
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Several concerns about aqueous zinc metal batteries: from components to unit cells
Mingzhu
Li
a,
Shuquan
Liang
 ab and
Guozhao
Fang
*ab
ab and
Guozhao
Fang
*ab
aSchool of Materials Science and Engineering, Central South University, Changsha 410083, P. R. China. E-mail: fg_zhao@csu.edu.cn
bKey Laboratory of Electronic Packaging and Advanced Functional Materials of Hunan Province, Central South University, Changsha, 410083, P. R. China
First published on 22nd December 2025
Abstract
Aqueous zinc metal batteries (AZMBs) remain highly promising for clean and safe large-scale energy storage. Currently, the pursuit of single performance indicators in laboratory research is often based on low-areal-capacity anodes, low-mass-loading cathodes, and excess electrolytes, which is insufficient to satisfy the stringent requirements of practical applications. There are also significant challenges in some promising strategies and performance evaluation models. This review highlights the key issues from electrodes and electrolytes to unit cells of AZMBs. Typical strategies for the high areal capacity and large-capacity batteries are systematically analyzed from the perspectives of the anode, cathode, and electrolyte components. Furthermore, the differences between laboratory and industrial batteries are discussed in terms of critical indicators such as energy density, energy efficiency, and self-discharge performance. Finally, critical perspectives are proposed for achieving large-capacity AZMBs, offering scientific value for their practical development.
Broader contextAqueous zinc metal batteries (AZMBs) have demonstrated strong competitiveness in large-scale energy storage owing to their intrinsic safety, low cost, and facile manufacturing process. In recent years, significant progress has been made in materials design and mechanistic understanding. However, most reported achievements still rely on conditions such as low-areal-capacity anodes, low-mass-loading cathodes, and excess electrolytes. Consequently, a substantial gap remains between component optimization and the realization of high-capacity unit cells, with the core issue being the lack of particular attention to the key parameters and influencing factors required for high-capacity batteries. This review calls for researchers to pay attention to several key challenges from components to unit cells, aiming at achieving large-capacity AZMBs. Furthermore, the differences between laboratory conditions and practical testing environments are compared in terms of critical performance indicators such as energy density, energy efficiency, and self-discharge behaviors. These insights will provide valuable references and guidance for the practical development of AZMBs. |
1. Introduction
Aqueous zinc metal batteries (AZMBs), as emerging energy storage devices, have become strong competitors in large-scale energy storage technologies due to their high intrinsic safety, low cost, and simple production environment.1 In recent years, there has been extensive fundamental research on their reaction mechanisms and the development of electrodes and electrolytes in AZMBs.2 However, the current evaluation criteria for the scalability of AZMBs tend to emphasize single-performance breakthroughs while neglecting their actual values.3 This is because most studies are conducted under simple laboratory conditions, such as low cathode loading, excessive zinc (Zn) anode, surplus electrolytes, and unrealistic charge/discharge rates. These testing conditions often lead to overestimated electrochemical performances for AZMBs.4,5 Consequently, most reported outstanding performances cannot be reproduced in large-capacity batteries.Despite the substantial advances in materials design and mechanistic understanding, a pronounced gap remains between fundamental research and practical application.6 The core issue lies in the lack of systematic attention to the key metrics and influencing factors required to achieve high-capacity cells.7 Nevertheless, even promising strategies and performance evaluation models still confront substantial challenges. To promote the practical development of AZMBs, it is essential to explore feasible engineering and application-oriented strategies on the basis of an in-depth mechanistic understanding. Specifically, electrode optimization and electrolyte design, evaluating the performance of unit cells under realistic operating conditions, and establishing assessment protocols that closely mimic practical scenarios are critical steps toward bridging the gap between laboratory research and actual application demands. Clarifying the disparities between fundamental studies and practical requirements points to potential research directions for realizing high-performance and long-life AZMBs.
This review outlines the key challenges encountered in AZMBs from fundamental research to practical development, encompassing critical aspects such as electrode materials, electrolytes, and unit-cell configuration. A systematic summary of diverse strategies for achieving high-capacity batteries is presented, including the construction of high-areal-capacity electrodes, electrolyte system design, and optimization of unit-cell parameters. Several promising technological approaches and optimization methods are scientifically analyzed. With a focus on industrialization, significant differences in key performance indicators including energy density, energy efficiency, and self-discharge performance are thoroughly compared between batteries tested under laboratory conditions and those deployed at a large-capacity scale. The discrepancies in evaluation standards that arise during the transition from laboratory research to practical development are highlighted. Finally, it is recommended that future development should focus on the following aspects: comprehensive evaluation of multiple performance indicators, synergistic optimization strategies, standardization of testing protocols, transparency in fabrication processes, and the establishment of in-depth scientific institution–enterprise collaboration models. These efforts will provide a scientific basis for promoting the transition of AZMBs from laboratory research to practical development.
2. Concerns about electrodes
2.1. Evaluation of coulombic efficiencies for high-areal-capacity Zn anodes
The reversibility and stability of the Zn anode are fundamental to determining the performance of AZMBs. Among various metrics, coulombic efficiency (CE) is particularly crucial,8 because it serves as the quantitative indicator for the plating/stripping reversibility of the Zn anode, reflecting the loss of active Zn during cycling.9,10 A low CE value indicates the considerable formation of inactive Zn species, such as isolated Zn and irreversible by-products, in each cycle. The continuous accumulation of these inactive species increases the interfacial resistance at the anode, ultimately undermining the long-term stability of AZMBs.11Taking practical performance targets as an example, if 80% capacity retention after 1000 cycles is required, the CE must be improved to 99.97%.8 Currently, the CE value of most coin cells reported exceeds 99.5% (Fig. 1a). However, these high CEs are mostly obtained under the optimized test parameters, including low areal capacity (<2 mAh cm−2), low Zn utilization (<10%), and high rate cycling (>1C), which leads to significant bias in CE evaluation.12 Under such arbitrary conditions, the results are completely unrealistic for practical applications.13 Compared with coin cells, achieving high CEs under realistic conditions that target high-capacity batteries is considerably more challenging. In ampere-hour (Ah) level pouch cells, the requirement for a low negative to positive capacity (N/P) ratio imposes a higher areal capacity of 5 mAh cm−2 or even higher for Zn anodes (Fig. 1b).4 Zhi et al. summarized the changes in the cycle life and CE values at different areal capacities and rates.14 Both the cycle life and CE values decrease as the areal capacity increases. Additionally, at a fixed total areal capacity, the average cycle life of the battery decreases as the rate is reduced (Fig. 1c). When tested under practical harsh conditions with a high areal capacity (>5 mAh cm−2), high Zn utilization (>50%), and low rate (<0.5C), the real performance often decays sharply. This is shown by a sudden drop in coulombic efficiency to around 97.5% and the cycle life may be drastically reduced to just tens of cycles. Therefore, rigorous evaluation under high areal capacity is essential for developing practical AZMBs with a long cycle life.
 | ||
| Fig. 1 Challenges and applications of high-areal-capacity anodes under high DOD conditions in AZMBs. (a) CE tests of current works. (b) Suggested electrode loadings and areal capacities for electrochemical testing of AZMBs at different stages.4 Copyright @ 2023 Elsevier. (c) Cycling performance of the Zn anode at various areal capacities.14 Copyright @ 2024 Elsevier. (d) Emim+ and I− co-oriented Zn electrodeposition.24 Copyright @ 2025 American Chemical Society. (e) The Zn plating process in nano-capsule electrolytes.25 Copyright @ 2025 Royal Society of Chemistry. (f) The favorable Zn plating/stripping behavior facilitated by the SEI.26 Copyright @ 2025 Royal Society of Chemistry. | ||
The absence of standardized CE measurement protocols under unified conditions hinders effective comparisons between different systems. To build a Zn anode suitable for AZMBs, it is necessary to refine CE evaluation strategies and emphasize certain key parameters. First, performance assessments should focus on the electrochemical behavior of the anode under a high areal capacity (>5 mAh cm−2) and high DOD (>50%) to better reflect practical application scenarios. Second, in addition to the rigorous control over the areal capacity, the influence of charge/discharge rates should not be overlooked. Notably, Ma et al.15 extended Adams’ methodology16 to AZMBs, underscoring the dependence of CEs on the current density. While a high current density is beneficial for monitoring morphological changes at high capacity, it may also obscure hydrogen evolution reactions at the Zn anode. Thus, it is recommended that cells for CE testing should be cycled at practical rates relevant to the application, specifically within the C/4 to C/6 range. Additionally, it is recommended to employ Zn reservoir CE tests, particularly under high areal capacity conditions, to enable a more comprehensive and practical assessment of the anode performance. Such systematic and realistic evaluation protocols will facilitate a deeper understanding of the dynamic electrochemical behavior of Zn anodes during cycling and ultimately guide the development of AZMBs suitable for practical applications.
2.2. Stability of Zn anodes under high DOD
High DOD should be regarded as the foundation criterion for evaluating the stability of Zn anodes. While AZMBs operating under low DOD conditions can achieve long cycling lifespans, primarily because only a minor fraction of Zn participates in side reactions, this often creates a misleading impression of excellent Zn reversibility. However, as the DOD increases, the Zn anode undergoes more frequent and intense plating/stripping processes. This imposes a substantial challenge on both the structural and interfacial stability of the electrode.17 Under high DOD conditions, significant volumetric changes occur within the Zn anode. Repeated Zn2+/Zn plating/stripping leads to expansion and contraction, which can easily result in electrode pulverization and fracture.18 At the microscopic level, Cheng19et al. revealed the fundamental mechanism. The inherently loose and porous nature of deposited Zn makes it prone to detach from the current collector, forming dead Zn, while also being readily corroded by the electrolyte into by-products. Consequently, the active material may detach, lose electrochemical activity, and ultimately cause a decline in the overall performance and cycling stability.20To address the stability issues under high DOD conditions, various effective strategies have been proposed in current research. One promising approach is the regulation of Zn anode crystallographic orientation, which can substantially enhance Zn plating behavior and, in turn, improve the overall battery stability under high DOD conditions.21 From a crystallographic perspective, the Zn (002) crystal plane exhibits the lowest surface energy,22 facilitating the even plating of Zn2+. Electrodeposition and optimization of anode processing methods can be employed to fabricate Zn anodes with a preferred (002) orientation,23 facilitating homogeneous Zn2+ nucleation and suppressing dendrite formation typically caused by locally excessive current densities. Li et al. engineered a (002)-textured Zn anode on an untextured substrate by exploiting the interactions of Emim+ and I− with distinct Zn crystal planes, achieving a remarkable (002) plane ratio (RTC) of 99% (approaching single-crystal Zn) (Fig. 1d). The ultrahigh (002) texture significantly enhanced the cycling performance of the Zn anode under 88.0% DOD with 25 mAh cm−2 over 500 h.24 In addition, the incorporation of functional additives, such as organic small molecules and polymers, enables the formation of a stable solid electrolyte interphase (SEI) on the Zn anode. This SEI effectively suppresses both electrolyte decomposition and dendrite growth, thereby offering enhanced protection under high DOD conditions. Chen et al. proposed the self-assembly of capsule-like solvation nanoclusters,25 which extended the cycle life to nearly 3500 h at a high current density of 50 mA cm−2 and over 1600 h even at a high DOD of 88.09% (Fig. 1e). Moreover, Zhang et al. applied electrostimulation to achieve a sequential process of initial enrichment followed by networking; an inside-out assembly approach was realized, resulting in a denser embedded SEI layer (Fig. 1f). With the protection of this SEI, the symmetric cell assembled with the Zn anode could still maintain stable cycling for up to 440 h even under extremely 90% DOD conditions.26
Future research on Zn anodes under high-capacity conditions should prioritize several industrial development directions. Compared to Zn foil, composite anodes fabricated by roll-bonding Zn foil with current collectors or electrodeposited Zn anodes on current collectors demonstrate superior controllability in industrial production due to their fixed current collector structure. These composite Zn anodes exhibit no perforation issues and achieve significantly improved consistency. Furthermore, scaling up orientation control represents a crucial objective. Although non-epitaxial electrodeposition methods have shown industrial potential for fabricating (002)-oriented Zn anodes, large-scale implementation still requires further optimization of operational parameters such as electrodeposition parameters and electrolyte composition. In addition, the alloying modification strategy utilizing elemental doping, which integrates microstructural regulation with interfacial engineering, effectively overcomes the inherent limitations of the hostless reaction mode in pure Zn anodes and demonstrates promising potential for industrial application. It is also worth noting that the integration of dynamic interfacial characterization and theoretical modeling will aid in better revealing the dynamic interfacial behaviors during Zn plating/stripping processes. Future studies could focus on integrating orientation control, electrolyte engineering, and interfacial modification to develop more efficient Zn anodes and further advance battery performance.
