 2d and Note S2).
To quantify the pH at the interface between the catalyst layer (CL) and membrane (pHCL/M), we carried out the CORR in a modified MEA cell equipped with a needle sensor that provides a metal/metal oxide redox couple (Note S3, Fig. S6 and S7). The observed pHCL/M value was 13.93 in the AEM system, and this increases rapidly to 14.37 at CEL:AEL = 0.2 and continues to 14.77 at 1:1 ratio (Fig. 2e and Fig. S7). This is consistent with the COMSOL study, in which a larger CEM:AEM ratio led to a higher pH at the CL-to-AEM interface than that of the anolyte. The collective effect of increasing pH and [K+] increases the production of ethanol (Fig. 2f) from FEethanol 21% in the AEMEA system to 51% in the CEMEA system, and we observe the highest FEethanol/FEethylene ratio at 0.5 CEL:AEL ratio (Fig. 2f).
Ethylene and ethanol shared similar intermediates in the stage immediately following C–C coupling.61–63 To study the mechanism of this shift in selectivity to ethanol and ethylene as a function of varying local pH and DK+, we used in situ Raman (SERS) across a bulk pH range from 13.5 to 15.0 to explore the adsorption behavior of reactive species on the Cu catalyst. The CuO pre-catalyst characterized by SEM, HRTEM, XRD and XPS showed ∼0.5 µm nanoplates (Fig. 3a and Fig. S8, S9). The oxidation state of the Cu in the pre-catalyst is mainly 2+, and this is electrochemically reduced to metallic copper during the CORR (Fig. 3b and Fig. S10). We tested the CORR on the CuO catalyst at different pH values with focus on the phonon modes at ca. 280, 360 and 535 cm−1 on SERS (Table S2), corresponding to the restricted rotation of *CO (COr), Cu–CO stretching (COs) and Cu–OH vibration (*OH).64–66 The ratio of these peaks is associated with the relative coverage of CO and OH on the copper surface, denoted as θCO and θOH, respectively.35,67,68 In the electrolyte pH range from 13.5 to 14.5, both θOH and θCO increase with higher pH, consistent with surface-enhanced infrared measurements (Fig. 3d), while the growth of θOH in low pH ranges may arise due to the lower energy barrier for Cu–OH formation from near neutral to moderate basic pH (Fig. 3e).37,38 The blue shifts of both COs and *OH bands indicate stronger binding of surface CO and OH, along with increased bulk pH values before reaching 14.5 (Fig. 3f).
 | ||
| Fig. 3 Cathode characterization and in situ Raman analysis. (a) Scanning electron microscopy image of the CuO pre-catalyst. (b) High-resolution XPS spectra of the catalyst-coated gas diffusion electrode (GDE) pre- and post-electrolysis. (c) Operando Raman spectra of CuNPs acquired during the CORR at bulk pH 13.5–15.0 at potential equal to −0.4 V vs. RHE; relative CO (d) and OH (e) coverage as a function of the electrolyte pH. (f) pH-Dependent Raman shifts of CO and OH adsorption peaks. | ||
At pH > 14.5, the growth of *CO leveled off, i.e. CO adsorption on the Cu surface saturated (Fig. 3d). We saw a significant decrease in θOH past bulk pH = 14.5 (Fig. 3e), a finding we correlate to the decreasing FEethanol/FEethylene after that pH (Fig. 2f). Previous reports agree on the trend of increased C2+ production upon higher CO coverage on the copper surface,38,67,69–72 particularly promoting oxygenate formation.6,32,73–76 The influence of the alkali cation concentration and pH on selectivity is itself a topic of ongoing discussion in the literature.1,22,28,31–33,38,62,71,76–78 Recent studies suggest that surface OH species is a key promoter of ethanol production.
