DBMLFF: linear scaling machine learning force fields via electron density decomposition for molecular electrolytes
Received
17th November 2025
, Accepted 19th January 2026
First published on 19th January 2026
Abstract
Machine learning force fields (MLFFs) are rapidly evolving to provide molecular dynamics simulations of molecules and materials with an accuracy comparable to ab initio methods, while significantly reducing the need for computational resources. However, conventional MLFFs are generally system-specific; introducing new chemical components requires assembling a new training dataset and retraining the entire model from scratch. To address this, we present a density-based machine learning force field (DBMLFF). The key advantage of DBMLFF lies in its modular parametrization strategy: by modeling each molecular species independently, the resulting force fields achieve seamless transferability across diverse chemical environments and retaining high accuracy without the need for retraining. This significantly improves model portability and cross-system applicability. Unlike most of the statistically based MLFFs, DBMLFF is a physics-based force field with machine learning components in it. It computes intermolecular interactions directly from electron density, enabling accurate descriptions of complex non-bonded behavior. In terms of computational efficiency, DBMLFF is three orders of magnitude faster than ab initio molecular dynamics and exhibits linear scaling with system size, allowing efficient simulations of large-scale systems. These features make DBMLFF a robust tool for multi-component electrolyte MD simulations, ideal for practical electrochemical systems with variable compositions and large scales.
1 Introduction
Under the global transition in energy infrastructure, lithium-ion batteries, as core components of next-generation energy storage systems, have become a critical research focus in computational materials science.1 While commercial lithium-ion batteries demonstrate satisfactory energy density and cycling stability, their rate capability and safety margins remain inadequate for rapidly evolving application demands.2 The key to overcoming this technical bottleneck lies in a fundamental understanding of electrolyte micro-mechanisms. As the ion-transport medium in battery systems, electrolytes not only govern lithium-ion migration kinetics but also critically influence electrode interfacial stability and thermal behavior through the dynamic evolution of solvation structures.3 Consequently, establishing precise multiscale characterization methods for electrolyte systems has emerged as a pivotal scientific challenge in developing high-safety, long-lifetime energy storage devices.4–6
Molecular dynamics (MD) simulations provide a unique perspective for elucidating microscopic interactions in electrolyte systems.7,8 By constructing atomic-scale interaction models, this technique enables the resolution of activation energy barriers for lithium-ion transport, dynamic reorganization of solvent coordination structures, and formation processes of interfacial electric double layers.9–11 However, conventional simulation approaches exhibit notable limitations: classical MD based on empirical potentials fails to accurately capture complex polarization effects between ions and solvents, often resulting in systematic deviations from experimental observations. Meanwhile, ab initio MD (AIMD), while offering higher accuracy, faces computational constraints that preclude simulations at practical operational timescales.12,13 This inherent trade-off between precision and efficiency significantly hinders in-depth investigations of electrolyte systems.
The emerging machine learning force field (MLFF) technology presents a novel solution to this dilemma.14–17 By integrating quantum chemical calculations with machine learning algorithms, MLFF achieves atomic-scale accuracy while dramatically enhancing computational efficiency.18–20 However, existing MLFF methods still face challenges in engineering applications.21,22 Current MLFFs typically exhibit a “system-specific nature,” wherein any change in system composition (such as the introduction of new components) necessitates computationally expensive retraining of the entire force field.23,24 This limited scalability poses a major bottleneck in the development of multi-component electrolyte systems.25,26 On the other hand, while general-purpose MLFFs (e.g., MACE, materials adaptive convolutional equivariants)27 demonstrate potential in terms of broad coverage, their predictive accuracy in complex molecular systems (such as electrolytes) still requires further improvement. For example, there are cases where the MD trajectories using such big models diverge, producing unphysical atomic configurations, and due to the large set of parameters, such models become slow and incapable of simulating large systems. This leaves the field grappling with the trade-off between generality and accuracy. Moreover, existing MLFFs typically assume that energy depends solely on local atomic environments, limiting their ability to accurately capture long-range electrostatic interactions.28,29 The predominant solution separates long-range electrostatic energy, commonly using point charge models, from the machine learning-fitted short-range energy. Other strategies such as environment-dependent charges30 or neural-network-predicted electronegativities31 have also been proposed. However, these still rely on the point charge approximation, resembling classical force fields.32 While the point charge model performs well at large intermolecular distances without electron density overlap, its accuracy degrades significantly at intermediate distances where electron cloud overlap becomes non-negligible. To address this limitation, our work introduces an explicit representation of electron charge density, aiming to overcome the inherent constraints of the point charge model in this regime.
To address these challenges, we developed the density-based machine learning force field (DBMLFF) simulation package for energy and force prediction tasks, as well as MD calculations.33 DBMLFF calculates intermolecular interactions based on fitted electron charge densities, while intramolecular energies are modeled using traditional atomic structure-based MLFF models. In addition, a local potential polarization model based on charge density calculations is introduced to describe polarization interactions between molecules. The methodology advances the field in two key aspects: first, it enables independent parametrization of individual molecular components in electrolyte systems. Following this, each molecular model can be seamlessly transferred across different chemical environments without retraining, significantly improving transferability and applicability. Second, by incorporating electron density distribution as a fundamental descriptor, DBMLFF achieves marked improvements in modeling non-bonded interactions, reaching quantum-chemical accuracy. Validations on typical electrolyte systems demonstrate that DBMLFF not only accurately captures atomic-scale solvation structures but also delivers a computational efficiency three orders of magnitude higher than AIMD simulations. This combination of accuracy, transferability, and efficiency opens a new technical pathway for investigating mesoscale electrolyte reaction kinetics.
2 Methods
2.1 Computational details
All AIMD, DBMLFF and density functional theory (DFT) calculations were carried out using the PWmat code,34,35 a plane-wave pseudopotential package optimized for GPU acceleration through algorithmic reconstruction. The norm-conserving SG15 pseudopotential36,37 was employed with the local density approximation (LDA)38 for exchange–correlation interactions. Although it will be straightforward to change the exchange–correlation functional to other semilocal functionals like the generalized gradient approximation (GGA),39 LDA offers the simplest functional to test our charge density based approach. Kohn–Sham wavefunctions were expanded in plane-wave basis sets, with a kinetic energy cutoff of 120 Rydberg for the charge density grid.
Classical molecular dynamics simulations utilized GROMACS40 with the optimized potential for liquid simulations for all atoms (OPLS-AA) force field.41 Force field parameters were generated using Ligpargen,42 and initial atomic configurations were constructed via PACKMOL.43 Long-range electrostatic interactions were treated by the particle–particle particle–mesh (PPPM) method,44 while van der Waals interactions were modeled with Lennard-Jones potentials. Energy minimization using the conjugate gradient algorithm was performed to eliminate unphysical configurations in initial structures.
The self-diffusion coefficients of ions were calculated from the uncorrelated mean square displacements (MSDs) of individual ions using the following equation.45
| |  | (1) |
Ionic conductivity from MD simulations is calculated using the Einstein relationship:46
| |  | (2) |
where
e is the electron charge,
V is the volume of the simulation box,
kB is Boltzmann's constant,
T is the temperature,
t is the time,
qi and
qj are the charges over ions
i and
j in electrons,
ri(
t) is the displacement of ion
i during time
t, the summation is performed over all ions, 〈〉 denotes the ensemble average, and
N is the number of cations plus anions in the simulation cell. By combining the Einstein relationship and the degree of uncorrelated motion
46–48 (
αd), the apparent ionic conductivity in the long-term equilibrium state is given by:
| | 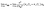 | (3) |
| | 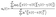 | (4) |
where
n+ and
n− are the number of cations and anions, respectively;
D+ and
D− denote the self-diffusion coefficients of cations and anions. For
αd(
t) = 1, the ion motion is completely uncorrelated, for
αd(
t) = 0, cations and anions move together as ion pairs, resulting in zero conductivity. Herein, the
αd was calculated using 4% of the total simulation run (Fig. S12).