2.3. Stability of high-loading cathodes
Constructing high-loading cathodes is a comprehensive test of the entire electrode system, the electrochemical interface, and the battery structure under real operating conditions. While low-loading cathodes (∼1 mg cm−2) commonly used in laboratories can reflect the intrinsic capacity of materials, the critical challenge is to maintain the energy density and cycling stability under high mass loadings.27 In order to meet the metric for practical AZMBs, the cathode loading typically needs to reach 10–20 mg cm−2, which is much higher than that of laboratory-test conditions.28 However, a high loading leads to limited active sites and poor electronic/ionic conductivity, which adversely affects the actual capacity and power output of the battery. Increased material thickness results in longer diffusion paths for Zn2+ and restricts electrolyte penetration, causing electrochemical reactions to be concentrated on the electrode surface and creating “dead zones” within the electrode.29 The increased slurry thickness can also reduce adhesion between the active layer and the current collector, resulting in cracking and delamination during cycling, further compromising battery reliability.Vanadium (V)-based materials undergo multi-step redox reactions (e.g., V5+/V4+/V3+) during cycling, involving phase changes and volume variations that can lead to layer collapse and lattice distortion.30 This transformation can convert active materials into non-conductive phases, significantly diminishing the battery performance. Lattice regulation of active materials can significantly enhance the ionic/electronic conductivity and structural stability. Yang et al. developed a high-loading Mn2.5V10O24·5.9H2O electrode with fast electron and ion transport properties,31 in which the interlayer Mn2+ doping and the presence of bound water alter the electronic structure, significantly enhancing its electronic conductivity (Fig. 2a). Additionally, the low-valent Mn2+ ions create more oxygen vacancies in the Mn2.5V10O24·5.9H2O crystal structure, weakening the strong electrostatic interactions between Zn2+ and the host material, thereby reducing the diffusion energy barrier. Under a high active material loading (>10 mg cm−2) and a high active material content (90%), this cathode exhibited exceptional capacity and cycling stability at moderate rates (0.25–2C) (Fig. 2b).
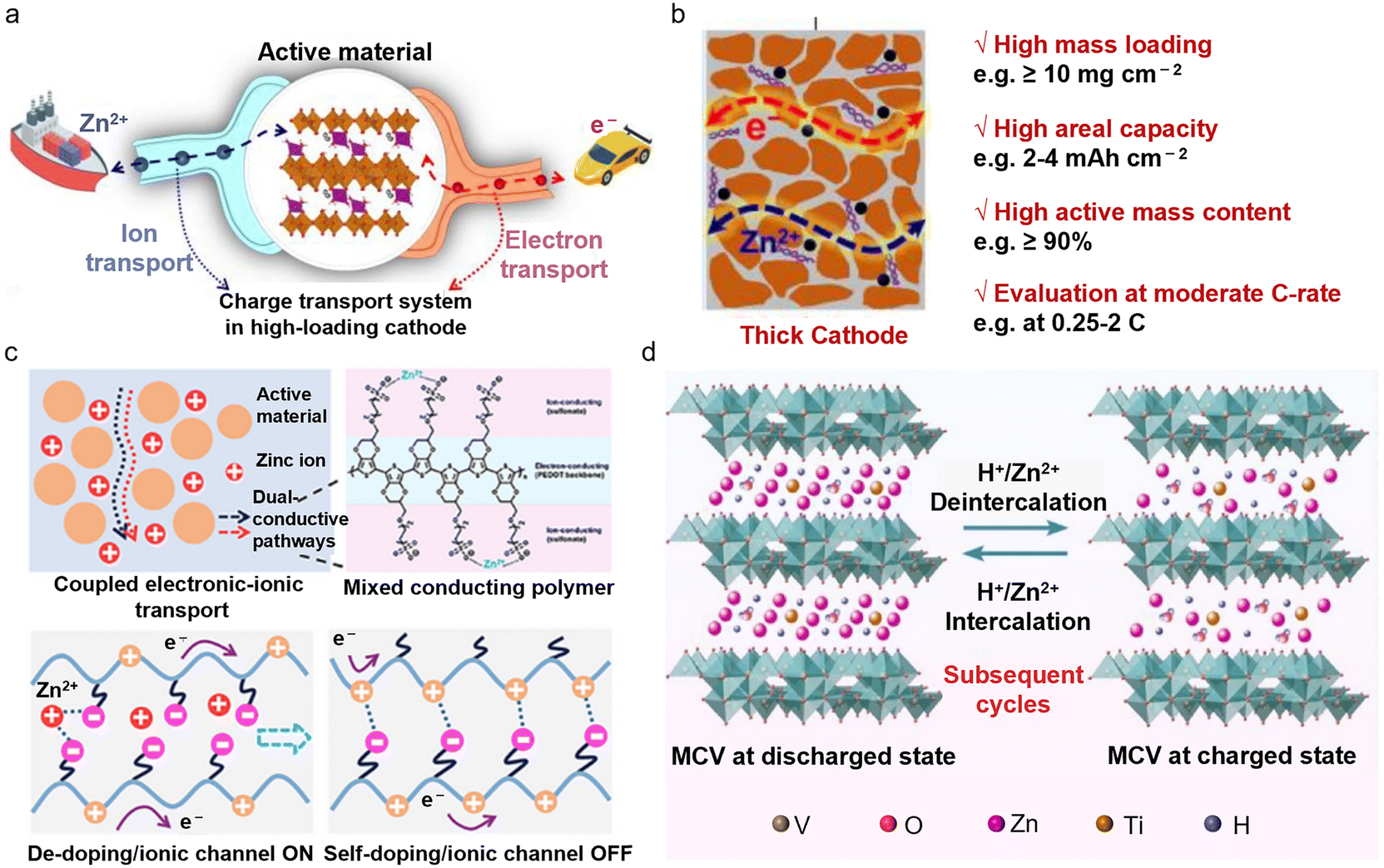 | ||
| Fig. 2 Challenges and applications of high-loading V-based cathodes in AZMBs. (a) The charge (electron/Zn2+) transport system in the high-loading cathode.31 (b) High-loading cathode for stationary energy storage systems, highlighting the key performance metrics.31 Copyright @ 2023 Elsevier. (c) An integrated mixed electronic–ionic conductor is developed by spatially coupling transport pathways, enabling the redistribution and fast transport of charges.32 Copyright @ 2024 Wiley VCH. (d) Schematic illustration of the electrochemical energy storage mechanism of the MCV@CC cathode.33 Copyright @ 2022 Elsevier. | ||
Combining V-based materials with highly conductive components to create composites with rapid ionic and electronic transport capabilities has shown to result in excellent performance and cycling stability under high-loading conditions. Zhu et al. proposed an integrated mixed electronic–ionic conductor that achieves charge redistribution and rapid transport through spatially coupled charge transport pathways32 (Fig. 2c). Importantly, this composite electrode achieved an ultra-high areal capacity of 6.0 mAh cm−2 at a mass loading of 21 mg cm−2 and exhibited excellent cycling stability, retaining 79.1% of the capacity after 100 cycles at a rate of 0.2 A g−1. This indicator is close to practical requirements, allowing further verification of the dispersion uniformity of multicomponent materials in the electrode at the pilot scale. Meanwhile, the increased thickness of the material leads to longer diffusion pathways for Zn2+ and restricts electrolyte penetration. This results in insufficient reaction depth and concentrating electrochemical reactions at the electrode surface, forming “dead zones” within the electrode. The design of binder-free electrodes can enhance the mechanical integrity and electrochemical performance of AZMBs by reducing the reliance on the electrode/electrolyte interface compatibility. Fang et al. developed a high-loading, binder-free vanadium oxide cathode (MCV@CC) through the bridging interaction between V2O5 and carbon cloth via an MXene.33 The MXene bridges facilitate the formation of a dense, interconnected active layer, preventing material agglomeration and detachment (Fig. 2d). Interwoven carbon nanotubes (CNTs) further improve charge transport. This electrode achieved an areal capacity of 2.36 mAh cm−2 at a mass loading of 10 mg cm−2 and retained 86.1% of the capacity after 1200 cycles, demonstrating remarkable cycling stability and application potential. However, the cost issue of the core material MXene and the difficulties in large-scale production remain major constraints for industrialization. Furthermore, most reported preparation methods currently rely on hydrothermal techniques, which are difficult to scale up for mass production under current conditions. Large-scale production methods such as ball milling are highly desirable.
Under such high-loading conditions, manganese-based (Mn-based) cathodes face not only common challenges similar to those in V-based systems, but also additional issues resulting from their complex electrochemical behavior. A unique feature of Mn-based cathodes is their energy storage mechanism, which involves both Zn2+ insertion/extraction and Mn2+ deposition/dissolution processes.34 The latter mechanism is highly dependent on a sufficient liquid-phase environment and mainly occurs at the electrode/electrolyte interface. However, due to limited electrolyte penetration in thick electrodes, the deposition/dissolution reactions are difficult to sustain effectively within the electrode bulk. Moreover, Mn-based cathodes are more severely affected by the Jahn–Teller effect, which leads to increased structural distortion and performance degradation. As a result, compared with V-based cathodes, Mn-based cathodes tend to suffer more significant performance decline under high mass loading conditions.
To address these challenges, researchers have proposed multidimensional strategies which mitigate the structural stability issues of Mn-based cathode materials under high mass loadings during long-term cycling. Through ion pre-intercalation, the interlayer spacing of the host material can be increased, creating more space for ion insertion and thereby enhancing the electrochemical performance of the material. Previous studies have confirmed that ion intercalation can promote the Mn2+/Mn4+ redox reaction of the cathode. However, the challenge of achieving a highly reversible Mn deposition/dissolution process under high mass loading while maintaining the stability of the active material remains to be addressed. Gao et al. adopted a synergistic strategy of co-intercalating Na+ and Cu2+ ions (NCMO). The pre-intercalation effect of Na+ amplifies the activation of the Mn2+/Mn4+ redox couple, while the smaller ionic radius of Cu2+ facilitates its rapid diffusion into the electrolyte, enabling the reversible Cu/Cu2+ redox reaction during charge and discharge. The Cu2+ generated during charging can accelerate the Jahn–Teller distortion of the intermediate ZnxMnO(OH)y phase, thereby promoting surface dissolution. The co-intercalation of Na+ and Cu2+ not only enhances the surface manganese conversion process of the cathode, but also ensures the structural stability of the host material during cycling (Fig. 3a).35 The NCMO cathode with a high loading of 10.9 mg cm−2 exhibited a reversible capacity of 2.10 mAh cm−2 after 50 stable cycles at 0.1 A g−1.