From our studies, we infer that θCO and θOH together may drive ethanol formation: the sharp decrease in θOH and a plateau of θCO suggest that the surface hydroxide determine product selectivity at high electrolyte concentrations and high pH regions, a model that is able to capture the nonmonotonic dependence of ethanol selectivity on the OH/CO coverage ratio35 and moderate surface OH species for optimal C2+ production as documented in previous reports.79 The absence of Raman shifts on COs and *OH above bulk pH = 14.5 suggests that the integrated effects of [K+] and [OH−] do not significantly affect the adsorption of *CO and *OH (Fig. 3f). Hence, the lower population of surface-bound hydroxide species can be explained by (1) a high density of K+ at the electrified interface minimizing the interfacial availability of H2O/OH;28,29,80 and (2) a balance between the higher consumption rate of surface hydroxide and its availability.36,68 This is also supported by a recent study that showed a negative correlation between ethanol/ethylene selectivity and concentrated cation (>6 M),28 as well as a nonmonotonic distribution of ethanol/ethylene selectivity along similar concentration ranges of pH values.33,76
Mechanistic studies
To study further the correlation between the surface hydroxide and ethanol selectivity, we used H218O solution for the anolyte 1 M KOH (18O atom% > 96% in total) and carried out in situ electrochemical mass spectrometry (EC-MS) to track the source of the oxygen in the ethanol (Fig. 4c). We tracked the isotope-labelled (48 amu) and unlabelled (46 amu) ethanol and recorded the percentage of labeled: which was 70%, 50% and 42% in f-BPMEA (AEL:CEL = 0.5 ratio), CEMEA and AEMEA, respectively (Fig. 4a). This trend aligns well with the nonmonotonic distribution of FEethanol/FEethylene upon pHCL/M on this system (Fig. 4a), consistent with the nonmonotonic distribution of θOH in the same pH range (Fig. 3b). The FEethanol/FEethylene selectivity is positively related to the ratio of Et18OH (Fig. 4b), from which we conclude that a favoured route to ethanol formation involves oxygen from the electrolyte instead of from carbon monoxide.
 | ||
| Fig. 4 EC-MS isotope analysis and proposed mechanism. (a) Ethanol/ethylene selectivity and 18O–ethanol percentage as functions of membrane type (corresponding pHCL/M) in the MEA setup. (b) Ethanol/ethylene selectivity versus18O–ethanol percentage in the MEA setup. (c) Key reaction pathways for the CO reduction reaction (CORR) to ethanol on Cu(111). (d) The reaction energies for the *CCH → *CHC18OH and *CCH → *CCH2 steps (left axis) and their corresponding energy differences (right axis, ΔG*CHCOH–*CCH2) at −0.4 V vs. SHE at various OH coverages. ΔG*CHCOH–*CCH2 represents the relative preference for *CHCOH or CCH2 formation, where a higher ΔG*CHCOH–*CCH2 value indicates more favorable *CHCOH formation and consequently higher ethanol selectivity. (e) Calculated Gibbs free-energy diagram for *CCH to *CHCO on Cu(111) at −0.4 V vs. SHE at various OH coverages. | ||
While the Et18OH can arise due to the acetaldehyde pathway81–83 or due to *CCH hydrolysis,62 the nonmonotonic distribution of 18O–ethanol percentage with monotonically increasing pH and the insensitivity of labelled/unlabelled propanol in different membrane configurations (Fig. S11) indicate little role for the acetaldehyde route (Fig. S18). We focus therefore on *CCH62 as the intermediate determining the selectivity between ethanol and ethylene, and found that increasing OH coverage lowers the energy of *CHCHOH formation (ethanol pathway) from 0.49 eV at 0/9 OH coverage to −0.09 eV at 2/9 OH coverage (Fig. S12). However, under ultra-high alkali conditions, the water concentration at the interface is expected to be much lower than OH−, suggesting that the oxygen in ethanol should originate mainly from surface-bound or solvated hydroxide. Hence, the observation of 18O-labeled ethanol indicates that one of the intermediates may be oxidized by the hydroxyl-containing species, also supported by a recent study that found that CO is oxidized by *OH under high coverage of CO and OH on copper.36 To account for the potential oxidation step, we propose the conversion of *CCH to *CHCOH to form 18O-labelled ethanol or to *CCH2, leading to ethylene (Fig. 4c). As shown in Fig. 4d, higher OH coverage enhances *CHCOH formation, while the formation energy of *CCH2 initially increases and then decreases. 1/9 ML OH coverage exhibits the lowest energy difference between *CHCOH and *CCH2 (Fig. S13–S15). Although *CHCOH selectivity does not increase monotonically with OH coverage, the negative ΔG*CHCOH–*CCH2 agrees with the preference for *CHCOH formation in the presence of surface *OH (Fig. 4d). DFT calculations herein also show that *OH coverage reduces the energy required for *CHCO formation (Fig. 4d), which is energetically favorable compared to the hydrolysis step of *CCH to *CHCHOH (Fig. S9),62 promoting ethanol formation over ethylene.