The enthalpy of vaporization consists of the gas phase enthalpy value and liquid phase enthalpy value, which we compute using the equation49,50
| | | ΔHvap = Hgas − Hliq = Ugas + PVgas − Uliq − PVliq | (5) |
where Δ
Hvap is the enthalpy of vaporization,
H is molar enthalpy,
U is molar energy,
P is pressure,
V is molar volume; the subscripts gas and liq mean the corresponding phase values of gas or liquid. Compared with
PVgas,
PVliq is usually neglected. Additionally,
PVgas ≈
RT, where
R is the ideal gas constant and
T is the temperature. Moreover, the average kinetic energy of the liquid phase is close to that of the gas phase at the same temperature. So the equation above is simplified to
| | | ΔHvap ≈ Ugas − Uliq + RT = Ep−gas − Ep−liq + RT | (6) |
where
Ep is the molar potential energy.
2.2 Material preparation
Ethylene glycol dimethyl ether (EGDME, 98%), lithium bis(fluorosulfonyl)imide (LiFSI, >98%), and 1,1,2,2-trifluoroethyl-2,2,3,3-tetrafluoropropyl ether (TTE, 98%) were all purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. Reagents were stored in Ar-filled glove boxes (H2O/O2 < 0.1 ppm) upon delivery and used without further purification for electrolyte preparation. The electrolyte was prepared by dissolving LiFSI in a TTE/EGDME co-solvent system (molar ratio 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2). Freshly prepared electrolytes were utilized within two weeks to minimize potential decomposition.
:2). Freshly prepared electrolytes were utilized within two weeks to minimize potential decomposition.
2.3 Electrochemical characterization
The ionic conductivity was calculated from the electrolyte resistance (Rb), the thickness of the electrolyte film (L), and the electrode area (A), according to eqn (7). The electrolyte resistance was measured via electrochemical impedance spectroscopy, employing symmetric SS (stainless steel)/electrolyte/SS cells, over a frequency range of 100 kHz to 0.1 Hz with a 10 mV voltage amplitude.
2.4 Pulsed-field-gradient-nuclear magnetic resonance (PFG NMR)
The diffusion coefficients were measured using a Bruker Avance III 600 MHz narrow-bore spectrometer equipped with a 5 mm DOTY VT 1H-19F/X PFG probe. The pulsed-gradient stimulated-echo (PGSTE) sequence was employed for diffusion measurements, with data analysis performed using the Stejskal–Tanner equation to correlate gradient strength (g) with NMR signal attenuation.51| | 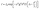 | (8) |
where I denotes the spectral peak signal intensity, I0 represents the initial intensity at g = 0, γ is the nuclear gyromagnetic ratio, δ the effective gradient pulse duration (2.0 ms), Δ the diffusion time (100 ms), and D the diffusion coefficient. Measurements employed 16 gradient steps with 16–32 scans per step, maintaining 2.0 ms gradient stabilization time. A double stimulated-echo sequence was implemented to mitigate convection artifacts.52,53
3 Results and discussion
Fig. 1 outlines the DBMLFF simulation workflow. The protocol is initiated with AIMD to sample molecular conformers, followed by machine learning model training on curated DFT datasets to establish conformation–energy relationships for intramolecular force field development. Intermolecular interactions are quantified through charge density reconstruction of individual molecules, complemented by polarization models capturing environmental induction effects. The complete DBMLFF framework systematically integrates three energy components: position-dependent intramolecular forces, electron density-derived intermolecular interactions, and polarization corrections, enabling high-fidelity molecular dynamics simulations.
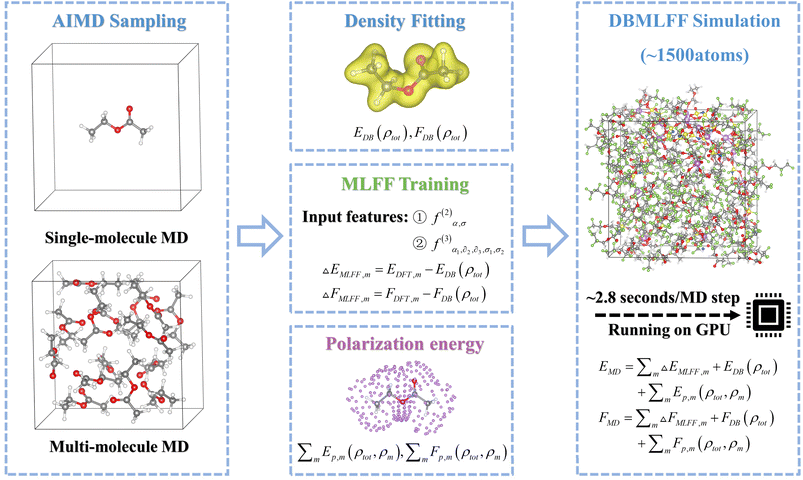 |
| | Fig. 1 Workflow of the DBMLFF training framework. | |
3.1 Dataset construction
To generate the ab initio data, we performed AIMD simulations at various temperatures. For each target molecule, we conducted room-temperature and high-temperature (e.g., 1000 K) AIMD simulations on both multimolecular systems (15-molecule clusters) and single-molecule systems, each spanning thousands of timesteps. Multimolecular trajectories extract diverse single-molecule configurations through conformational sampling, with subsequent self-consistent field calculations yielding system energies and atomic forces. Single-molecule trajectories directly provide corresponding energetics and atomic forces from AIMD results. Both multimolecular systems and elevated-temperature simulations cooperatively expand conformational space sampling, substantially broadening molecular–atomic configuration coverage. This systematic sampling strategy ensures MLFF-MD simulations remain strictly within well-trained configuration spaces throughout. During parameter optimization, room-temperature conformations received enhanced weighting in the loss function to prioritize their prediction accuracy.
3.2 Intramolecular machine learning force field (MLFF)
For a given molecule, its energy as a function of its configuration will be modeled with an intramolecular MLFF. To this aim, we have used 2-body and 3-body local atomic structure descriptors (features) as our MLFF input.54 These features have been successfully applied to covalent bonding silicon systems,55 metallic systems,56 and multi-element systems. More specifically, a 2-body feature for an atom at Ri can be specified as:| | 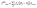 | (9) |
where Rj is the atomic position for atom j within a cutoff radius to the center atom position Ri, and σj is the element type of the atom j. The feature is specified by two indexes, α indicates the shape of the fα(x) function and σ specifies the neighbor atom element type. We have used truncated sinusoidal peak functions at different locations and widths as a function fα(x). Similarly, for 3-body features, we have:| | 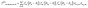 | (10) |
where  ,
,  and
and  are
are 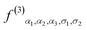 functions with different parameters. Due to the different element types and the number of index α used, we typically have one to two hundred features for a given center atom.
functions with different parameters. Due to the different element types and the number of index α used, we typically have one to two hundred features for a given center atom.