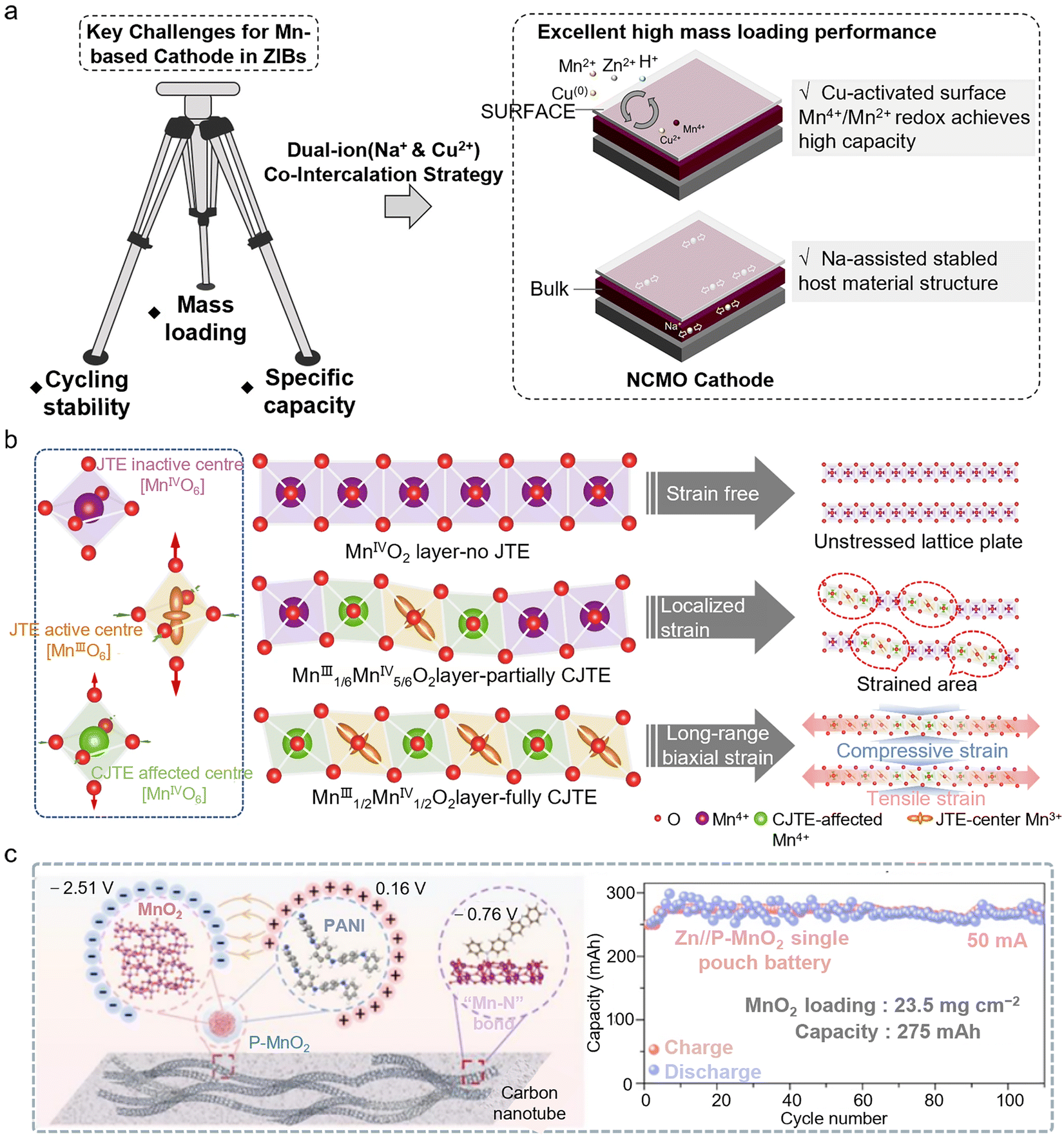 | ||
| Fig. 3 Challenges and applications of high-loading Mn-based cathodes in AZMBs. (a) Schematic of the MnO2 cathode with dual-ion co-intercalation, enhancing Mn2+/Mn4+ two-electron transfer and structural stability to address challenges in mass loading, cycling, and capacity.35 Copyright @ 2024 Royal Society of Chemistry. (b) Schematic illustration depicting the distinct structural scenarios in layered MnO2, including no Jahn–Teller effect (JTE), partially cooperative Jahn–Teller effect (CJTE), and fully CJTE.37 Copyright @ 2025 Springer Nature. (c) The PANI-modified MnO2 (P-MnO2) cathode exhibits excellent structural stability, and the proton-rich environment effectively stimulates the efficient reversible dissolution of deposited MnO2.38 Copyright @ 2024 Royal Society of Chemistry. | ||
Traditional MnO2 electrodes often suffer from self-aggregation of active materials due to the use of insulating and hydrophobic binders, which inhibits Zn2+ diffusion at the interface and promotes MnO2 dissolution, thereby limiting the development of high-capacity Zn–Mn batteries. Andreu Cabot et al. addressed this issue by integrating the active material with multi-walled carbon nanotubes (ZMOC) to form a binder-free, self-supporting architecture, which directly addresses key challenges in traditional electrode manufacturing, such as “dead zones” and low active material utilization caused by insulating binders and solvents, while also enabling the streamlining of the process flow. High-mass-loading ZMOC electrodes, with loadings of up to ∼20 mg cm−2, maintained capacities close to 200 mAh g−1 (corresponding to approximately 4 mAh cm−2).36 Furthermore, alleviating the Jahn–Teller distortions of materials and constructing a stable cathode structure are also important research directions for high-loading processes. The eg orbital of high-spin Mn3+ ions contains an unpaired electron, inducing the Jahn–Teller distortion and causing elongation of the [MnO6] octahedra. Such distortions tend to accumulate locally within the crystal, leading to lattice strain, distortion, and eventual degradation of the layered MnO2 structure. Wang et al. utilized the electronic modulation capability of graphene layers to precisely adjust the oxidation state of Mn. By tuning the Mn3+/Mn4+ ratio to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and adopting a superlattice structure, they achieved a long-range and uniform distribution of the resulting distortions37 (Fig. 3b). Meanwhile, interfacial engineering can effectively address the stability and capacity fading issues of high-mass-loading MnO2 cathode materials over 5000 cycles. Liu et al. introduced a polyaniline (PANI) layer to construct a proton-rich interfacial microenvironment on the surface of MnO2, which not only facilitated the proton insertion/extraction reactions of MnO2/Mn3+, but also successfully activated the reversible dissolution/deposition process of MnO2 formed by the electrodeposition of Mn2+ from the electrolyte.38 High-mass-loading (23.5 mg cm−2) pouch cells assembled with this cathode retained a high capacity of 275 mAh after 110 cycles (Fig. 3c). The raw materials and synthesis processes entail relatively low costs, are highly compatible with existing electrode material production lines, and hold significant potential for practical application.
:1 and adopting a superlattice structure, they achieved a long-range and uniform distribution of the resulting distortions37 (Fig. 3b). Meanwhile, interfacial engineering can effectively address the stability and capacity fading issues of high-mass-loading MnO2 cathode materials over 5000 cycles. Liu et al. introduced a polyaniline (PANI) layer to construct a proton-rich interfacial microenvironment on the surface of MnO2, which not only facilitated the proton insertion/extraction reactions of MnO2/Mn3+, but also successfully activated the reversible dissolution/deposition process of MnO2 formed by the electrodeposition of Mn2+ from the electrolyte.38 High-mass-loading (23.5 mg cm−2) pouch cells assembled with this cathode retained a high capacity of 275 mAh after 110 cycles (Fig. 3c). The raw materials and synthesis processes entail relatively low costs, are highly compatible with existing electrode material production lines, and hold significant potential for practical application.
Aqueous-based Zn–iodine (Zn–I2) batteries are gaining increasing attention due to their low cost, safety, and environmental friendliness. However, as the loading of I2 in the cathode increases (≥80–100 mg cm−2), the batteries face a series of complex challenges that significantly restrict their electrochemical performance in practical applications.39 Firstly, the inherent poor electronic conductivity of iodine, combined with the increased electrode thickness associated with higher loading, significantly reduces the charge and ion transport efficiency within the depth of the cathode. This low transport efficiency leads to severe polarization, resulting in insufficient utilization of the active material and slow redox reaction kinetics. Secondly, the elevated iodine concentration exacerbates the generation and diffusion of soluble polyiodide species (such as I3−, I5−, etc.), which can shuttle to the Zn anode and participate in detrimental parasitic reactions, resulting in self-discharge, Zn corrosion, dendrite growth, and rapid capacity loss.40
The conventional slurry processing method relies on solvent evaporation, during which I2 is prone to sublimation losses, leading to a reduction in the actual I2 loading and potential misjudgment of the electrode capacity. In order to suppress the polyiodide shuttle effect, a large amount of conductive carbon is typically added, which further dilutes the proportion of active materials and results in a low tap density, making it difficult to meet the requirement for a high energy density. Wu et al. employed a solvent-free dry powder mixing and hot-rolling/calendering process,39 which not only avoids the use of solvents and the sublimation of I2 but also enables the fabrication of dense, crack-free, and self-supporting I2 cathodes with a mass loading of up to 100 mg cm−2 (Fig. 4a). Dry coating technology demonstrates significant potential for fabricating electrodes with a uniform microstructure and excellent electrochemical performance. This process currently remains distant from industrial-scale commercialization. Several key challenges require resolution, including enhancing equipment availability, optimizing process parameters, and developing novel binder systems. With continued research advancements and equipment improvements, dry coating is positioned to achieve broader adoption in battery manufacturing.
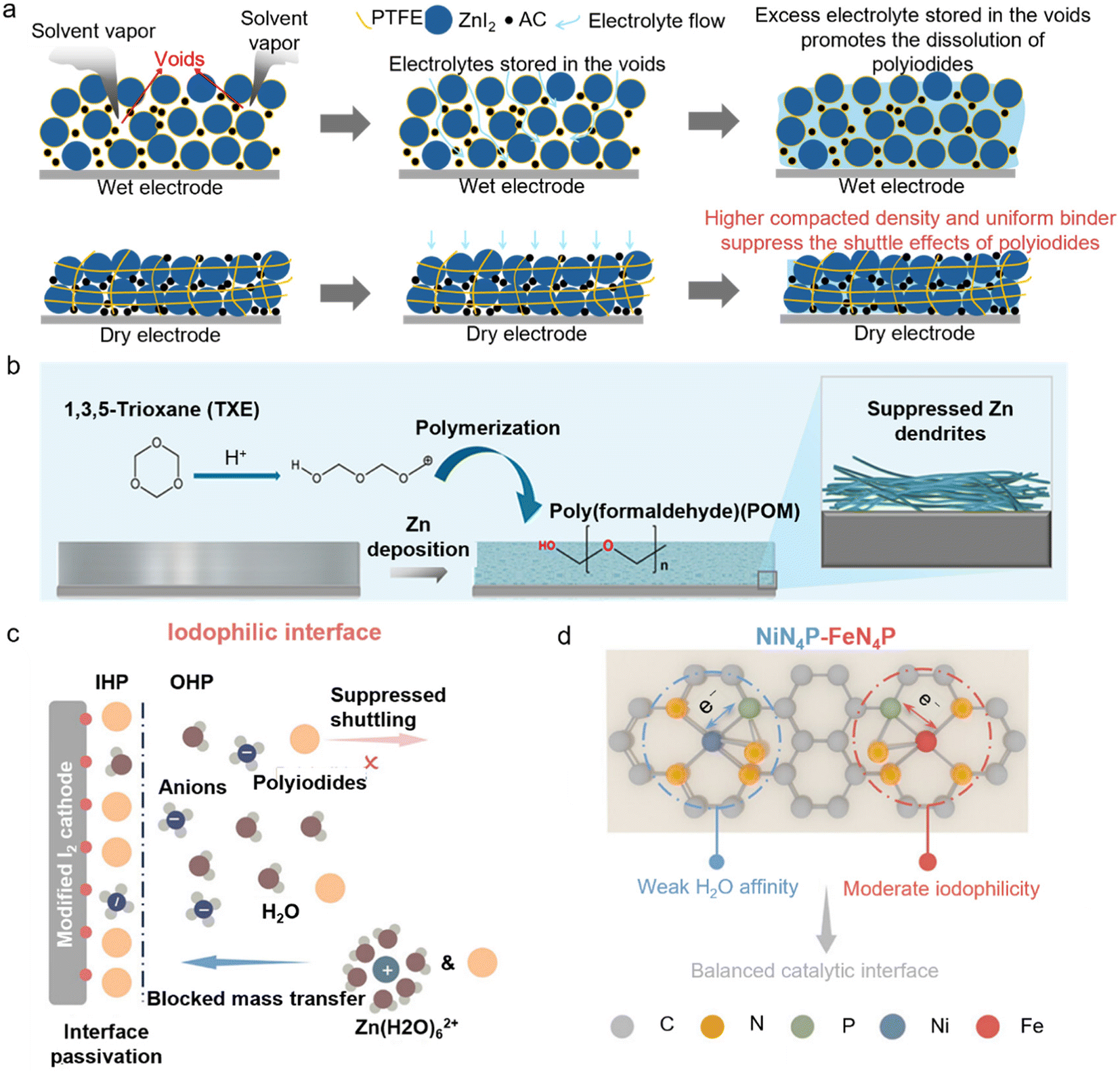 | ||
| Fig. 4 Challenges and applications of high-loading I2-based cathodes in AZMBs. (a) Schematic illustration of structural differences in wet and dry electrodes and impacts on the shuttle effects of polyiodides.39 (b) Schematic diagram of the structural evolution of the anode surface during Zn plating with TXE.39 Copyright @ 2025 Elsevier. (c) Iodophilic interface of the cathode with strong iodine affinity.41 (d) Schematics of the proposed optimal catalytic interface with balanced iodophilicity and solvophilicity enabled by NiN4P–FeN4P.41 Copyright @ 2025 American Chemical Society. | ||
In addition, to accommodate a high-loading cathode, it is essential to achieve interfacial stability of the Zn anode under high current densities. In traditional electrolyte systems, the Zn anode tends to form dendrites and “dead Zn” during cycling, which reduces the CE and induces short circuits. To achieve synergistic high-capacity cycling, 1,3,5-trioxane (TXE) is introduced as an electrolyte additive, enabling the in situ formation of a flexible polyoxymethylene (POM)-based solid electrolyte interphase (SEI) film on the anode surface, which effectively suppresses dendrite growth and side reactions (Fig. 4b). Compared with conventional inorganic SEI films (such as ZnF2 or ZnS), which are prone to fracture, the POM-based SEI exhibits a higher elastic modulus and superior stability, better accommodating the volume changes of the Zn anode and significantly enhancing the interfacial stability and cycling life. Notably, this approach has achieved impressive results, with 88.6% of the initial capacity retained after 750 cycles at a high areal capacity of 15.8 mAh cm−2 and a DOD of 53.3% in pouch cells. In addition, Xiao et al. emphasized the importance of balancing polyiodide adsorption and interfacial mass transfer kinetics at the cathode surface.41 Excessive adsorption of iodine species can impede ion transport, a problem that becomes particularly pronounced under conditions of lean-electrolyte and high mass loading (Fig. 4c). They employed a dual single-atom catalyst encompassing NiN4P and FeN4P sites to promote a catalytic interface with well-balanced solvophilicity and iodophilicity. The P ligands further strengthen these functions by tuning Fe sites from low to medium spin states and creating an anion-rich inner Helmholtz plane at Ni sites (Fig. 4d). Benefiting from this dual-metal atomic catalyst, a 100 cm2 Zn–I2 pouch cell with a high I2 loading (30 mg cm−2) delivers a stable areal capacity of about 5.6 mAh cm−2 over 400 cycles.