Conclusions
This work investigated how membrane configurations tune product selectivity in the CORR. The correlation between the system and reaction selectivity was linked through membrane kinetics and interfacial speciation at the molecular level. Among the configurations tested, a f-BPM delivered >93% selectivity for liquid production and 70-to-1 ethanol selectivity over ethylene by maintaining optimal local pH and potassium ion availability at the catalyst surface. The higher CEL-to-AEL ratio will lead to elevated pH values between the CL and the membrane in the reported systems. These conditions favor pathways that suppress gaseous products, ethylene and hydrogen. Operando techniques, coupled with computational modeling, indicate that *CO and *OH surface coverage drive selectivity toward ethanol over ethylene. Future work could beneficially probe the interfacial pH in such MEA systems.84Overall, the present findings highlight the importance of membrane design in catholyte-free MEA systems for efficient carbon utilization and the potential of f-BPM for liquid production.
This work focuses on the use of f-BPMEA for the CORR, although the CO2RR could, in principle, be performed in the same device. In this case, the CO2RR in f-BPMEA will need to tackle the challenge of salt precipitation at the cathode, this being the result of the high local concentration of cations near the catholyte interface; and CO2 bubble formation at the CEL/AEL junction, and its potential to produce delamination of the f-BPM, would also need to be addressed.45,85
Methods
Chemicals
Copper(II) sulfate pentahydrate (CuSO4·5H2O), methanol (ACS reagent grade, >99.8%), dimethyl sulfoxide (ACS reagent grade >99.9%), lithium hydroxide (ACS reagent grade 98%), sodium hydroxide (ACS reagent grade 98%), potassium hydroxide (semiconductor grade 99.99%), and water–18O (97 atom% 18O) were purchased from Sigma Aldrich. Nafion solution (D520, 5 wt% and D2020, 20 wt%), carbon paper (Freudenberg H23C6), iridium oxide, Nafion 212 and Nafion 211 were purchased from the fuel cell store. Sustainion (X37-50 grade 60) was purchased from Dioxide Materials. All chemicals were used as received or pretreated under standard procedures. The aqueous solutions were prepared using de-ionized water with a resistivity of 18.2 MΩ cm.Synthesis of Cu catalysts
The CuO nanoplate material was prepared by a hydrothermal method modified from a previous protocol.32 In a typical procedure, 1.0 g CuSO4·5H2O were dissolved in 50 ml deionized water to form a homogeneous blue solution. The solution was placed in an ice–water bath with vigorous magnetic stirring. 10 ml 1.2 M NaOH solution was dropped into the above solution slowly and stirred half an hour continuously. Then, keep the mixture refrigerated at 3 °C for 24 hours before transferring it to the Teflon-lined autoclave. The hydrothermal program was set to 130 °C for 18 hours, followed by cooling to room temperature. The Cu treated material was collected via centrifugation, washed several times with deionized water and dried in a vacuum at 60 °C overnight.Materials characterization
The morphologies of the samples were characterized by SEM (Hitachi SU8030). TEM characterization was conducted using a JEOL ARM-200F microscope equipped with a JEOL delta aberration corrector and a 200 kV cold field emission gun with powder samples. X-ray powder diffractometer (XRD) measurements were performed in a MiniFlex600 with Cu-Kα radiation. XPS measurements were conducted using a ThermoFisher Scientific NEXSA G2, equipped with a monochromated, micro-focused, low-power Al K-alpha X-ray source. Catalyst coated electrodes were used directly for measurements.Preparation of gas diffusion electrodes (GDEs)
Cu NPs were dispersed in a mixture of methanol and Nafion solution and left under ultrasonication for at least 30 min. The catalyst ink composition was 25 mgCuNPs mlMeOH−1 and 100 µlNafion 5 wt% mlMeOH−1. The ink was spray coated on a carbon paper GDL with a Cu nanoparticle loading of 3 mg cm−2 to prepare the Cu electrode. The electrodes were left under room conditions overnight before the electrochemical measurements were conducted.Preparation of bipolar membranes
