
Synergistic regulation of electrolyte environment and anode interface for constructing ultralong-life Zn–metal batteries with high depth discharge
Cheng-Lin
Miao†
ab,
Lu
Feng†
a,
Xiao-Xue
Wang
a,
De-Hui
Guan
*a,
Xin-Yuan
Yuan
a and
Ji-Jing
Xu
 *ab
*ab
aState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, College of Chemistry, Jilin University, Changchun 130012, P. R. China
bInternational Center of Future Science, Jilin University, Changchun 130012, P. R. China
First published on 17th November 2025
Abstract
Organic additives are effective in solving the intractable problems of aqueous zinc–metal batteries (AZMBs). However, the structural design principles and structure–activity relationship of the additives are still elusive. Herein, a multifunctional bioorganic molecule additive, asparaginate (Asn), was introduced into the electrolyte to regulate the electrolyte environment and electrode/electrolyte interface synergistically. The hydrogen evolution side reactions are effectively restrained by the well-controlled electrolyte pH with acid–base functional groups carried by Asn and the optimized solvation structures of Zn2+ with multiple zincophilic sites in Asn, as well as removing the excess active H2O from the inner Helmholtz plane to reconstruct the electric double layer with the preferential adsorption of Asn on anode, while the formation of Zn dendrites is also effectively inhibited with the Asn on anode. Zn||Zn symmetric batteries delivered an ultra-long cycle life of 5700 h and sustained reversible Zn plating/stripping for 1000 h even with an extremely high depth of discharge of 85%. Furthermore, Zn||MnO2 batteries and Zn||AC capacitors with designed electrolytes exhibited high capacity and cycling stability, providing a new design concept for AZMBs with excellent performance and promising practical application potential.
Broader contextAs the world's energy structure is gradually transforming from fossil fuels into new energy, rechargeable Zn–metal batteries (ZMBs) are considered promising energy storage systems due to their high theoretical capacity and intrinsic safety. However, highly active water molecules in common aqueous electrolytes are easy to cause side reactions. Introducing additives into the electrolytes of ZMBs has become a strategy that is widely adopted. Therefore, exploring the structure–activity relationship among the molecular structure, electrolyte environment and electrode/electrolyte interface from a more microscopic perspective is crucial for the selection and structural design of additives. In this report, a synergistic regulation of the electrolyte environment and electrode/electrolyte interface to achieve favorable ZMBs is proposed using the multifunctional bioorganic molecule additive of asparaginate. The symmetric batteries with the designed electrolytes exhibit ultra-long-term reversible Zn plating/stripping at an extremely high depth of discharge. The assembled Zn metal batteries and Zn-ion capacitors achieve favorable cycling stability and capacity retention. |
Introduction
Lithium-ion batteries (LIBs) have brought great convenience to industrial production and daily life over the past few decades.1–3 However, serious concerns about the high manufacturing cost, scarce lithium resources, and security risks of LIBs make people search for a suitable replacement for next-generation large-scale electrochemical energy storage.4–6 Aqueous zinc–metal batteries (AZMBs), utilizing water as a safe electrolyte solvent and zinc (Zn) metal as the anode, are promising among many candidates.7 Zn metal has several unique advantages as the anode of a battery, such as a low redox potential (−0.76 V versus standard hydrogen electrode), high theoretical capacity (a volumetric capacity of 5855 mA h cm−3, surpassing 2061 mA h cm−3 of Li, and a mass capacity of 820 mA h g−1), environmental friendliness, and intrinsic safety.8,9Nevertheless, the commercialization of AZMBs has been severely hampered by the thermodynamic instability of Zn metal anodes in traditional aqueous electrolytes, leading to serious side reactions. Excess dipolar H2O molecules in the inner Helmholtz plane (IHP) of the electric double layer (EDL) construct an unstable anode/electrolyte interface, triggering unevenly distributed interfacial electric fields, Zn anode corrosion, and dendrite formation.10–15 Along with the issues with the anode/electrolyte interface, issues with the electrolyte cannot be ignored. The active H2O molecules desolvated from the [Zn(H2O)6]2+ solvation structure during the Zn deposition process are capable of triggering the hydrogen evolution reaction (HER) and further increasing the pH value of the electrolyte.16–19 Undesired alkaline Zn4SO4(OH)6·xH2O (ZSH) is generated on the anode along with the Zn ions and redundant OH−, resulting in the accumulation of by-products,20–23 which not only leads to non-ideal operation under high current and capacity but also causes inferior cycling stability and battery life due to the continuous consumption of the Zn anode and electrolyte.24
Recently, introducing organic additives into the traditional aqueous electrolytes of AZMBs has emerged as a promising strategy due to its simplicity and practicability. To date, a variety of organic additives have been reported, including organic solvents,25,26 ionic liquids,27,28 polymers,29 and biomolecules.30,31 These additives are capable of improving the battery performance through various mechanisms, such as modulating the solvation structure,32,33 providing electrostatic shielding,34 regulating EDL,35 forming a solid electrolyte interphase (SEI) layer,36,37 and reconstructing a hydrogen-bond network.38 However, most of the previous studies do not focus on solving the problems in AZMBs and do not explore the relationship between molecular structure and the electrolyte environment/anode interface from a more microscopic perspective, which is very important for the selection and structural design of additives.
Amino acids are organic compounds containing elements such as C, H, N, and O, and exhibit characteristics of both cyclic (indole and imidazole rings) and straight-chain structures. They are commonly found in nature or can be easily obtained from commercial sources. Herein, a multifunctional bioorganic molecule, zwitterionic amino acid asparaginate (Asn) with multiple zincophilic sites, was used to explore the relationship between the molecular structure and its function in AZMBs. The acid–base functional groups contained in Asn dynamically regulate the pH of the electrolyte, avoiding the generation of ZSH by-products and inhibiting the HER. Additionally, experiments and theoretical calculations demonstrated that the interaction between H2O and Zn ions was effectively weakened, and the amount of H2O in the initial solvation structures of Zn ions was reduced through multiple zincophilic sites in Asn, improving the desolvation kinetics. Furthermore, because of its preferential adsorption, Asn adsorbed onto the Zn anode via functional groups. Similar to the principle of wind prevention and sand fixation by planting shelterbelts, Asn adsorbed on the anode surface can prevent further erosion by reconstructing the EDL. In addition, through a competitive adsorption mechanism and electrostatic shielding effect, Asn induces uniform Zn deposition to inhibit the formation of Zn dendrites (Fig. 1). Benefiting from the synergistic effect caused in the bulk electrolyte and electrode/electrolyte interface, Zn||Zn symmetric batteries within Asn additive achieve highly stable and reversible Zn plating/stripping of 5700 h and an ultra-high cycle life of over 1000 h even with an extremely high depth of discharge (DOD) of 85%. Moreover, the modified electrolyte enables Zn||MnO2 batteries and Zn||AC capacitors to deliver superior cycling stability and rate performance. The design principle applied in this study opens up a new way to realize high-performance AZMBs and promote their commercial applications. Overall, this study outlines transferable selection principles, including zwitterionic backbone, balanced coordination, and strong hydrogen bonding, which support the wide applicability of a class of biomolecule-based additives and guide practical ZMB systems.