3. Concerns about electrolytes
3.1. Dual interface functionality of electrolytes
Currently, most electrolyte optimizations are focused on a single electrode interface which are effective in laboratory-scale coin cells. However, these deceptively favorable testing conditions obscure potential problems that may arise as battery systems move toward practical application. Commercial AZMBs may require high active material loadings (>10 mg cm−2), high areal capacities (>3 mAh cm−2), and low E/C ratios (<2.5 g Ah−1).6 Under such harsh conditions, single-interface optimization strategies struggle to meet the comprehensive requirements spanning from fundamental research to practical large-scale battery applications.The development of AZMBs faces the fundamentally different requirements that the electrolyte must satisfy different electrodes (Fig. 5). The Zn anode is prone to corrosion and dendrite growth in aqueous environments, which significantly undermines the reversibility of Zn plating/stripping, thereby compromising the battery safety and cycle life. In contrast, cathode materials such as Mn-based compounds rely on the presence of H2O to facilitate the efficient insertion and extraction of Zn2+ and H+, enabling high capacity and structural stability. For V-based materials, it is necessary to minimize the activity of water as much as possible to suppress the dissolution of active materials and maintain structural stability. This intrinsic contradiction in electrolyte requirements indicates that approaches focusing solely on modifying the electrolyte for a single electrode are inherently flawed. It is therefore essential to pay greater attention to the different operating mechanisms of various electrode materials and to further investigate the microscopic interfaces between different electrodes and electrolytes. By tailoring the electrolyte to the specific electrochemical characteristics of each electrode, the battery performance can be optimized.
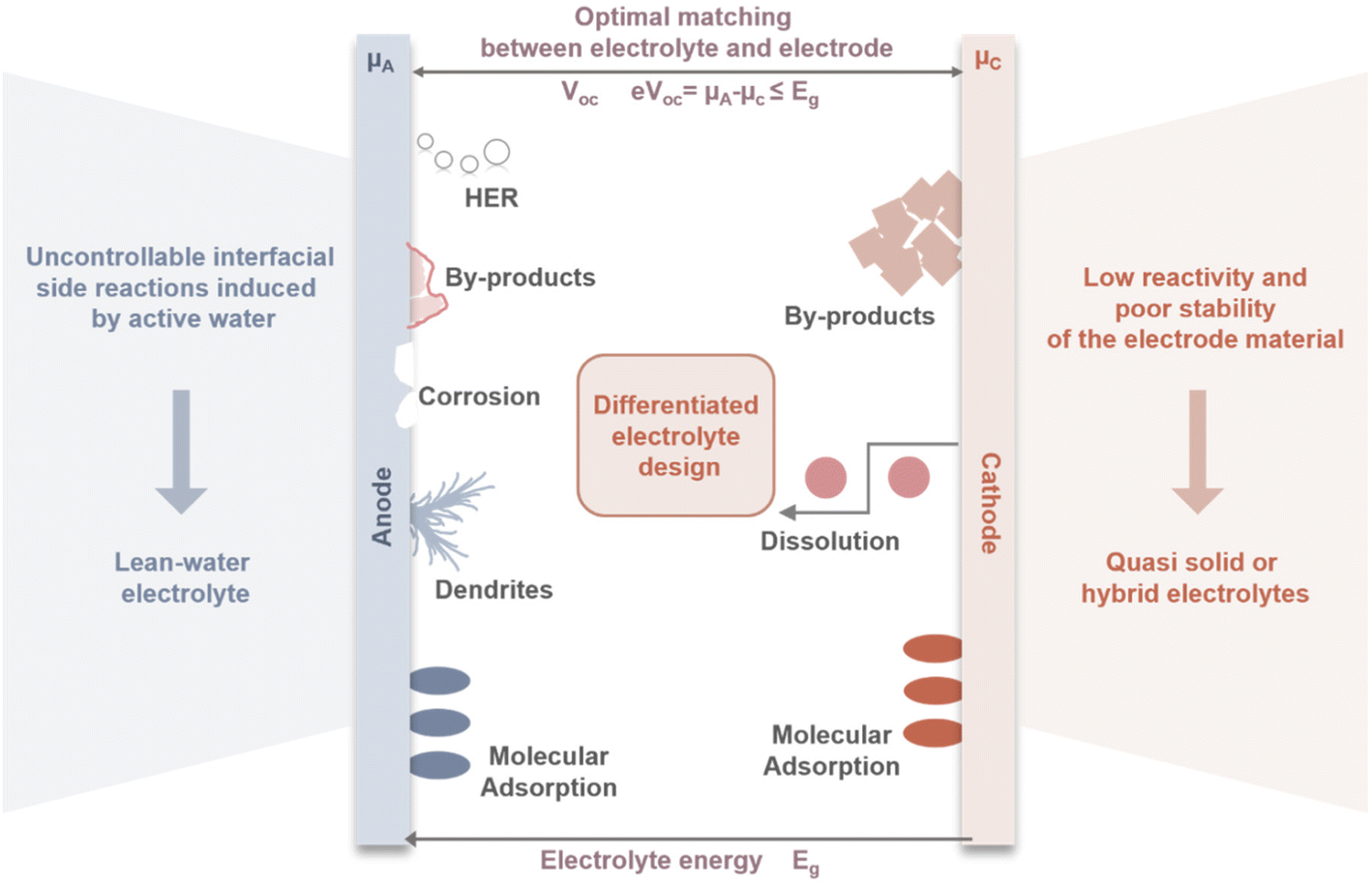 | ||
| Fig. 5 Schematic diagram of differentiated electrolyte design for different electrodes. | ||
In Ah-level batteries, the thick electrodes operate at high current densities, and active reactions between the circulating electrolyte and active materials are more intense. Single-interface optimization only protects a single electrode interface (solely suppressing Zn dendrites or only mitigating cathode dissolution), whereas dual-interface strategies can maintain an overall balance in the electrolyte environment, alleviating instability caused by mutual perturbations between the two electrode interfaces under extreme conditions.42 This greatly improves reaction coordination and cycling life in large-capacity batteries. Against this background, biphasic electrolytes leverage physical spatial separation and the targeted enrichment of functional components to form a multidimensional protection system characterized by “zonal adaptability and interfacial synergy”. Typically, the phase adjacent to the Zn anode is an organic solvent-rich phase, which can effectively suppress dendrite growth, reduce hydrogen evolution, and promote steady Zn plating. Meanwhile, the phase near the cathode is usually an additive-containing aqueous phase that ensures rapid redox kinetics at the cathode interface while simultaneously reducing side reactions. The interfaces can also block by-products, greatly mitigating the interface imbalance and chain-reaction amplification that often occur in high-loading cathodes.
Unlike conventional single-interface additives, bilateral in situ functional additives can continuously self-assemble at the charge/discharge interface and adaptively adjust the compactness of the protective layer. This ensures that even under severe internal environment fluctuations in high-loading batteries, the adsorption layer can promptly cover newly exposed active surfaces, effectively limiting the spread of side reactions. He et al. introduced a multi-functional electrolyte, KAl(SO4)2 (KASO).43 The high-valence Al3+ plays dual roles of competing with Zn2+ for solvation and forming a Zn–Al alloy layer with a homogeneous electric field on the anode surface to mitigate side reactions and dendrite generation. The Al-containing cathode–electrolyte interface (CEI) considerably alleviates the irreversible dissolution of the VOx cathode and the accumulation of byproducts (Fig. 6a). Consequently, the Zn||VO2 full cell exhibits excellent cycling stability at a low N/P ratio of 2.83 and a high mass loading (∼16 mg cm−2). Meanwhile, based on the total mass of the cathode and anode, the energy density of the Zn||VO2 pouch cell is 43.1 Wh kg−1. Similarly, Song et al. proposed a dual-sided in situ functionalization strategy to address the challenges of high-mass-loading and high-areal-capacity commonly encountered in AZMBs.44 By introducing an organic sodium salt electrolyte additive, directional adsorption and in situ decomposition were achieved, forming a gradient interface on the Zn anode. Simultaneously, the cathode interface was modified in situ by the electrolyte additive, which not only promoted ion intercalation but also inhibited cathode dissolution through interfacial adsorption, resulting in improved cycling stability under high loading (Fig. 6b). As a result, the Ah-scale pouch cell can last more than 680 cycles with an accumulated capacity of 319.6 Ah.
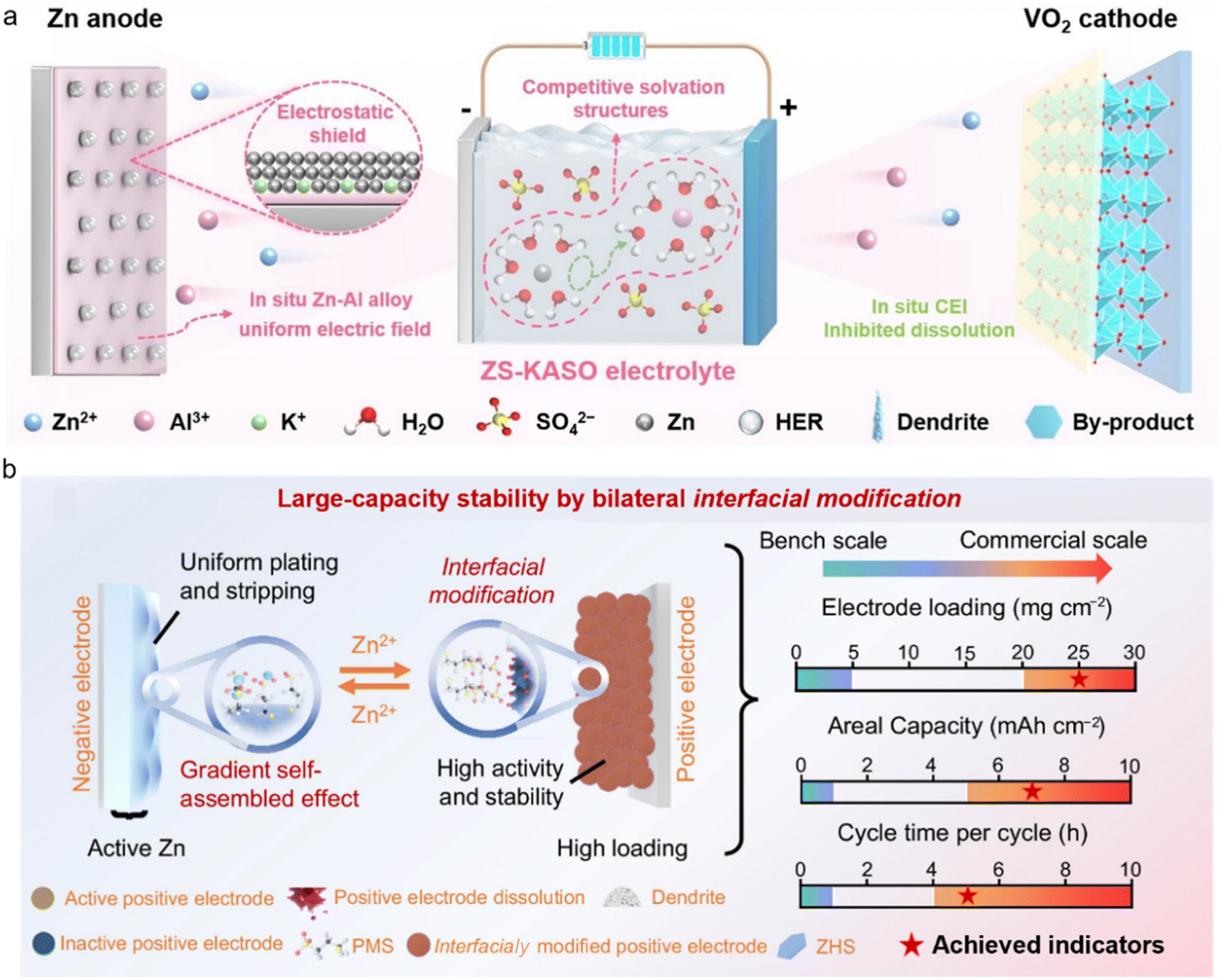 | ||
| Fig. 6 Application of dual-interface modified electrolytes. (a) The evolution process of Zn||VOx cells in ZS-KASO systems.43 Copyright @ 2025 Wiley VCH. (b) Large capacity stability by bilateral in situ functionalization regulation.44 Copyright @ 2025 Springer Nature. | ||
The synergistic modification of biphasic electrolytes not only enables the optimization of electrode interfaces, but also ensures the overall stability and high-performance of the batteries. However, to achieve this goal, electrolyte additives must be compatible with both electrode materials and the electrolyte system and must form beneficial electrochemical environments at both interfaces without inducing new side reactions. This imposes stricter requirements on the molecular design, concentration control, and mechanistic understanding of the additives. In the future, it is necessary to design multifunctional and tailored additives for different electrode materials and to combine in situ analytical techniques to thoroughly investigate the interfacial regulation mechanisms.