The CEM with different thicknesses was fabricated following a reported method.53 0.5 ml Nafion® D2020 dispersion was dispensed and spin-coated onto pre-cleaned, and dust-free Si wafer chips (∼6 cm2) at 500–5000 rpm. The samples were then dried on a hot stage at 80 °C for 60 s. Alternatively, thicker membranes were fabricated by casting polymer solutions on Si wafers by a homemade system using a doctor blade with glass rod and double sided tape. The samples were then transferred into a tube furnace and annealed in N2 for 4 h at 110 °C. The thickness and uniformity of each membrane were measured with a profilometer on the center and different edges to obtain <5% thickness difference. The edges of the CEM on the Si were removed with a razor, allowing the film to fully detach from the substrate in the next step. The substrate with the thin film were slowly immersed into water and the Sustainion AEM was carefully pressed onto the layer. Then the thin-CEL BPM was removed from the water bath and transferred to the MEA. This method was used to produce BPMs with a 2–15 µm CEM, while the Nafion 211 and Nafion 212 were used for BPMs with 25- and 50 mm thickness CEMs. The BPMs were used immediately after assembly.Electrochemical measurements
A homemade titanium MEA electrolyzer was used, consisting of a gas chamber and an anodic chamber. The MEA was composed of a cathode electrode, an Ni foam (or NiFe-P) anode electrode and an ion-exchange membrane (Sustainion for AEMEA, Nafion for CEMEA, and BPM for BPMEA). The electrochemical testing cell was assembled by placing the membrane between the cathode and anode electrode, in which the catalyst layers of both electrodes were facing the membrane. The electrodes were protected and surrounded by 0.01-inch-thick gaskets for electrical insulation. The reaction area was regulated by the pore area in the gasket, which was set at 1 cm2. This assembly was then inserted into the homemade fuel cell device, which had serpentine flow channels that were equally compressed with torque applied to the bolts. A constant rate of 20 ml min−1 of the KOH anolyte flowed through the anodic channel, and a digital mass flow meter supplied the humidified CO feed gas to the cathodic channel at a constant rate of 25 sccm. After the electrolyzer assembly, a galvanostatic method was used to apply electrolysis. For the stability test, the MEA was operated at a constant full-cell current density of 100 mA cm−2 with the NiFe-P anode.Electrochemical reduction product measurement
The gas products were collected from the gas outlet of the MEA and injected into a gas chromatograph (PerkinElmer Clarus 600) coupled with a flame ionization detector (FID) for the detection of CH4 and C2H4 and a thermal conductivity detector (TCD) for the detection of H2, and CO signals. The faradaic efficiency of gas products was calculated using the following equation:where FEi is the faradaic efficiency of product “i”, yi is the volumetric fraction of the gas product, V is the outlet gas flow rate in sccm, zi is the number of electrons associated with producing one molecule of product from CO, F is the Faraday constant, P0 is the atmospheric pressure, R is the ideal gas constant, T is the temperature and jtotal is the total current density.

The liquid products were analyzed using 1H NMR spectroscopy (500 MHz Bruker Avance III HD) with water suppression. Dimethyl sulfoxide was used as the reference standard and deuterium oxide as the lock solvent. The faradaic efficiency of liquid products was calculated using the equation:
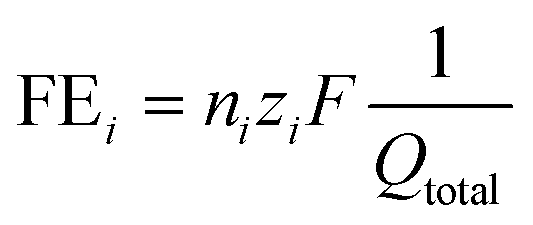
Operando spectroscopic analysis
Operando Raman spectroscopy was performed with a water immersion objective (×63) using a Renishaw inVia Raman microscope. The spectra were collected using a 785 nm laser at 0.1% intensity. Each spectrum was recorded using the Renishaw WiRE (v.4.4) software by integrating three times, with each integration lasting 10 s. An open-structured flow cell was utilized for the measurements. CO was continuously supplied to the gas chamber during the measurement in a modified flow cell set up with standard Cu NPs on GDE as the working electrode. An Ag/AgCl electrode (filled with saturated aqueous KCl solution) and a platinum wire were used as the reference and counter electrode, respectively (Fig. S16). While −0.4 V vs. RHE was applied to the Raman cell, electrolyte with different pH was exchanged by flow to monitor the surface species on the same laser spot. Actual pH was measured using a pH meter. The potentials from the Raman measurements were converted to values versus RHE.EC-MS