The parameterization of our intramolecular MLFF incorporates a threefold constraint based on total energy, atomic forces, and atomic energy decomposition. A distinctive aspect of our approach is the decomposition of the DFT total energy into atomic contributions, which are then used as fitting targets; this strategy significantly increases the amount of valuable data available during the fitting process and reduces the need for lengthy ab initio simulations to generate training data. While these features could serve as inputs for fully connected neural networks, we find that a linear regression model is sufficient to produce highly accurate intramolecular MLFF. This can be attributed to two main reasons: first, our focus is solely on molecular configurations near equilibrium, without chemical reactions or bond breaking/formation, resulting in a relatively low-dimensional configuration space; and second, the combination of the index α and the extended element type σ provides a large number of features, a scenario in which linear models often perform well. Furthermore, linear regression models are easy to train and can be learned on-the-fly with minimal computational cost, making them well-suited as accelerators for MD simulations. The accuracy of this approach is demonstrated by the fitting results for ethyl acetate (EA) molecules in Fig. 2a and b, where total energy predictions yield the root-mean-square error (RMSE), mean absolute error (MAE), and coefficient of determination (R2) values of 0.045 eV, 0.027 eV, and 0.998, respectively, while atomic force predictions demonstrate RMSE, MAE, and R2 values of 0.142 eV Å−1, 0.092 eV Å−1, and 0.999, respectively. It should be noted that the intrinsic molecular properties presented in Fig. 2a and b, as described by the intramolecular MLFF trained on isolated molecules in vacuum DFT data, do not include intermolecular interactions or polarization effects, which will be incorporated in subsequent stages.
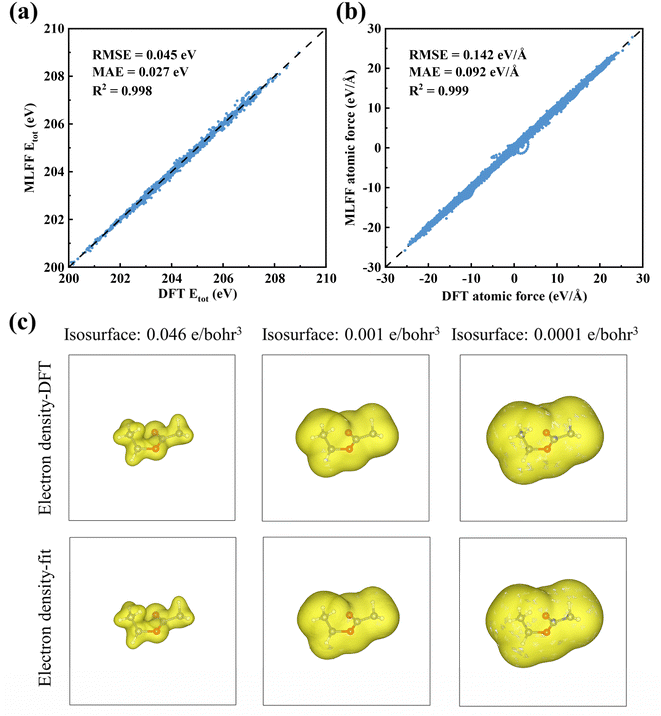 |
| | Fig. 2 (a) Total energy and (b) atomic force comparisons between DFT calculations and MLFF predictions for EA molecules in various configurations. (c) Electron density isosurfaces of EA molecule from DFT (upper) and spherical charge center model fittings (lower), with isovalues of 0.046, 0.001, and 0.0001 e bohr−1.3 | |
3.3 Fitting the density of single molecules
Following the development of intramolecular MLFF for individual molecules, this work focuses on modeling intermolecular interactions. The protocol is initiated with single-molecule charge density reconstruction. Computational efficiency is prioritized through atom-centered spherical charge density models, augmented by bond-centered components to capture non-spherical electron distribution characteristics. The resultant molecular electron charge density expression is formulated as:33| |  | (11) |
where j is the index for the centers (either atomic positions or bond centers), k is the index for the basis function fk(r), and β(j) is the type of the center. This type is determined by the element type of the atom (if the center is an atom) and the bonding situation inside the molecule. We have used the Gaussian function exp[−(r − Rk)2/ωk2] as the basis fk(r). To complete the model, we only need to get the coefficients C(k,β) for a given molecule. About 30 Gaussian basis functions are used per atom and bond center, leading to a total number of coefficients on the order of hundreds.
We randomly selected 50 distinct atomic configurations from AIMD simulations at different temperatures and fitted their DFT calculated charge density ρDFT(r) with the above model ρm(r). These 50 configurations were fitted collectively, with each contributing thousands of volumetric grid points to the fitting process. A linear fitting to minimize ∫[ρDFT(r) − ρm(r)]2d3r was used to yield the coefficient C(k,β). We have shown a comparison of the model fitted charge density and DFT calculated charge density in Fig. 2c for EA molecules. It shows that the charge density is well fitted not only in the large density areas but also in the small density tail regions. The small tail regions are important for intermolecular interactions as they modify the electrostatic interaction, as well as induce exchange coupling between the molecules. Note that the charge density tail extends 2 Å from atoms, a range comparable to molecule–molecule or molecule–ion distances; thus, density overlaps exert a notable influence, with the induced exchange coupling serving as a major source of intermolecular attraction. Notably, such overlap-induced interactions cannot be adequately captured by the simple point charge models used in most classical force fields. Residual errors, particularly in tail regions, may propagate to affect subsequent predictions. For instance, subtle mismatches in tail density (e.g., deviations in density decay 1–2 Å from atoms) could distort intermolecular electrostatic fields, leading to Coulomb energy biases. These, in turn, alter atomic forces and molecular trajectories, distorting properties like diffusion coefficients and viscosity. Similarly, fitting errors in density overlap regions (critical for exchange coupling) may modify intermolecular attraction, impacting liquid-phase cohesive energy and causing deviations in thermodynamic properties such as enthalpy of vaporization. While Fig. 2c only shows one atomic configuration, it is important to note that the same C(k,β) coefficients can generate electron charge densities for other configurations of the same molecule. Future work may test configuration-dependent C(k,β) coefficients in eqn (11) for optimization, but overall, interaction energies derived from the fitted charge densities are satisfactory.
Having developed a fit for the electron charge density of individual molecules for any given molecular configuration, the next step is to calculate intermolecular interactions. Our approach assumes that the total charge density of a system is the sum of the individual molecular charge densities, even if there are overlaps. This assumption has been confirmed in other theories, such as symmetry-adapted perturbation theory to calculate intermolecular interaction energies,57 and in the charge patching method.58 Given this assumption, to describe the total charge density of an aggregate of closed-shell molecules, it only needs to provide the charge densities for each molecule before polarization effects are considered. Conversely, we define the polarization electron charge as the difference from the simple summation result after an ab initio self-consistent field (SCF) calculation of the whole system. To further demonstrate this point, we tested two dimethyl ether (DME) molecules, by calculating the total charge density when they are close to each other and compared it to the sum of the individual molecule densities. The results are shown in Fig. 3a–c when they approach each other. To estimate the overlap of the two molecule charge densities, we have defined an overlap charge density as:
| |  | (12) |
where
ρ1(
r) and
ρ2(
r) are the charge densities of the two individual molecules in isolation. This overlapping charge density is compared with the charge density summation error:
| | | ρe(r) = ρ12(r) − ρ1(r) − ρ2(r) | (13) |
where
ρ12(
r) is the DFT calculated charge density of the supermolecule (the whole system). Herein,
ρe(
r) mostly concentrates in regions where
ρ0(
r) is maximum. However,
ρe(
r) is more complex, especially with some atomic characteristics, which can be a consequence of atomic-level polarization. Although the dipole moment of the DME molecule is small, when two of them are only 2–3 Å apart, there could still be local electric fields and polarization effects. Overall,
ρe(
r) is about an order of magnitude smaller than that of
ρ0(
r), which is in turn also an order of magnitude smaller than the total density itself. All these demonstrate the overall correctness of the charge density summation assumption.