3.2. Electrochemical in situ polymerization of hydrogel electrolytes
Gel polymer electrolytes (GPEs), owing to their structural and functional advantages, are considered promising electrolyte candidates for AZMBs.45 GPEs aim to significantly reduce or even eliminate free water in the solvent system, thereby preventing active water molecules from reaching the Zn anode surface and triggering the HER.46 This design ensures that functional ions or molecules in the solute can directly interact with the Zn anode, rather than with water molecules. However, traditional ex situ GPE fabrication processes, including mold casting, polymerization, soaking, and assembly, are often complex, costly, and difficult to integrate with mainstream battery production lines, posing significant challenges for large-scale applications. Moreover, prefabricated gel membranes struggle to achieve intimate contact with irregular electrode surfaces, which easily leads to increased interfacial resistance. Particularly, the static interfacial design of ex situ GPEs makes it difficult to dynamically regulate Zn plating behavior and thus fails to fundamentally address the coupled failure issues associated with dendrites and side reactions. More importantly, these methods lack precise control over gel thickness and are generally incompatible with current industrial battery manufacturing processes (Fig. 7a).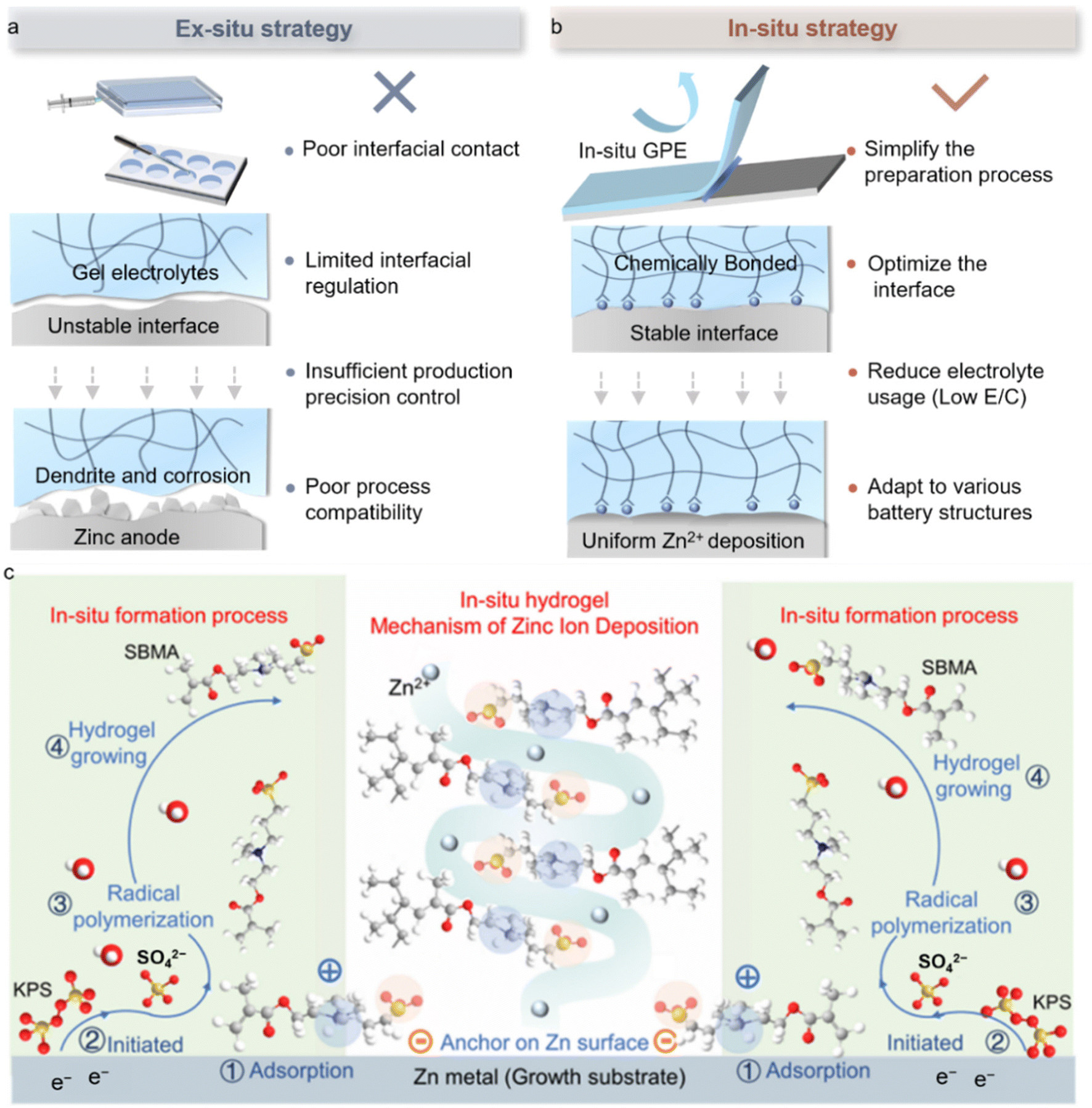 | ||
| Fig. 7 In situ polymerized gel electrolytes for high-performance AZMBs. (a) Evaluation of the ex situ polymerization mechanism of the GPE at the interface. (b) Evaluation of the in situ polymerization mechanism of the GPE at the interface. (c) In situ polymerization mechanism of the GPE at the interface.49 Copyright @ 2025 American Chemical Society. | ||
An in situ GPE offers an efficient and scalable solution to the aforementioned interfacial challenges in AZMBs. This method involves injecting a low-viscosity precursor solution into a pre-assembled cell, followed by in situ polymerization. The technique not only produces mechanically stable gel electrolytes with high ionic conductivity, but also enables adaptive chemical bonding at the electrode/electrolyte interface, significantly enhancing interfacial adhesion and dynamic stability (Fig. 7b). Notably, this technology has demonstrated clear advantages in the field of lithium-ion batteries. For example, Li et al. induced in situ cationic ring-opening polymerization of 1,3-dioxolane (DOL) with LiPF6, forming a dense gel layer on the lithium anode surface, which both suppressed dendrite growth and was compatible with high-voltage anodes.47 Xue et al. constructed continuous ion channels using a COF-based in situ strategy, increasing the ionic conductivity to 10.5 mS cm−1 and achieving dendrite-free plating for 1800 h.48 These studies indicate that in situ gelation, through the synergistic design of ion transport pathways and interfacial chemistry, can markedly improve the kinetic stability and cycling longevity of batteries.
The significance of in situ electrochemical polymerization lies in its ability to bridge fundamental scientific principles and practical battery manufacturing, thereby providing a solid foundation for the realization of high-performance batteries. For instance, Yang et al. developed an electronically-triggered self-forming in situ hydrogel electrolyte, which enables controllable vertical orientation of the gel electrolyte through interfacial chemical reconstruction.49 This unique formation mechanism results in axially aligned polymer chains, with zwitterionic functional groups precisely oriented to create highly efficient Zn2+ transport channels. In addition, the pre-adsorption effect of the in situ hydrogel electrolyte effectively suppresses excessive and disordered decomposition of electrolyte salts, forming a robust and compatible electrode/electrolyte interface with a progressive protective layer (Fig. 7c). Even under a low E/C of 17.5 μL mAh−1, the full cell retains a specific capacity of 180 mAh g−1, demonstrating the practical potential of this in situ GPE strategy in AZMBs.
In situ polymerized electrolyte technology demonstrates significant advantages in enhancing the electrode/electrolyte interface stability and safety. Its industrialization remains in the transitional phase from laboratory research to mass production. The technology faces several critical challenges in large-scale applications including precise control over polymerization processes, achieving batch-to-batch consistency, ensuring compatibility with existing production lines and addressing key technical bottlenecks in cathode material compatibility. Future breakthroughs will require collaborative innovation across the industrial chain particularly in monomer purification, standardization of polymerization processes and development of low-cost alternative materials to fully realize its application potential in advanced battery systems.
3.3. Possibility of quasi-solid or hybrid electrolytes
Introducing quasi-solid-state or hybrid electrolytes by incorporating inorganic solids such as clays or polymer hydrogels into liquid electrolytes has emerged as an effective strategy to enhance the performance of AZMBs.50 The resultant three-dimensional network structure encapsulates electrolyte molecules, which not only substantially suppresses the uncontrolled growth of Zn dendrites but also mitigates direct contact between Zn and water molecules, thereby reducing corrosion and parasitic reactions. Moreover, the porous architecture and high specific surface area of quasi-solid-state and hybrid electrolytes facilitate the orderly transport and distribution of ions, minimizing concentration polarization and improving both the rate performance and the cycling stability.51 The robust network also effectively immobilizes water molecules and ions, preventing leakage, suppressing volatilization, and reducing compositional loss, which together contribute to the improved safety and reliability of the battery system.Lead-acid batteries (LABs) employ electrode materials that are prone to corrosion by the electrolyte, leading to relatively high corrosion rates.52 In LABs, the addition of gel-forming materials such as silicon dioxide to the sulfuric acid electrolyte creates a colloidal network that decreases the activity of free acid and water, forms a physical barrier, thereby effectively curbing the loss of active material, reduces corrosion rates, and inhibits dendrite formation53 (Fig. 8a). This successful experience provides valuable inspiration for optimizing the electrolyte systems of AZMBs. The design concept of quasi-solid-state and hybrid electrolytes is highly analogous to the use of gel electrolytes in LABs to address electrode corrosion and dendrite growth. Through spatial confinement and regulation of free water, the stability and safety of the electrolyte are enhanced, the corrosion rate of the anode is reduced, and the overall cycling and energy storage performance of the system are improved.
 | ||
| Fig. 8 Application and modification mechanisms of quasi-solid-state and hybrid electrolytes in AZMBs. (a) Schematic diagram of LABs and NaAlg-based gel electrolytes with additives for LABs.52,53 Copyright @ 2020 American Association for the Advancement of Science. Copyright @ 2024 Royal Society of Chemistry. (b) Schematic illustration of the Zn2+ diffusion and transport across the PLCZ polymer electrolyte.54 Copyright @ 2024 Springer Nature. (c) Schematic of ion distribution states.55 Copyright @ 2025 Royal Society of Chemistry. (d) Water-poor quasi-solid electrolyte with proton transfer pathways in Zn–Mn batteries.56 Copyright @ 2024 Wiley VCH. | ||
Hybrid electrolytes are typically composed of a combination of liquid electrolytes, inorganic solids (such as clays, oxides, or silicates), and organic/polymer gels, forming a three-dimensional framework that effectively confines free water molecules and lowers water activity. This confinement markedly decreases direct contact between the Zn anode and water molecules, thereby significantly suppressing hydrogen evolution, Zn self-corrosion, and other side reactions, which in turn extends the lifespan of the battery. Furthermore, active functional groups on the surface of the solid components, along with their layered or porous structures, provide favorable interfaces for the uniform plating of Zn2+, preventing uncontrolled dendrite growth. For instance, Huang et al. developed a multifunctional quasi-solid-state polymer electrolyte by molecularly crosslinking sodium polyacrylate, magnesium lithium silicate, and cellulose nanofibers, yielding highly selective ion transport channels. The abundant negatively charged ion channels modulate the desolvation process of Zn2+ and promote ion transport. In addition, the in situ formation of a Zn–Mg–Si high-entropy alloy on the Zn anode improves nucleation kinetics and uniform Zn plating54 (Fig. 8b). Owing to the comprehensive advantages of the polymer electrolyte, the Zn anode exhibited an average CE of 99.7% over 2400 cycles and highly reversible cycling for up to 600 h with a DOD of 85.6%. Similarly, Zhao et al. reported a Janus-type quasi-solid-state electrolyte. The spontaneously generated built-in electric field between PVDF-HFP and PMMA polymer layers induces an ionic double layer (IDL), which effectively addresses the inherent limitations in ionic transport kinetics within solid-state systems operating under low-salt-concentration conditions.55 Benefiting from the electrolyte-constructed IDL and the derived organic outer–inorganic inner gradient SEI, effective ion rectification and transport have been achieved (Fig. 8c). Thus, Zn||Zn symmetric cells exhibited highly reversible Zn plating/stripping without dendrite growth, achieving cycle lifetimes exceeding 13300 h at 25 °C and 3000 h at 60 °C.
Moreover, the hybrid electrolyte structure optimizes ion pathways and enhances both the migration rate and uniform distribution of Zn2+. The surface charges and nanoscale channels on the inorganic/organic framework can induce directional movement and uniform deposition of ions at the electrode interface, reducing local polarization and improving the stability and controllability of electrode/electrolyte interfacial reactions. Liu et al. reported a lean-water quasi-solid-state electrolyte based on montmorillonite and Pr3+ additives. Montmorillonite, with its excellent adsorption capacity, can effectively encapsulate water molecules and cations within its layered structure, favoring the formation of an interlayer proton-conducting network56 (Fig. 8d). In this system, Pr3+ ions preferentially adsorb onto the surface and between the layers of montmorillonite, and even in a lean-water state, the strong hydration shell of Pr3+ provides a significant hydrogen-bonding network among water molecules, enabling proton conduction via the Grotthuss mechanism. This charge carrier transport mode not only avoids sluggish kinetics in water-poor states, but also suppresses other potential side reactions caused by excessively active ions or molecules. Furthermore, Pr3+ participates in interfacial Mn deposition reactions, reduces the surface potential, and helps construct interconnected pathways for proton transfer via abundant interfacial water molecules, thereby increasing the interfacial dielectric constant and enhancing the reactivity. As a result, the Cu@Zn∥α-MnO2 full cell delivers a high specific capacity of 433 mAh g−1 and excellent cycling stability over 800 cycles. The assembled Ah-level pouch cell also achieves 100 cycles after initial activation, demonstrating promising commercial potential.
Quasi-solid or hybrid electrolytes demonstrate high compatibility with existing liquid battery production lines. Their higher costs and unpredictable process stability remain bottlenecks for large-scale application. Continuous investment in material system optimization and process maturation is still required. In the transition toward all-solid-state batteries it is essential to first overcome key technical barriers including interface stability and process controllability to fully leverage their dual advantages in safety and energy density.