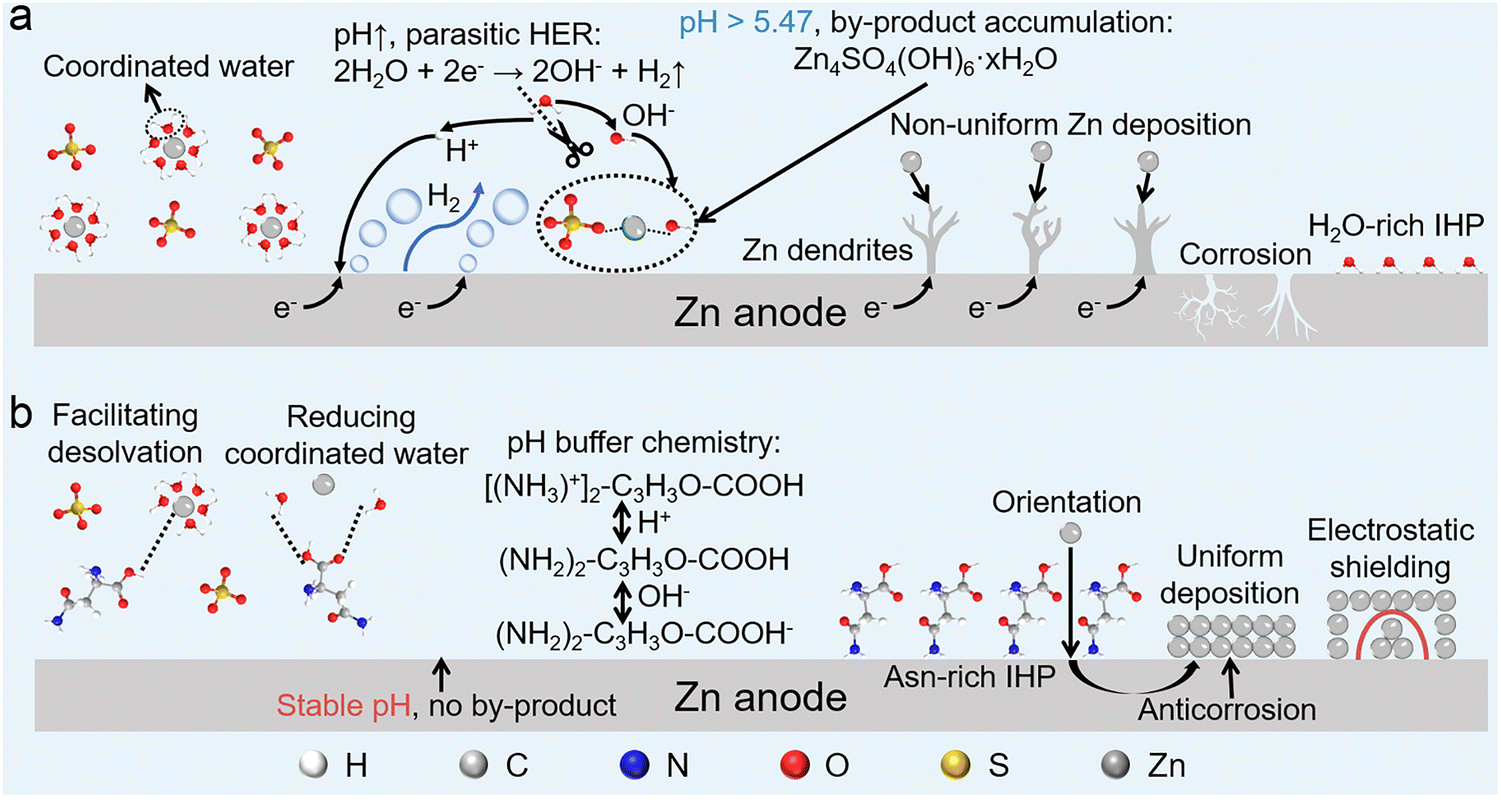 | ||
| Fig. 1 Schematic of the (a) challenges in BE and (b) working mechanism of the Asn additive. | ||
Results and discussion
As a common amino acid in living organisms, asparaginate (Asn) plays an important role in sustaining life and is widely used in the food and pharmaceutical industries. It not only exhibits an excellent binding affinity for zinc (Zn), an essential element in living organisms, but also possesses other advantages, such as being non-toxic, environmentally friendly, and cost-effective. The unique molecular structure of Asn contains multiple oxygen-containing and nitrogen-containing functional groups, such as carboxyl, amino, and amide groups, which can provide lone-pair electrons for the solvated hydrated Zn ions in the electrolyte to form coordinate bonds. Different Asn contents were dissolved in a typical 2 M ZnSO4 baseline electrolyte (denoted as BE) to investigate the optimal concentration. According to the ionic conductivities of different electrolytes calculated from electrochemical impedance spectroscopy (EIS), the electrolyte with 0.2 M Asn obtains the highest ionic conductivity of 39.55 mS cm−1, which indicates that it possesses superior ion transmission capability (Fig. S1). In addition, the electrolyte contains 0.2 M Asn and achieves a maximal Zn2+ transference number of 0.62 among them (Fig. S2), which is conducive to reducing the concentration polarization on the electrode surface and inhibiting the formation of dendrites, thereby improving the overall performance of the battery. Therefore, an electrolyte with 0.2 M Asn + 2 M ZnSO4 (denoted as Asn) was identified as the target electrolyte and further studied in subsequent experiments. The proposed properties of Asn are illustrated in Fig. 2(a), in which the Asn molecule consists of carboxyl, amino, and amide functional groups, which are known for their zincophilic nature and ability to regulate pH. Specifically, the alkaline amino group can neutralize excess H+ under low pH conditions, while the carboxyl group can release H+ to prevent the electrolyte from becoming over-alkaline. To evaluate the stability of the different electrolytes, electrochemical windows were measured using linear scanning voltammetry (LSV). As shown in Fig. 2(b), the HER onset potential of the Asn electrolyte was negatively shifted compared with that of BE, indicating the effective role that Asn played in suppressing the HER. Moreover, the increased overpotential of the oxygen evolution reaction (OER) in the Asn electrolyte confirmed the positive effect of the Asn additive in inhibiting the side reactions. The pH buffer capacities of the two electrolytes were tested by adding 0.5 M NaOH solution dropwise. It can be observed from Fig. 2(c) that the pH of the Asn electrolyte increases more slowly than that of BE.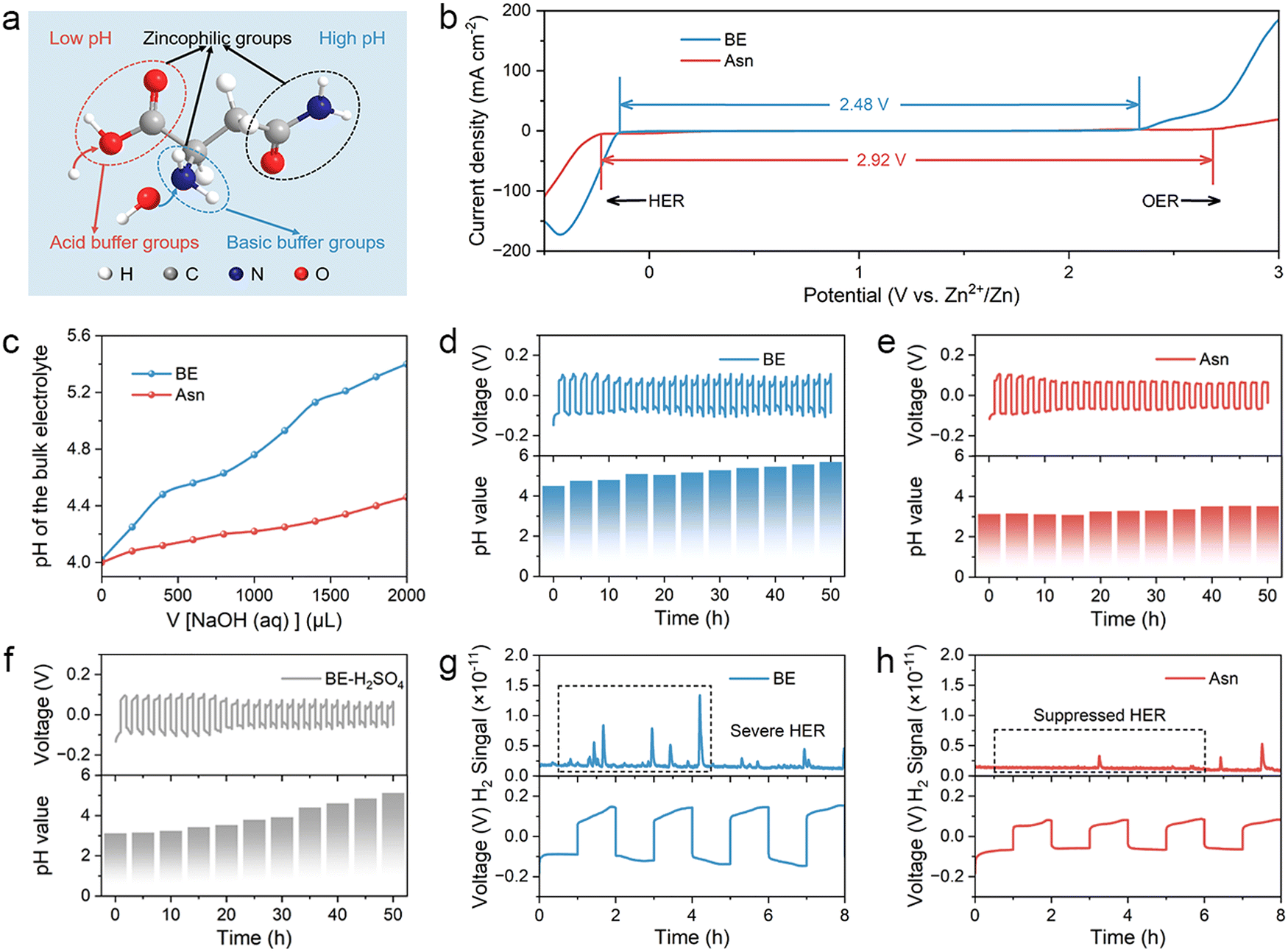 | ||
| Fig. 2 Positive effect of the Asn additive in pH buffering. (a) Schematic of the Asn molecular structure. (b) Electrochemical windows of different electrolytes. (c) Titration of 50 mL BE and Asn electrolyte with 0.5 mol L−1 NaOH as the titrant. In situ pH monitoring of (d) BE, (e) Asn electrolyte, and (f) BE + H2SO4 within Zn||Zn symmetric batteries upon cycling at 5 mA cm−2. In situ DEMS profiles and corresponding galvanostatic time curves of (g) BE and (h) Asn electrolytes. | ||
To further explore the positive pH buffering effect of Asn in the bulk electrolyte, in situ pH monitoring was carried out during the Zn plating/stripping process of Zn||Zn symmetric batteries upon cycling at 5 mA cm−2. The pH value of BE gradually increased from 4.3 to 5.8 after 50 h, indicating serious HER (Fig. 2(d)). However, the Asn additive can alleviate the increase in pH and finally stabilize the pH value at 3.5 (Fig. 2(e)). Since this pH value is lower than the isoelectric point of Asn, it indicates that Asn was protonated in the electrolyte and existed in the form of Asn cations enriched within the compact double layer, thereby reducing interfacial proton activity, partially blocking H+-formation sites, and increasing the desolvation/Volmer-step barrier. Then, we make the initial pH value of BE the same as that of the Asn electrolyte by adding a dilute sulfuric acid solution. After cycling under the same conditions for 50 h, the pH value of BE + H2SO4 was still higher than that of the Asn electrolyte (Fig. 2(f)). In addition, according to the SEM images and optical photographs of Zn metal after immersion in different electrolytes for one month, it is demonstrated that simply lowering the initial pH value of the electrolyte can not inhibit the HER and further lead to anode corrosion (Fig. S3). This phenomenon suggests that the amphoteric Asn additive acts not only as a favorable pH buffer but also as an interfacial regulator in aqueous electrolytes. In the H2O-splitting pathway, the carboxylate groups of Asn scavenge HER-generated OH−, restraining bulk and interfacial alkalization and suppressing the precipitation of Zn4SO4(OH)6·xH2O (ZSH). Meanwhile, the amino groups can receive excess H+ at a low pH. In situ differential electrochemical mass spectrometry (DEMS) was adopted to dynamically monitor the HER process in symmetric batteries.39 For BE, obvious H2 signals were observed during battery operation, indicating that the corrosion reaction is self-generated and intensifies during the charge/discharge process (Fig. 2(g)). Remarkably, the symmetric batteries with Asn electrolyte showed obviously suppressed H2 signals throughout the process, indicating the positive role of Asn in suppressing the HER (Fig. 2(h)).
The presence of Asn in the electrolyte affects the solvation structures of Zn ions through intermolecular interactions. To deeply explore the solvation interaction behavior, a series of theoretical calculations and spectroscopic characterizations were performed. We first investigated the binding energies between Zn2+–H2O and Zn2+–Asn in the electrolyte by density functional theory (DFT) calculations.40 The complexation ability of H2O with Zn2+ (−4.6 eV) is smaller than that of Asn (−6.2 eV), revealing a stronger combination between Zn2+ and Asn additives (Fig. S4). The high binding energy endows the Asn additives with the ability to optimize the solvation structures of Zn ions to achieve regular Zn plating/stripping behavior. Due to the existence of polar oxygen-containing functional groups, Asn displays a stronger binding ability than H2O. Fourier transform infrared spectroscopy (FTIR), Raman spectra, and nuclear magnetic resonance (NMR) were conducted to determine the impact of the Asn additive on the solvation structure.41–43 As shown in Fig. S5, the FTIR peak of the symmetric stretching vibration at 1400 cm−1 of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O can be utilized as a probe of the bonding configuration of Zn2+ and Asn. The shift in the bending vibration (Vb–OH) and vibrational stretching (Vs–OH) of O–H in H2O reveals a weakening of the O–H bond in H2O due to the interaction between H2O and Asn. The S–O symmetric stretching band of SO42− in the range of 960–1000 cm−1 in the Raman spectra can be divided into contact ion pairs (CIP, [Zn2+(H2O)5–OSO32−]) and solvent-separated ion pairs (SSIP, [Zn2+ (H2O)n(Asn)m]–SO42−). A decrease in the percentage of CIP occurs with the addition of Asn, indicating that the substitution of inner-sphere complexes in the solvation structure is regulated (Fig. S6a). The O–H stretching vibration within the region from 3000 to 3500 cm−1 can be divided into strong and medium H-bonds, respectively. The proportion of strong H-bonds increases with the addition of Asn, while the medium H-bonds show a downward trend (Fig. S6b). In addition, the 1H signal of BE shifts upfield, reaching 4.70 ppm in the Asn electrolyte, indicating an increased electron cloud density due to the release of confined water molecules for further interaction with Asn (Fig. S7). These findings confirm the superior ability of Asn in optimizing the solvation structures of Zn ions, enabling stable Zn2+ transport kinetics and weakening the initial H-bond network to reduce the activity of water, further inhibiting side reactions.