In situ EC-MS analysis was conducted using a Hiden HPR-20 QIC capillary system coupled with a customized electrochemical cell. The gaseous outlet of regular MEA setup with different membrane configurations was connected to the EC-MS. 1 M KOH solution was prepared using water–18O (97 atom% 18O). The gaseous products were continuously monitored simultaneously under electrochemical potential protocols that varied in real time. All gaseous products entered the capillary inlet and were then ionized. Ionization was performed at an electron energy of 70 V. The resulting ions were detected using a secondary electron multiplier (SEM) detector operating at 800 V.Computation
Spin-polarized density functional theory (DFT) calculations86 were performed using the Vienna ab initio Simulation Package (VASP).87,88 The exchange–correlation interactions in the Kohn–Sham equations were modeled using the generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) functional.89,90 Core–valence interactions were described through the projector-augmented wave (PAW) method,91 while long-range dispersion effects were incorporated using Grimme's DFT-D3(BJ) scheme.92,93 An energy cutoff of 450 eV was employed, and the Brillouin zone was sampled with a 4 × 4 × 1 Monkhorst–Pack k-point grid.94 Structural optimization was carried out until the residual forces on atoms were below 0.03 eV Å−1 and energy changes were less than 10−4 eV. The free energies were calculated based on the computational hydrogen electrode (CHE) model.95A 3 × 3 Cu(111) supercell comprising four atomic layers was constructed as the surface model, with the bottom two layers fixed in their bulk lattice positions. To account for solvation effects, three layers of water containing 18 water molecules were placed on the Cu(111) surface. The local pH on the Cu(111) surfaces with OH coverages of 0/9, 1/9, and 2/9 ML were thus approximated as 7, 14.47, and 14.74, respectively. Additionally, a vacuum layer of at least 15 Å was introduced perpendicular to the surface to minimize interactions between periodic images.
Conflicts of interest
There are no conflicts to declare.Data availability
Supplementary information (SI) including measurements of cation concentration and pH, pre- and post-electrolysis material characterization, additional in-situ measurements, and calculation results is available. See DOI: https://doi.org/10.1039/d5ee04672f.All data are available from the authors upon reasonable request.
Acknowledgements
This work was supported by the Bill & Melinda Gates Foundation under the investment # INV-061828 and INV-079283. This work made use of facilities at IMSERC at Northwestern University.References
- M. Jouny, W. Luc and F. Jiao, Nat. Catal., 2018, 1, 748–755 Search PubMed.
- P. Zhu, C. Xia, C. Y. Liu and K. Jiang, et al. , Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2010868118 CrossRef CAS PubMed.
- W. Niu, J. Feng, J. Chen and L. Deng, et al. , Nat. Commun., 2024, 15, 7070 CrossRef CAS PubMed.
- A. Ozden, Y. Wang, F. Li and M. Luo, et al. , Joule, 2021, 5, 706–719 CrossRef CAS.
- H. Shin, K. U. Hansen and F. Jiao, Nat. Sustainability, 2021, 4, 911–919 Search PubMed.
- F. Li, Y. C. Li, Z. Wang and J. Li, et al. , Nat. Catal., 2019, 3, 75–82 CrossRef.
- M. Luo, Z. Wang, Y. C. Li and J. Li, et al. , Nat. Commun., 2019, 10, 5814 CrossRef CAS PubMed.
- J. Ding, H. Bin Yang, X.-L. Ma and S. Liu, et al. , Nat. Energy, 2023, 8, 1386–1394 CrossRef CAS.
- L. Zhang, J. Feng, L. Wu and X. Ma, et al. , J. Am. Chem. Soc., 2023, 145, 21945–21954 CrossRef CAS PubMed.
- Z. Liu, L. Song, X. Lv and M. Liu, et al. , J. Am. Chem. Soc., 2024, 146, 14260 Search PubMed.
- C. Long, K. Wan, Y. Chen and L. Li, et al. , J. Am. Chem. Soc., 2024, 146, 4632–4641 CrossRef CAS PubMed.