4. Concerns about unit cells
4.1. Balance between stability and energy density
As discussed above, conventional coin-type cells can achieve relatively long cycling stability under laboratory conditions, owing to their low areal capacities and the compensation provided by the excess electrolyte. However, in many typical cases, high-capacity pouch cells still suffer from a limited cycle life, often less than 300 cycles (Fig. 9a). Furthermore, the overall energy density of high-capacity pouch cells remains far below the requirements for practical applications (Fig. 9b). When using a high cathode loading of 20 mg cm−2 combined with typical parameters for high-capacity pouch cells (N/P ratio of 5 and E/C ratio of 10 g Ah−1), the overall energy density reaches approximately 50 Wh kg−1. Under extreme conditions with an N/P ratio of 1.05, an E/C ratio of 2.5 g Ah−1, and a maximum reported cathode loading of 50 mg cm−2, the energy density can reach about 110 Wh kg−1, approaching that of Li-LiFePO4 batteries (Fig. 9c). Current pouch cells still exhibit pronounced deficiencies in both stability and energy density, leaving a considerable gap from practical implementation. The practical challenges underlying these idealized targets, as well as the stability of cells under harsh operating conditions, have rarely been thoroughly investigated.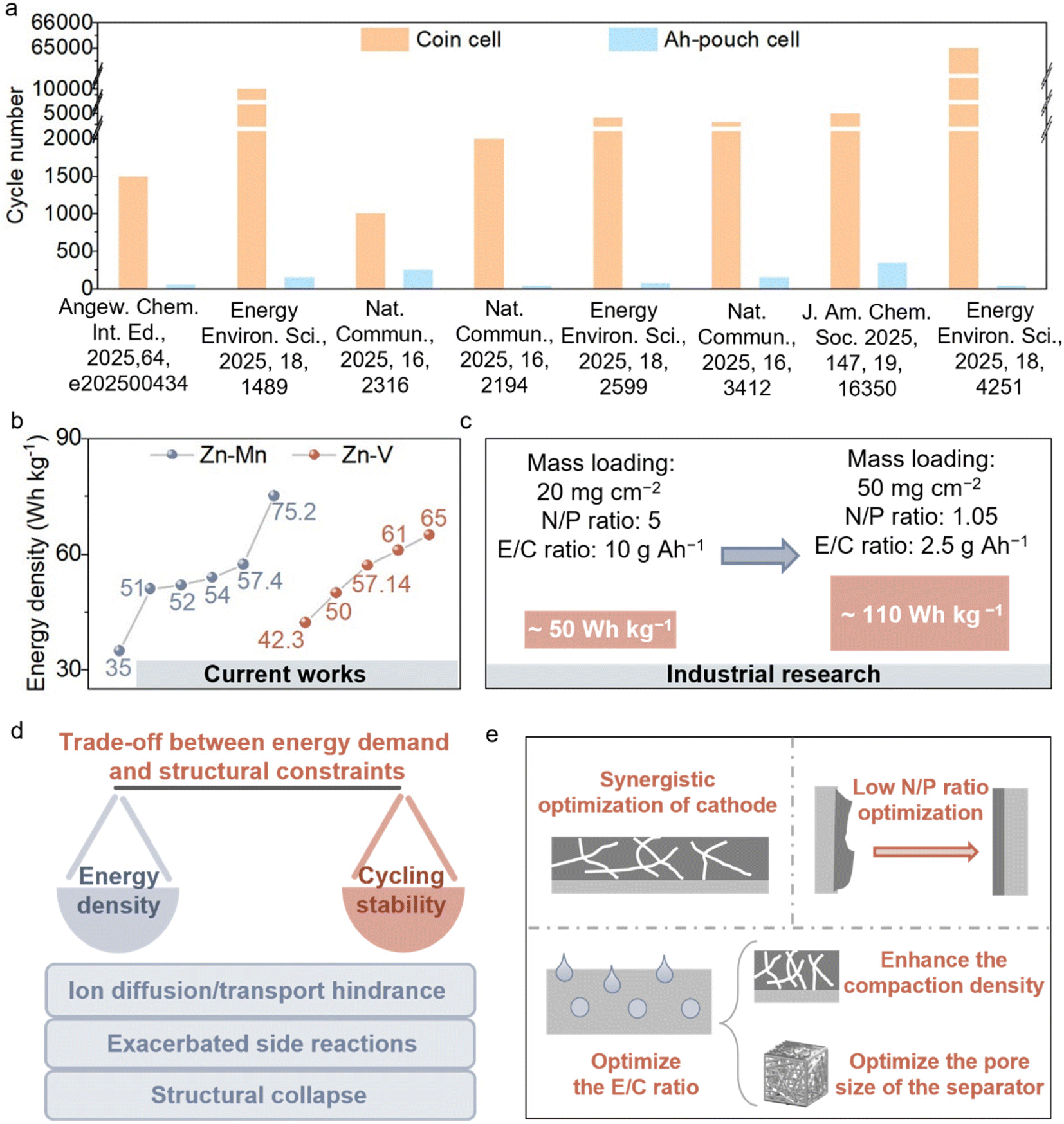 | ||
| Fig. 9 Challenges in the cycling stability of AZMBs with high energy density. (a) Summary and comparison of the stable cycle numbers of coin cells and Ah-pouch cells in current works. (b) Summary of the energy density for pouch cells in current works. (c) The theoretical energy density of AZMBs in different situations. (d) The trade-off between energy density and stability. (e) The multidimensional and synergistic optimization for achieving a balance between energy density and stability. | ||
At present, research efforts often overemphasize the pursuit of high energy density, while insufficient attention is paid to the stability of high-capacity, high-energy-density cells.57,58 In fact, stability is the prerequisite for realizing practical application of batteries. Only when reliable cycling stability of high-capacity cells under realistic working conditions is ensured does further optimization and enhancement of the energy density become meaningful. If stability is overlooked and the energy density is pursued blindly, the resulting performance is often irreproducible under practical conditions and thus cannot satisfy the requirements of engineering deployment and industrialization. Therefore, we suggest that the research focus of current AZMBs should shift toward ensuring the cycling stability of high-capacity batteries. And then, further optimization of energy densities is subsequently executed.
In practical applications, the contradiction between high energy density and cycling stability mainly arises from the conflict between energy output requirements and structural design limitations of the battery. On the one hand, enhancing the energy density often requires increasing the areal capacity and compaction density of the electrodes while minimizing the proportion of inactive components such as the electrolyte and current collector. Although this strategy can theoretically improve the gravimetric and volumetric energy output, it inevitably results in longer ion diffusion pathways, higher transport resistance, and more severe polarization. These factors exacerbate side reactions at the electrode/electrolyte interface, ultimately accelerating capacity decay. On the other hand, under high-loading conditions, electrode materials experience more pronounced volume expansion and stress concentration, which lead to structural collapse of the active materials and disruption of the conductive network, thereby further compromising the cycling stability (Fig. 9d).
Against this backdrop, achieving a balance between energy density and stability requires multidimensional and synergistic optimization (Fig. 9e). Achieving high areal capacity generally requires high-mass-loading cathodes. However, thicker electrodes inevitably hinder ion transport, decrease the ionic conductivity, and aggravate polarization. Therefore, structural and processing optimizations are essential to balance the energy density and cycling stability. Designing cathodes with hierarchical porosity, flexible frameworks, or layered architectures, combined with the optimization of conductive agents, binders, and current collectors, can effectively shorten ion diffusion pathways, reduce interfacial polarization, and maintain structural integrity. Gaberscek et al. demonstrated that adsorbing carbon black onto the surface of active material particles improved the uniformity of interfacial charge distribution, while simultaneously reducing the amount of conductive carbon and binder required, thereby enhancing the overall energy density of the cell.59 On the anode side, uneven Zn plating/stripping often leads to perforation of the Zn foil, limited Zn utilization, and severe instability at low N/P ratios. Collectively, these issues represent a critical bottleneck for improving both the energy density and cycle life. To address this, structural design and interfacial modification play vital roles. Yu et al. regulated the hydrogen-bonding network and Zn2+ coordination structure to promote more uniform Zn plating, thereby significantly enhancing the reversibility of the anode.60 They successfully assembled cells that can operate for extended periods under practical conditions (lean electrolyte: 2.95 μL mAh−1, low N/P ratio) and fabricated Zn||LiFePO4 pouch cells with a LiFePO4 loading of 506.49 mg. In addition, optimizing the E/C ratio is also crucial for maximizing the practical energy density.61 Increasing electrode compaction reduces porosity, thereby lowering electrolyte consumption.62 The rational optimization of the separator pore architecture can suppress interfacial side reactions and dendrite growth, ensuring stable operation even at high energy density.63 Notably, the introduction of solid-state or quasi-solid-state electrolytes offers new opportunities for separator-free, high-energy-density designs. Benefiting from their higher mechanical modulus, such electrolytes can more effectively suppress dendrite formation and further enhance the cycling stability.
4.2. Energy efficiency
Despite the substantial progress in extending the cycle life and achieving a high CE, the discussion of energy efficiency in AZMBs remains relatively limited. Energy efficiency is typically defined as the ratio of the energy output during discharge to the energy input during charge. It is a critical metric for assessing how effectively the energy stored in a battery or energy storage system is utilized during charge/discharge processes, reflecting how much of the input energy can be efficiently stored and subsequently released.The poor diffusivity of Zn2+ in many common cathodes and high desolvation energy result in significant overpotential issues, which are adverse for energy efficiencies.64 Taking V3O7·H2O as an example, the voltage difference between charge and discharge exceeds 0.3 V, with a charge voltage of about 0.9 V, and this voltage difference increases with rising current density (Fig. 10a). This results in an energy efficiency below 64%, decreasing further as the current density increases. In contrast, layered MnO2 performs better in terms of voltage difference (0.2 to 0.3 V) and has a relatively high charging voltage (above 1.5 V), but its energy efficiency typically remains below 86%, dropping to 81% under high current density conditions (Fig. 10b). Notably, there is almost a linear relationship between the energy efficiency and the voltage difference of a specific battery. Exploring new host materials to minimize the voltage difference between charging and discharging curves and to improve charging voltage is an effective way to enhance the energy efficiency.
 | ||
| Fig. 10 Evaluation enhancement mechanisms of energy efficiency for AZMBs. The voltage difference and energy efficiency calculated from the GCD curves together with the CEs of (a) V3O7·H2O and (b) MnO2.64 (c) Illustration of the discharge process showing the intercalation of metal ions into the cathode.64 (d) Energy efficiencies of the Zn||FeMnHCF batteries obtained with the Zn + K electrolyte, compared with energy efficiencies achieved in other AZMBs.64 Copyright @ 2022 Elsevier. (e) Mechanistic diagram of the in situ mineralization strategy.69 (f) Energy efficiency and voltage efficiency of Zn||I2@AC full cells using CMCS + KBr and IME + KBr.69 Copyright @ 2025 Wiley VCH. | ||
Specifically, the energy efficiencies of Zn–Mn, Zn–V, and Zn–I2 systems typically range from 60% to 85%, whereas lead-acid batteries have an energy efficiency of 70% to 85%. Metal Prussian blue analogues (PBAs), with their unique three-dimensional frameworks and large cavities for hosting a variety of ions, are promising candidates.65,66 The presence of tunnels facilitates ion insertion and diffusion,67 while guest ions occupying cavity or vacancy sites in the framework can yield diverse electrochemical properties including voltage plateaus and overpotential (Fig. 10c). Based on this concept,68 Wang et al. developed a rhombohedral Fe0.35Mn0.65 [Fe(CN)6] (FeMnHCF) cathode that exhibits a small overpotential within 0.2 V and a high charge voltage above 1.9 V, resulting in an energy efficiency exceeding 89% (Fig. 10d).
Electrolytes play a critical role in battery systems, especially as interfacial polarization between electrolytes and electrodes directly impacts both the energy density and efficiency. Precise tuning of electrolyte composition can dynamically regulate the electrode/electrolyte interface. The goal of electrolyte optimization is to enhance the ionic conductivity while simultaneously reducing water activity and polarization. Yao et al. implemented an in situ mineralization strategy to uniformly self-assemble electrochemically active PBAs within a hydrogel electrolyte, thus regulating the interfacial reaction kinetics69 (Fig. 10e). Specifically, ZnHCF served as a mediator to effectively suppress the shuttle effect of multi-halide and bromine species, further improving the cycling stability and energy efficiency. The Zn||I2@AC full cell employing IME + KBr achieved an average energy efficiency of up to 86.73% and an average voltage efficiency of 89.40% over 1000 cycles (Fig. 10f).