O can be utilized as a probe of the bonding configuration of Zn2+ and Asn. The shift in the bending vibration (Vb–OH) and vibrational stretching (Vs–OH) of O–H in H2O reveals a weakening of the O–H bond in H2O due to the interaction between H2O and Asn. The S–O symmetric stretching band of SO42− in the range of 960–1000 cm−1 in the Raman spectra can be divided into contact ion pairs (CIP, [Zn2+(H2O)5–OSO32−]) and solvent-separated ion pairs (SSIP, [Zn2+ (H2O)n(Asn)m]–SO42−). A decrease in the percentage of CIP occurs with the addition of Asn, indicating that the substitution of inner-sphere complexes in the solvation structure is regulated (Fig. S6a). The O–H stretching vibration within the region from 3000 to 3500 cm−1 can be divided into strong and medium H-bonds, respectively. The proportion of strong H-bonds increases with the addition of Asn, while the medium H-bonds show a downward trend (Fig. S6b). In addition, the 1H signal of BE shifts upfield, reaching 4.70 ppm in the Asn electrolyte, indicating an increased electron cloud density due to the release of confined water molecules for further interaction with Asn (Fig. S7). These findings confirm the superior ability of Asn in optimizing the solvation structures of Zn ions, enabling stable Zn2+ transport kinetics and weakening the initial H-bond network to reduce the activity of water, further inhibiting side reactions.
Molecular dynamics (MD) simulations were further performed to analyze the bulk electrolyte and elucidate the inside mechanism of the Asn additive in optimizing the solvation structures of Zn ions.44–46 As shown in Fig. 3(a), six water molecules coordinate with one Zn ion in the BE to form the initial solvation structure of Zn ions. In contrast, one Asn can access the initial solvation structure of the Zn ions and replace one original water molecule in the Asn electrolyte, demonstrating the effective regulation of the solvation structures (Fig. 3(b)). Radial distribution functions (RDFs) and average coordination number (CN) distribution functions were used to investigate the solvation structures of Zn ions in different electrolytes. For the BE, the sharp peak of the Zn–O (H2O) pair is about 2 Å, indicating the occupancy of water molecules around the Zn ion (Fig. 3(c)). However, in the Asn electrolyte, three new peaks assigned to Zn–O2 (Asn), Zn–O3 (Asn), and Zn–N2 (Asn) appeared at the same position as Zn–O (H2O), nearly 2 Å, indicating the successful substitution of Asn for water within the initial solvation structure of the Zn ion (Fig. 3(d)).
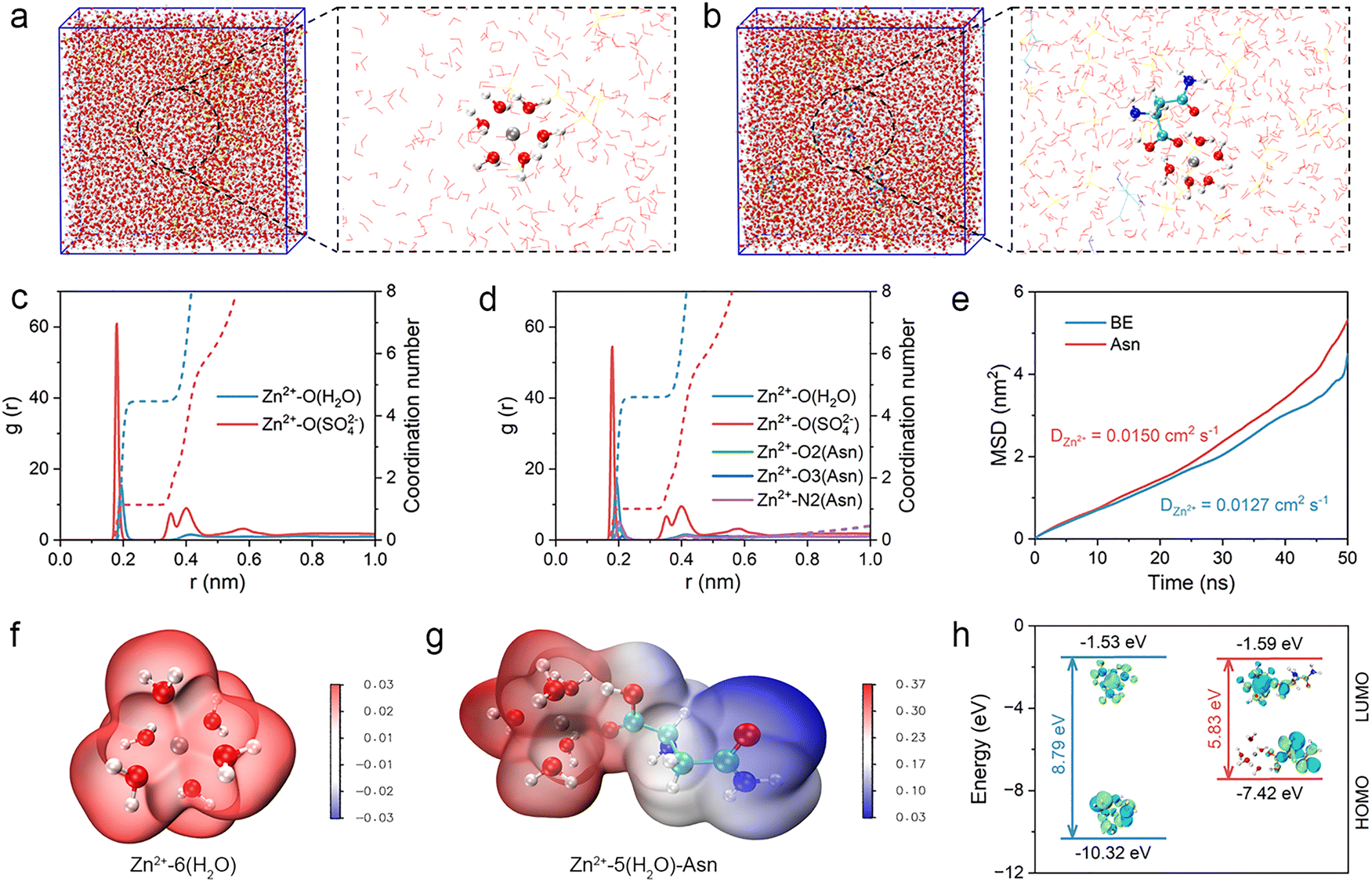 | ||
| Fig. 3 Optimization of the solvation structure. 3D snapshots calculated from MD simulations and partially enlarged snapshots of the Zn2+ solvation structure (white: H; green: C; blue: N; red: O; and grey: Zn) for (a) BE and (b) Asn electrolytes. RDFs and the CN in (c) BE and (d) Asn electrolyte. (e) MSD of Zn ions in different electrolytes. ESP mapping of the solvated structures of (f) Zn2+–6(H2O) and (g) Zn2+–5(H2O)–Asn. (h) HOMO and LUMO energy levels of Zn2+–6(H2O) and Zn2+–5(H2O)–Asn solvation structures. | ||
We subsequently investigated the diffusion coefficient of Zn ions in different electrolytes using the results calculated from the mean square displacement (MSD). As shown in Fig. 3(e), compared with BE, Zn ion exhibits a stronger diffusion ability in the Asn electrolyte due to a more reasonable solvation structure, indicating the effectiveness of the Asn additive in reshaping the solvation structures of Zn ions. The electrostatic potential (ESP) mapping distribution was carried out to investigate the intrinsic driving force for the formation of different solvation structures (Fig. 3(f) and (g)). Asn is found to reduce the ESP distribution of [Zn(H2O)6]2+. This trend is attributed to the abundant coordination sites provided by Asn (–COOH and –NH2), which disrupt the hydrogen bond networks of H2O, promote the desolvation of [Zn(H2O)6]2+, and facile Zn2+ transport. The atomic charge was further used to reveal the electron transfer among the solvation structures of the Zn ions. As shown in Fig. S8, the atomic charge of Zn in [Zn(H2O)6]2+ is +0.505, which is owing to electron transfer from water molecules to the Zn ion. For the [(Zn(H2O)5Asn)]2+ structure, Asn entered the solvation structures of Zn ions through competitive coordination, reducing the number of water molecules with a high shielding effect in the first solvation shell of Zn ions, which causes a higher atomic charge of Zn. The introduction of Asn leads to an increase in the atomic charge of Zn, which is a crucial microscopic characteristic for the successful regulation of solvation structures. A higher atomic charge of Zn implies a stronger interaction between Zn and the O atoms in the remaining coordinated water molecules, which weakens the polarization of the O–H bonds, making them less likely to break down and form H2. Subsequently, DFT calculations were conducted to determine the energy levels of the lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO) for Asn. The results reveal a narrower LUMO–HOMO energy gap for Asn compared to H2O, indicating enhanced chemical reactivity (Fig. 3(h)). Specifically, the LUMO energy level of Asn (−1.59 eV) is lower than that of H2O (−1.53 eV), suggesting a greater propensity for electron acceptance by Asn. This characteristic facilitates electrochemical decomposition and promotes SEI formation.
The Asn additive in the solvated coordination is preferentially adsorbed on the Zn anode, enriching the EDL by strong electrostatic adsorption between the positively charged sites in Asn and the negatively charged anode surface under the electric field. Thus, we systematically explored the impact of Asn on the EDL. First, a contact angle experiment was performed to examine the wettability of the two electrolytes on the Zn anode. As shown in Fig. 4(a), the angle decreases from 84.7° to 74.9° after introducing Asn. Enhanced wettability is verified according to the decreased droplet surface tension on the macroscopic scale, which originates from the reduction in surface energy due to the adsorption of Asn. Additionally, zeta potential measurements of the Zn powders were employed to reveal the interaction between Asn and the surface of the Zn anode. The test results show that the potential values positively shift from −8.58 to 39.28 mV after the addition of Asn, which suggests the spontaneous absorption of Asn with strong zincophilic properties on the Zn surface, further contributing to the Zn surface charge regulation via the strong electrostatic interaction (Fig. 4(b)). The positive shift in the zeta potential also reveals that Asn is ionized in the electrolyte to generate the Asn cation. Simultaneously, the positive shift in the zeta potential also suggests that the generated Asn cations are prone to regulate the surface charge field via the rearrangement of molecules and ions on the surface of the Zn anode, further excluding the active water molecules from the IHP.