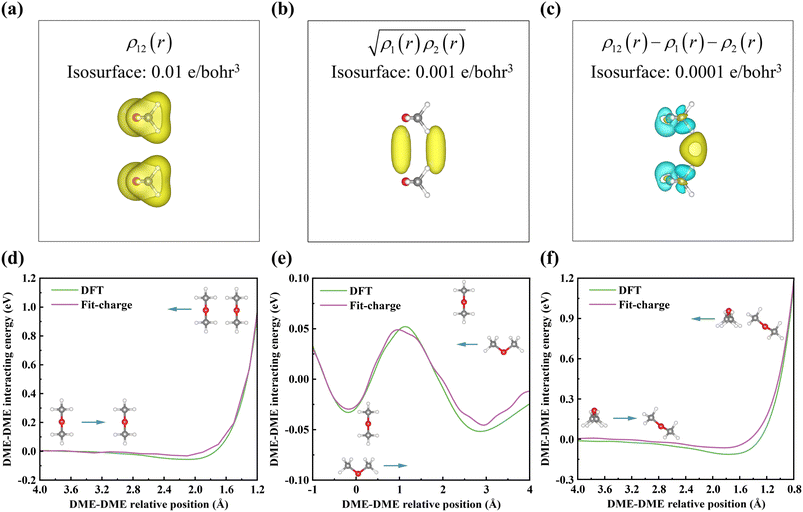 |
| | Fig. 3 DFT-calculated (a) charge density distribution, (b) charge density overlap region, and (c) superposition approximation error of DME dimer supermolecule at 2.5 Å parallel separation. (d)–(f) DME–DME intermolecular interaction energy comparison for three configurations: (d) parallel aligned contact; (e) perpendicular staggered contact; (f) tilted slip contact. | |
Using the sum of the individual molecule charge density ρm(r) to yield the total charge density ρtotal(r), we can now calculate the intermolecular interaction as follows:
| | 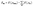 | (14) |
where
m index stands for the molecules, and
. The functional
E(
ρ) uses the charge density to calculate the following energies:
| | 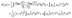 | (15) |
| | | ρtotal(r) = ρe(r) + ρnucl(r) | (16) |
where
ρnucl(
r) is the nuclear charge, and
εxc is the DFT local or semilocal exchange–correlation functional. In
eqn (15), the first term is the Coulomb interaction. The last two terms are Thomas–Fermi von Weizsäcker kinetic energy functional.
59 We keep
α as a general fitting parameter (
e.g., when we have used different
α with and without polarization energy).
In summary, we have expressed intermolecular interactions by constructing the system's charge density via superposition of pre-fitted molecular densities and computing the interaction energy using an energy density functional. As shown in Fig. 3d–f, the DME–DME molecule interaction energies calculated from this approach are in close agreement with direct DFT calculations. In the direct DFT calculation, the binding energy is calculated from:
| | | Etot(DFT) − Em1(DFT) − Em2(DFT) | (17) |
where each DFT calculation is a self-consistent calculation. We tested the DME–DME interactions as the two molecules approached each other in various orientations and alignments. The interaction energy calculated from the fitted charge density was found to deviate by no more than 20 meV from the original DFT result. Therefore, the interaction energy using the fitted charge density is satisfactory. So far, we have not included the polarization term and have used
α = 0.04 for the van Weizsäcker kinetic energy term in
eqn (15).
Herein, we combine the intramolecular MLFF with an intermolecular density-based interaction model to construct a complete force field for MD simulations. Notably, polarization potential is not incorporated in the current model. However, for systems without strong electrostatic interactions (e.g., those containing no charged ions), the polarization effect is expected to be relatively weak, rendering the model reasonable. To validate this, we present the energy profile of two EA molecules during AIMD simulations, while the DBMLFF was employed to compute the potential energy (excluding nuclear kinetic energy) along these trajectories. Specifically, independent ab initio molecular dynamics simulations were performed for five pairs of EA molecules with distinct orientations and positions, each lasting 100 fs at a time step of 0.5 fs. Subsequently, the DBMLFF was used to infer the energy of all 1000 snapshots extracted from these AIMD simulations. As illustrated in Fig. 4, the energies computed by the DBMLFF exhibit quantitative agreement with the AIMD reference data (RMSE = 0.068 eV, MAE = 0.055 eV, R2 = 0.950), accurately capturing both global trends and local features.
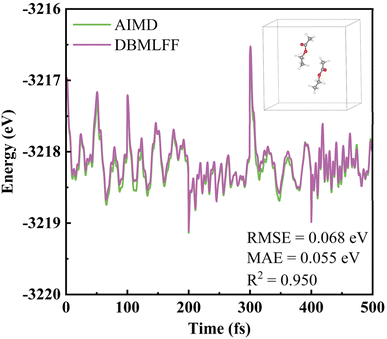 |
| | Fig. 4 Comparison of the potential energies along the AIMD simulation trajectory. The reference potential energy from direct AIMD calculation (green curve) is shown versus the potential energy predicted by the DBMLFF without the polarization term (purple curve). A representative structural snapshot of an EA molecular pair from the simulation is shown in the inset. | |
3.4 Polarization energy
While existing models may suffice for systems with weak electrostatic interactions, accurate modeling of ionic species like Li+ and FSI− necessitates explicit consideration of polarization effects. This requirement becomes critical in solvent electrolytes where Li+-O/F atomic distances approach 2 Å, generating substantial local electrostatic interactions despite the absence of covalent bonding.60 Classical force field treatments of polarization involve two principal aspects: (1) charge redistribution under external potentials generating induced dipole moments, and (2) intermolecular polarization arising from these induced dipoles.61,62 In disordered systems such as organic solvents, the weak coherence of induced dipoles permits neglect of higher-order coupling terms. Our approach preserves physical rigor while circumventing computationally intensive self-consistent field iterations through explicit modeling of first-order polarization mechanisms.
To accurately capture electronic relaxation under local external potentials, we implement a spatially resolved local electric field model, superseding conventional uniform field approximations. The polarization energy for individual molecules is formulated as:
| |  | (18) |
where the numbers
i,
j denote the atoms, and
h,
k denote the three spatial components of the electric field
Eh(
i) and
Ek(
j). The parameter
P(
i,
h;
j,
k) is the model coefficient to be fitted. To make the model transferrable, we have used internal coordinate orientation for
h and
k,
e.g., directions along bonds related to atom
i and
j. The local electric field
Eh(
i) is calculated as:
| | | Eh(i) = ∫Vext(r)exp[−(r − Ri)2/ω2]rhd3r | (19) |
where
Vext(
r) is the Coulomb and exchange correlation potential caused by other molecules. Having established the polarization model of
eqn (18) and
(19), DFT calculations were performed to fit the coefficients
P(
i,
h;
j,
k). Specifically, the polarization response of the molecule is probed by adding an external potential
Vext(
r) at different points
Rp as a perturbation for a given molecule:
| | | Vext(r) = aerf(d)/d + bexp(−d2/ω2) | (20) |
where
d = |
r −
Rp|, the error function erf(
x) and the Gaussian width
ω = 1 are used to model the actual potential near the nucleus. For a given molecule, about a thousand positions of
Rp are used, as shown in
Fig. 5a, to probe different situations surrounding the molecule. The distance between
Rp and the nearest atom in the molecule varies from 2 Å to 8 Å. In the test set, we have also included cases where multiple probing charges exist simultaneously. All these are used to represent different external potential situations in real solvents. The total energy difference
Epol before and after wave function relaxation was recorded by DFT self-consistent calculations, and
Eh(
k) was calculated according to
eqn (19). Using the least squares linear fitting, we obtained the coefficients
P(
i,
h;
j,
k). The results are shown in
Fig. 5b, which shows that the polarization model can accurately predict the polarization energy calculated by DFT within a few percentage points.