4.3. Self-discharge performance
Recently, a range of cutting-edge technologies and advanced tools have been developed to promote the design of high-performance cathodes, long-life Zn metal anodes, and functional electrolytes for AZMBs. However, in laboratory environments, cells are often subjected to continuous charging in a constant discharge state, a method that obscures the negative impact of self-discharge on battery stability. In reality, cells frequently exist in an open-circuit static state. If the self-discharge rate of a cell is too high, it can lead to significant energy loss during dormancy which not only affects the efficiency of cells but also complicates capacity management in cell packs. Thus, the self-discharge index is crucial in practical applications. The intrinsic challenge of refractory self-discharge has emerged as a neglected drawback of AZMBs, imposing severe limitations on practical applications.Self-discharge represents the natural energy loss of an idle cell, leading to declines in voltage and capacity. Thermodynamically, a fully charged cell exists in a state of high Gibbs free energy, and over time, the energy decreases, resulting in spontaneous self-discharge. The general electrochemical self-discharge mechanism proposed by Conway indicates that the self-discharge of energy storage devices (EESDs) primarily arises from ohmic leakage, charge redistribution, and faradaic reactions.70 Ohmic leakage occurs due to incomplete isolation between the cathode and anode. The charge redistribution is related to the concentration gradient of adsorbed ions. The faradaic reaction pertains to the redox reactions between the electrolyte and electrodes. The self-discharge characteristics of different EESDs are influenced by various factors,71,72 including the composition of cathode and anode materials, the type of electrolyte used, crosstalk effects between components, and operating conditions.73,74
On the cathode side, the dissolution of active materials and uncontrollable side reactions can lead to the formation of unstable electrode/electrolyte interfaces, resulting in severe self-discharge and greatly shortening the calendar life of the battery. This, in turn, undermines the long-term feasibility of AZMBs in demanding applications.75,76 By modulating the electrolyte to restrict the activity of free water molecules and forming a highly concentrated electrolyte or a water/organic hybrid electrolyte, the risk of calendar aging can be effectively reduced. Chen et al. developed a hybrid electrolyte by introducing PEG 400 as a co-solvent.77 PEG 400, which contains numerous nucleophilic groups, can immobilize free water molecules in the electrolyte, thereby alleviating water-induced side reactions. A reliable testing mode encompassing cycling performance and anti-self-discharge resistance was employed to simulate the stability of Zn||HVO batteries in actual application environments with intermittent usage characteristics. During continuous charge/discharge cycling, the cells with an aqueous electrolyte exhibit high-capacity retention. However, their capacity drops sharply after a resting period (Fig. 11a). In contrast, batteries with the hybrid electrolyte maintain good capacity retention both during continuous cycling and after resting. Notably, after the first 24 h shelving period, the CE of the hybrid electrolyte battery reaches 97.1%, which is much higher than that of the cell with the aqueous electrolyte (Fig. 11b). It is worth noting that the voltage–time profile of the battery with the hybrid electrolyte initially shows a steep drop, followed by a subsequent flat trend during shelving. However, the batteries with the aqueous electrolyte exhibit a sloped decrease corresponding to the self-discharge process, which can be attributed to cathode dissolution and self-discharge.
 | ||
| Fig. 11 The self-discharge mechanism and modification strategy of AZMBs. Storage performance comparison of Zn||HVO batteries evaluated by resting for 24 h at 100% state of charge (SOC) after 8 cycles at 0.5 A g−1, followed by full discharging: (a) 1 M ZOT-H2O and (b) 1 M ZOT-H2O-60% PEG.77 Insets show the charge–discharge curves during resting for 24 h in the fully charged state after the first 8 cycles at 0.5 A g−1. Copyright @ 2022 Wiley VCH. (c) The ratio of calendar aging-induced loss to total capacity loss (Rcld, left Y-axis) and the intrinsic hydrogen generation rate (HGR, right Y-axis) after 12 h of aging in Zn||Cu half-cells.78 Copyright @ 2025 Wiley VCH. (d) Comparison between laboratory and industrial evaluation tests of self-discharge and testing indicators. | ||
Moreover, the low electrode potential of Zn and the narrow ESW of aqueous electrolytes can trigger corrosion processes during self-discharge and calendar aging. Pu et al. investigated the Zn anode in weakly acidic 2 M ZnSO4 electrolyte during calendar aging. Their research found that battery aging leads to significant capacity loss of the Zn anode, with the efficiency loss exceeding 70% after just 24 h of aging in some cases.78 Gas bubbles formed and accumulated at the Zn anode/electrolyte interface displace the electrolyte from certain regions, causing a loss of ionic contact and thus substantial capacity loss in AZMBs. The cumulative effects of corrosion and “dead Zn” formation were shown to be detrimental to cycling stability.79 Furthermore, Wang et al. demonstrated that the influence of anions on plating morphology plays a crucial role in capacity degradation induced by calendar aging.80 The ratio of capacity loss caused by calendar aging to the total capacity loss, defined as Rcld, was used to further compare several common electrolytes containing representative anions. After 12 h of aging, the OTf− electrolyte exhibited a capacity loss of 1.29 mAh cm−2 (32.3%), whereas the Br− electrolyte showed a moderate capacity loss of 0.21 mAh cm−2 (5.3%). In contrast, the OAc− electrolyte demonstrated the least capacity loss, with only 0.02 mAh cm−2 (0.5%). For most electrolytes, Rcld exceeded 50%, with the OTf−-based electrolyte displaying a particularly high Rcld of 1.21 mAh cm−2 (93.8%). In addition, the intrinsic corrosion rate was quantified by measuring the hydrogen generation rate (HGR) using polished Zn foils with comparable surface areas. The results revealed that, although the OTf− electrolyte had the lowest HGR, it exhibited a significant capacity loss. In contrast, the SO42− electrolyte showed the most severe intrinsic corrosion, yet it did not lead to substantial capacity loss. Notably, the acetate electrolyte not only presented the lowest capacity loss, but also maintained a moderate intrinsic HGR (Fig. 11c).
Researchers should concentrate on investigating the self-discharge performance and optimizing testing metrics to assess the industrial stability of batteries (Fig. 11d). For instance, conducting random self-discharge tests during battery cycling can simulate real conditions. Current self-discharge tests in laboratories typically involve cycling a new cell several times, followed by a prolonged period in a charged state (24–48 h), and then discharging. Most self-discharge tests conducted today are independent and do not capture the self-discharge performance of batteries after a certain usage period.81 Additionally, this type of testing tends to use coin cells, and for pouch cells with high mass loadings, especially Ah-level pouch cells, the reference value is limited due to the more complex degradation dynamics associated with high mass loadings.
5. Summary and perspectives
This review systematically summarizes the core challenges encountered in AZMBs in terms of electrodes, electrolytes, and unit cells. Diverse strategies for achieving high-capacity batteries are discussed, including the construction of high-areal-capacity Zn anodes and high-loading cathodes, innovations in interface engineering, and the development of advanced electrolyte systems, with scientific analysis of several promising technological routes and optimization approaches. Furthermore, a detailed comparison of key performance indicators such as energy density, energy efficiency, and self-discharge is provided, highlighting the significant differences between laboratory batteries and those designed for practical deployment. In the end, the following insights may offer scientific value for facilitating the advancement of large-capacity AZMBs.Synergistic strategies
In the pursuit of enhanced stability for high-loading, high-areal-capacity AZMBs, synergistic strategies are particularly crucial. By simultaneously optimizing materials, electrolytes, and processing techniques across multiple dimensions, the limitations of single modification approaches can be effectively overcome. For instance, the combination of electrode material structural engineering and the rational design of electrolytes that form compatible and stable interfaces with electrodes further extends the cycle life and enhances the energy efficiency of the overall battery system. In addition, the proper integration of cell component architectures with manufacturing processes also plays a vital role in ensuring performance and stability under high-areal-capacity conditions. Therefore, the future development of AZMBs should emphasize multi-disciplinary and multi-scale collaborative innovation, promoting the integrated design of materials, electrolytes, structures, and manufacturing techniques. Such a comprehensive approach will provide a solid foundation for achieving high-performance batteries suitable for practical applications.Establishing unified evaluation protocols
Currently, whether for high-capacity coin cells or pouch cells, the evaluation standards for electrode materials, electrolyte functions, battery performance, and testing conditions remain limited and lack uniformity. There is no unified standard for assessment ranges and performance testing conditions. We recommend establishing a standardized evaluation framework that incorporates high-loading cathodes exceeding 20 mg cm−2, thin Zn foil anodes below 50 μm, N/P ratios under 5, E/C ratios below 10 g Ah−1, and practically relevant current rates ranging from C/4 to C/6. Researchers should prioritize the systematic investigation of the cycling performance under these combined parameters as a critical research direction. Developing such a comprehensive and practical evaluation system is essential for advancing AZMBs from laboratory research to practical applications.Process transparency for Ah-scale batteries
With the increasing focus on the development of Ah-scale AZMBs, the transparency and standardization of fabrication processes have become common requirements for both academia and industry. However, most of the current literature lacks detailed information on the production of Ah-scale AZMBs. Therefore, we strongly advocate and encourage research articles to share non-commercially sensitive details of their fabrication procedures, including explicit parameters for material processing, electrode preparation, and cell assembly. Such transparency will enable meaningful cross-comparison of technologies, scientific validation, and continuous innovation, ultimately accelerating the practical application and industrialization of next-generation high-performance AZMBs.Establishing a scientific institution–enterprise cooperation model
At present, scientific institution research on Ah-scale AZMBs primarily focuses on fundamental science and laboratory-scale validation. This approach often lacks an in-depth understanding of practical application scenarios and has limited access to industrial-grade materials and equipment, thereby restricting the efficiency of technology transfer. At the same time, universities or research institutes also lack corresponding industrialization conditions. To overcome these bottlenecks and accelerate the implementation of large-scale energy storage, it is recommended to strengthen collaboration between scientific institutions and enterprises. The in-depth scientific institution–enterprise cooperation model not only facilitates the industrialization of research outcomes, but also enables the adjustment of research directions based on practical needs, thereby enhancing both the relevance and applicability of research work. Ultimately, such collaboration will promote the technological advancements and industrial development of AZMBs for large-scale energy storage applications.Author contributions
M. Li gathered information and wrote the manuscript. S. Liang and G. Fang supervised the research. All authors contributed to the paper.Conflicts of interest
There are no conflicts to declare.Data availability
All data supporting the findings of this review are available within the article.Acknowledgements
This work was supported by the National Natural Science Foundation of China (92472116) and Songshan Lake Materials Laboratory Open Research Topics (2023SLABFK05).References
- J. F. Parker, J. S. Ko, D. R. Rolison and J. W. Long, Joule, 2018, 2, 2519–2527 CrossRef.
- L. E. Blanc, D. Kundu and L. F. Nazar, Joule, 2020, 4, 771–799 CrossRef.
- Y. D. Hu, P. Y. Wang, M. Z. Li, Z. X. Liu, S. Q. Liang and G. Z. Fang, Energy Environ. Sci., 2024, 17, 8078–8093 RSC.
- S. W. D. Gourley, R. Brown, B. D. Adams and D. Higgins, Joule, 2023, 7, 1415–1436 CrossRef.
- C. Li, S. Jin, L. A. Archer and L. F. Nazar, Joule, 2022, 6, 1733–1738 CrossRef.
- Y. Shang and D. Kundu, Joule, 2023, 7, 244–250 CrossRef.
- J. Feng, X. Li, Y. Ouyang, H. Zhao, N. Li, K. Xi, J. Liang and S. Ding, Angew. Chem., Int. Ed., 2024, 63, e202407194 CrossRef PubMed.
- L. Ma, M. A. Schroeder, O. Borodin, T. P. Pollard, M. S. Ding, C. Wang and K. Xu, Nat. Energy, 2020, 5, 743–749 CrossRef.
- Q. Li, A. Chen, D. Wang, Y. Zhao, X. Wang, X. Jin, B. Xiong and C. Zhi, Nat. Commun., 2022, 13, 3699 CrossRef PubMed.
- H. Jiang, L. Tang, Y. Fu, S. Wang, S. K. Sandstrom, A. M. Scida, G. Li, D. Hoang, J. J. Hong, N.-C. Chiu, K. C. Stylianou, W. F. Stickle, D. Wang, J. Li, P. A. Greaney, C. Fang and X. Ji, Nat. Sustain., 2023, 6, 806–815 CrossRef.
- Z. Wu, Y. Li and J. Liu, Small Methods, 2024, 8, 2300660 CrossRef.
- J. Sun, X. Zheng, Z. Zhu, M. Wang, Y. Xu, K. Li, Y. Yuan, M. Chuai, Z. Liu, T. Jiang, H. Hu and W. Chen, Nano Lett., 2025, 25, 7266–7275 CrossRef.
- N. Dong, F. Zhang and H. Pan, Chem. Sci., 2022, 13, 8243–8252 RSC.
- Z. Wu, Y. Wang and C. Zhi, Joule, 2024, 8, 2442–2448 CrossRef.
- L. Ma, M. A. Schroeder, T. P. Pollard, O. Borodin, M. S. Ding, R. Sun, L. Cao, J. Ho, D. R. Baker, C. Wang and K. Xu, Energy Environ. Mater., 2020, 3, 516–521 CrossRef.
- B. D. Adams, J. Zheng, X. Ren, W. Xu and J.-G. Zhang, Adv. Energy Mater., 2018, 8, 1702097 CrossRef.
- X. Zhang, L. Zhang, X. Jia, W. Song and Y. Liu, Nano-Micro Lett., 2024, 16, 75 CrossRef PubMed.
- S. Chen, Y. Xia, R. Zeng, Z. Luo, X. Wu, X. Hu, J. Lu, E. Gazit, H. Pan, Z. Hong, M. Yan, K. Tao and Y. Jiang, Sci. Adv., 2024, 10, eadn2265 CrossRef PubMed.
- H. Gan, H. Li, M. Xu, C. Han and H.-M. Cheng, Joule, 2024, 8, 3054–3071 CrossRef.
- H. Yan, S. Li, J. Zhong and B. Li, Nano-Micro Lett., 2023, 16, 15 CrossRef PubMed.