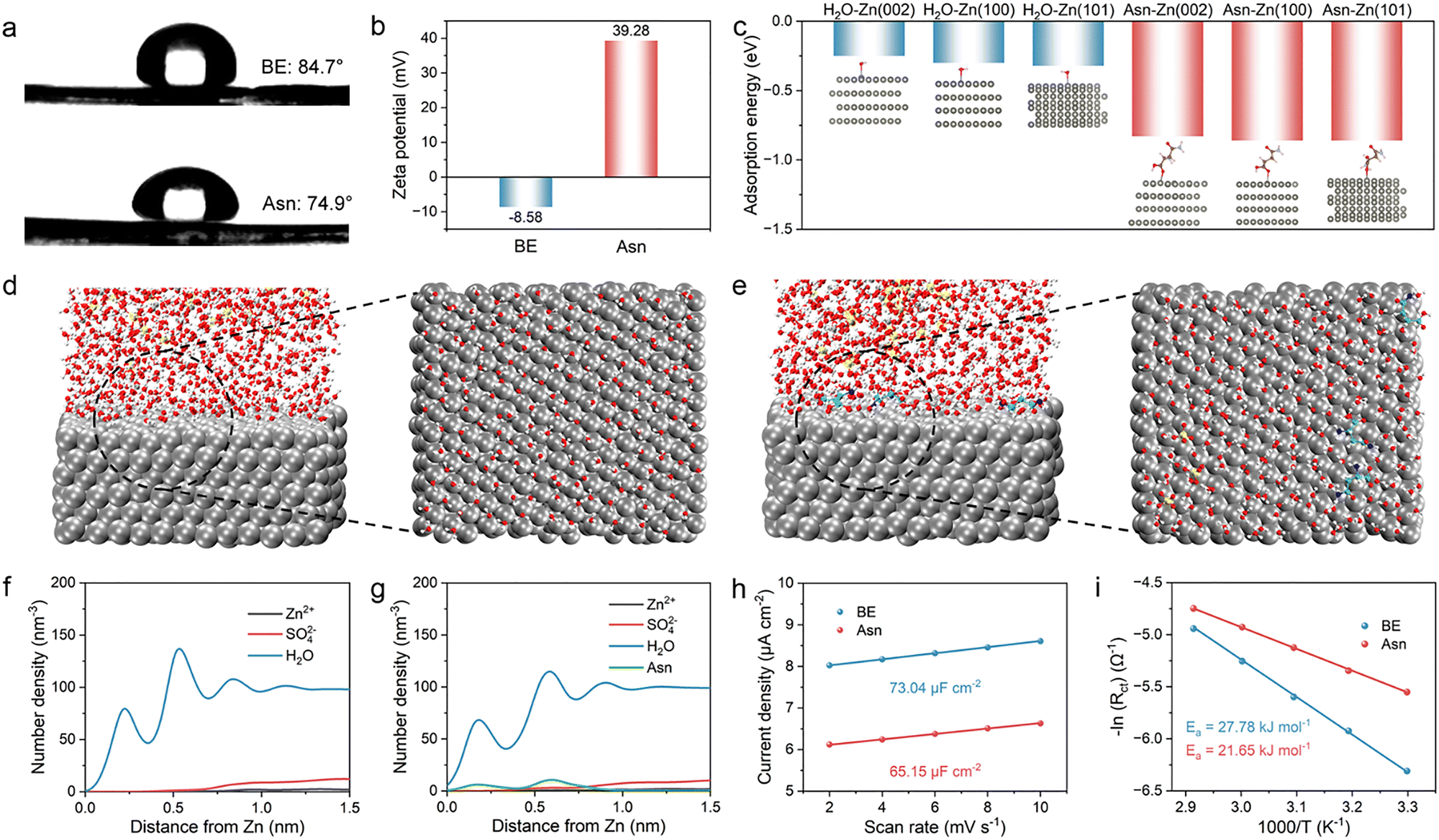 | ||
| Fig. 4 Illustration of the reconstruction of EDL based on the theoretical calculations and characterizations. (a) Wettability tests of different electrolytes on the surface of the Zn anode. (b) Zeta potential values of Zn powder in different electrolytes. (c) Adsorption energies of H2O and Asn on different crystal planes of Zn. 3D snapshots calculated from MD simulations and partially enlarged snapshots of the anode/electrolyte interfaces for (d) BE and (e) Asn electrolyte. (f) Density profiles as a function of distance from the Zn anode for the Zn2+, SO42−, and H2O in BE. (g) Density profiles as a function of distance from the Zn anode for Zn2+, SO42−, H2O, and Asn in the Asn electrolyte. (h) EDL measurements for the Zn anode with different electrolytes. (i) Activation energies in different electrolytes calculated using the Arrhenius equation. | ||
The intrinsic mechanism of Asn-induced Zn deposition was studied in detail using density functional theory (DFT) calculations. The adsorption energies of Asn and H2O on different crystal planes of Zn were calculated. The results are displayed in Fig. 4(c) and Fig. S9. The adsorption energy of Asn on all Zn crystal planes is much lower than that of H2O, indicating that Asn is preferentially adsorbed on the surface of the Zn anode in the Asn electrolyte. This result is consistent with the previous experimental phenomenon in which Asn can replace the H2O dipoles absorbed at the electrode/electrolyte interface to induce a H2O-poor EDL structure. In addition, the adsorption energy between Asn and the (002) plane is −0.83 eV, which is higher than that of the (100) plane (−0.86 eV) and the (101) plane (−0.86 eV). Therefore, Asn is prone to be selectively adsorbed on the (100) and (101) planes, leaving the exposed (002) plane. The Zn ions deposited along the (100) and (101) planes are hindered by the adsorbed Asn on them. In this situation, Zn ions tend to be deposited on the exposed (002) plane, and the deposited Zn grows along the (002) orientation. Due to the lowest surface energy of the Zn (002) plane, its electrochemical activity for HER and corrosion reactions is relatively low. Therefore, a Zn anode with more exposed (002) planes eliminates the threat of dendrites and suppresses side reactions. The presence of the N+ peak in the N 1s spectra also demonstrates the stable electrostatic adsorption of Asn on the electrode surface during the charge/discharge process (Fig. S10).
To understand the differences between BE and Asn electrolytes in manipulating the EDL structure, MD simulations were employed to elucidate the chemical structure of the EDL in different electrolytes. The snapshots of EDL in the simulated electrolytes and the representative top-view snapshots of the adsorbed species in the Helmholtz layer are displayed in Fig. 4(d) and (e). Clear residual H2O in BE is observed compared with the Asn electrolyte to form a H2O-enriched interface, which can draw more Zn ions to gather for charge compensation. In the Asn electrolyte, more Asn aggregates on the surface of the Zn anode. Thus, some highly active water molecules can be removed from the original EDL because Asn has a higher affinity with the Zn ion and the Zn anode surface. The EDL structures in the two electrolytes are shown in Fig. S11, which demonstrates the enhanced chemical environment due to the rearrangement of interfacial constituents. Furthermore, the normalized density profiles were recorded to show the distribution of interfacial ions along the distance from the Zn anode surface within the EDL. The closer distance and higher density of H2O are accompanied by Zn2+ in greater proximity to the Zn anode, contributing to a more condensed Helmholtz layer (Fig. 4(f)), which is in good accordance with the significantly enhanced EDL in the Asn electrolyte. Specifically, the strong polarity of Asn features a strong coordination and adsorption capability that can generate a more condensed interface micro-environment, which is anticipated to tackle multiple issues for Zn metal anodes (Fig. 4(g)). Electric double layer capacitance (EDLC) measurements of Zn anodes were carried out in different electrolytes (Fig. 4(h) and Fig. S12). A lower EDLC in the Asn electrolyte (65.15 µF cm−2) than in BE (73.04 µF cm−2) indicates that the adsorption of Asn on the anode is beneficial to the diffusion of Zn ions without sacrificing the reaction kinetics of the Zn anodes. The differential capacitance curves show that the surface capacitance of the Zn anode was significantly reduced in the presence of Asn (Fig. S13). This decrease indicates the thickening of the EDL by the adsorption of Asn on the Zn surface. The activation energy (Ea) for the desolvation kinetics of Zn ions based on the Arrhenius equation in different electrolytes was compared, as depicted in Fig. 4(i), where the Rct was obtained from the (Fig. S14). The Ea values are 27.78 and 21.65 kJ mol−1 in the BE and Asn electrolytes, respectively, indicating a lower desolvation energy barrier in the Asn. This is due to the modulated solvation structure, which is beneficial for interfacial charge transfer and ion transport.
To explore the impact of the Asn additive on Zn plating/stripping behavior, Zn||Cu asymmetric batteries with BE and Asn electrolytes were assembled. The initial nucleation overpotential (NOP) of Zn||Cu batteries in the two electrolytes was obtained by cyclic voltammetry (CV) tests. As shown in Fig. 5(a), compared with BE, the NOP of Asn is reduced, indicating faster nucleation dynamics and a lower energy barrier to be overcome for Zn ions during the nucleation process. This can be attributed to the multiple zincophilic functional groups on Asn, which are prone to attract Zn ions through the strong interaction between them to induce nucleation. This perspective is further confirmed by the galvanostatic test of Zn||Cu asymmetric batteries. The voltage–capacity profiles of the first discharge display a sharp voltage drop and then gradually tend to a steady state, representing the two stages of instantaneous nucleation and piecemeal growth of the Zn nucleation process. It can be observed that after the addition of Asn, the NOP relative to BE decreases from 72.6 mV to 48.1 mV, contributing to an easier nucleation process (Fig. 5(b)). To investigate the continuous effectiveness of Asn in the Zn plating/stripping process, Zn||Cu asymmetric batteries were cycled at 1 mA cm−2 and 1 mA h cm−2. The battery with BE experienced a sudden death only after 163 cycles with a voltage hysteresis of 127.7 mV due to the short circuit caused by the grievous dendrites (Fig. 5(c)). On the contrary, the battery with the optimized electrolyte exhibits a steady operation of over 500 cycles, accompanied by a lower voltage hysteresis of 74.8 mV (Fig. 5(d)). Coulombic efficiency (CE) is a key parameter for evaluating the cycling stability and reversibility of Zn plating/stripping in batteries. As shown in Fig. S15, an extremely disordered trend for BE is displayed due to dendrite growth and the formation of by-products. On the contrary, Zn||Cu asymmetric batteries using Asn electrolytes exhibit an average CE of 99.17% for 500 cycles.