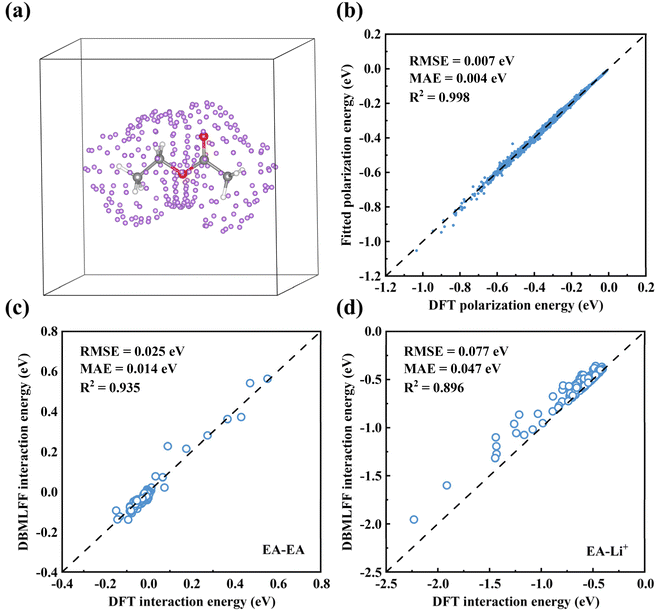 |
| | Fig. 5 (a) Statistical distribution of probe charge position Rp for EA molecule at a representative probe–molecule distance. (b) Correlation between DFT-calculated and fitted polarization energies for the EA molecule across multiple probe configurations. Comparison of intermolecular interaction energies of (c) EA–EA and (d) EA-Li+ with random displacements and orientations. The DBMLFF model incorporates coulombic, exchange-kinetic, and polarization-induced interactions. | |
By integrating the polarization model, we coupled the polarization energy from eqn (18) with coulombic, exchange, and kinetic energy terms in eqn (14) to construct the total interaction energy. Systematic evaluations were performed for EA dimer systems (Fig. 5c) and EA-Li+ complexes (Fig. 5d), covering 200 distinct configurations for each system with varying displacements and orientations. Note that we have revised the prefactor α of the van Weizsäcker kinetic energy term in eqn (15) from 0.04 to 0.025. This adjustment is based on our observation that the contribution of this kinetic energy term is consistently negative and sometimes compensates for the polarization energy to some degree. Thus, in subsequent work where the polarization energy is included, we will adopt α = 0.025. As shown in Fig. 5c and d, we calculated the intermolecular and molecule–ion interactions (obtained by subtracting the energies of isolated components from the total system energy) in two systems using both DBMLFF and DFT methods. The results demonstrate excellent agreement between DBMLFF and DFT in both systems, with RMSE of 25 meV and 77 meV, respectively. Remarkably, Li+-containing systems exhibit interaction energies reaching −2 eV magnitude. The low RMSE values, particularly in the strongly interacting Li+ system, demonstrate that our polarization model effectively captures ion–molecule interactions. Any residual error in polarization treatment inherently disrupts the delicate balance between charge-transfer attraction and short-range repulsion. Such deviations propagate through atomic forces to molecular dynamics simulations, thereby impairing the predictive accuracy of dynamic properties like diffusion coefficients and viscosity. Particularly in strongly coupled systems (e.g., concentrated electrolytes), errors tend to amplify, exerting a more pronounced impact on the prediction of key properties such as ion-pair structures, free ion concentration, and ionic conductivity.
While the LDA functional was chosen for its computational efficiency and simplicity to test our charge-density-based approach, we acknowledge its inherent limitations. The use of LDA exchange–correlation functional and the spherical charge density approximation may limit accuracy for systems with significant charge transfer or anisotropic interactions. To quantitatively assess these limitations, we compared the performance of our DBMLFF model against GGA using a model charge-transfer system (the Li+–EA complex), as shown in Fig. S9. The results indicate that while LDA underestimates the binding energy by approximately 4.3% compared to PBE-D3, the deviation between the DBMLFF predicted values and the PBE-D3 benchmarks remains within an acceptable margin. Furthermore, for anisotropic systems such as DME molecular pairs with varying orientations and positions, the spherical density approximation introduces minor deviations in intermolecular interaction energies. Therefore, the relative interaction trends predicted by our model remain consistent with those obtained from higher-level functionals.
3.5 DBMLFF molecular dynamics simulations
By integrating intramolecular MLFF with intermolecular interactions derived from electron density and polarization models, we have developed the complete DBMLFF methodology for molecular dynamics simulations. Initial tests demonstrate that each MD step requires approximately one second per molecule on a single CPU. GPU acceleration substantially enhances computational efficiency. Fig. 6a compares the system time per MD step for DBMLFF, PWmat and VASP software under four-GPU acceleration across systems containing varying numbers of EA molecules. The construction of the electron density is a local operation, resulting in computational costs that scale linearly with system size, as illustrated in Fig. 6a. The sole exception is the fast Fourier transform (FFT) required to solve the global Poisson equation, which scales as Nlog(N), where N is the system size. However, this global FFT constitutes only a minor fraction of the total computational cost and is therefore a secondary contribution. Notably, even classical force fields that employ methods like the fast multipole63 or particle–particle particle–mesh44 for long-range electrostatics exhibit the same Nlog(N) scaling for Coulomb interactions. For typical electrolyte systems comprising over 200 molecules, DBMLFF achieves three orders of magnitude acceleration compared to AIMD methods, as quantitatively demonstrated in Fig. 6b.