- L. Re, Z. Hu, C. Peng, L. Zhang, N. Wang, F. Wang, Y. Xia, S. Zhang, E. Hu and J. Luo, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2309981121 CrossRef PubMed.
- M. Zhou, S. Guo, J. L. Li, X. B. Luo, Z. X. Liu, T. S. Zhang, X. X. Cao, M. Q. Long, B. G. Lu, A. Q. Pan, G. Z. Fang, J. Zhou and S. Q. Liang, Adv. Mater., 2021, 33, 2100187 CrossRef PubMed.
- X. T. Zhang, J. X. Li, Y. F. Liu, B. A. Lu, S. Q. Liang and J. Zhou, Nat. Commun., 2024, 15, 2735 CrossRef PubMed.
- X. Li, W. Yuan, Y. Wang, X. Bao, T. Bi, D. Cui, Z. Yang, G. Ma, Y. Wang, Z. Shen and N. Zhang, ACS Nano, 2025, 19, 14484–14498 CrossRef CAS PubMed.
- Q. Li, D. Luo, Q. Ma, Z. Zheng, S. Li, Y. Xie, L. Xue, M. Lin, Y. Nie, G. Feng, H. Dou, J. Chen, X. Wang and Z. Chen, Energy Environ. Sci., 2025, 18, 1489–1501 RSC.
- R. Zhang, T. Shui, A. Li, H. Xia, G. Xu, L. Ji, C. Lu, W. Zhang and Z. Sun, Energy Environ. Sci., 2025, 18, 1011–1026 RSC.
- Z.-H. Huang, Y. Song, D.-Y. Feng, Z. Sun, X. Sun and X.-X. Liu, ACS Nano, 2018, 12, 3557–3567 CrossRef CAS.
- S. Liu, R. Zhang, C. Wang, J. Mao, D. Chao, C. Zhang, S. Zhang and Z. Guo, Angew. Chem., Int. Ed., 2024, 63, e202400045 CrossRef CAS PubMed.
- J. Wan, H. Song, J. Tian, S. Zhong and J. Liu, Energy Mater., 2025, 5, 500090 CAS.
- X. Dou, X. Xie, S. Liang and G. Fang, Sci. Bull., 2024, 69, 833–845 CrossRef CAS PubMed.
- L. Yang, Y.-J. Zhu, F. Zeng, H.-P. Yu, L.-Y. Dong, J. Tao, G. He and H. Li, Energy Storage Mater., 2024, 65, 103162 CrossRef.
- M. Zhu, R. Gao, Q. Ran, S.-g. Gong, Q. Li, S.-P. Zeng, H. Huang, L. Hu, D. Yang, T. Dai, Y. Wang, D. Chao, M. Feng and Z. Chen, Angew. Chem., Int. Ed., 2025, 64, e202425080 CrossRef CAS PubMed.
- K. Fang, F. Li, G.-G. Wang, Y.-L. Liu, M.-L. Tan, D.-Q. Zhao, H.-Y. Zhang and J.-C. Han, J. Mater. Sci. Technol., 2023, 143, 84–92 CrossRef.
- Q. Zhang, J. Zhao, X. Chen, R. Yang, T. Ying, C. Cheng, B. Liu, J. Fan, S. Li and Z. Zeng, Adv. Funct. Mater., 2024, 34, 2306652 CrossRef.
- X. Gao, C. Shen, H. Dong, Y. Dai, P. Jiang, I. P. Parkin, H. Zhang, C. J. Carmalt and G. He, Energy Environ. Sci., 2024, 17, 2287–2297 RSC.
- J. Chacón-Borrero, X. Chang, Z. Min, J. Yu, G. Montaña-Mora, K. V. Mejia-Centeno, Y. Sun, X. Zhou, S. Tunmee, P. Kidkhunthod, J. Li, J. Llorca, J. Arbiol and A. Cabot, Energy Storage Mater., 2025, 81, 104486 CrossRef.
- S. Wang, X. Guo, K. Huang, A. Achari, J. Safaei, Y. Lei, D. Li, Q. Gu, C. Sun, L. Gloag, S. Langford, A. Geim, R. R. Nair and G. Wang, Nat. Commun., 2025, 16, 5191 CrossRef.
- Q. Li, M. Xu, S. Wei, A. Kumar, K. K. Abdalla, Y. Wang, L. Yu, M. Liu, X. Jin, J. Li, L. Song, Y. Zhao and X. Sun, Energy Environ. Sci., 2025, 18, 7939–7949 RSC.
- H. Wu, S.-J. Zhang, J. Vongsvivut, M. Jaroniec, J. Hao and S.-Z. Qiao, Joule, 2025, 9, 102000 CrossRef.
- Y. Wang, Y. Cui, M. Zhao, J. Wang, X. Liu, Y. Zhu and Y. Yang, Nat. Commun., 2025, 16, 5565 CrossRef PubMed.
- T. Xiao, J.-L. Yang, R. J. Xu, H. Xu, H. Liu, J. Li, H. Bao, X. Jin, S.-J. Hwang, Z. Wang and H. J. Fan, J. Am. Chem. Soc., 2025, 147, 28820–28830 CrossRef.
- X. Yang, X. Wang, Y. Xiang, L. Ma and W. Huang, Nano-Micro Lett., 2023, 16, 51 CrossRef PubMed.
- L. He, C. Lin, L. Zeng, F. Xiao, H. Lin, P. Xiong, Q. Qian, Q. Chen, Z. Yan and J. Chen, Angew. Chem., Int. Ed., 2025, 64, e202415221 CrossRef PubMed.
- Y. Song, M. Chen, Z. Zhong, Z. Liu, S. Liang and G. Fang, Nat. Commun., 2025, 16, 3142 CrossRef.
- Q. He, Y. Zhong, J. Li, S. Chai, Y. Yang, S. Liang, Z. Chang, G. Fang and A. Pan, Adv. Energy Mater., 2024, 14, 2400170 CrossRef.
- Q. He, Z. Chang, Y. Zhong, S. Chai, C. Fu, S. Liang, G. Fang and A. Pan, ACS Energy Lett., 2023, 8, 5253–5263 CrossRef.
- S. Li, H. Hong, X. Yang, D. Li, Q. Xiong, D. Zhang, S. Wang, Z. Huang, H. Lv and C. Zhi, Adv. Mater., 2025, 37, 2504333 CrossRef PubMed.
- J. Meng, M. Yin, K. Guo, X. Zhou and Z. Xue, Nano-Micro Lett., 2025, 17, 248 CrossRef PubMed.
- Y. Yang, Q. He, C. Hu, X. Xie, S. Liang, Z. Zeng and G. Fang, ACS Nano, 2025, 19, 21717–21728 CrossRef PubMed.
- Y. Hu, Z. Liu, L. Li, S. Guo, X. Xie, Z. Luo, G. Fang and S. Liang, Natl. Sci. Rev., 2023, 10, nwad220 CrossRef PubMed.
- Y. Pan, Z. Liu, S. Liu, L. Qin, Y. Yang, M. Zhou, Y. Sun, X. Cao, S. Liang and G. Fang, Adv. Energy Mater., 2023, 13, 202203766 Search PubMed.
- P. P. Lopes and V. R. Stamenkovic, Science, 2020, 369, 923–924 CrossRef CAS PubMed.
- K. Pan, G. Shi, A. Li, H. Li, R. Zhao, F. Wang, W. Zhang, Q. Chen, H. Chen, Z. Xiong and D. Finlow, J. Power Sources, 2012, 209, 262–268 CrossRef CAS.
- C. Yang, P. Woottapanit, S. Geng, R. Chanajaree, Y. Shen, K. Lolupiman, W. Limphirat, T. Pakornchote, T. Bovornratanaraks, X. Zhang, J. Qin and Y. Huang, Nat. Commun., 2025, 16, 183 CrossRef PubMed.
- S. Zhao, S. Yang, X. Huang, X. Wang, H. Xu, Q. Ma, Y. Chen, G. Wang and S. Chen, Energy Environ. Sci., 2025, 18, 8618–8630 RSC.
- Z. Liu, M. Qin, B. Fu, M. Li, S. Liang and G. Fang, Angew. Chem., Int. Ed., 2025, 64, e202417049 CrossRef CAS.
- C.-C. Kao, C. Ye, J. Hao, Y. Chen, S.-J. Zhang and S.-Z. Qiao, Adv. Energy Mater., 2025, 15, 2501201 CrossRef CAS.
- M. Tang, Q. Liu, X. Zou, B. Zhang and L. An, Adv. Mater., 2025, 37, 2501361 CrossRef.
- R. Dominko, M. Gaberscek, J. Drofenik, M. Bele, S. Pejovnik and J. Jamnik, J. Power Sources, 2003, 119, 770–773 CrossRef.
- J. Wu, X. Zhang, Z. Ju, L. Wang, Z. Hui, K. Mayilvahanan, K. J. Takeuchi, A. C. Marschilok, A. C. West, E. S. Takeuchi and G. Yu, Adv. Mater., 2021, 33, 2101275 CrossRef PubMed.
- J. Wang, Y. Liu, Q. Cai, A. Dong, D. Yang and D. Zhao, Adv. Mater., 2022, 34, 2107957 CrossRef.
- Y. Ma, Q. Zhang, L. Liu, Y. Li, H. Li, Z. Yan and J. Chen, Natl. Sci. Rev., 2022, 9, nwac051 CrossRef.
- R. Guo, Y. Bao, X. Zheng, W. Zhang, C. Liu, J. Chen, J. Xu, L. Wang and J. Ma, Adv. Funct. Mater., 2023, 33, 2213283 CrossRef.
- D. Wang, C. Li, Q. Li, H. Li, J. Rehman, C. Zhi and L. Zhu, Nano Energy, 2022, 104, 107990 CrossRef.
- V. Renman, D. O. Ojwang, M. Valvo, C. P. Gómez, T. Gustafsson and G. Svensson, J. Power Sources, 2017, 369, 146–153 CrossRef.
- M. S. Chae, J. W. Heo, H. H. Kwak, H. Lee and S.-T. Hong, J. Power Sources, 2017, 337, 204–211 CrossRef.
- L. Jiang, Y. Lu, C. Zhao, L. Liu, J. Zhang, Q. Zhang, X. Shen, J. Zhao, X. Yu, H. Li, X. Huang, L. Chen and Y.-S. Hu, Nat. Energy, 2019, 4, 495–503 CrossRef.
- W. Deng, Z. Li, Y. Ye, Z. Zhou, Y. Li, M. Zhang, X. Yuan, J. Hu, W. Zhao, Z. Huang, C. Li, H. Chen, J. Zheng and R. Li, Adv. Energy Mater., 2021, 11, 2003639 CrossRef.
- L. Yao, L. Jiang, Y. Wang, X. Chi and Y. Liu, Adv. Mater., 2025, 37, 2505342 Search PubMed.
- B. E. Conway, W. G. Pell and T. C. Liu, J. Power Sources, 1997, 65, 53–59 CrossRef.
- C.-J. Huang, B. Thirumalraj, H.-C. Tao, K. N. Shitaw, H. Sutiono, T. T. Hagos, T. T. Beyene, L.-M. Kuo, C.-C. Wang, S.-H. Wu, W.-N. Su and B. J. Hwang, Nat. Commun., 2021, 12, 1452 CrossRef PubMed.
- R. Yazami and Y. F. Reynier, Electrochim. Acta, 2002, 47, 1217–1223 CrossRef.
- P. Gerlach and A. Balducci, Electrochim. Acta, 2021, 377, 138070 CrossRef.
- S.-E. Chun, B. Evanko, X. Wang, D. Vonlanthen, X. Ji, G. D. Stucky and S. W. Boettcher, Nat. Commun., 2015, 6, 7818 CrossRef PubMed.
- M. Yan, P. He, Y. Chen, S. Wang, Q. Wei, K. Zhao, X. Xu, Q. An, Y. Shuang, Y. Shao, K. T. Mueller, L. Mai, J. Liu and J. Yang, Adv. Mater., 2018, 30, 1703725 CrossRef.
- J. Janek and W. G. Zeier, Nat. Energy, 2016, 1, 16141 CrossRef.
- Y. Chen, S. Guo, L. Qin, Q. Wan, Y. Pan, M. Zhou, M. Long, G. Fang and S. Liang, Batteries Supercaps, 2022, 5, e202200001 CrossRef.
- S. D. Pu, B. Hu, Z. Li, Y. Yuan, C. Gong, Z. Ning, C. Chau, S. Yang, S. Zhang, L. Pi, Y. T. Tang, J. Yue, T. J. Marrow, X. Gao, P. G. Bruce and A. W. Robertson, Joule, 2023, 7, 366–379 CrossRef.
- B. Liu, X. Yuan and Y. Li, ACS Energy Lett., 2023, 8, 3820–3828 CrossRef.
- J. Wang, B. Zhang, S. Luo, X. Huang, A. Duan, H. Chen and W. Sun, Angew. Chem., Int. Ed., 2025, 64, e202510354 CrossRef PubMed.
- W. M. Seong, K.-Y. Park, M. H. Lee, S. Moon, K. Oh, H. Park, S. Lee and K. Kang, Energy Environ. Sci., 2018, 11, 970–978 RSC.
| This journal is © The Royal Society of Chemistry 2026 |