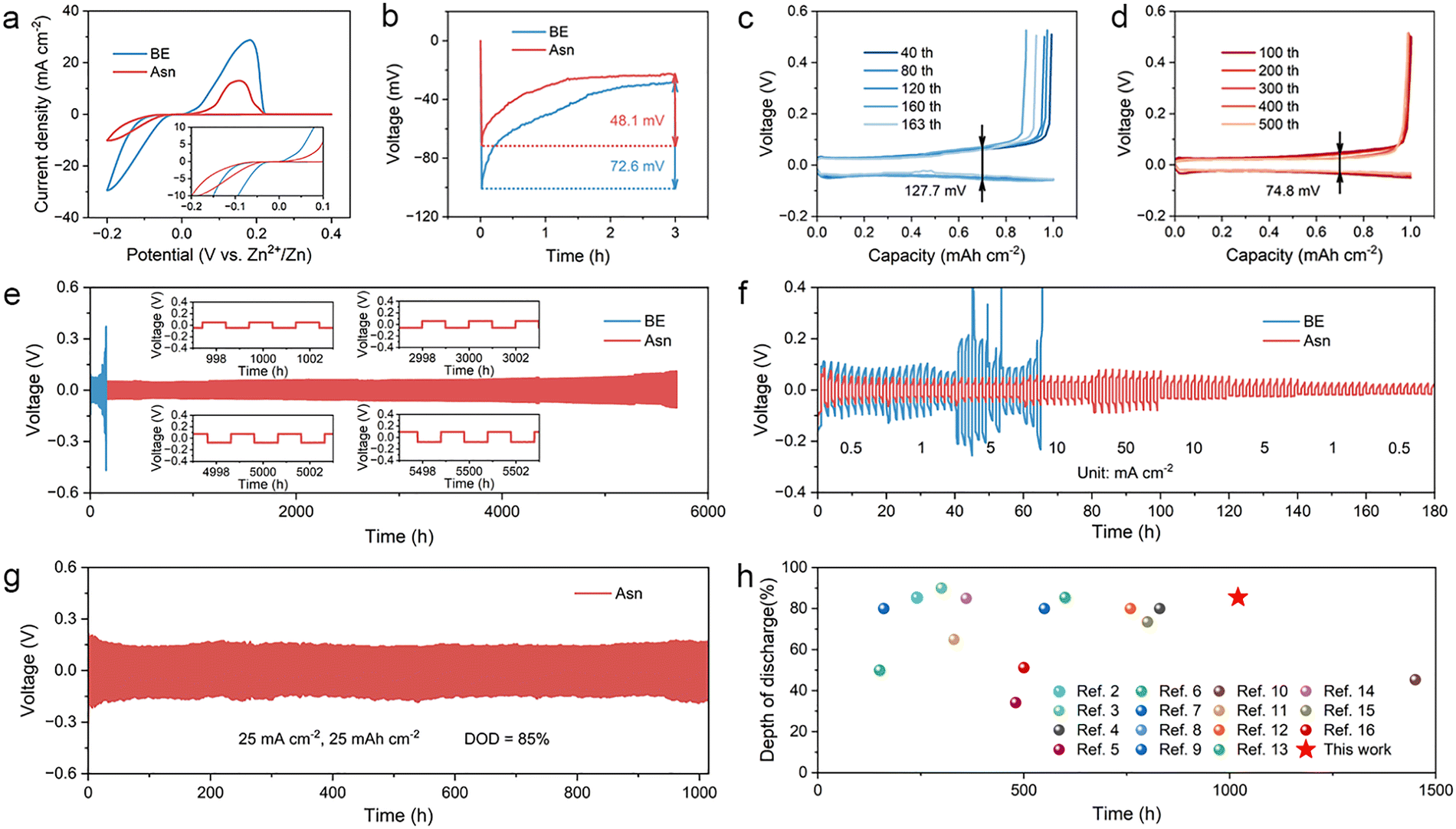 | ||
| Fig. 5 Electrochemical performance evaluation of Zn plating/stripping. (a) CV curves of Zn||Cu asymmetric batteries in different electrolytes. (b) Initial nucleation overpotential in different electrolytes. Voltage profiles at various cycles in (c) BE and (d) Asn electrolytes at 1 mA cm−2 and 1 mA h cm−2. (e) Galvanostatic cycling performance of Zn||Zn symmetric batteries in different electrolytes at 1 mA cm−2 and 1 mA h cm−2. (f) Rate performance of Zn||Zn symmetric batteries in different electrolytes. (g) Galvanostatic cycling performance of Zn||Zn symmetric batteries in different electrolytes at 25 mA cm−2 and 25 mA h cm−2. (h) Comparison of the electrochemical performance of Zn||Zn symmetric batteries between this study and other previously reported studies with DOD and cycling lifespan. | ||
Zn||Zn symmetric batteries were assembled to evaluate the cycling stability of Zn anodes with different electrolytes through long-term galvanostatic charge/discharge (GCD) measurements. Under routine test conditions of 1 mA cm−2 and 1 mA h cm−2, the Asn guaranteed stable Zn plating/stripping of 5700 hours (near 8 months) without an obvious increase in the voltage polarization throughout the whole operating process, which is almost 37 times longer than that of the battery with BE (Fig. 5(e)). Subsequently, the improvement by Asn was investigated in the rate performance of Zn||Zn symmetric batteries with different electrolytes at various current densities ranging from 0.5 to 50 mA cm−2 for 10 cycles under each condition. As illustrated in Fig. 5(f), the voltage profile of the Asn-based battery displays a steady and regular variation with varied current densities and a stable operation even at 50 mA cm−2. Conversely, the battery using BE encountered dramatic voltage fluctuation as the current density increased from 1 mA cm−2 to 5 mA cm−2. In addition, to further explore the limit of Zn plating/stripping and the possibility of practical application, Zn||Zn symmetric batteries with Asn electrolyte were tested under an extreme condition of high depth of discharge (DOD) to evaluate the utilization efficiency of the Zn anodes with a thickness of 50 µm (Fig. 5(g)). Testing at such a high DOD directly interrogates Zn utilization and exposes the failure modes, dendrite-induced shorting, shape evolution/voiding, passivation by basic salts, and parasitic hydrogen evolution, which typically accelerate when most of the active Zn is repeatedly stripped and replated. Surprisingly, even running at 25 mA cm−2 and 25 mA h cm−2 with a rigorous DOD of 85%, the batteries still maintain stable cycling and an ultralong lifespan of 1000 h, which is superior to most of the previously reported work (Fig. 5(h)). The results confirm that Asn in the electrolyte not only enhances cycling stability but also ensures durability at high rates and areal capacities.
According to the above studies, the surface adsorption of Asn displays multiple pivotal roles. To further investigate the protective effect of the Asn additive, a series of anti-corrosion performance evaluations and related characteristic tests were conducted. The corrosion resistance towards different electrolytes is depicted in Fig. 6(a). Obviously, compared to the electrolyte without Asn, the corrosion potential of Zn with the Asn-modified electrolyte increased from −27 to 22 mV, reflecting a reduced tendency towards corrosion. Furthermore, the Asn additive reduced the corrosion current density from 0.587 mA cm−2 to 0.574 mA cm−2, indicating a retardant corrosion rate. Chronoamperometry (CA) is further performed to investigate the Zn deposition behavior and nucleation mechanism by applying an overpotential of −150 mV (Fig. 6(b)). For BE, the current density continuously decreases within 200 s, indicating a rampant 2D diffusion process and inhomogeneous Zn deposition. During the plating process, Zn ions tend to accumulate at the anode surface, and the exposed area decreases, leading to the accumulation of Zn ions on the tips to form dendrites. Interestingly, for the Asn electrolyte, the current density decreases rapidly within 50 s, demonstrating a rapid 2D diffusion process. Then, the current density tends to stabilize due to the electrostatic shielding effect of the Asn additive, which restrains the plane diffusion of Zn ions in the 2D range. This may be attributed to the fact that surface-absorbed Asn suppresses the growth of Zn dendrites and facilitates the formation of a uniform Zn surface. In addition, this result was further verified by finite element method (FEM) simulation, which is adopted to describe the distribution of the electric field and Zn dendrite growth during the electroplating process. In the case of BE, the current density at the tip of the protrusions is higher than that at other locations due to the tip charge accumulation effect (Fig. 6(c) and Fig. S16a). Consequently, a large number of Zn ions quickly aggregate at the protruding tip, while a weak signal of Zn ions can be observed at adjacent flat areas (the bottom of the protrusion). In contrast, the electric field at the tip of the protrusions is significantly suppressed through the adsorption of Asn and displays a more uniform distribution in the non-tip region due to electrostatic shielding (Fig. 6(d) and Fig. S16b). With the thermodynamically stable Zn anode in the Asn-modified electrolyte, the Zn ion transport kinetics of the anode/electrolyte interface were synchronously boosted. This conclusion was derived from the results of the in situ EIS technique. Here, in situ EIS related to the impedance spectra collection of Zn||Zn symmetric batteries after continuous Zn plating/stripping was carried out. When cycling in the BE, the passivation of the Zn anode becomes serious with the generation of Zn4SO4(OH)6·xH2O (ZSH), resulting in a continuous increase in the interface impedance (Fig. 6(e)). Conversely, the interface impedance of the Asn Asn-modified anode/electrolyte interface remained stable after a drastic reduction, indicating greatly enhanced interface transport kinetics (Fig. 6(f)). Therefore, a highly uniform distribution of Zn ions is achieved in the Asn electrolyte, effectively suppressing the growth of Zn dendrites.