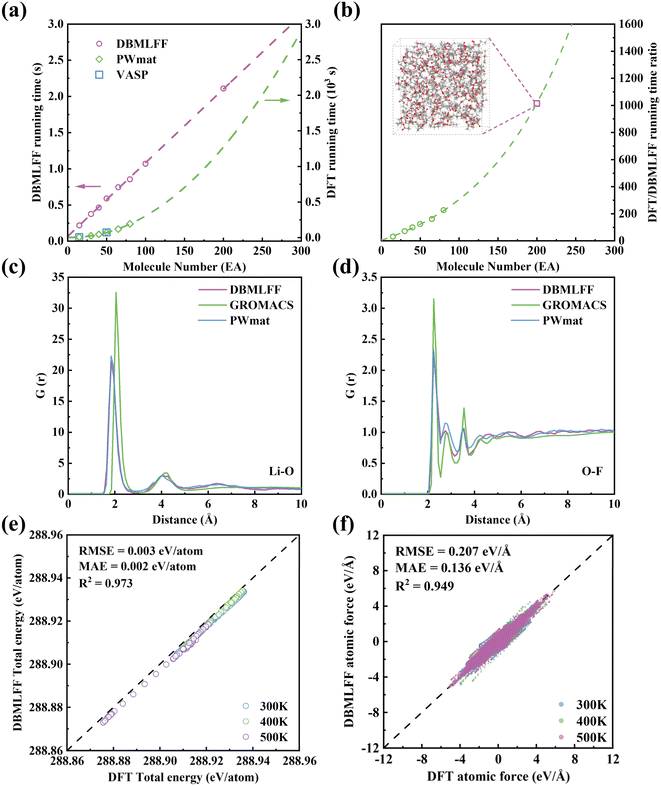 |
| | Fig. 6 (a) Running time performance and (b) running time ratio for systems with increasing numbers of EA molecules, comparing AIMD and DBMLFF. (c) Li–O and (d) O–F pair distribution functions from DBMLFF and AIMD simulations of the LiFSI/EA/TTE system at 500 K. (e) Total energy and (f) atomic forces predicted by DBMLFF against DFT benchmarks at 300 K, 400 K, and 500 K. | |
To confirm the performance of DBMLFF for actual liquid aggregated states, we therefore conducted comparative simulations on an electrolyte system containing 10 EA, 15 TTE, and 5 LiFSI, with a total atomic number of 460 for the whole system. This system, characterized by strong electrostatic/dipole interactions and polarization effects, serves as an exemplary platform for methodological validation. In modeling Li+, we only performed charge density fitting (Fig. S1) without including a polarization energy term. This is because Li+, as a closed-shell ion, has highly localized electron density and negligible polarizability, and is thus treated as a source of the polarizing field rather than a polarizable species. TTE (Fig. S2) and FSI− (Fig. S3) were processed using the same fitting protocol as EA. All simulations were performed at 500 K for 5 ps under identical initial conditions to ensure sufficient mixing and mobility, with representative configurations shown in Fig. S10a. Radial distribution function analyses (Fig. 6c and d) demonstrate excellent agreement between DBMLFF and AIMD results, whereas GROMACS simulations show shortened Li–O bond lengths with intensified first peaks. To quantitatively assess the accuracy and temperature transferability of the DBMLFF methodology, we performed DFT single-point energy calculations on 150 configuration snapshots systematically sampled from three independent DBMLFF-generated molecular dynamics trajectories at 300 K, 400 K, and 500 K (50 snapshots from each 5 ps trajectory). A comprehensive comparison was conducted on potential energy surface distributions (including the polarization term) and atomic force characteristics across this thermally diverse dataset. As shown in Fig. 6e and f, the DBMLFF calculations maintain excellent agreement with DFT benchmark data across all temperature regimes: the consolidated analysis yields an RMSE of 0.003 eV per atom (MAE = 0.002 eV per atom, R2 = 0.973) for total energy, while atomic forces exhibit an RMSE of 0.207 eV Å−1 (MAE = 0.136 eV Å−1, R2 = 0.949). The consistent performance across this 200 K temperature range demonstrates the robustness of the DBMLFF approach for molecular simulations under varying thermodynamic conditions. In comparison, the MACE shows improved force prediction accuracy (RMSE = 0.354 eV Å−1, Fig. S11b) over conventional GROMACS simulations (RMSE = 1.412 eV Å−1, Fig. S11a), yet still falls short of the DBMLFF performance, further underscoring the marked superiority of the DBMLFF approach in modeling atomic-scale interactions.
To assess reliability and transferability in extended electrolyte simulations, we selected the well-studied LiFSI/EGDME/TTE system. Since DBMLFF models for LiFSI and TTE were already established in our previous work, only the newly introduced EGDME molecule required parametrization (fitting procedure shown in Fig. S4). We constructed an initial configuration by randomly packing 24 EGDME, 60 TTE, and 20 LiFSI molecules (totaling 1664 atoms) at the experimental density of 1.49 g ml−1, with a representative snapshot provided in Fig. S10b. Subsequently, we performed 700 ps MD simulations at 300 K using both DBMLFF and the classical OPLS-AA force field in GROMACS. Ion diffusion coefficients were calculated from MSD analysis during the final 550 ps following adequate equilibration (Fig. 7a and b). The MSD curves exhibit clear linear regimes throughout this period, confirming well-converged diffusive behavior. It should be noted that all reported diffusion coefficients, for both DBMLFF and GROMACS, are presented without finite-size corrections, as our primary objective was to conduct a direct comparison under identical simulation conditions. To validate the simulations against experiment, we conducted pulsed field gradient nuclear magnetic resonance (PFG-NMR) measurements on the same electrolyte. The self-diffusion coefficients derived from Stejskal–Tanner equation analysis were 1.377 × 10−10 m2 s−1 for Li+ and 1.180 × 10−10 m2 s−1 for FSI− (see NMR spectra in Fig. 7c and d). The experimental ionic conductivity was obtained from Nyquist plots (Fig. S13). As quantified in Table S1, DBMLFF demonstrates significantly better agreement with the experimental diffusion coefficients and ionic conductivity than the classical force field, confirming its superior predictive accuracy.
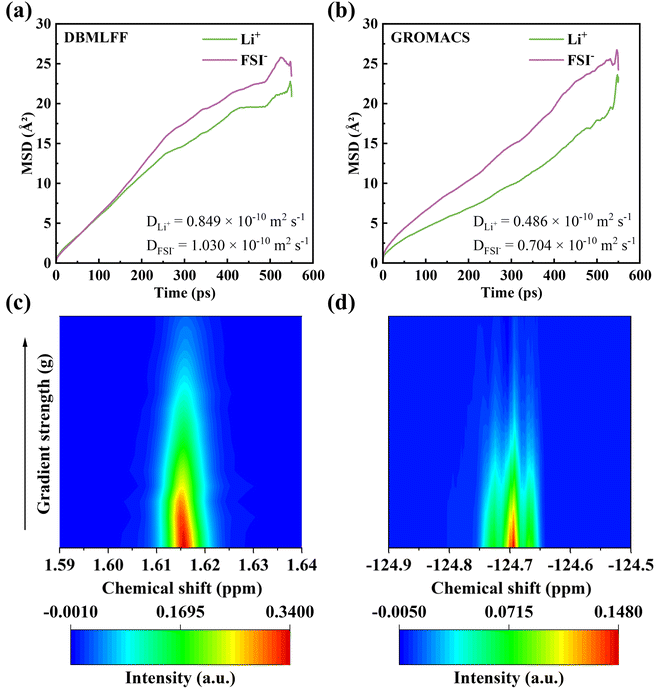 |
| | Fig. 7 Mean square displacement (MSD) of Li+ and FSI− in the LiFSI/EGDME/TTE system from (a) DBMLFF and (b) GROMACS simulations. Experimental PFG-NMR spectra of Li+ (c) and FSI− (d) diffusion in the LiFSI/EGDME/TTE system. Color corresponds to peak intensity; increasing number indicates increasing gradient strength. | |
We further extended our assessment to ionic mobility in a lithium hexafluorophosphate (LiPF6)/ethylene carbonate (EC)/dimethyl carbonate (DMC) system (80 EC, 60 DMC, 10 LiPF6) via 2.5 ns MD simulations at 300 K (representative configuration in Fig. S10c). The fitting procedures for EC, DMC, and PF6− are detailed in Fig. S5–S7. The diffusion coefficients from MSD analysis (Fig. S14) again show that DBMLFF quantitatively aligns the established experimental trends64 and offers a marked improvement over classical force field prediction (Table S2). The observed systematic deviations may originate from the simulation errors due to limited simulation times and system sizes, the intrinsic error of the DBMLFF model, as well as the possible experimental uncertainties. In the future more systematic studies might be necessary to analyze the source of remaining errors.
To evaluate force prediction stability in extended systems, we randomly selected 20 snapshots from the DBMLFF molecular dynamics trajectories of the LiFSI/EGDME/TTE and LiPF6/EC/DMC electrolyte systems and performed a comparative analysis of atomic forces using DFT calculations and MACE predictions. The results (Fig. S15) demonstrate that in both electrolyte systems, DBMLFF achieves significantly better agreement with DFT reference forces than MACE, along with markedly lower prediction errors. More critically, the same LiFSI and TTE models were employed in both the LiFSI/EA/TTE system (Fig. 6d) and the LiFSI/EGDME/TTE system (Fig. S15a). Despite the change in solvent from EA to EGDME, which alters the chemical environment, the forces predicted by the DBMLFF models remain in excellent agreement with DFT. This robust performance, maintained without the need for retraining, strongly demonstrates the outstanding transferability of the DBMLFF framework.