- X. Wang, P. Ou, A. Ozden and S.-F. Hung, et al. , Nat. Energy, 2022, 7, 170–176 Search PubMed.
- K. V. Petrov, C. I. Koopman, S. Subramanian and M. T. M. Koper, et al. , Nat. Energy, 2024, 9, 932–938 CrossRef CAS.
- D. A. Salvatore, C. M. Gabardo, A. Reyes and C. P. O’Brien, et al. , Nat. Energy, 2021, 6, 339–348 Search PubMed.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 Search PubMed.
- M. A. Blommaert, R. Sharifian, N. U. Shah and N. T. Nesbitt, et al. , J. Mater. Chem. A, 2021, 9, 11179–11186 Search PubMed.
- K. Yang, M. Li, S. Subramanian and M. A. Blommaert, et al. , ACS Energy Lett., 2021, 6, 4291–4298 CrossRef CAS PubMed.
- D. A. Henckel, P. Saha, S. Rajana and C. Baez-Cotto, et al. , ACS Energy Lett., 2024, 9, 3433–3439 Search PubMed.
- F. P. Garcia de Arquer, C. T. Dinh, A. Ozden and J. Wicks, et al. , Science, 2020, 367, 661–666 Search PubMed.
- E. Perez-Gallent, G. Marcandalli, M. C. Figueiredo and F. Calle-Vallejo, et al. , J. Am. Chem. Soc., 2017, 139, 16412–16419 Search PubMed.
- A. S. Malkani, J. Li, N. J. Oliveira and M. He, et al. , Sci. Adv., 2020, 6, eabd2569 CrossRef CAS PubMed.
- S. J. Shin, H. Choi, S. Ringe and D. H. Won, et al. , Nat. Commun., 2022, 13, 5482 CrossRef CAS PubMed.
- Y. Xu, Z. Xia, W. Gao and H. Xiao, et al. , Nat. Catal., 2024, 7, 1120–1129 Search PubMed.
- S. Yu, H. Yamauchi, S. Wang and A. Aggarwal, et al. , Nat. Catal., 2024, 7, 1000–1009 CrossRef CAS.
- Z.-M. Zhang, T. Wang, Y.-C. Cai and X.-Y. Li, et al. , Nat. Catal., 2024, 7, 807–817 CrossRef CAS.
- Q. Zhu, C. L. Rooney, H. Shema and C. Zeng, et al. , Nat. Catal., 2024, 7, 987–999 Search PubMed.
- J. M. Yoo, J. Ingenmey, M. Salanne and M. R. Lukatskaya, J. Am. Chem. Soc., 2024, 146, 31768 CrossRef CAS PubMed.
- H. Zhang, J. Gao, D. Raciti and A. S. Hall, Nat. Catal., 2023, 6, 807–817 CrossRef CAS.
- N. T. Nesbitt and W. A. Smith, J. Phys. Chem. C, 2021, 125, 13085–13095 Search PubMed.
- H. Xiao, T. Cheng, W. A. Goddard, 3rd and R. Sundararaman, J. Am. Chem. Soc., 2016, 138, 483–486 Search PubMed.
- C. T. Dinh, T. Burdyny, M. G. Kibria and A. Seifitokaldani, et al. , Science, 2018, 360, 783–787 Search PubMed.
- X. Wang, Y. Chen, F. Li and R. K. Miao, et al. , Nat. Commun., 2024, 15, 616 CrossRef CAS PubMed.
- W. Luc, X. Fu, J. Shi and J.-J. Lv, et al. , Nat. Catal., 2019, 2, 423–430 CrossRef CAS.
- J. Zhang, G. Zeng, S. Zhu and H. Tao, et al. , Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2218987120 CrossRef CAS PubMed.
- A. Herzog, M. Lopez Luna, H. S. Jeon and C. Rettenmaier, et al. , Nat. Commun., 2024, 15, 3986 Search PubMed.
- H. Zhang, Y. Cui, C. Ren and Q. Li, et al. , ACS Catal., 2024, 14, 10737–10745 Search PubMed.
- X. Chang, Y. Zhao and B. Xu, ACS Catal., 2020, 10, 13737–13747 Search PubMed.