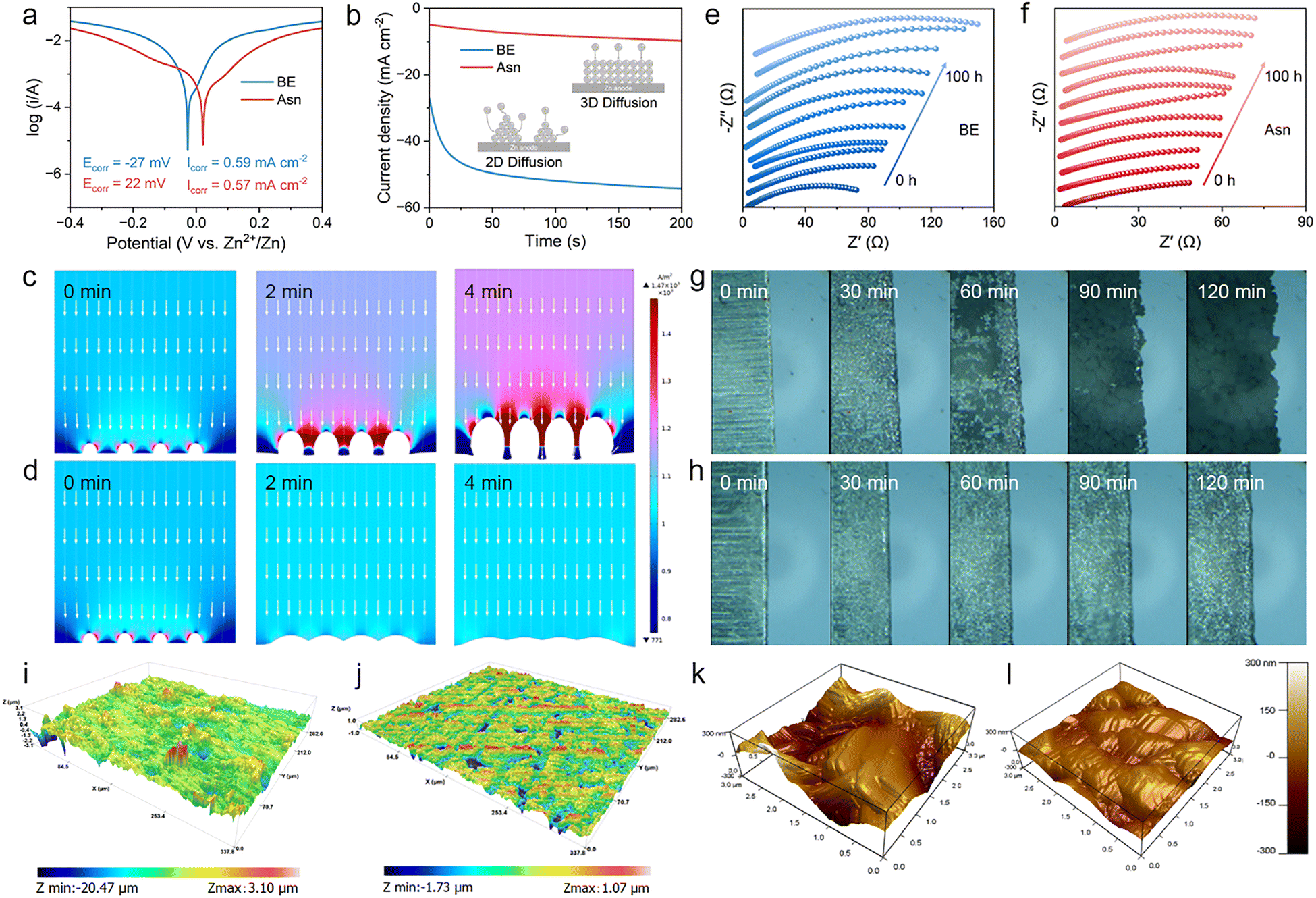 | ||
| Fig. 6 Simulated and experimental research on Zn deposition. (a) Tafel curves of Zn||Zn symmetric batteries with different electrolytes at a scan rate of 1 mV s−1. (b) CA test of Zn deposition in different electrolytes with a polarization voltage of −150 mV. COMSOL simulation of the electric field distribution on Zn anode in (c) BE and (d) Asn electrolyte. In situ EIS of Zn||Zn symmetric batteries using (e) BE and (f) Asn electrolytes during Zn2+ electroplating. In situ optical microscope images of the Zn deposition behavior in (g) BE and (h) Asn electrolyte at a current density of 10 mA cm−2. 3D CLSM images of Zn anodes in (i) BE and (j) Asn electrolytes after 50 cycles. 3D AFM images of Zn anodes in (k) BE and (l) Asn electrolytes after 50 cycles. | ||
Additionally, a systematic study was conducted on the deposition behavior of Zn anodes in different electrolytes to reveal the regulation of Asn on the anode/electrolyte interface. The Zn plating/stripping behavior in Zn||Zn symmetric batteries was monitored using an in situ optical microscope.47 Only after 30 min, plate-like dendrites emerge on the anode surface (Fig. 6(g)) and eventually lead to mossy Zn dendrites and bubbles after 2 h. Regarding Asn (Fig. 6(h)), homogeneous and dendrite-free Zn deposition is identified, demonstrating the positive impact of Asn on Zn plating/stripping. The SEM images of Zn anodes after cycling in different electrolytes also exhibit significant differences in Zn deposition behavior (Fig. S17). Compared with the obviously disordered and loose sheet-like dendrites observed on the anode surface in BE after 50 cycles, under the same experimental conditions, the Zn anode after cycling in Asn electrolyte presents a smooth and dense surface morphology. This confirms that the introduction of Asn can effectively regulate the deposition behavior of Zn and promote uniform deposition on the anode. The corresponding X-ray diffraction (XRD) patterns reveal that the Zn anode after cycling in the Asn electrolyte shows a higher intensity of the Zn (002) plane compared with the pristine Zn anode (Fig. S18a). Conversely, when cycled in BE, the intensity of the Zn (002) plane significantly decreases. This result demonstrates that Asn at the anode/electrolyte interface can inhibit the growth of Zn dendrites by promoting the deposition of Zn ions along the (002) direction. In addition, X-ray photoelectron spectroscopy (XPS) of the retrieved Zn anodes likewise shows no discernible N 1s signal or other nitrogen-containing species beyond the BE baseline (Fig. S18b), excluding additive-derived interphase formation. Together, these morphological, structural, and spectroscopic results demonstrate that Asn modulates interfacial deposition without detectable oxidation or decomposition under our cycling conditions. Confocal laser scanning microscopy (CLSM) was applied to investigate the morphological evolution of the cycled Zn anodes assembled with different electrolytes. As shown in Fig. 6(i) and Fig. S19a, S20a, the 3D CLSM image exhibited an obvious coarse surface with a high surface roughness of 3.10 µm for Zn metal cycled within BE. In sharp contrast, when Zn deposition occurred in the Asn electrolyte, the corresponding value of surface roughness was remarkably reduced to 1.07 µm, reflecting the capability of the Asn-containing electrolyte in suppressing side reactions and dendritic Zn (Fig. 6(j) and Fig. S19b, S20b). The surface potential and geometry of Zn anodes cycled in different electrolytes were further measured by 3D atomic force microscopy (AFM) and Kelvin probe force microscopy (KPFM).48 As shown in Fig. 6(k) and Fig. S21a, an uneven electric field distribution can be detected in the Zn anode cycled in BE because the local charge accumulates at the tip, further leading to uncontrolled Zn plating. Correspondingly, the Zn anode cycled in BE shows fluctuating and rough morphology. In contrast, a relatively flat surface morphology and uniform electric field distribution are observed for the Zn anode in the Asn electrolyte (Fig. 6(l) and Fig. S21b). A comparison of the 2D small-angle X-ray scattering (SAXS) patterns on the deposited Zn clearly demonstrates the facet preference (Fig. S22). Zn deposition in the BE exhibits an almost invisible Debye ring of the Zn (002) plane, which corresponds to the formation of severe Zn dendrites. In contrast, in the electrolyte with Asn, the Debye ring corresponding to the Zn (002) plane presents conspicuous diffraction intensity, indicating the preferred orientation of the Zn deposition. This observation is consistent with the contrasting crystal facet orientations and the seamless alignment conclusion based on the DFT results.
To assess how the Asn additive affects cathode stability, we combined an ultraviolet-visible (UV/Vis) spectrum, high-resolution transmission electron microscopy (HRTEM), and inductively coupled plasma optical emission spectroscopy (ICP-OES) (Fig. S23–S25). After 10 days of cathode immersion, the BE electrolyte exhibited a pronounced absorption band in the 250–400 nm region, whereas the Asn-containing electrolyte showed no obvious signal (Fig. S23). BE absorption is likely attributed to the electron transfer from O to Mn4+, which results in the formation of a soluble Mn(OH)42− species, indicating spontaneous MnO2 dissolution in the absence of Asn. Complementary high-resolution transmission electron microscope (HR-TEM) analysis revealed ZSH-related crystalline byproducts on the cathode cycled in BE (Fig. S24a). In contrast, electrodes cycled in the Asn electrolyte preserved a regular lattice with fast Fourier transform (FFT) patterns, evidencing suppressed byproduct accumulation (Fig. S24b). Consistent results were obtained by XRD and XPS (Fig. S24c and d), which further demonstrated that the cathode cycled in the Asn-containing electrolyte largely retains the structural stability. Quantitative inductively coupled plasma (ICP) emission spectrometer analysis further showed that Mn concentration in BE increased rapidly with cycle number, particularly beyond ∼30 cycles, whereas Mn levels in the Asn electrolyte remained comparatively stable (Fig. S25). Notably, both systems displayed similar Mn increased over the first 1–10 cycles, after which the Asn electrolyte exhibited a sharp deceleration, which is consistent with the progressive formation of a cathode–electrolyte interphase (CEI) that mitigates MnO2 dissolution. Taken together, these results demonstrate that Asn, in addition to regulating the anode/electrolyte interface, also protects the cathode by suppressing Mn leaching and by-product formation.
The excellent contribution of the Asn additive to constructing a stable electrode/electrolyte interface and bulk electrolyte inspires us to verify the practical feasibility of the synergistic effect induced by the designed Asn electrolyte in electrochemical energy storage devices. Therefore, Zn–MnO2 (energy type) batteries and Zn-activated carbon (Zn-AC, power type) capacitors were fabricated with different electrolytes, and their electrochemical performance was fully studied.49–51 As shown in the CV curves of Zn–MnO2 batteries of Fig. 7(a), both batteries exhibit analogous redox peaks, indicating that the utilization of Asn does not alter the insertion/extraction mechanism of H+ and Zn2+ in the MnO2 cathode. Remarkably, a smaller voltage hysteresis between charging and discharging was observed in the Asn-based battery with a lower initial impedance than in the battery with BE (Fig. S26), which benefits from the higher current densities ascribed to the enhanced redox reaction kinetics owing to the addition of Asn. To verify the effect and stability of Asn in the full batteries, in situ DEMS was applied to monitor the H2 and CO2 signals generated during the charge/discharge processes of the Zn–MnO2 batteries with different electrolytes. As shown in Fig. S27, Asn can still effectively inhibit HER that occurs in the full batteries, and it did not decompose within the operating voltage range of full batteries, demonstrating its excellent stability. Then, the rate performance of the Zn–MnO2 batteries over a wide range of current densities from 0.5 to 10 A g−1 was further examined. The enhanced reaction kinetics due to fast charge transfer markedly improve the rate performance. As depicted in Fig. 7(b), the Asn-modified battery exhibited much higher specific capacities at various current densities and almost recovered to its initial level when the current density returned to 0.5 A g−1. Nevertheless, the unmodified battery displays fast capacity decay at high current densities due to its instability and poor reversibility. The cycling performance of the Zn–MnO2 batteries is evaluated at a current density of 1.0 A g−1. As depicted in Fig. 7(c), the battery with Asn exhibited long-term stability for 800 cycles, while a dramatic degradation of capacity appeared in BE. Additionally, the practical application of rechargeable batteries requires storage stability to minimize irreversible capacity loss during rest. Therefore, battery self-discharge behavior was monitored to evaluate the stability of full batteries by full charging and standing for 48 h. A higher CE of 92.29% is still realized in the Asn-modified electrolyte, while only 74.91% CE is obtained for BE (Fig. S28). Thus, the Zn–MnO2 batteries with Asn-modified electrolytes display better cycling performance and capacity retention, indicating the crucial roles of Asn in stabilizing the anode/electrolyte interface and electrolyte system. In practical applications, larger-sized electrodes are usually used for assembling batteries, which exacerbates the side reactions in the batteries. To further verify the practicality of the Asn additive, we assembled a 3 × 5 cm2 Zn||MnO2 pouch cell using a high-mass-loading MnO2 cathode (12 mg cm−2) with Asn electrolyte, which maintained stable cycling for nearly 80 cycles at a current density of 1 mA cm−2. In contrast, the pouch cells of the same size using the same electrodes with BE exhibited accelerated capacity decay and only survived 34 cycles (Fig. S29). What is more, two pouch cells with an Asn electrolyte connected in series can power a circuit board assembled with 35 LED lights successfully. Even under severe mechanical deformation, such as bending and cutting, they can still normally power the LED lights, demonstrating the potential of the Asn additive for practical applications in AZMBs (Fig. S30).