Beyond electrolyte applications, we envision that this machine learning potential based on electron density descriptors will pioneer new directions in force field development for molecular simulations, particularly for systems where intermolecular interactions govern macroscopic properties. To validate the accuracy and general applicability of DBMLFF in describing intermolecular interactions, we computed evaporation enthalpies for five representative molecules. Specifically, for each molecule, we constructed a liquid system containing 100 molecules at experimental density and performed 2 ns molecular dynamics simulations at 298 K using DBMLFF and GROMACS, respectively. Each system was independently simulated three times to obtain statistical averages. The potential energy of gaseous molecules was derived from 20 ps molecular dynamics simulations at 298 K in large-sized simulation boxes (ensuring no intermolecular interactions). The fitting process of DME is shown in Fig. S8. As shown in Table S3, the evaporation enthalpies calculated using DBMLFF and the traditional force field were compared with experimental values. The results demonstrate that DBMLFF significantly outperforms the traditional force field (although traditional force fields often use such experimental vaporization energies as fitting targets), indicating its superior ability to accurately describe intermolecular interactions.
4 Conclusion
Although we have demonstrated the feasibility of the transferable DBMLFF, with predicted dynamic diffusion and enthalpy of evaporation in good agreement with experimental values, several key issues remain to be explored. First, our current approach only handles intermolecular interactions via charge density, which is effective for small molecules but notably insufficient for large systems such as polymers and proteins. Incorporating density-based intramolecular interactions is straightforward, simply by reusing the formal framework of intermolecular interaction energy. This modification can also simplify the computational workflow of MD simulations: obtain the charge density, calculate the charge-density-based interaction energy and polarization energy, subtract these energies/forces from DFT results, and then fit the residual energies/forces with an MLFF. Second, dispersion interactions are not included in this study and can be supplemented by empirical methods (e.g., DFT-D2,65 DFT-D3 (ref. 66)) or non-local integrals dependent on charge density (e.g., rVV10 (ref. 67)), both of which are straightforward and direct. Finally, the assumption that the total system charge density equals the sum of molecular densities essentially ignores many-body interactions beyond first order polarization effect treatment. For systems with strong and coherent polarization effects, e.g., under a strong external electric field, the charge density change (polarization charge density) might need to be described, and a self-consistent calculation might be needed to calculate the screened electric field.
Author contributions
Jie Shen: investigation, theoretical calculations, data curation, and writing – original draft; Chenyu Wang: experimental verification; Libin Chen and Jianhui Chen: data curation; Cuilian Wen and Bo Wu: writing – review & editing; Shaoqin Jiang: data discussion and suggestions; Baisheng Sa: supervision, writing – review & editing, and project administration; Lin-Wang Wang: conceptualization and methodology; writing – review & editing – all authors.
Conflicts of interest
The authors declare no competing financial interest.
Data availability
Data for this article are available at Figshare, AIMD trajectories at https://doi.org/10.6084/m9.figshare.30487070, and DBMLFF trajectories at https://doi.org/10.6084/m9.figshare.30487109. The code for DBMLFF is available on GitHub at https://github.com/Sj-ai-ikahgi/DBMLFF.git and has been archived on Zenodo (DOI: https://doi.org/10.5281/zenodo.17974171).
Supplementary information (SI): figures and tables that support the key findings reported in this work. See DOI: https://doi.org/10.1039/d5dd00508f.
Acknowledgements
This work was supported by the Advanced Materials-National Science and Technology Major Project (2025ZD0618403), the National Natural Science Foundation of China (No. 52571005), and the Natural Science Foundation of Fujian Province (Nos. 2024J01262 and 2025J02008).
References
- N. Yao, X. Chen, Z.-H. Fu and Q. Zhang, Chem. Rev., 2022, 122, 10970–11021 CrossRef CAS PubMed.
- Y. Ou, P. Zhou, W. Hou, X. Ma, X. Song, S. Yan, Y. Lu and K. Liu, J. Energy Chem., 2024, 94, 360–392 CrossRef CAS.
- S. Wang, J. Shi, Z. Liu and Y. Xia, Adv. Energy Mater., 2024, 14, 2401526 CrossRef CAS.
- F. Zhai, Q. Zhou, Z. Lv, Y. Wang, X. Zhou and G. Cui, EnergyChem, 2022, 4, 100082 CrossRef CAS.
- J. Hou, L. Wang, X. Feng, J. Terada, L. Lu, S. Yamazaki, A. Su, Y. Kuwajima, Y. Chen, T. Hidaka, X. He, H. Wang and M. Ouyang, Energy Environ. Mater., 2023, 6, e12297 CrossRef CAS.
- L. Kong, Y. Li and W. Feng, Electrochem. Energy Rev., 2021, 4, 633–679 CrossRef CAS.
- Y. Ou, W. Hou, D. Zhu, C. Li, P. Zhou, X. Song, Y. Xia, Y. Lu, S. Yan, H. Zhou, Q. Cao, H. Zhou, H. Liu, X. Ma, Z. Liu, H. Xu and K. Liu, Energy Environ. Sci., 2025, 18, 1464–1476 RSC.
- M. Sun, Y. Xie, C. Zhong, Y. Huang, H. Chen, H. Huang, P. Dai, S. Liu, W. Zheng, C. Liu, S. Liao, L. Huang, S. Sun and X. Wang, Energy Storage Mater., 2024, 65, 103166 Search PubMed.
- L. Yu, X. Chen, N. Yao, Y.-C. Gao and Q. Zhang, J. Mater. Chem. A, 2023, 11, 11078–11088 RSC.
- X. Wang, H. Peng, K. Sun, F. Yang, Z. Liu, S. Cui, X. Xie and G. Ma, Energy Storage Mater., 2024, 66, 103208 Search PubMed.
- W. Zhang, T. Yang, X. Liao, Y. Song and Y. Zhao, Energy Storage Mater., 2023, 57, 249–259 Search PubMed.
- H. Jeong, H. Wang and L. Cheng, J. Mater. Chem. A, 2024, 12, 33150–33161 RSC.
- J. Huang, S.-J. Shin, K. Tolborg, A. M. Ganose, G. Krenzer and A. Walsh, Mater. Horiz., 2023, 10, 2883–2891 RSC.
- Y. Shi, R. Wang, Z. Zhong, Y. Wu, S. Liu, L. Si and R. He, Mater. Genome Eng. Adv., 2025, 3, e70012 CrossRef CAS.
- S. Röcken and J. Zavadlav, npj Comput. Mater., 2024, 10, 1–10 Search PubMed.
- B. Cui and J. Xu, J. Mater. Chem. A, 2025, 13, 8223–8245 Search PubMed.
- D. Hedman, B. McLean, C. Bichara, S. Maruyama, J. A. Larsson and F. Ding, Nat. Commun., 2024, 15, 4076 CrossRef CAS PubMed.
- S. Wu, S. Zheng, W. Zhang, M. Zhang, S. Li and F. Pan, J. Mater. Inf., 2025, 5, 14 CAS.
- S. Wu, X. Yang, X. Zhao, Z. Li, M. Lu, X. Xie and J. Yan, J. Chem. Inf. Model., 2023, 63, 6972–6985 CrossRef CAS PubMed.
- M. Chen, X. Jiang, L. Zhang, X. Chen, Y. Wen, Z. Gu, X. Li and M. Zheng, Med. Res. Rev., 2024, 44, 1147–1182 Search PubMed.