- J. Li, X. Chang, H. Zhang and A. S. Malkani, et al. , Nat. Commun., 2021, 12, 3264 Search PubMed.
- M. Etzi Coller Pascuzzi, A. J. W. Man, A. Goryachev and J. P. Hofmann, et al. , Catal. Sci. Technol., 2020, 10, 5593–5601 RSC.
- Y. Han, J. Wang, Y. Liu and T. Li, et al. , Carbon Neutralization, 2024, 3, 172–198 CrossRef CAS.
- D. Prando, A. Brenna, M. V. Diamanti and S. Beretta, et al. , J. Appl. Biomater. Funct. Mater., 2017, 15, e291–e302 CAS.
- R. M. Katona, J. Carpenter, E. J. Schindelholz and R. F. Schaller, et al. , J. Electrochem. Soc., 2021, 168, 071509 Search PubMed.
- Q. Ye, X. Zhao, R. Jin and F. Dong, et al. , J. Mater. Chem. A, 2023, 11, 21498–21515 RSC.
- C. P. O'Brien, R. K. Miao, A. Shayesteh Zeraati and G. Lee, et al. , Chem. Rev., 2024, 124, 3648–3693 Search PubMed.
- X. She, L. Zhai, Y. Wang and P. Xiong, et al. , Nat. Energy, 2024, 9, 81–91 CrossRef CAS.
- Y. C. Li, Z. Yan, J. Hitt and R. Wycisk, et al. , Adv. Sustainable Syst., 2018, 2, 1700187 Search PubMed.
- K. Xie, R. K. Miao, A. Ozden and S. Liu, et al. , Nat. Commun., 2022, 13, 3609 Search PubMed.
- C. P. O’Brien, R. K. Miao, S. Liu and Y. Xu, et al. , ACS Energy Lett., 2021, 6, 2952–2959 Search PubMed.
- T. Alkayyali, A. S. Zeraati, H. Mar and F. Arabyarmohammadi, et al. , ACS Energy Lett., 2023, 8, 4674–4683 Search PubMed.
- J. C. Bui, I. Digdaya, C. Xiang and A. T. Bell, et al. , ACS Appl. Mater. Interfaces, 2020, 12, 52509–52526 Search PubMed.
- B. Mayerhöfer, D. McLaughlin, T. Böhm and M. Hegelheimer, et al. , ACS Appl. Energy Mater., 2020, 3, 9635–9644 CrossRef.
- J. A. Wrubel, Y. Chen, Z. Ma and T. G. Deutsch, J. Electrochem. Soc., 2020, 167, 114502 Search PubMed.
- S. Z. Oener, L. P. Twight, G. A. Lindquist and S. W. Boettcher, ACS Energy Lett., 2020, 6, 1–8 Search PubMed.
- W. L. Toh, H. Q. Dinh, A. T. Chu and E. R. Sauvé, et al. , Nat. Energy, 2023, 8, 1405–1416 CrossRef CAS.
- T. Alkayyali, M. Zargartalebi, A. Ozden and F. Arabyarmohammadi, et al. , Joule, 2024, 8, 1478–1500 CrossRef CAS.
- C. McCallum, C. M. Gabardo, C. P. O’Brien and J. P. Edwards, et al. , Cell Rep. Phys. Sci., 2021, 2, 100522 CrossRef CAS.
- T. Yamanaka, T. Takeguchi, H. Takahashi and W. Ueda, J. Electrochem. Soc., 2009, 156, B831 Search PubMed.
- Z. Jing, Y. Zhou, T. Yamaguchi and K. Yoshida, et al. , J. Phys. Chem. Lett., 2023, 14, 6270–6277 CrossRef CAS PubMed.
- J. C. Bui, E. W. Lees, D. H. Marin and T. N. Stovall, et al. , Nat. Chem. Eng., 2024, 1, 45–60 Search PubMed.
- G. A. El-Nagar, F. Haun, S. Gupta and S. Stojkovikj, et al. , Nat. Commun., 2023, 14, 2062 CrossRef CAS PubMed.
- F. Calle-Vallejo and M. T. Koper, Angew. Chem., Int. Ed., 2013, 52, 7282–7285 Search PubMed.
- Y. Lum, T. Cheng, W. A. Goddard, 3rd and J. W. Ager, J. Am. Chem. Soc., 2018, 140, 9337–9340 CrossRef CAS PubMed.
- R. Kortlever, J. Shen, K. J. Schouten and F. Calle-Vallejo, et al. , J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed.
- C. M. Gunathunge, X. Li, J. Li and R. P. Hicks, et al. , J. Phys. Chem. C, 2017, 121, 12337–12344 CrossRef CAS.
- G. Niaura, Electrochim. Acta, 2000, 45, 3507–3519 Search PubMed.
- B. D. Smith, D. E. Irish, P. Kedzierzawski and J. Augustynski, J. Electrochem. Soc., 1997, 144, 4288–4296 CrossRef CAS.
- C. Zhan, F. Dattila, C. Rettenmaier and A. Bergmann, et al. , ACS Catal., 2021, 11, 7694–7701 CrossRef CAS PubMed.
- C. Zhan, F. Dattila, C. Rettenmaier and A. Herzog, et al. , Nat. Energy, 2024, 9, 1485 CrossRef CAS PubMed.
- R. B. Sandberg, J. H. Montoya, K. Chan and J. K. Nørskov, Surf. Sci., 2016, 654, 56–62 CrossRef CAS.
- X. Chang, H. Xiong, Q. Lu and B. Xu, JACS Au, 2023, 3, 2948–2963 CrossRef CAS PubMed.
- A. Wuttig, C. Liu, Q. Peng and M. Yaguchi, et al. , ACS Cent. Sci., 2016, 2, 522–528 Search PubMed.
- M. Schreier, Y. Yoon, M. N. Jackson and Y. Surendranath, Angew. Chem., Int. Ed., 2018, 57, 10221–10225 Search PubMed.
- Y. Ji, Z. Chen, R. Wei and C. Yang, et al. , Nat. Catal., 2022, 5, 251–258 CrossRef CAS.
- J. Jin, J. Wicks, Q. Min and J. Li, et al. , Nature, 2023, 617, 724–729 CrossRef CAS PubMed.
- P. Wei, D. Gao, T. Liu and H. Li, et al. , Nat. Nanotechnol., 2023, 18, 299–306 CrossRef CAS PubMed.
- J. Li, Z. Wang, C. McCallum and Y. Xu, et al. , Nat. Catal., 2019, 2, 1124–1131 CrossRef CAS.
- J. Li, D. Wu, A. S. Malkani and X. Chang, et al. , Angew. Chem., Int. Ed., 2020, 59, 4464–4469 Search PubMed.
- Y. Cao, Z. Chen, P. Li and A. Ozden, et al. , Nat. Commun., 2023, 14, 2387 CrossRef CAS PubMed.
- M. Sun, A. Staykov and M. Yamauchi, ACS Catal., 2022, 12, 14856–14863 Search PubMed.
- X. Yang, H. Ding, S. Li and S. Zheng, et al. , J. Am. Chem. Soc., 2024, 146, 5532–5542 Search PubMed.
- Y. Hori, R. Takahashi, Y. Yoshinami and A. Murata, J. Phys. Chem. B, 1997, 101, 7075–7081 CrossRef CAS.
- E. Bertheussen, A. Verdaguer-Casadevall, D. Ravasio and J. H. Montoya, et al. , Angew. Chem., Int. Ed., 2016, 55, 1450–1454 CrossRef CAS PubMed.
- E. L. Clark and A. T. Bell, J. Am. Chem. Soc., 2018, 140, 7012–7020 CrossRef CAS PubMed.
- Y. Y. Wang, H. Shi, Y. Gong and B. Zhang, et al. , Langmuir, 2023, 39, 16807–16811 CrossRef CAS PubMed.
- J. Disch, S. Ingenhoven and S. Vierrath, Adv. Energy Mater., 2023, 13, 2301614 Search PubMed.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B:Condens. Matter Mater. Phys., 1999, 59, 1758–1775 Search PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 Search PubMed.
- J. P. Perdew, J. A. Chevary, S. H. Vosko and K. A. Jackson, et al. , Phys. Rev. B:Condens. Matter Mater. Phys., 1992, 46, 6671–6687 Search PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B, 1976, 13, 5188 Search PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir and L. Lindqvist, et al. , J. Phys. Chem. B, 2004, 108, 17886–17892 Search PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2026 |