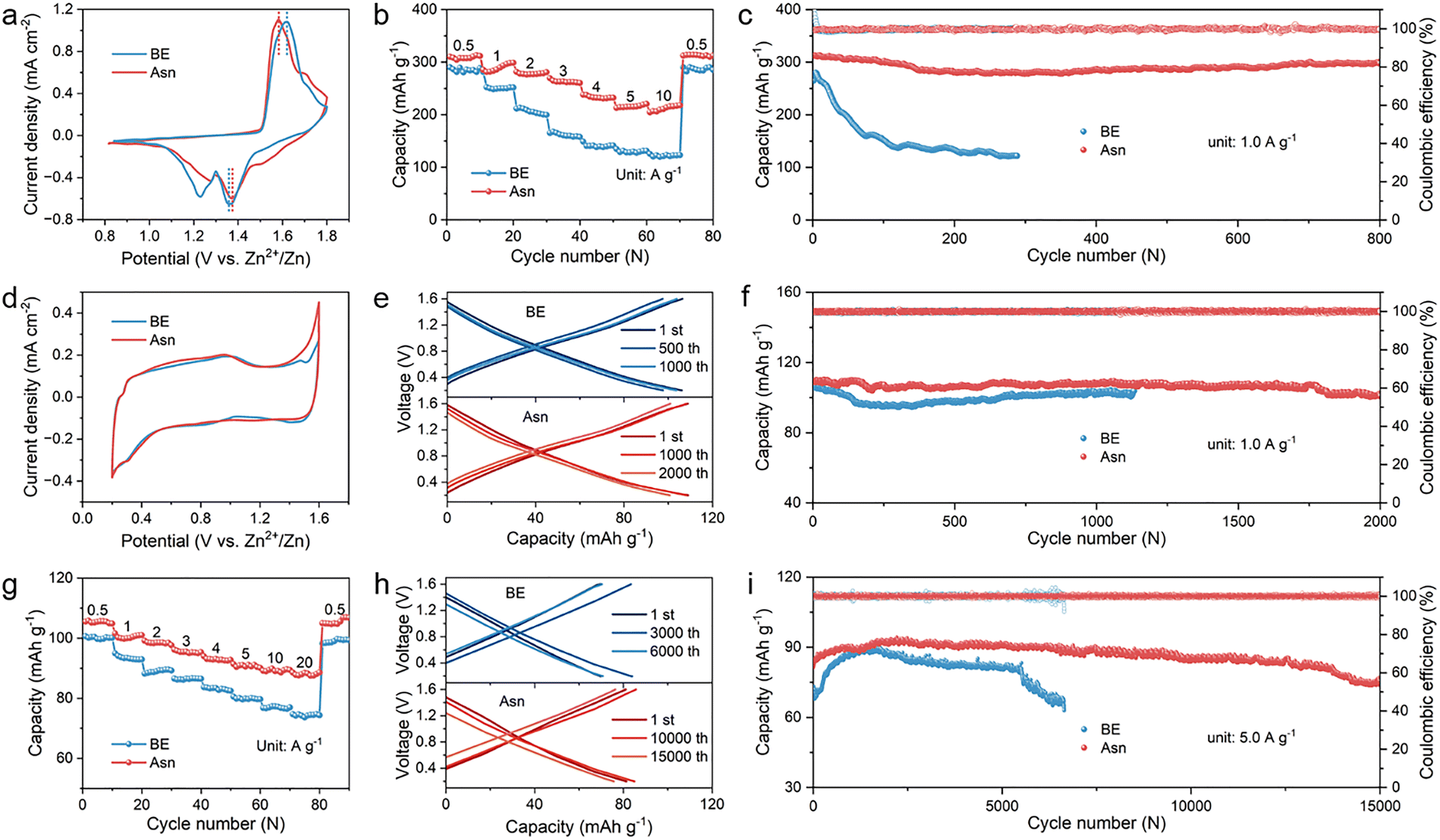 | ||
| Fig. 7 Electrochemical performance of ZMBs and ZICs. (a) CV curves of Zn||MnO2 batteries at a scan rate of 0.2 mV s−1 in different electrolytes. (b) Rate performance of Zn||MnO2 batteries in different electrolytes. (c) Cycling performance of Zn||MnO2 batteries at a current density of 1 A g−1. (d) CV curves of Zn||AC capacitors tested at a scan rate of 0.2 mV s−1 in different electrolytes. (e) GCD curves of Zn||AC capacitors at various cycles at a current density of 1 A g−1. (f) Cycling performance of Zn||AC capacitors at a current density of 1 A g−1. (g) Rate performance of Zn||AC capacitors in different electrolytes. (h) GCD curves of Zn||AC capacitors at various cycles at a current density of 5 A g−1. (i) Cycling performance of Zn||AC capacitors at a current density of 5 A g−1. | ||
To certify the universality of the designed Asn electrolyte in other electrochemical energy storage devices, Zn ion capacitors (ZICs) were fabricated by coupling the Zn metal anode with the AC cathode. As shown in Fig. 7(d), typical quasi-rectangular-shaped CV curves can be observed for ZICs with different electrolytes, revealing the highly reversible physical adsorption/desorption process of electrolyte ions on the AC cathode. Higher peak current density and larger electrochemically active area demonstrate greater electrochemical reactivity with better reversibility and faster reaction kinetics in the Asn electrolyte. The semicircles of the EIS spectra in the high-frequency region demonstrated that the Asn electrolyte is more conducive to enabling rapid charge transfer than the BE electrolyte. Besides, the slope in the low-frequency regions is inversely proportional to ion diffusion resistance. The Asn electrolyte enables a lower ion diffusion resistance than the BE electrolyte. The lower initial impedance of Asn compared with BE also confirmed the faster reaction kinetics in the ZICs (Fig. S31). The overlap comparison of galvanostatic charge/discharge curves based on different electrolytes at 1 A g−1 is shown in Fig. 7(e), confirming that the ZICs with Asn exhibited a specific capacity of 108.84 mA h g−1, whereas their counterpart with BE achieved only a specific capacity of 102.88 mA h g−1. The corresponding cycling performance obviously revealed both higher specific capacity and much better cycling stability after 2000 cycles due to the advantages of Asn (Fig. 7(f)). Then, the rate performance of the assembled ZICs with different electrolytes was measured. As shown in Fig. 7(g), the ZICs with Asn harvest a superior rate capability under various current densities from 0.5 to 20 A g−1, and a capacity of 108 mA h g−1 can be recovered as the current density is set back to 0.5 A g−1, indicating high reversibility of the supercapacitor with Asn. Besides, 43.5% of the initial capacity can be obtained as the current density further increases to 5 A g−1. Moreover, the charge/discharge curves and the corresponding cycling performance at a higher current density of 5 A g−1 are consistent with the above results (Fig. 7(h) and (i)). As expected, the ZICs with Asn maintained a stable specific capacity of 82 mA h g−1 with a capacity retention of 97.10% over 15 000 cycles. In contrast, the BE ZICs showed a sharp decay in capacity only after 6000 cycles, facilitating the faster reaction kinetics with the utilized Asn. These results further demonstrate that the addition of Asn can significantly improve cycling performance and inhibit side reactions during operation. The excellent electrochemical performance in the Zn–MnO2 batteries and Zn-AC capacitors verifies the significance of the Asn additive in achieving high reversible capacities and a long lifespan.
Conclusions
In this study, a bioorganic molecule, Asn, was selected as a multifunctional additive in the electrolyte for AZMBs to explore the relationship between molecular structure and bulk/interface. The acid–base functional groups carried by Asn can dynamically adjust the pH of the bulk electrolyte, restraining the HER and generating by-products. Meanwhile, Asn present in the electrolyte can change the solvation structures of Zn ions through its multiple zincophilic sites to accelerate desolvation kinetics. The DFT calculations and experimental results confirm that the preferential adsorption of Asn compared with water removes the excess active water molecules from the IHP to reconstruct the EDL, thus alleviating the side reactions. Asn adsorbed on the anode surface can also inhibit the formation of Zn dendrites by inducing uniform Zn deposition. The regulation of the bulk electrolyte and electrode/electrolyte interface synergistically improves battery performance. Ultimately, Zn||Zn symmetric batteries with Asn realize highly stable and reversible Zn plating/stripping for 5700 h at 1 mA cm−2 and 1 mA h cm−2 and 1000 h at 25 mA cm−2 and 25 mA h cm−2 (85% DOD). Furthermore, the assembled Zn||MnO2 batteries are capable of cycling stably under a current density of 1 A g−1. The designed electrolyte for Zn||AC capacitors exhibits a long lifespan of 2000 cycles at 1 A g−1 and 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles at 5 A g−1. The design strategy in this study provides theoretical guidance for constructing high-performance and long-term AZMBs with practical commercialization potential.
000 cycles at 5 A g−1. The design strategy in this study provides theoretical guidance for constructing high-performance and long-term AZMBs with practical commercialization potential.
Author contributions
J. J. X., D. H. G., and C. L. M. developed the concept, designed the experiments, and co-wrote the manuscript. C. L. M. and L. F. prepared materials and performed the experimental measurements and experimental data analysis. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.All data supporting the findings in this study are available within the paper and the supplementary information (SI). Supplementary information: Methods, Fig. S1–S31. See DOI: https://doi.org/10.1039/d5ee04445f.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (grant 22425902, 22309060), Jilin Province Science and Technology Development Program (Grant YDZJ202501ZYTS293), the 111 Project (B17020), the Postdoctoral Fellowship Program of CPSF (GZB20240260), the Jilin Youth Growth Science and Technology Plan Project (grant 20250602010RC), and the Fundamental Research Funds for the Central Universities.Notes and references
- M. Kim, J. Lee, Y. Kim, Y. Park, H. Kim and J. W. Choi, J. Am. Chem. Soc., 2023, 145, 15776–15787 CrossRef CAS PubMed.
- X. Wang, K. Feng, B. Sang, G. Li, Z. Zhang, G. Zhou, B. Xi, X. An and S. Xiong, Adv. Energy Mater., 2023, 13, 2301670 CrossRef CAS.
- S. Chen, Y. Xia, R. Zeng, Z. Luo, X. Wu, X. Hu, J. Lu, E. Gazit, H. Pan, Z. Hong, M. Yan, K. Tao and Y. Jiang, Sci. Adv., 2024, 10, eadn2265 CrossRef CAS PubMed.
- N. Hu, W. Lv, H. Tang, H. Qin, Y. Zhou, L. Yi, D. Huang, Z. Wu, J. Liu, Z. Chen, J. Xu and H. He, J. Mater. Chem. A, 2023, 11, 14921–14932 RSC.
- X. Yang, Y. Zhao, S. Lv, L. Zhong, C. Yue, S. Zhan, L. Zhao, C. Wang, X. Li, X. Liu, Z. Tang, C. Zhang, C. Zhi and H. Lv, Energy Environ. Sci., 2024, 17, 4758–4769 RSC.
- P. Xu, M. Xu, J. Zhang, J. Zou, Y. Shi, D. Luo, D. Wang, H. Dou and Z. Chen, Angew. Chem., Int. Ed., 2024, 63, e202407909 CrossRef CAS PubMed.
- R. Zhao, H. Wang, H. Du, Y. Yang, Z. Gao, L. Qie and Y. Huang, Nat. Commun., 2022, 13, 3252 CrossRef CAS PubMed.
- H. Dou, X. Wu, M. Xu, R. Feng, Q. Ma, D. Luo, K. Zong, X. Wang and Z. Chen, Angew. Chem., Int. Ed., 2024, 63, e202401974 CrossRef CAS PubMed.
- J. Luo, L. Xu, Y. Zhou, T. Yan, Y. Shao, D. Yang, L. Zhang, Z. Xia, T. Wang, L. Zhang, T. Cheng and Y. Shao, Angew. Chem., Int. Ed., 2023, 62, e202302302 CrossRef CAS PubMed.
- H. Lyu, S. Zhao, C. Liao, G. Li, J. Zhi and F. Huang, Adv. Mater., 2024, 36, 2400976 CrossRef CAS PubMed.
- T. C. Li, C. Lin, M. Luo, P. Wang, D.-S. Li, S. Li, J. Zhou and H. Y. Yang, ACS Energy Lett., 2023, 8, 3258–3268 CrossRef CAS.
- N. Hu, W. Lv, W. Chen, H. Tang, X. Zhang, H. Qin, D. Huang, J. Zhu, Z. Chen, J. Xu and H. He, Adv. Funct. Mater., 2024, 34, 2311773 CrossRef CAS.
- H. Peng, C. Wang, D. Wang, X. Song, C. Zhang and J. Yang, Angew. Chem., Int. Ed., 2023, 62, e202308068 CrossRef CAS PubMed.
- Y. Lv, M. Zhao, Y. Du, Y. Kang, Y. Xiao and S. Chen, Energy Environ. Sci., 2022, 15, 4748–4760 RSC.
- X. Shi, J. Wang, F. Yang, X. Liu, Y. Yu and X. Lu, Adv. Funct. Mater., 2023, 33, 2211917 CrossRef CAS.
- Q. Ma, R. Gao, Y. Liu, H. Dou, Y. Zheng, T. Or, L. Yang, Q. Li, Q. Cu, R. Feng, Z. Zhang, Y. Nie, B. Ren, D. Luo, X. Wang, A. Yu and Z. Chen, Adv. Mater., 2022, 34, 2207344 CrossRef CAS PubMed.
- K. Ouyang, S. Chen, W. Ling, M. Cui, Q. Ma, K. Zhang, P. Zhang and Y. Huang, Angew. Chem., Int. Ed., 2023, 62, e202311988 CrossRef CAS PubMed.
- H. Li, L. Yang, S. Zhou, J. Li, Y. Chen, X. Meng, D. Xu, C. Han, H. Duan and A. Pan, Adv. Funct. Mater., 2024, 34, 2313859 CrossRef CAS.
- C. Huang, J. Mao, S. Li, W. Zhang, X. Wang, Z. Shen, S. Zhang, J. Guo, Y. Xu, Y. Lu and J. Lu, Adv. Funct. Mater., 2024, 34, 2315855 CrossRef CAS.
- D. Xu, X. Ren, H. Li, Y. Zhou, S. Chai, Y. Chen, H. Li, L. Bai, Z. Chang, A. Pan and H. Zhou, Angew. Chem., Int. Ed., 2024, 63, e202402833 CrossRef CAS PubMed.
- J. Dong, L. Su, H. Peng, D. Wang, H. Zong, G. Wang and J. Yang, Angew. Chem., Int. Ed., 2024, 63, e202401441 CrossRef CAS PubMed.
- Q. Deng, S. You, W. Min, Y. Xu, W. Lin, J. Lu and C. Yang, Adv. Mater., 2024, 36, 2312924 CrossRef CAS PubMed.
- S. Zhou, X. Meng, Y. Chen, J. Li, S. Lin, C. Han, X. Ji, Z. Chang and A. Pan, Angew. Chem., Int. Ed., 2024, 63, e202403050 CrossRef CAS PubMed.
- Y. Dai, R. Lu, C. Zhang, J. Li, Y. Yuan, Y. Mao, C. Ye, Z. Cai, J. Zhu, J. Li, R. Yu, L. Cui, S. Zhao, Q. An, G. He, G. I. N. Waterhouse, P. R. Shearing, Y. Ren, J. Lu, K. Amine, Z. Wang and L. Mai, Nat. Catal., 2024, 7, 776–784 CrossRef CAS.
- Q. Ma, R. Gao, Y. Liu, H. Dou, Y. Zheng, T. Or, L. Yang, Q. Li, Q. Cu, R. Feng, Z. Zhang, Y. Nie, B. Ren, D. Luo, X. Wang, A. Yu and Z. Chen, Adv. Mater., 2022, 34, 2207344 CrossRef CAS PubMed.
- L. Zhou, F. Wang, F. Yang, X. Liu, Y. Yu, D. Zheng and X. Lu, Angew. Chem., Int. Ed., 2022, 61, e202208051 CrossRef CAS PubMed.
- H. Zhang, Y. Zhong, J. Li, Y. Liao, J. Zeng, Y. Shen, L. Yuan, Z. Li and Y. Huang, Adv. Energy Mater., 2023, 13, 2203254 CrossRef CAS.
- L. Yu, J. Huang, S. Wang, L. Qi, S. Wang and C. Chen, Adv. Mater., 2023, 35, 2210789 CrossRef CAS PubMed.
- D. Luo, X. Ma, P. Du, Z. Chen, Q. Lin, Y. Liu, B. Niu, X. He and X. Wang, Angew. Chem., Int. Ed., 2024, 63, e202401163 CrossRef CAS PubMed.
- H. Li, Y. Ren, Y. Zhu, J. Tian, X. Sun, C. Sheng, P. He, S. Guo and H. Zhou, Angew. Chem., Int. Ed., 2023, 62, e202310143 CrossRef CAS PubMed.
- Q. Wu, J. Huang, J. Zhang, S. Yang, Y. Li, F. Luo, Y. You, Y. Li, H. Xie and Y. Chen, Angew. Chem., Int. Ed., 2024, 63, e202319051 CrossRef CAS PubMed.
- M. Yang, J. Zhu, S. Bi, R. Wang, H. Wang, F. Yue and Z. Niu, Angew. Chem., Int. Ed., 2024, 63, e202400337 CrossRef CAS PubMed.
- M. Wu, Y. Sun, Z. Yang, S. Deng, H. Tong, X. Nie, Y. Su, J. Li and G. Chai, Angew. Chem., Int. Ed., 2024, 63, e202407439 CAS.
- G. Ma, W. Yuan, X. Li, T. Bi, L. Niu, Y. Wang, M. Liu, Y. Wang, Z. Shen and N. Zhang, Adv. Mater., 2024, 36, 2408287 CrossRef CAS PubMed.
- R. Huang, J. Zhang, W. Wang, X. Wu, X. Liao, T. Lu, Y. Li, J. Chen, S. Chen, Y. Qiao, Q. Zhao and H. Wang, Energy Environ. Sci., 2024, 17, 3179–3190 RSC.
- Y. Huang, H. Yan, W. Liu and F. Kang, Angew. Chem., Int. Ed., 2024, 63, e202409642 CrossRef CAS PubMed.
- Q. Zong, R. Li, J. Wang, Q. Zhang and A. Pan, Angew. Chem., Int. Ed., 2024, 63, e202409957 CrossRef CAS PubMed.
- Y. Liang, M. Qiu, P. Sun and W. Mai, Chem. Sci., 2024, 15, 1488–1497 RSC.
- Z. Yang, Y. Sun, J. Li, G. He and G. Chai, Adv. Sci., 2024, 11, 2404513 CrossRef CAS PubMed.
- D.-Q. Cai, M. Chen, H. Cheng, J.-L. Yang, H. Liu, T. Xiao, X. Liu, M. Chen and H. J. Fan, Energy Environ. Sci., 2024, 17, 8349–8359 RSC.
- C. Liu, Q. Li, Y. Lin, Z. Wei, Y. Yang, C. Han, M. Zhu, H. Zhang and H. Li, Nano Res. Energy, 2023, 2, e9120064 CrossRef.
- Y. Ji, Q. Hu, J. Zhao, C. Han and H.-M. Cheng, Angew. Chem., Int. Ed., 2025, 64, e202412853 CrossRef CAS PubMed.
- M. Xu, B. Zhang, Y. Sang, D. Luo, R. Gao, Q. Ma, H. Dou and Z. Chen, Energy Environ. Sci., 2024, 17, 8966–8977 RSC.
- H. Ge, X. Feng, D. Liu and Y. Zhang, Nano Res. Energy, 2023, 2, e9120039 CrossRef.
- Z. Peng, L. Tang, S. Li, L. Tan and Y. Chen, Angew. Chem., Int. Ed., 2025, 64, e202418242 CrossRef CAS PubMed.
- K. Ouyang, S. Chen, L. Yu, H. Qin, A. Liu, Y. Liu, Q. Wu, B. Ran, S. Wei, F. Gao, K. Zhang, J. Hu and Y. Huang, Energy Environ. Sci., 2025, 18, 4416–4430 RSC.
- K. Liu, M. Sun, Y. Wu, T. Zhang, A. Zhu, S. Bu, C. Luan, K. Liu, Y. Zhou, D. Lin, S. Wu, C. S. Lee, B. Huang, G. Hong and W. Zhang, Adv. Mater., 2025, 37, 2420079 CrossRef CAS PubMed.
- Y. Lv, C. Huang, M. Zhao, M. Fang, Q. Dong, W. Tang, J. Yang, X. Zhu, X. Qiao, H. Zheng, C. Sun, L. Zheng, M. Zheng, Y. Xu and J. Lu, J. Am. Chem. Soc., 2025, 147, 8523–8533 CrossRef CAS PubMed.
- C. Lin, L. Zeng, M. Liu, F. Xiao, H. Lin, L. Chen, Y. Lu, Q. Qian, Q. Chen, K. Zhang, Z. Yan and J. Chen, Adv. Mater., 2025, e11484 CrossRef CAS PubMed.
- Y. Wang, R. Zhao, M. Liu, J. Yang, A. Zhang, J. Yue, C. Wu and Y. Bai, Adv. Energy Mater., 2023, 13, 2302707 CrossRef CAS.
- H. Lin, L. Zeng, C. Lin, J. Wu, H. He, C. Huang, W. Lai, P. Xiong, F. Xiao, Q. Qian, Q. Chen and J. Lu, Energy Environ. Sci., 2025, 18, 1282–1293 RSC.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2026 |