- A. Kabylda, V. Vassilev-Galindo, S. Chmiela, I. Poltavsky and A. Tkatchenko, Nat. Commun., 2023, 14, 3562 CrossRef CAS PubMed.
- J. T. Frank, O. T. Unke, K.-R. Müller and S. Chmiela, Nat. Commun., 2024, 15, 6539 CrossRef CAS PubMed.
- X. He, Y. Chen, S. Wang and G. Zhang, ACS Appl. Mater. Interfaces, 2024, 16, 24494–24501 Search PubMed.
- M. Ha, A. Hajibabaei, D. Y. Kim, A. N. Singh, J. Yun, C. W. Myung and K. S. Kim, Adv. Energy Mater., 2022, 12, 2201497 CrossRef CAS.
- A. Kabylda, V. Vassilev-Galindo, S. Chmiela, I. Poltavsky and A. Tkatchenko, Nat. Commun., 2023, 14, 3562 CrossRef CAS PubMed.
- T. Morawietz and N. Artrith, J. Comput. Aided Mol. Des., 2021, 35, 557–586 Search PubMed.
-
I. Batatia, D. P. Kovács, G. N. C. Simm, C. Ortner and G. Csányi, arXiv, 2023, preprint, arXiv:2206.07697, DOI:10.48550/arXiv.2206.07697.
- S. Duangdangchote, D. S. Seferos and O. Voznyy, Digital Discovery, 2024, 3, 2177–2182 RSC.
- S. Y. Willow, A. Hajibabaei, M. Ha, D. C. Yang, C. W. Myung, S. K. Min, G. Lee and K. S. Kim, Chem. Phys., 2024, 5, 041307 CAS.
- N. Artrith, T. Morawietz and J. Behler, Phys. Rev. B:Condens. Matter Mater. Phys., 2011, 83, 153101 Search PubMed.
- T. W. Ko, J. A. Finkler, S. Goedecker and J. Behler, Nat. Commun., 2021, 12, 398 CrossRef CAS PubMed.
- R. E. Duke, O. N. Starovoytov, J.-P. Piquemal and G. A. Cisneros, J. Chem. Theory Comput., 2014, 10, 1361–1365 Search PubMed.
-
L.-W. Wang, ChemRxiv, 2025, preprint, DOI:10.26434/chemrxiv-2025-83grg.
- W. Jia, J. Fu, Z. Cao, L. Wang, X. Chi, W. Gao and L.-W. Wang, J. Comput. Phys., 2013, 251, 102–115 CrossRef.
- W. Jia, Z. Cao, L. Wang, J. Fu, X. Chi, W. Gao and L.-W. Wang, Comput. Phys. Commun., 2013, 184, 9–18 CrossRef CAS.
- D. R. Hamann, Phys. Rev. B:Condens. Matter Mater. Phys., 2013, 88, 085117 CrossRef.
- M. Schlipf and F. Gygi, Comput. Phys. Commun., 2015, 196, 36–44 Search PubMed.
- J. P. Perdew and A. Zunger, Phys. Rev. B:Condens. Matter Mater. Phys., 1981, 23, 5048–5079 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 Search PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 Search PubMed.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- L. S. Dodda, I. Cabeza de Vaca, J. Tirado-Rives and W. L. Jorgensen, Nucleic Acids Res., 2017, 45, W331–W336 CrossRef CAS PubMed.
- L. Martínez, R. Andrade, E. G. Birgin and J. M. Martínez, J. Comput. Chem., 2009, 30, 2157–2164 Search PubMed.
- B. A. Luty and W. F. van Gunsteren, J. Phys. Chem., 1996, 100, 2581–2587 CrossRef CAS.
- Th. D. N. Reddy and B. S. Mallik, J. Phys. Chem. B, 2020, 124, 6813–6824 Search PubMed.
- O. Borodin and G. D. Smith, J. Phys. Chem. B, 2009, 113, 1763–1776 CrossRef CAS PubMed.
- B. Ghalami Choobar, H. Modarress, R. Halladj and S. Amjad-Iranagh, J. Phys. Chem. C, 2019, 123, 21913–21930 Search PubMed.
- R. Sasaki, B. Gao, T. Hitosugi and Y. Tateyama, npj Comput. Mater., 2023, 9, 48 Search PubMed.
- S. Wang, K. Huang, S. Jia, H. Wang and Z. Liu, J. Phys. Chem. A, 2025, 129, 1342–1347 CrossRef CAS PubMed.
- J. Pulido, L. Macaya and E. Vöhringer-Martinez, J. Chem. Eng. Data, 2024, 69, 2917–2926 CrossRef.
- E. O. Stejskal and J. E. Tanner, J. Chem. Phys., 1965, 42, 288–292 CrossRef CAS.
- W. Mei, D. Yu, C. George, L. A. Madsen, R. J. Hickey and R. H. Colby, J. Mater. Chem. C, 2022, 10, 14569–14579 RSC.
- J. E. Bostwick, C. J. Zanelotti, D. Yu, N. F. Pietra, T. A. Williams, L. A. Madsen and R. H. Colby, J. Mater. Chem. C, 2022, 10, 947–957 RSC.
- Y. Huang, J. Kang, W. A. Goddard and L.-W. Wang, Phys. Rev. B, 2019, 99, 064103 CrossRef.
- L. Miao and L.-W. Wang, J. Chem. Phys., 2020, 153, 074501 CrossRef CAS PubMed.
- Q. Hu, M. Weng, X. Chen, S. Li, F. Pan and L.-W. Wang, J. Phys. Chem. Lett., 2020, 11, 1364–1369 CrossRef CAS PubMed.
- K. Patkowski, WIREs Comput. Mol. Sci., 2020, 10, e1452 CrossRef CAS.
- Z. Song, X. Sun and L. Wang, J. Phys. Chem. Lett., 2020, 11, 9224–9229 CrossRef CAS PubMed.
- A. Chandrasekaran, D. Kamal, R. Batra, C. Kim, L. Chen and R. Ramprasad, npj Comput. Mater., 2019, 5, 22 CrossRef.
- O. N. Starovoytov, J. Phys. Chem. B, 2021, 125, 11242–11255 Search PubMed.
- H. Yu, T. W. Whitfield, E. Harder, G. Lamoureux, I. Vorobyov, V. M. Anisimov, A. D. J. Mackerell and B. Roux, J. Chem. Theory Comput., 2010, 6, 774–786 CrossRef CAS PubMed.
- Z. Jing, C. Liu, S. Y. Cheng, R. Qi, B. D. Walker, J.-P. Piquemal and P. Ren, Annu. Rev. Biophys., 2019, 48, 371–394 CrossRef CAS PubMed.
- V. Rokhlin, J. Comput. Phys., 1985, 60, 187–207 CrossRef.
- S. Uchida and T. Kiyobayashi, Phys. Chem. Chem. Phys., 2021, 23, 10875–10887 RSC.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- R. Sabatini, T. Gorni and S. de Gironcoli, Phys. Rev. B:Condens. Matter Mater. Phys., 2013, 87, 041108 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Bo
Wu
a,
Bo
Wu








 ,
,  and
and  are
are  functions with different parameters. Due to the different element types and the number of index α used, we typically have one to two hundred features for a given center atom.
functions with different parameters. Due to the different element types and the number of index α used, we typically have one to two hundred features for a given center atom.





 . The functional E(ρ) uses the charge density to calculate the following energies:
. The functional E(ρ) uses the charge density to calculate the following energies:




