
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
shnitsel-tools: a toolkit for the full lifecycle of surface hopping trajectory data
Kevin
Höllring†
 ,
Theodor E.
Röhrkasten†
and
Carolin
Müller
*
,
Theodor E.
Röhrkasten†
and
Carolin
Müller
*
Friedrich-Alexander-Universität Erlangen-Nürnberg, Department Chemistry and Pharmacy, Computer-Chemistry-Center, Nägelsbachstraße 25, 91052 Erlangen, Germany. E-mail: carolin.cpc.mueller@fau.de
First published on 27th March 2026
Abstract
Trajectory surface hopping (SH) simulations are essential for modeling excited-state dynamics, yet their analysis remains complex and fragmented. Existing tools provide access to basic quantities, but extracting deeper mechanistic insight, especially across multiple molecules, often requires extensive manual work and custom scripting. To address this gap, we present  (
( ), an integrated software package for the streamlined analysis, filtering, and visualization of SH data. Supporting automated and reproducible workflows, it enables efficient interpretation of excited-state simulations across diverse systems. As a demonstration, we apply
), an integrated software package for the streamlined analysis, filtering, and visualization of SH data. Supporting automated and reproducible workflows, it enables efficient interpretation of excited-state simulations across diverse systems. As a demonstration, we apply  in a comparative analysis of the photoinduced dynamics across chemical compound space spanned by a series of alkenes (CnH2n, with n = {2,3,4}) and CH2NH+2. This highlights how the toolkit facilitates comparative studies and mechanistic discovery across the chemical compound space of molecules with similar molecular scaffolds.
in a comparative analysis of the photoinduced dynamics across chemical compound space spanned by a series of alkenes (CnH2n, with n = {2,3,4}) and CH2NH+2. This highlights how the toolkit facilitates comparative studies and mechanistic discovery across the chemical compound space of molecules with similar molecular scaffolds.
1 Introduction
Mechanistic understanding of photoinduced phenomena, such as internal conversion or intersystem crossing, and photochemical reactions, relies on accurately modeling the coupled dynamics of nuclei and electrons in electronically excited states. To this end, non-adiabatic molecular dynamics (NAMD) simulations have emerged as a powerful class of computational approaches capable of capturing these intricate interactions.1,2 Over the years, a wide range of NAMD approaches has been developed, spanning from fully quantum to mixed quantum-classical frameworks.Fully quantum approaches, such as Multiconfiguration Time-Dependent Hartree (MCTDH)3 or variational Multiconfigurational Gaussian (vMCG) propagation,4 provide high accuracy but are typically restricted to model potentials or systems with only a few degrees of freedom due to their high computational cost. In contrast, trajectory-based methods, like ab initio multiple spawning (AIMS),2,5,6 Ehrenfest dynamics,7,8 and trajectory surface hopping (SH),8–10 treat nuclear motion using classical mechanics, while describing the electronic states quantum mechanically. These methods rely on the approximation that the nuclear wavefunction can be represented by a swarm of classical trajectories. This mixed treatment allows them to strike a practical balance between accuracy and computational efficiency. Among these methods, SH has emerged as the most widely used approach for simulating NAMD.7,11 Its widespread adoption is also supported by the availability of open-source and user-friendly software packages such as SHARC,12,13 Newton-X,14,15 PyRAI2MD,16,17 MLatom18 and JADE-NAMD.19
SH simulations generate intrinsically complex and high-dimensional datasets. Each trajectory corresponds to a time series of molecular geometries, accompanied by a set of electronic properties computed on-the-fly. These properties include state-specific quantities, such as electronic energies (Ei), atomic forces (Fi), and vertical transition dipole moments (µij), as well as interstate couplings like non-adiabatic (NACij) and spin–orbit (SOCij) couplings.
To interpret such rich datasets, a variety of analysis strategies is necessary. In typical workflows, both individual trajectories and ensembles thereof are analyzed to extract physical insight. Single-trajectory analysis commonly involves assessing energy conservation, monitoring the evolution of state populations, relative energies and oscillator strengths of all states relative to the current or active state; as well as evaluating selected internal coordinates such as bond lengths or torsional angles.12,20,21 Due to the stochastic character of SH, however, mechanistic interpretation generally requires ensemble analysis. This typically includes computing averaged state populations, identifying collective nuclear motions, and performing statistical analyses of predefined geometric parameters.12,20,21
Beyond conventional single-trajectory or ensemble-based analyses, more sophisticated strategies have emerged that leverage unsupervised learning techniques to uncover patterns in SH simulations and classify distinct types of reactive behavior.21–23 A typical first step is dimensionality reduction, using approaches such as principal component analysis (PCA)21,24 or diffusion maps,25,26 which project the high-dimensional nuclear motion data into a lower-dimensional space that captures the main structural changes. Clustering algorithms, as implemented for example in ULaMDyn21 or DONKEY,27 are employed to group trajectories based on similarities in their nuclear motion over time. This allows for the identification of representative trajectories that correspond to distinct reaction pathways.21,22,24,27–30
This automated categorization not only reduces the number of trajectories that require detailed manual inspection, but also facilitates a more systematic and interpretable understanding of the system's photochemical reactivity.21,23,27 Despite these advancements, many current SH analysis strategies remain highly system-specific and often demand significant manual intervention. While established packages such as SHARC12,13 and Newton-X14,15,20 support the extraction of fundamental observables from SH simulations, more advanced tasks, such as identifying population dynamics within trajectory subsets that exhibit specific nuclear motions (e.g., bond dissociation or torsional rotation relevant to photoisomerization), typically require custom scripting, manual sorting, or external post-processing.
In contrast, ground-state molecular dynamics simulations have long benefited from integrated analysis environments like TRAVIS,31,32 MDAnalysis,33 and the Visual Molecular Dynamics (VMD) program package,34 which centralize many essential analysis steps. A step toward similar integration for excited-state dynamics is represented by ULaMDyn,21 an open-source Python package that automates unsupervised analysis of SH trajectories. Despite this advancement, its scope and format compatibility remain limited, as it primarily supports Newton-X20 outputs and requires additional scripts or converters to process data from other widely used packages such as SHARC12,13 or PyRAI2MD.16,17 Other existing methods and studies22,23,27 concentrate on individual analysis tasks, such as PCA or trajectory clustering, and are often implemented through case-specific scripts.
This fragmented landscape poses a barrier to reproducibility, modularity, and cross-platform compatibility, not only hindering comprehensive analysis of individual systems but also limiting the ability to conduct systematic comparisons across molecular systems or simulation conditions, such as varying excitation wavelengths.
Herein, we introduce  (
( ), a versatile open-source Python toolkit designed to unify and streamline processing, storage, management, analysis, and visualization of SH simulation data. By supporting data from widely used packages such as SHARC, Newton-X, and PyRAI2MD,
), a versatile open-source Python toolkit designed to unify and streamline processing, storage, management, analysis, and visualization of SH simulation data. By supporting data from widely used packages such as SHARC, Newton-X, and PyRAI2MD,  standardizes complex excited-state trajectory outputs into a consistent format, enabling efficient and reproducible exploratory analysis.
standardizes complex excited-state trajectory outputs into a consistent format, enabling efficient and reproducible exploratory analysis.  also serves as the core analysis extension of the
also serves as the core analysis extension of the  dataset (Surface Hopping Nested Instances Training Set for Excited-State Learning) recently published by some of us.35,36
dataset (Surface Hopping Nested Instances Training Set for Excited-State Learning) recently published by some of us.35,36
In the following sections, we first present key features of the  package, focusing on data curation, including the loading and writing of trajectory data, followed by advanced processing and visualization methods centered on filtering and exploratory analysis in geometric and property spaces. Finally, we demonstrate the capabilities of
package, focusing on data curation, including the loading and writing of trajectory data, followed by advanced processing and visualization methods centered on filtering and exploratory analysis in geometric and property spaces. Finally, we demonstrate the capabilities of  by a showcase application analyzing trajectories across chemical compound space for a series of alkenes (CnH2n, with n = {2,3,4}) and CH2NH+2,36,37 showcasing its versatility and power to handle diverse molecular datasets.
by a showcase application analyzing trajectories across chemical compound space for a series of alkenes (CnH2n, with n = {2,3,4}) and CH2NH+2,36,37 showcasing its versatility and power to handle diverse molecular datasets.
2
In this work, we introduce 
 (
( ), a Python package developed for the post-processing, statistical analysis, and visualization of surface hopping (SH) trajectory data. Designed to support comprehensive data analysis workflows,
), a Python package developed for the post-processing, statistical analysis, and visualization of surface hopping (SH) trajectory data. Designed to support comprehensive data analysis workflows,  builds on the
builds on the  ,38
,38 ,39 and
,39 and  40 frameworks, and is compatible with trajectory outputs from commonly used engines such as SHARC, Newton-X and PyRAI2MD.
40 frameworks, and is compatible with trajectory outputs from commonly used engines such as SHARC, Newton-X and PyRAI2MD.
At the core of  is a hierarchical data model for organizing SH trajectory ensembles. Data is stored in a tree-like structure that allows trajectories to be grouped and subdivided according to molecular system, electronic structure reference method, and other simulation settings (e.g. time step or hopping scheme, see Fig. S1). This enables comparison of different methods for a given molecule, analysis of multiple molecules at a common level of theory, and combinations of these views within a single dataset. In this way, complex collections of SH simulations can be analyzed in a consistent and systematic manner, supporting ensemble level statistics and cross-method and -system studies without the need for manual data restructuring. It is specifically designed to support all stages of the SH trajectory data lifecycle following data generation – including processing and storage, management, analysis, and visualization (cf.Fig. 1) – thereby facilitating the efficient interpretation of SH simulations through streamlined data handling and interactive analysis.
is a hierarchical data model for organizing SH trajectory ensembles. Data is stored in a tree-like structure that allows trajectories to be grouped and subdivided according to molecular system, electronic structure reference method, and other simulation settings (e.g. time step or hopping scheme, see Fig. S1). This enables comparison of different methods for a given molecule, analysis of multiple molecules at a common level of theory, and combinations of these views within a single dataset. In this way, complex collections of SH simulations can be analyzed in a consistent and systematic manner, supporting ensemble level statistics and cross-method and -system studies without the need for manual data restructuring. It is specifically designed to support all stages of the SH trajectory data lifecycle following data generation – including processing and storage, management, analysis, and visualization (cf.Fig. 1) – thereby facilitating the efficient interpretation of SH simulations through streamlined data handling and interactive analysis.
 | ||
Fig. 1 Schematic overview of the  ( ( ) data lifecycle and analysis workflow. ) data lifecycle and analysis workflow.  reads surface-hopping (SH) trajectory data produced by various simulation engines (such as SHARC, PyRAI2MD, and Newton-X) via a unified parsing layer, which standardizes their diverse outputs into a hierarchical data model enriched with metadata. A sanity-check stage filters non-physical data based on energetic and geometric criteria. The selection stage then allows restriction of the dataset to specific electronic states or molecular substructures, so that all subsequent operations act only on the chosen subset (or on all states and the full structure if no selection is applied). The analysis stage provides tools for computing geometric descriptors (e.g., bond lengths, angles, dihedrals, pyramidalization) as well as their dimensionality-reduced representations and calculation of statistical observables such as state populations, spectra, and distributions of electronic and geometric properties. The respective results are explored in the visualization stage using time-dependent plots, spectra, scatter plots, and ensemble statistics. Finally, the write stage exports curated datasets in NetCDF format for reproducible storage and sharing, and generates PDF or SVG datasheets containing standardized summary plots of the geometry and property space of a certain dataset (e.g., histograms, time plots, absorption spectra) for the selected states and molecular substructures. reads surface-hopping (SH) trajectory data produced by various simulation engines (such as SHARC, PyRAI2MD, and Newton-X) via a unified parsing layer, which standardizes their diverse outputs into a hierarchical data model enriched with metadata. A sanity-check stage filters non-physical data based on energetic and geometric criteria. The selection stage then allows restriction of the dataset to specific electronic states or molecular substructures, so that all subsequent operations act only on the chosen subset (or on all states and the full structure if no selection is applied). The analysis stage provides tools for computing geometric descriptors (e.g., bond lengths, angles, dihedrals, pyramidalization) as well as their dimensionality-reduced representations and calculation of statistical observables such as state populations, spectra, and distributions of electronic and geometric properties. The respective results are explored in the visualization stage using time-dependent plots, spectra, scatter plots, and ensemble statistics. Finally, the write stage exports curated datasets in NetCDF format for reproducible storage and sharing, and generates PDF or SVG datasheets containing standardized summary plots of the geometry and property space of a certain dataset (e.g., histograms, time plots, absorption spectra) for the selected states and molecular substructures. | ||
The following sections start by describing the internal data structure of  (see Section 2.1) and subsequently turns to a detailed overview of the core modules and functionality features of
(see Section 2.1) and subsequently turns to a detailed overview of the core modules and functionality features of  in Sections 2.2–2.6, which cover (cf.Fig. 1):
in Sections 2.2–2.6, which cover (cf.Fig. 1):
• Collection, management & storage: the  module of
module of  supports the parsing of SH trajectories with varying simulation parameters (e.g., initial active states, state types, simulation length, and atom counts) into structured
supports the parsing of SH trajectories with varying simulation parameters (e.g., initial active states, state types, simulation length, and atom counts) into structured  datasets, where observables are annotated with metadata, converted to standardized units, and made available for export to NetCDF, supporting reproducibility and data sharing37 (see Section 2.2).
datasets, where observables are annotated with metadata, converted to standardized units, and made available for export to NetCDF, supporting reproducibility and data sharing37 (see Section 2.2).
• Data processing: at the postprocessing stage, the  module supports the systematic identification and removal of non-physical or undesired trajectory segments based on energetic and geometric criteria (see Section 2.3). Furthermore, the
module supports the systematic identification and removal of non-physical or undesired trajectory segments based on energetic and geometric criteria (see Section 2.3). Furthermore, the  module supports the restriction of the analysis to selected electronic states and chemically relevant molecular substructures (see Section 2.4).
module supports the restriction of the analysis to selected electronic states and chemically relevant molecular substructures (see Section 2.4).
• Analysis: for analysis,  offers tools for the statistical evaluation and post-processing of computed properties, including e.g. the calculation of relative energies, averaged oscillator strengths, and state populations. It further supports the identification of collective geometric descriptors and the statistical analysis of user-defined internal coordinates (see Section 2.5).
offers tools for the statistical evaluation and post-processing of computed properties, including e.g. the calculation of relative energies, averaged oscillator strengths, and state populations. It further supports the identification of collective geometric descriptors and the statistical analysis of user-defined internal coordinates (see Section 2.5).
• Visualization & interpretation:  supports exploratory visualization of selected substructures and their associated geometric features as well as the distribution of certain electronic properties or spectra for selected states, and geometric features at both the single trajectory and ensemble level. A datasheet module enables rapid generation of summary plots that highlight trends across geometry and property space, both streamlining and standardizing common post-processing and analysis tasks (see Section 2.6).
supports exploratory visualization of selected substructures and their associated geometric features as well as the distribution of certain electronic properties or spectra for selected states, and geometric features at both the single trajectory and ensemble level. A datasheet module enables rapid generation of summary plots that highlight trends across geometry and property space, both streamlining and standardizing common post-processing and analysis tasks (see Section 2.6).
2.1 Data structure
Given the multidimensional nature of SH data, leverages the
leverages the  package38 for effective and user-friendly data handling. In
package38 for effective and user-friendly data handling. In  , data is stored in memory as an
, data is stored in memory as an  (see performance benchmarks in Tables. S2 and S3). These objects function as labeled dictionaries of arrays, where different properties can share common axes – referred to as dimensions. Key dimensions for both static and dynamic data include the number of electronic states (state), couplings (statecomb), atoms (atom) and time (time), the latter of which only exhibits a single tick in static datasets.
(see performance benchmarks in Tables. S2 and S3). These objects function as labeled dictionaries of arrays, where different properties can share common axes – referred to as dimensions. Key dimensions for both static and dynamic data include the number of electronic states (state), couplings (statecomb), atoms (atom) and time (time), the latter of which only exhibits a single tick in static datasets.
To retain information of different trajectories with potentially different metadata in regards to parameter configuration, SH software versions and setup parameters used, or to keep track of varying length of trajectories, we employ a hierarchical tree structure similar to a file system hierarchy to store multiple datasets in a shared database-like structure. This structure, which we refer to as  in
in  , can group datasets by distinct compounds but additionally supports further subdivision at an arbitrary number of levels into groups to, e.g., differentiate different uses of the respective member datasets or different time resolution of trajectories. For analysis and postprocessing purposes, this enables filtering for certain features like the version of the SH software used, the EST level or additional derived features like whether a state transition has occurred. This not only allows for comparison of results between different setups but also efficient retention of information for potential future applications in machine learning, where the accidental combination of data from different levels of theory or different setups can limit the ability of a network to produce highly accurate results.
, can group datasets by distinct compounds but additionally supports further subdivision at an arbitrary number of levels into groups to, e.g., differentiate different uses of the respective member datasets or different time resolution of trajectories. For analysis and postprocessing purposes, this enables filtering for certain features like the version of the SH software used, the EST level or additional derived features like whether a state transition has occurred. This not only allows for comparison of results between different setups but also efficient retention of information for potential future applications in machine learning, where the accidental combination of data from different levels of theory or different setups can limit the ability of a network to produce highly accurate results.
On a technical level, the current implementation of  employs a custom, lightweight tree structure in memory, which is mapped to an
employs a custom, lightweight tree structure in memory, which is mapped to an  structure to support easy mapping to and loading from NetCDF files in a way that allows all relevant meta-data to be preserved for reproducibility purposes contrary to other tools in the field. Furthermore,
structure to support easy mapping to and loading from NetCDF files in a way that allows all relevant meta-data to be preserved for reproducibility purposes contrary to other tools in the field. Furthermore,  offers a range of API functions for the purpose of parallel processing and filtering of its hierarchically structured datasets that should be used to interact with the contained data, similar to the API of other data management systems. For the analysis of ensembles (i.e., collections of trajectories corresponding to the same reaction or molecular system),
offers a range of API functions for the purpose of parallel processing and filtering of its hierarchically structured datasets that should be used to interact with the contained data, similar to the API of other data management systems. For the analysis of ensembles (i.e., collections of trajectories corresponding to the same reaction or molecular system),  can introduce an additional synthetic dimension (frame) along which frames (i.e. system states at an arbitrary time step), can be continuously indexed. Naturally, a single static dataset only contains a single frame, whereas a dynamic dataset may contain hundreds or thousands. This transformation introduces a structure similar to that of SPaiNN databases,41 where properties are stored in a series of arbitrary snapshots of a system to better support ML applications. In technical terms, the frame axis associates each index with a trajectory and a timestep within that trajectory using a
can introduce an additional synthetic dimension (frame) along which frames (i.e. system states at an arbitrary time step), can be continuously indexed. Naturally, a single static dataset only contains a single frame, whereas a dynamic dataset may contain hundreds or thousands. This transformation introduces a structure similar to that of SPaiNN databases,41 where properties are stored in a series of arbitrary snapshots of a system to better support ML applications. In technical terms, the frame axis associates each index with a trajectory and a timestep within that trajectory using a  with trajectory index (
with trajectory index ( ) and time (
) and time ( ) as levels (see Fig. 2) allowing for mapping between either a frame-sequential or hierarchical representation. Functions for the conversion from hierarchical to the frame-sequential representation are provided by
) as levels (see Fig. 2) allowing for mapping between either a frame-sequential or hierarchical representation. Functions for the conversion from hierarchical to the frame-sequential representation are provided by  . However, the retention of all relevant metadata is only guaranteed in the hierarchical data structure and frame-segmentalization is only recommended for data-processing steps, where the knowledge of detailed metadata is no longer essential, but not for long-term data storage and data exchange.
. However, the retention of all relevant metadata is only guaranteed in the hierarchical data structure and frame-segmentalization is only recommended for data-processing steps, where the knowledge of detailed metadata is no longer essential, but not for long-term data storage and data exchange.
 | ||
Fig. 2 Schematic overview of the data access pipeline for retrieving permanent dipole moments of a specific frame (geometry) from a trajectory  , illustrating two selection paths: (left) selecting a specific frame using the. , illustrating two selection paths: (left) selecting a specific frame using the. method of method of  , which returns a reduced , which returns a reduced  while retaining all other dimensions; and (right) selecting a specific data variable (here permanent dipoles, while retaining all other dimensions; and (right) selecting a specific data variable (here permanent dipoles,  ) using dictionary-like syntax, which returns a ) using dictionary-like syntax, which returns a  . These selection operations can be applied in either order to isolate the permanent dipoles corresponding to a specific single frame. . These selection operations can be applied in either order to isolate the permanent dipoles corresponding to a specific single frame. | ||
Through the use of 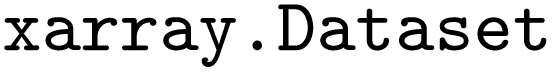 features, users can either extract individual variables, such as energies, from an ensemble yielding an
features, users can either extract individual variables, such as energies, from an ensemble yielding an  (see SI Section 2) or a collection of variables through selection across shared axes between individual trajectories within the same time step, yielding a view of the dataset (see Fig. S1–S3). This simplifies both the implementation of provided functionality in the
(see SI Section 2) or a collection of variables through selection across shared axes between individual trajectories within the same time step, yielding a view of the dataset (see Fig. S1–S3). This simplifies both the implementation of provided functionality in the  package as well as future custom extensions by the user. For variables supported and parsed by the default
package as well as future custom extensions by the user. For variables supported and parsed by the default  input routines, annotations for the individual variables will be provided, adding a description of their contents and units if the variable may need to be converted to be processed together with other data for which
input routines, annotations for the individual variables will be provided, adding a description of their contents and units if the variable may need to be converted to be processed together with other data for which  also provides appropriate tooling.
also provides appropriate tooling.
2.2 Data curation ( )
)

 supports the processing of geometries and their corresponding quantum chemical properties relevant to excited-state dynamics. These include state-specific properties such as energies (Ei), forces (Fi), phases (pi) and permanent dipole moments (µi) of electronic states i (see overview in Table. S1). Additionally, properties arising from interactions between states, such as transition dipole moments (µij), non-adiabatic couplings (NACij) and spin–orbit couplings (SOCij) are processed for pairs of electronic states (ij) with j > i. Each of these properties is associated with a specific molecular geometry, which is stored as Cartesian coordinates upon parsing/loading of the data. The module additionally tracks the active (diabatic) electronic state, state combination indices (ij), as well as trajectory-specific metadata such as the number of time steps and time step size.
supports the processing of geometries and their corresponding quantum chemical properties relevant to excited-state dynamics. These include state-specific properties such as energies (Ei), forces (Fi), phases (pi) and permanent dipole moments (µi) of electronic states i (see overview in Table. S1). Additionally, properties arising from interactions between states, such as transition dipole moments (µij), non-adiabatic couplings (NACij) and spin–orbit couplings (SOCij) are processed for pairs of electronic states (ij) with j > i. Each of these properties is associated with a specific molecular geometry, which is stored as Cartesian coordinates upon parsing/loading of the data. The module additionally tracks the active (diabatic) electronic state, state combination indices (ij), as well as trajectory-specific metadata such as the number of time steps and time step size.
 package provides dedicated functionality for parsing trajectory data from standard output files generated by the widely used SH programs SHARC12,13 (versions 2.0 to 4.0), Newton-X14,15 (versions 2.0 to 2.6), and PyRAI2MD16,17 (version 2.4) via the
package provides dedicated functionality for parsing trajectory data from standard output files generated by the widely used SH programs SHARC12,13 (versions 2.0 to 4.0), Newton-X14,15 (versions 2.0 to 2.6), and PyRAI2MD16,17 (version 2.4) via the  module. Furthermore, this module supports direct parsing of initial condition files from SHARC simulations through the same interface. Input routines have been extensively tested to ensure reliable operation across a large range of configurations with test data for several versions provided in the
module. Furthermore, this module supports direct parsing of initial condition files from SHARC simulations through the same interface. Input routines have been extensively tested to ensure reliable operation across a large range of configurations with test data for several versions provided in the  release on Zenodo.42 Specifically, we have tested the parsing of data from versions 2.0 to 4.0 of SHARC, versions 2.0 to 2.6 of Newton-X and Version 2.4 of PyRAI2MD. Additional versions can easily be supported in the future as they become available (see SI Section 3.1). For this purpose but also more general extensibility to other formats, we provide the
release on Zenodo.42 Specifically, we have tested the parsing of data from versions 2.0 to 4.0 of SHARC, versions 2.0 to 2.6 of Newton-X and Version 2.4 of PyRAI2MD. Additional versions can easily be supported in the future as they become available (see SI Section 3.1). For this purpose but also more general extensibility to other formats, we provide the  abstract base class, which can be implemented for different formats in their own sub-module of
abstract base class, which can be implemented for different formats in their own sub-module of  (see e.g.
(see e.g. for the SHARC implementation). This guarantees the transferability of the
for the SHARC implementation). This guarantees the transferability of the  pipeline and extensibility to newly emerging tools, e.g. associated with novel ML approaches.
pipeline and extensibility to newly emerging tools, e.g. associated with novel ML approaches.
The parsing of all supported formats is supported via the format-agnostic  function, e.g.
function, e.g. . It automatically attempts to detect the format of the input directory and will search for subdirectories of
. It automatically attempts to detect the format of the input directory and will search for subdirectories of  that may be of an appropriate input format in case the targeted directory itself holds subdirectories containing different trajectories that should be loaded in parallel (see also SI Section 3.1). This functionality has been employed in the construction of the
that may be of an appropriate input format in case the targeted directory itself holds subdirectories containing different trajectories that should be loaded in parallel (see also SI Section 3.1). This functionality has been employed in the construction of the  dataset,35,36 where the
dataset,35,36 where the  module was used to extract and organize trajectory data from SHARC and Newton-X simulations. Finely granular configuration options and controls for the parsing function are documented for the
module was used to extract and organize trajectory data from SHARC and Newton-X simulations. Finely granular configuration options and controls for the parsing function are documented for the  function, allowing, e.g., to control the patterns for subdirectories that should be considered, specifying formats that should be checked, the units of input variables or whether multiple trajectories should be read in parallel or sequentially, which can help with the debugging of newly added format interfaces.
function, allowing, e.g., to control the patterns for subdirectories that should be considered, specifying formats that should be checked, the units of input variables or whether multiple trajectories should be read in parallel or sequentially, which can help with the debugging of newly added format interfaces.
For ease of use and to promote data sharing and reproducibility, we also provide a simple CLI script  that performs the conversion of a set of input directories into a
that performs the conversion of a set of input directories into a  -style format, to support standardized data publication processes without requiring any coding by the user.
-style format, to support standardized data publication processes without requiring any coding by the user.
 can be exported in the standardized NetCDF43 format using the
can be exported in the standardized NetCDF43 format using the  function, e.g.,
function, e.g.,  . Each dataset retains both original and processed data, with derived properties stored as additional data variables, if so desired. This structure aligns with the FAIR principles by ensuring interoperability and enabling reproducible workflows through transparent tracking of data transformations. In principle, the generic nature of NetCDF file formats also allows for the storage of licensing and publication information, which
. Each dataset retains both original and processed data, with derived properties stored as additional data variables, if so desired. This structure aligns with the FAIR principles by ensuring interoperability and enabling reproducible workflows through transparent tracking of data transformations. In principle, the generic nature of NetCDF file formats also allows for the storage of licensing and publication information, which  is intended to support in the future to facilitate programmatic reuse of published data with appropriate attribution of the original sources.
is intended to support in the future to facilitate programmatic reuse of published data with appropriate attribution of the original sources.
Thus, the data structure of  provides a consistent and extensible format for long-term storage and exchange of SH simulation data in addition to a user-friendly foundation for further processing in-memory. Notably, the recently introduced
provides a consistent and extensible format for long-term storage and exchange of SH simulation data in addition to a user-friendly foundation for further processing in-memory. Notably, the recently introduced  dataset,37 that aims to provide a growing platform of SH data, is based on this format. Users can access the respective dataset or any NetCDF file generated with
dataset,37 that aims to provide a growing platform of SH data, is based on this format. Users can access the respective dataset or any NetCDF file generated with  by loading it as an
by loading it as an  object via the
object via the  function.
function.
In addition,  supports the export to the Atomic Simulation Environment (ASE) database format,44,45 which is widely used in the training and development of machine learning potentials for excited states.41,46,47 The import from such databases is supported given appropriate internal structure of the observables (shape and consistent dimension sizes) and sufficient provided metadata like units for each property.
supports the export to the Atomic Simulation Environment (ASE) database format,44,45 which is widely used in the training and development of machine learning potentials for excited states.41,46,47 The import from such databases is supported given appropriate internal structure of the observables (shape and consistent dimension sizes) and sufficient provided metadata like units for each property.
2.3 Data cleaning ( )
)
In SH simulations, trajectory ensembles may contain non-physical or otherwise undesired segments arising from numerical instabilities, limitations of the electronic-structure description, or rare but chemically irrelevant reaction pathways. Such segments must be excluded prior to further analysis to ensure meaningful interpretation of ensemble-based observables.13 To this end, the 
 module provides filtering procedures based on multiple energetic criteria, commonly employed in SH simulations (e.g. those provided in the diagnosis functionality of SHARC13) as well as bond-length thresholds as geometric criteria, combined with different filtering strategies.
module provides filtering procedures based on multiple energetic criteria, commonly employed in SH simulations (e.g. those provided in the diagnosis functionality of SHARC13) as well as bond-length thresholds as geometric criteria, combined with different filtering strategies.
To account for problems with time step consistency, total energy conservation, potential/kinetic energy smoothness or hopping energy restriction, the core energetic descriptors are derived from the kinetic, potential and total energies, and include the deviation (drift) of the total energy from its initial value (t = 0), frame-to-frame changes (steps) in each energy component, and state-hopping events tracked explicitly. The respective default thresholds of the resulting five energy-based criteria are listed in Table. S4 (note that a respective unit conversion is handled automatically). The geometric filtration is based on interatomic distances, where by default, any bond exceeding 300 pm is flagged as violation, while a stricter threshold of 200 pm is by default applied to C–H and N–H bonds, to take into account their dissociation that might be triggered based only on the selection of initial conditions. Any other bond criterion can be defined and added by the user.
The core concept of the filtering framework is the definition of trajectory- and frame-resolved diagnostic quantities, referred to here as filtranda, whose excursions beyond user-defined thresholds indicate the onset of unreliable or undesired behavior. By applying the criteria, for each trajectory and each criterion, a maximum usable time is determined, corresponding either to the full simulation length or to the time at which the first violation occurs. Because all filtranda and thresholds are stored alongside the raw trajectory data within the same  -based data structure, they are directly accessible for inspection. Dedicated visualizations are provided to help users assess how changes in threshold values affect the retained ensemble in terms of the number of trajectories, the number of frames, and the accessible simulation time (cf. Fig. S4). Importantly, different filtering choices can be explored without modifying the original trajectory data on disk.
-based data structure, they are directly accessible for inspection. Dedicated visualizations are provided to help users assess how changes in threshold values affect the retained ensemble in terms of the number of trajectories, the number of frames, and the accessible simulation time (cf. Fig. S4). Importantly, different filtering choices can be explored without modifying the original trajectory data on disk.
In analogy to the maximum usable time concept employed in SHARC diagnostics,13 filtering in  involves an inherent trade-off between ensemble size and the temporal range over which ensemble-averaged observables can be interpreted reliably. This trade-off is illustrated by sequential filtering of SH trajectories of ethene (A01) using one energetic and one geometric criterion (see Fig. 3). When both criteria are applied using the truncation strategy, most trajectories contribute at least to the early part of the dynamics, resulting in a large number of retained frames (see Fig. 3a). However, because trajectories are truncated at different times, ensemble-averaged observables are only statistically meaningful up to the earliest truncation times, analogously to analyses based on the maximum analysis time in SHARC. A stricter filtering is exemplarily achieved by combining truncation for energetic criteria with omission for geometric criteria, such that trajectories exceeding any C–H bond-length of 200 pm are removed entirely from the ensemble (see Fig. 3b). This reduces the number of retained trajectories and frames but still shares the limitation that reliable ensemble statistics are confined to early simulation times (e.g. circa 10 fs in the example). In contrast, the transection strategy explicitly enforces a common analysis window by defining a minimum acceptable trajectory length. Only trajectories that remain free of violations up to this time are retained, and all retained trajectories are cut to the same temporal extent (see Fig. 3c). This ensures that ensemble-averaged observables are statistically consistent throughout the selected time interval, at the cost of excluding a larger fraction of trajectories. As with SHARC's maximum usable time criterion, choosing a longer transection time improves temporal coverage but reduces ensemble size, while shorter transection times admit more trajectories but limit the accessible simulation window.
involves an inherent trade-off between ensemble size and the temporal range over which ensemble-averaged observables can be interpreted reliably. This trade-off is illustrated by sequential filtering of SH trajectories of ethene (A01) using one energetic and one geometric criterion (see Fig. 3). When both criteria are applied using the truncation strategy, most trajectories contribute at least to the early part of the dynamics, resulting in a large number of retained frames (see Fig. 3a). However, because trajectories are truncated at different times, ensemble-averaged observables are only statistically meaningful up to the earliest truncation times, analogously to analyses based on the maximum analysis time in SHARC. A stricter filtering is exemplarily achieved by combining truncation for energetic criteria with omission for geometric criteria, such that trajectories exceeding any C–H bond-length of 200 pm are removed entirely from the ensemble (see Fig. 3b). This reduces the number of retained trajectories and frames but still shares the limitation that reliable ensemble statistics are confined to early simulation times (e.g. circa 10 fs in the example). In contrast, the transection strategy explicitly enforces a common analysis window by defining a minimum acceptable trajectory length. Only trajectories that remain free of violations up to this time are retained, and all retained trajectories are cut to the same temporal extent (see Fig. 3c). This ensures that ensemble-averaged observables are statistically consistent throughout the selected time interval, at the cost of excluding a larger fraction of trajectories. As with SHARC's maximum usable time criterion, choosing a longer transection time improves temporal coverage but reduces ensemble size, while shorter transection times admit more trajectories but limit the accessible simulation window.
 | ||
| Fig. 3 Comparison of three trajectory-filtering strategies applied to an trajectory ensemble of ethene (A01; taken from ref. 35 and 36). Filtering is performed sequentially using two criteria: (i) a maximum allowed change in potential energy between consecutive frames in the active state of ΔEactivepot ≤ 0.7 eV; and (ii) a maximum C–H bond length of 250 pm. Each trajectory is represented by a horizontal bar; the vertical ordering is arbitrary. Segments removed by the energetic filter are shown in pale yellow, those removed by subsequent geometric filtering in white, and segments retained for the final analysis are shown in yellow. (a) Both criteria are applied by truncation, yielding trajectories that are first cut by the energy threshold (blue) and then further truncated on the basis of the geometric threshold (yellow). (b) The energy criterion is applied by truncation, whereas the geometric criterion is applied by omission, so that trajectories exceeding the C–H threshold at any time are removed entirely (white). (c) Both criteria are applied by truncation, followed by a transection at 50 fs (black dashed line), which removes all trajectories shorter than this time and cuts the remaining ones accordingly. | ||
2.4 Structure and state selection ( )
)
SH simulations are frequently used to investigate specific photochemical pathways, so that often only certain electronic states or structural motifs are of direct mechanistic interest. Focusing the analysis on key states and structures enables a more direct and interpretable characterization of the underlying reaction pathways, avoiding dilution of mechanistic insight by geometric motions that are not directly coupled to the reactive process or state interactions that are irrelevant to a photochemical reaction of interest.48,49

 provides the
provides the  class. In
class. In  , structure selection is implemented as a dedicated pre-processing step based on an index-labeled molecular graph constructed from a suitable (i.e., the energetically lowest or the first) frame of a respective trajectory. Substructures may be specified using SMARTS patterns, explicit atom indices, or combinations thereof. SMARTS queries are matched against the molecular graph to identify pattern matching structure fragments, while atom indices enable the selection of arbitrary atom sets. If no structure is specified, the full molecular geometry is retained for analysis. Selected substructures can be visualized using built-in drawing routines (see Fig. S5), as demonstrated in the accompanying tutorial notebooks.42
, structure selection is implemented as a dedicated pre-processing step based on an index-labeled molecular graph constructed from a suitable (i.e., the energetically lowest or the first) frame of a respective trajectory. Substructures may be specified using SMARTS patterns, explicit atom indices, or combinations thereof. SMARTS queries are matched against the molecular graph to identify pattern matching structure fragments, while atom indices enable the selection of arbitrary atom sets. If no structure is specified, the full molecular geometry is retained for analysis. Selected substructures can be visualized using built-in drawing routines (see Fig. S5), as demonstrated in the accompanying tutorial notebooks.42
Multiple selections can be combined using standard set operations, including union, intersection, difference, and complement, enabling flexible definitions of substructures of interest. The resulting selected set is then passed to downstream analysis stages (cf. Section 2.5), where users may specify which internal coordinates, including bond lengths, bond angles, dihedral angles, or pyramidalization angles, should be evaluated.  automatically determines which coordinates are well-defined based on the size and connectivity of the selected fragment; for example, distances require two (bonded) atoms, dihedral angles require four atoms sequentially connected by three bonds, and pyramidalization angles require a central atom bonded to three neighbors. In this way, all geometrically meaningful descriptors are generated consistently and automatically for the chosen molecular substructure.
automatically determines which coordinates are well-defined based on the size and connectivity of the selected fragment; for example, distances require two (bonded) atoms, dihedral angles require four atoms sequentially connected by three bonds, and pyramidalization angles require a central atom bonded to three neighbors. In this way, all geometrically meaningful descriptors are generated consistently and automatically for the chosen molecular substructure.
Beyond the analysis of a single molecular system,  also supports the comparison of structurally related compounds through the extraction of analogous substructures across multiple molecules. This enables cross-compound analyses in which equivalent reactive or functional motifs are examined on a common geometric footing. To perform such an analysis, trajectory datasets must first be grouped within the
also supports the comparison of structurally related compounds through the extraction of analogous substructures across multiple molecules. This enables cross-compound analyses in which equivalent reactive or functional motifs are examined on a common geometric footing. To perform such an analysis, trajectory datasets must first be grouped within the  , e.g. according to molecular identity, for example into separate branches for the methylenimmonium cation (I01) and a retinal model system (I02) (see Fig. S3). For each branch, the molecular graph is constructed and used as input to a maximum common substructure (MCS) search, which yields a canonicalized SMARTS representation of the shared chemical motif. Alternatively, a user-defined SMARTS pattern may be specified (see Fig. 4). The resulting substructure pattern is then passed to the structure selection to generate a consistent atom mapping for each compound. This mapping is applied to all trajectory datasets within the respective branch, restricting all coordinate-dependent properties to the atoms belonging to the matched substructure. In this way, properties such as atomic positions, forces, or geometric descriptors are reduced to a common set of atoms with identical ordering across compounds. As an example, the maximum common substructure of I01 and I02 is described by the SMARTS pattern
, e.g. according to molecular identity, for example into separate branches for the methylenimmonium cation (I01) and a retinal model system (I02) (see Fig. S3). For each branch, the molecular graph is constructed and used as input to a maximum common substructure (MCS) search, which yields a canonicalized SMARTS representation of the shared chemical motif. Alternatively, a user-defined SMARTS pattern may be specified (see Fig. 4). The resulting substructure pattern is then passed to the structure selection to generate a consistent atom mapping for each compound. This mapping is applied to all trajectory datasets within the respective branch, restricting all coordinate-dependent properties to the atoms belonging to the matched substructure. In this way, properties such as atomic positions, forces, or geometric descriptors are reduced to a common set of atoms with identical ordering across compounds. As an example, the maximum common substructure of I01 and I02 is described by the SMARTS pattern  , corresponding to a five-atom substructure motif. Applying this mapping reduces the atom position shaped data of I01 by one atom and that of I02 by nine atoms, yielding an aligned five-atom representations for both systems (see Fig. S3).
, corresponding to a five-atom substructure motif. Applying this mapping reduces the atom position shaped data of I01 by one atom and that of I02 by nine atoms, yielding an aligned five-atom representations for both systems (see Fig. S3).
 | ||
| Fig. 4 Schematic illustration of structure selection across compounds, using the alkenes ethene (A01), propene (A02), butene (A03), and the methylene immonium cation (I01) as illustrative examples. A maximum common substructure analysis across these molecules yields a carbon atom bearing one hydrogen and two explicit additional connections (SMARTS: [#6;H1;D2]). As a result, the corresponding substructure matches occur twice in the alkenes but only once in I01, as indicated by the gray boxes on the left. Structure selection using the SMARTS pattern [#6,#1][#6;H1] = [#6,#7;H1][#6,#1] identifies an analogous four-atom substructure in all compounds, highlighted in purple on the right. For this substructure, the projections of the trajectories onto the first two principal components obtained from a principal component analysis performed on pairwise distances of A03 are shown on the right-hand side. In each subplot, the full trajectory ensemble of all molecules is shown in gray, while the trajectories of the respective highlighted compound are overlaid in color. For A03, photoisomerization outcomes are distinguished by final torsion angle: Z → Z (blue, φ < 80°) and Z → E (yellow, φ > 100°). White and yellow dots indicate S1 → S0 and S2 → S1 hopping points, respectively. Furthermore, the distributions of the associated pyramidalisation and torsion angles of the hopping points are shown. For I01 the pyramidalisation angles refer to S2 → S1 hops, while the dihedrals are shown for the S1 → S0 hops. For the alkenes, all properties are shown for S1 → S0 hops. The underlying trajectory data is taken from SHNITSEL-data;35,36 trajectories exhibiting any bond length exceeding 200 pm were excluded from the analysis. | ||
 provides a dedicated
provides a dedicated  framework to restrict ensemble analysis to specific electronic states and state combinations. This enables chemically targeted investigation of electronic processes such as internal conversion or intersystem crossing by focusing the analysis on the relevant parts of the electronic state space.
framework to restrict ensemble analysis to specific electronic states and state combinations. This enables chemically targeted investigation of electronic processes such as internal conversion or intersystem crossing by focusing the analysis on the relevant parts of the electronic state space.
State selection in  is hierarchical. Users may first specify subsets of electronic states, for example restricting the analysis to singlet or triplet manifolds, and may subsequently (independently) define state combinations to isolate specific types of transitions. For instance, singlet–singlet or triplet–triplet combinations allow focused analysis of internal conversion processes, whereas singlet–triplet combinations enable the investigation of intersystem crossing events. These selections can be applied either independently or jointly within a single call. Electronic states are specified using a term symbol-like notation. Classes of states are defined by their spin multiplicity (
is hierarchical. Users may first specify subsets of electronic states, for example restricting the analysis to singlet or triplet manifolds, and may subsequently (independently) define state combinations to isolate specific types of transitions. For instance, singlet–singlet or triplet–triplet combinations allow focused analysis of internal conversion processes, whereas singlet–triplet combinations enable the investigation of intersystem crossing events. These selections can be applied either independently or jointly within a single call. Electronic states are specified using a term symbol-like notation. Classes of states are defined by their spin multiplicity ( ,
,  , or
, or  ). Individual states can be identified by their index or their assigned names. By default, in
). Individual states can be identified by their index or their assigned names. By default, in  , this is their multiplicity followed by a state index, such as
, this is their multiplicity followed by a state index, such as  for the singlet ground state or
for the singlet ground state or  for the first electronically excited singlet state. Triplet manifolds are represented by their degenerate components (e.g.,
for the first electronically excited singlet state. Triplet manifolds are represented by their degenerate components (e.g.,  ,
,  , and
, and  ). If a label for a multiplicity class is given, all the corresponding singlet, doublet or triplet states are selected for further analysis. Alternatively, states may be addressed directly by their numerical indices in the adiabatic representation provided by the underlying SH engines.
). If a label for a multiplicity class is given, all the corresponding singlet, doublet or triplet states are selected for further analysis. Alternatively, states may be addressed directly by their numerical indices in the adiabatic representation provided by the underlying SH engines.
State combinations are specified using arrow notation. By default, combinations such as  are treated as undirected and include both directions of population transfer, thus selecting trajectory segments before and after hopping events between the two states. When required, selections can be made directional, for example to isolate trajectory segments preceding the first directed
are treated as undirected and include both directions of population transfer, thus selecting trajectory segments before and after hopping events between the two states. When required, selections can be made directional, for example to isolate trajectory segments preceding the first directed  transition. In this case, bidirectional transitions are indicated by
transition. In this case, bidirectional transitions are indicated by  . This flexible formalism allows concise definitions of complex electronic subsets; for example, a selection such as
. This flexible formalism allows concise definitions of complex electronic subsets; for example, a selection such as  restricts the analysis to all singlet states and undirected singlet–triplet transitions, enabling postprocessing focused on intersystem crossing while retaining access to singlet-state dynamics.
restricts the analysis to all singlet states and undirected singlet–triplet transitions, enabling postprocessing focused on intersystem crossing while retaining access to singlet-state dynamics.
The hop-selection framework of  allows subsets of hopping events to be extracted by specifying, similar to the state combination selection, initial-final state pairs (e.g.,
allows subsets of hopping events to be extracted by specifying, similar to the state combination selection, initial-final state pairs (e.g.,  or
or  ), enabling directional filtering, e.g., forward, backward, or both (see Fig. 5a and S7). The selection is applied consistently to all stored observables, including user-defined and derived quantities, enabling direct comparison of hopping geometries, such as distributions of torsion and pyramidalization angles as illustrated for A01, A02, A03 and I01 in Fig. 4.
), enabling directional filtering, e.g., forward, backward, or both (see Fig. 5a and S7). The selection is applied consistently to all stored observables, including user-defined and derived quantities, enabling direct comparison of hopping geometries, such as distributions of torsion and pyramidalization angles as illustrated for A01, A02, A03 and I01 in Fig. 4.
 | ||
| Fig. 5 Schematic illustration of two concatenated trajectories (yellow and blue) on an absolute time axis (a), with surface hops indicated by white markers. Panels (b) and (c) illustrate two approaches to hop-centered analysis. In the trajectory-focused representation (b), trajectories are aligned to a hop-centered time coordinate and each trajectory is included once; when multiple hops are present, a single event is selected for alignment (here the last hop of the yellow trajectory). In the hop-focused representation (c), each hop is treated as an independent event, such that trajectories containing multiple hops are duplicated and restricted to a defined time window around each hop (here −0.5 ≤ t ≤ 0.5 fs, indicated by the double arrow). | ||
For the analysis of trends around hops,  supports hop-aligned time coordinates, defined as time offsets relative to a selected hopping event. Two alignment modes are available: (i) In the trajectory-focused mode (see Fig. 5b), either the first or the last hop is selected per trajectory, and the entire trajectory is aligned to that event, facilitating the identification of structural motifs associated with the hopping event. (ii) In the hop-focused mode (see Fig. 5c), each hop is treated as an independent event; trajectories containing multiple selected hops are duplicated and aligned to each hop individually. This is particularly useful for defining hop-centered time windows, for example to generate trajectory movies or to increase sampling density near these regions. Missing frames introduced by time shifting are padded with
supports hop-aligned time coordinates, defined as time offsets relative to a selected hopping event. Two alignment modes are available: (i) In the trajectory-focused mode (see Fig. 5b), either the first or the last hop is selected per trajectory, and the entire trajectory is aligned to that event, facilitating the identification of structural motifs associated with the hopping event. (ii) In the hop-focused mode (see Fig. 5c), each hop is treated as an independent event; trajectories containing multiple selected hops are duplicated and aligned to each hop individually. This is particularly useful for defining hop-centered time windows, for example to generate trajectory movies or to increase sampling density near these regions. Missing frames introduced by time shifting are padded with  values and are ignored by standard aggregation operations.
values and are ignored by standard aggregation operations.
2.5 Analyze ( )
)
The phase space sampled in SH simulations comprises a wide range of observables describing both electronic and nuclear dynamics. In 
 , these observables are organized into three complementary categories: (i) per-state properties associated with individual electronic states, such as state energies Ei and permanent dipole moments µi; (ii) inter-state properties that describe couplings between pairs of states, including non-adiabatic couplings NACij, transition dipole moments µij, and energy gaps Eij; and (iii) over-state properties that are independent of electronic state, such as Cartesian coordinates or trajectory-level labels. This classification provides a consistent framework for organizing, transforming heterogeneous simulation data, which further guides the visualization and analysis strategies used throughout the package.
, these observables are organized into three complementary categories: (i) per-state properties associated with individual electronic states, such as state energies Ei and permanent dipole moments µi; (ii) inter-state properties that describe couplings between pairs of states, including non-adiabatic couplings NACij, transition dipole moments µij, and energy gaps Eij; and (iii) over-state properties that are independent of electronic state, such as Cartesian coordinates or trajectory-level labels. This classification provides a consistent framework for organizing, transforming heterogeneous simulation data, which further guides the visualization and analysis strategies used throughout the package.
Building on this representation,  supports the analysis of trajectory ensembles, individual trajectories, and static datasets through a set of modular postprocessing tools. These include the computation of geometric descriptors, ensemble-level statistical evaluation (such as averages and confidence intervals), dimensionality-reduction techniques, and the construction of derived electronic observables, for example absorption spectra and population dynamics. All operations are performed in a unit-consistent manner, with automatic conversion between different unit systems (if needed).
supports the analysis of trajectory ensembles, individual trajectories, and static datasets through a set of modular postprocessing tools. These include the computation of geometric descriptors, ensemble-level statistical evaluation (such as averages and confidence intervals), dimensionality-reduction techniques, and the construction of derived electronic observables, for example absorption spectra and population dynamics. All operations are performed in a unit-consistent manner, with automatic conversion between different unit systems (if needed).
While many of these capabilities apply to both nuclear and electronic observables,  offers specialized tools for their respective analysis. Accordingly, the following sections highlight two complementary views: the geometry space, which focuses on structural descriptors and collective nuclear motion, and the property space, which addresses per-state and inter-state electronic observables and their (statistical) postprocessing.
offers specialized tools for their respective analysis. Accordingly, the following sections highlight two complementary views: the geometry space, which focuses on structural descriptors and collective nuclear motion, and the property space, which addresses per-state and inter-state electronic observables and their (statistical) postprocessing.
 provides tools for computing structural descriptors and for identifying and visualizing the dominant modes of structural variation using principal component analysis (PCA). These analyses can be performed using geometric descriptors such as pairwise distances, internal coordinates, or collective variables including the bond-length alternation (BLA).
provides tools for computing structural descriptors and for identifying and visualizing the dominant modes of structural variation using principal component analysis (PCA). These analyses can be performed using geometric descriptors such as pairwise distances, internal coordinates, or collective variables including the bond-length alternation (BLA).
A central component of this workflow is the automated generation of geometric descriptors.  supports a broad range of internal coordinates, including bond lengths, dihedral (torsion) angles, and pyramidalization angles. During the substructure selection step (see Section 2.4), users may specify which atoms or fragments are of interest (e.g., a reactive moiety or a particular dihedral motif). For the selected atoms, or for the full molecule if no selection is applied,
supports a broad range of internal coordinates, including bond lengths, dihedral (torsion) angles, and pyramidalization angles. During the substructure selection step (see Section 2.4), users may specify which atoms or fragments are of interest (e.g., a reactive moiety or a particular dihedral motif). For the selected atoms, or for the full molecule if no selection is applied,  automatically determines which geometric features are well defined based on molecular connectivity and size (see Section 2.4) all applicable geometric descriptors (bond lengths, bond angles, pyramidalization angles, and torsion angles) are then generated automatically and consistently for the chosen substructure. Combinations of these descriptors enable the characterization of collective motions such as hydrogen out-of-plane distortions, and
automatically determines which geometric features are well defined based on molecular connectivity and size (see Section 2.4) all applicable geometric descriptors (bond lengths, bond angles, pyramidalization angles, and torsion angles) are then generated automatically and consistently for the chosen substructure. Combinations of these descriptors enable the characterization of collective motions such as hydrogen out-of-plane distortions, and  additionally provides explicit support for the selection and calculation of BLA in conjugated systems. If only specific features (e.g., only dihedral angles) are required, these can be requested directly in the structure selection step (see Section 2.4).
additionally provides explicit support for the selection and calculation of BLA in conjugated systems. If only specific features (e.g., only dihedral angles) are required, these can be requested directly in the structure selection step (see Section 2.4).
The resulting geometric descriptors can be used as input for dimensionality reduction techniques, including PCA, Linear Discriminant Analysis (LDA) and Partial Least Squares (PLS). However,  provides extensive support for PCA in combination with the selection and filtering capabilities described above, enabling the identification of the key geometric degrees of freedom that govern excited-state dynamics (see Fig. S6). PCA can be performed either on translation-invariant descriptors like pairwise distance matrices |rij| or on any chosen set of geometric features. The
provides extensive support for PCA in combination with the selection and filtering capabilities described above, enabling the identification of the key geometric degrees of freedom that govern excited-state dynamics (see Fig. S6). PCA can be performed either on translation-invariant descriptors like pairwise distance matrices |rij| or on any chosen set of geometric features. The  module visualizes the low-dimensional embedding (i.e. the projection onto the first two principal components), highlights density-based clusters using kernel density estimation, and maps the dominant PCA loadings back onto the molecular structure. This representation directly reveals which geometry descriptor drive the principal geometric rearrangements and data clustering.
module visualizes the low-dimensional embedding (i.e. the projection onto the first two principal components), highlights density-based clusters using kernel density estimation, and maps the dominant PCA loadings back onto the molecular structure. This representation directly reveals which geometry descriptor drive the principal geometric rearrangements and data clustering.
As an example, Fig. 6 shows the PCA biplot for SH trajectories of 1,3-cyclohexadiene (R02),36,52 using pairwise distances as descriptors. Upon photoexcitation, this molecule undergoes an electrocyclic ring-opening reaction following population of a σσ* state, yielding hexatriene.52 The PCA projection reveals two well-separated clusters, colored by the distance between the two sp3 carbon atoms, which ranges from approximately 150 pm (blue) to 650 pm (yellow). These atoms form a bond in the closed-ring structure, and its cleavage during ring opening gives rise to the photoproduct. The separation in PCA space therefore provides a direct distinction between reactive and non-reactive trajectories.
 | ||
Fig. 6 PCA biplot for the SH data of 1,3-cyclohexadiene (R02). (Left) PCA representation of R02 (ref. 36 and 52) showing the projection onto the first two principal components. Data points are color-coded by the distance between the two sp3 carbon atoms that are cleaved during the photoinduced electrocyclic 6π ring-opening reaction. (Right) Representative molecular structures highlighting atom pairs associated with the dominant PCA loadings (a and b). Loading a corresponds to the primary reaction coordinate and captures the C–C bond breaking accompanied by a coupled change in the C–H distance, defined between one of the cleaving sp3 carbon atoms and a hydrogen atom bound to the opposite cleaving carbon. Loading (b) is dominated by distances between the hydrogen atoms attached to the sp3 carbons and the central sp2 carbons, indicating an out-of-plane ring distortion (puckering). The plot was generated using the data of R02 reported as  datatree on Zenodo.37 datatree on Zenodo.37 | ||
This interpretation is further supported by the PCA loadings mapped onto the molecular structure. Loading a highlights the breaking C–C bond and an associated C–H distance involving the two sp3 carbon atoms (Fig. 6, subset a), consistent with the dominant reaction coordinate. In contrast, for non-reactive trajectories, loading b is dominated by distances between hydrogen atoms attached to the sp3 carbons and the central sp2 carbons (subset b), reflecting out-of-plane ring distortions rather than bond cleavage.21,52 In addition,  can provide a textual summary of PCA results that identifies the most important features contributing to the overall projection and to individual principal components, as demonstrated in the accompanying tutorial.
can provide a textual summary of PCA results that identifies the most important features contributing to the overall projection and to individual principal components, as demonstrated in the accompanying tutorial.
 provides an integrated framework for transforming, visualizing, and statistically evaluating electronic and nuclear observables across trajectories and time. In close analogy to the structural selection described above,
provides an integrated framework for transforming, visualizing, and statistically evaluating electronic and nuclear observables across trajectories and time. In close analogy to the structural selection described above,  allows users to restrict the analysis to selected electronic states of interest (see Section 2.4). All subsequent processing steps, including, for example, the construction of inter-state quantities and ensemble-level statistics, are then carried out consistently for this chosen subset of states and state combinations, enabling a targeted analysis of the photoinduced dynamics.
allows users to restrict the analysis to selected electronic states of interest (see Section 2.4). All subsequent processing steps, including, for example, the construction of inter-state quantities and ensemble-level statistics, are then carried out consistently for this chosen subset of states and state combinations, enabling a targeted analysis of the photoinduced dynamics.
Generally, the analysis and visualization is adapted to the type of observable. Per-state quantities are typically represented as state-colored one-dimensional histograms, whereas inter-state observables are explored using two-dimensional correlation plots that reveal relationships between quantities such as NACij, µij, and Eij. For high-dimensional tensorial quantities, including forces, non-adiabatic couplings, and dipole moments,  employs the Frobenius norm to reduce dimensionality while preserving physical interpretability (see property distributions in Fig. S8). A schematic overview of property-space visualization strategies is provided in Fig. S11 and in the accompanying tutorials on Zenodo.42
employs the Frobenius norm to reduce dimensionality while preserving physical interpretability (see property distributions in Fig. S8). A schematic overview of property-space visualization strategies is provided in Fig. S11 and in the accompanying tutorials on Zenodo.42
Beyond the inspection of raw observables,  supports the computation of derived, experimentally relevant quantities. Electronic absorption spectra are constructed from vertical excitation energies Eij and transition dipole moments µij by evaluating oscillator strengths, for all selected state pairs. These transitions are separated into ground-state (i = 0) and excited-state (i > 0) contributions and subjected to Gaussian broadening to yield continuous spectral line shapes (see Fig. S14).
supports the computation of derived, experimentally relevant quantities. Electronic absorption spectra are constructed from vertical excitation energies Eij and transition dipole moments µij by evaluating oscillator strengths, for all selected state pairs. These transitions are separated into ground-state (i = 0) and excited-state (i > 0) contributions and subjected to Gaussian broadening to yield continuous spectral line shapes (see Fig. S14).

Time-dependent relaxation kinetics are obtained from electronic state populations, computed as the fraction of trajectories occupying a given active state at each time step (see Fig. S9 and S10).  evaluates ensemble-averaged population curves together with statistical confidence intervals, allowing the reliability of kinetic features to be assessed, in particular at longer simulation times where trajectory filtering may reduce ensemble size (see Section 2.3).
evaluates ensemble-averaged population curves together with statistical confidence intervals, allowing the reliability of kinetic features to be assessed, in particular at longer simulation times where trajectory filtering may reduce ensemble size (see Section 2.3).
2.6 Visualization ( )
)
In 
 , the postprocessing and analysis of data is closely integrated with visualization. In general,
, the postprocessing and analysis of data is closely integrated with visualization. In general,  provides tools for handling both time-dependent and static data in property and conformational space, employing techniques such as dimensionality reduction, slicing, and aggregation. Frequently-used functions are made available as methods of every
provides tools for handling both time-dependent and static data in property and conformational space, employing techniques such as dimensionality reduction, slicing, and aggregation. Frequently-used functions are made available as methods of every  and
and  by registering an
by registering an  accessor for users familiar with the
accessor for users familiar with the  data model. Users can generate individual plots for detailed inspection or employ the
data model. Users can generate individual plots for detailed inspection or employ the  class to automatically compile comprehensive summary visualizations of trajectory datasets for a quick analysis (see accompanying tutorial on Zenodo42 and Github53).
class to automatically compile comprehensive summary visualizations of trajectory datasets for a quick analysis (see accompanying tutorial on Zenodo42 and Github53).
3 Example application
To illustrate the capabilities of , we present a cross-compound analysis of four structurally related molecules from the SHNITSEL-data set:36,37 ethene (A01), propene (A02), Z-but-2-ene (A03), and the methylenimmonium cation (I01, CH2NH+2). All scripts used for this analysis are provided in the SI (cf. SI section 4). These molecules were chosen because they exhibit closely related excited-state relaxation pathways. In all cases, population transfer from S1 to S0 occurs via twisted and pyramidalized geometries around the central double bond, associated with S1/S0 conical intersections (Fig. 7). This makes them an ideal test case for evaluating whether
, we present a cross-compound analysis of four structurally related molecules from the SHNITSEL-data set:36,37 ethene (A01), propene (A02), Z-but-2-ene (A03), and the methylenimmonium cation (I01, CH2NH+2). All scripts used for this analysis are provided in the SI (cf. SI section 4). These molecules were chosen because they exhibit closely related excited-state relaxation pathways. In all cases, population transfer from S1 to S0 occurs via twisted and pyramidalized geometries around the central double bond, associated with S1/S0 conical intersections (Fig. 7). This makes them an ideal test case for evaluating whether  can identify shared reaction coordinates across chemically distinct systems.
can identify shared reaction coordinates across chemically distinct systems.
 | ||
| Fig. 7 Schematic representation of the potential energy surfaces of the ground state (S0) and first excited state (S1) along a torsional coordinate defined by a dihedral angle. The inset structures correspond to optimized geometries of conical intersections between S0 and S1 of the methylenimmonium cation (I01) and the alkenes A01–A03, computed using spin-flip time-dependent density functional theory (SF-TDDFT)54 at the BHANDHLYP/def2-TZVPD level.55–58 | ||
The trajectory data for all four compounds were first loaded into a common  , with each molecule assigned to a separate branch. Standard energetic and geometric sanity filters were applied, and to enable direct structural comparison across molecules with different sizes, we selected a common chromophoric substructure defined by the SMARTS pattern
, with each molecule assigned to a separate branch. Standard energetic and geometric sanity filters were applied, and to enable direct structural comparison across molecules with different sizes, we selected a common chromophoric substructure defined by the SMARTS pattern  . This yields a six-atom fragment representing the central double-bond unit, which serves as a common geometric reference for all compounds (Fig. 4). All subsequent geometric analyses were restricted to this aligned substructure.
. This yields a six-atom fragment representing the central double-bond unit, which serves as a common geometric reference for all compounds (Fig. 4). All subsequent geometric analyses were restricted to this aligned substructure.
To explore how this shared motif evolves during the excited-state dynamics,  was used to compute torsion and pyramidalization angles as well as pairwise distances for the selected atoms. The pairwise distances were then subjected to PCA, where the PCA basis was constructed from the trajectories of A03, and the trajectories of all four molecules were subsequently projected into this two-dimensional space (see right panel in Fig. 4). In this representation, hopping events are highlighted, with S1 → S0 transitions shown in white and S2 → S1 transitions in yellow (note, trajectories of I01 were initialized in S2, while all other trajectories started in S1 as active state).
was used to compute torsion and pyramidalization angles as well as pairwise distances for the selected atoms. The pairwise distances were then subjected to PCA, where the PCA basis was constructed from the trajectories of A03, and the trajectories of all four molecules were subsequently projected into this two-dimensional space (see right panel in Fig. 4). In this representation, hopping events are highlighted, with S1 → S0 transitions shown in white and S2 → S1 transitions in yellow (note, trajectories of I01 were initialized in S2, while all other trajectories started in S1 as active state).
For A03, the trajectories were colored by means of the dihedral of the heavy atom backbone, where yellow refers to trajectories showing Z → E isomerization and blue to the photophysical Z → Z processes. In the respective PCA projection these two photoinduced reaction channels are clearly separated, appearing as two distinct clusters. Importantly, the S1 → S0 hopping events are concentrated between these clusters, at torsion angles near ±90° (see hopping point dihedral distributions in Fig. 4), consistent with the geometry of the underlying conical intersection. A similar pattern is observed for A02 and A01, whose hopping points likewise cluster around φ ≈ 90°, demonstrating that  captures a common reaction coordinate across the alkene series. Remarkably, the trajectories of I01 overlap strongly with those of A01 in the PCA space, indicating closely related nuclear dynamics despite the different chemical composition. For I01, the additional S2 → S1 transitions form a shifted but parallel distribution, reflecting analogous distortions along pyramidalization coordinates, in agreement with previously reported conical-intersection geometries.2,41,59,60
captures a common reaction coordinate across the alkene series. Remarkably, the trajectories of I01 overlap strongly with those of A01 in the PCA space, indicating closely related nuclear dynamics despite the different chemical composition. For I01, the additional S2 → S1 transitions form a shifted but parallel distribution, reflecting analogous distortions along pyramidalization coordinates, in agreement with previously reported conical-intersection geometries.2,41,59,60
Overall, the geometric analysis reveals the following key trends: (i) hopping geometries for both alkenes and I01 predominantly occur near φ ≈ 90°; (ii) A01 and I01 exhibit well-separated signatures of pyramidalization during the dynamics; and (iii) all molecules show torsional motion around the central double bond, with A03 trajectories clearly separating into reactive and non-reactive pathways. Taken together, this analysis demonstrates that  facilitates a comprehensive, cross-compound exploration of SH trajectory data. By integrating automated substructure alignment with postprocessing of the respective geometric features by means of dimensionality reduction and a systematic interrogation of the underlying geometrical space, the toolkit elucidates shared reaction coordinates, discriminates between reactive and non-reactive pathways, and uncovers subtle mechanistic differences among related photochemical systems within a unified, data-driven framework.
facilitates a comprehensive, cross-compound exploration of SH trajectory data. By integrating automated substructure alignment with postprocessing of the respective geometric features by means of dimensionality reduction and a systematic interrogation of the underlying geometrical space, the toolkit elucidates shared reaction coordinates, discriminates between reactive and non-reactive pathways, and uncovers subtle mechanistic differences among related photochemical systems within a unified, data-driven framework.
4 Conclusion
We have presented (
( ), a Python-based framework for the organization, analysis, and visualization of surface-hopping trajectory ensembles. By introducing a hierarchical, metadata-rich data model for SH simulations,
), a Python-based framework for the organization, analysis, and visualization of surface-hopping trajectory ensembles. By introducing a hierarchical, metadata-rich data model for SH simulations,  enables trajectories from different molecules, electronic-structure methods, and simulation protocols to be combined and analyzed within a unified representation, providing a practical foundation for ensemble-level statistics and cross-system comparisons.
enables trajectories from different molecules, electronic-structure methods, and simulation protocols to be combined and analyzed within a unified representation, providing a practical foundation for ensemble-level statistics and cross-system comparisons.
Designed to support all stages of the SH trajectory data lifecycle following trajectory generation,  streamlines cleaning, filtering, storage, management, analysis, and visualization of SH data. Its integrated tools facilitate chemically meaningful post-processing, including state-resolved population analysis, characterization of hopping geometries, and identification of collective nuclear motions. The capabilities of
streamlines cleaning, filtering, storage, management, analysis, and visualization of SH data. Its integrated tools facilitate chemically meaningful post-processing, including state-resolved population analysis, characterization of hopping geometries, and identification of collective nuclear motions. The capabilities of  are demonstrated using a cross-compound analysis of structurally related systems, including a series of alkenes (CnH2n, n = 2,3,4) and CH2NH+2, illustrating how diverse datasets can be analyzed consistently to gain insight into excited-state dynamics.
are demonstrated using a cross-compound analysis of structurally related systems, including a series of alkenes (CnH2n, n = 2,3,4) and CH2NH+2, illustrating how diverse datasets can be analyzed consistently to gain insight into excited-state dynamics.
By providing a flexible and extensible framework for exploring complex SH data across individual molecules and simulation settings,  offers researchers a powerful resource for reproducible analysis, benchmarking, and data-driven discovery. In this context, natural future directions could involve extending the framework to QM/MM surface hopping simulations and coupling it more closely to machine learning workflows, for example to support active learning strategies or the targeted generation of training data from dynamically important regions of configuration space.
offers researchers a powerful resource for reproducible analysis, benchmarking, and data-driven discovery. In this context, natural future directions could involve extending the framework to QM/MM surface hopping simulations and coupling it more closely to machine learning workflows, for example to support active learning strategies or the targeted generation of training data from dynamically important regions of configuration space.
Author contributions
The development of was a collaborative effort involving all authors. C.M. conceptualized and supervised this work. All authors contributed to the writing of the manuscript.
was a collaborative effort involving all authors. C.M. conceptualized and supervised this work. All authors contributed to the writing of the manuscript.
Conflicts of interest
There are no conflicts to declare.Data availability
The code of the (
( ) Python package is available on Zenodo.42 To run
) Python package is available on Zenodo.42 To run  , essential Python libraries such as
, essential Python libraries such as  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  , and
, and  must be available. All required dependencies are specified in the provided
must be available. All required dependencies are specified in the provided  environment file,42 which can be used to recreate the computational environment via tools like uv. Instructions for installing any additional libraries are included in the accompanying Zenodo repository.42 It is recommended to use the code within a Jupyter Notebook environment.
environment file,42 which can be used to recreate the computational environment via tools like uv. Instructions for installing any additional libraries are included in the accompanying Zenodo repository.42 It is recommended to use the code within a Jupyter Notebook environment.
As part of the  Python package, a set of interactive Jupyter notebook tutorials is provided to guide users through data import, processing, and visualization using small tutorial datasets available alongside the code on Zenodo.42 Larger surface-hopping datasets for the
Python package, a set of interactive Jupyter notebook tutorials is provided to guide users through data import, processing, and visualization using small tutorial datasets available alongside the code on Zenodo.42 Larger surface-hopping datasets for the  36 molecules A01, A02, A03, I01, and R02 are available on Zenodo in both stacked and unstacked tree formats.37 The tutorials demonstrate core
36 molecules A01, A02, A03, I01, and R02 are available on Zenodo in both stacked and unstacked tree formats.37 The tutorials demonstrate core  workflows, including principal component analysis of geometric descriptors, as well as visualization and statistical analysis of electronic observables such as state populations, property distributions, and simulated absorption spectra. By providing these resources, we aim to make
workflows, including principal component analysis of geometric descriptors, as well as visualization and statistical analysis of electronic observables such as state populations, property distributions, and simulated absorption spectra. By providing these resources, we aim to make  accessible to assist users in utilizing the package for data analysis and the development of ML models in excited-state dynamics research.
accessible to assist users in utilizing the package for data analysis and the development of ML models in excited-state dynamics research.
Supplementary information (SI): data structure and usage of  . See DOI: https://doi.org/10.1039/d5dd00299k.
. See DOI: https://doi.org/10.1039/d5dd00299k.
Acknowledgements
We thank Tobias Werner for his support in data plotting and code testing. C.M. gratefully acknowledges financial support by the Emerging Talents Initiative (eti) of the Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), the FAU Competence Center Engineering of Advanced Materials (FAU EAM) for funding with the EAM Starting Grant; and the Bayerische Akademie der Wissenschaften (Junges Kolleg (C. M.)). Furthermore, this work was funded by Deutsche Forschungsgemeinschaft (DFG) within the Collaborative Research Center ‘ChemPrint’ (project 538767711, CRC 1719, project: M02, C.M. and K.H.). All high-performance computational work was performed at the Erlangen National High Performance Computing Center (NHR@FAU).References
- S. Gómez, I. F. Galván, R. Lindh and L. González, in Motivation and Basic Concepts, John Wiley & Sons, Ltd, 2020, ch. 1, pp. 1–12 Search PubMed.
- B. F. E. Curchod and T. J. Martínez, Chem. Rev., 2018, 118, 3305–3336 CrossRef CAS PubMed.
- M. H. Beck, A. Jäckle, G. A. Worth and H. D. Meyer, Phys. Rep., 2000, 324, 1–105 CrossRef CAS.
- G. A. Worth and B. Lasorne, in Quantum Chemistry and Dynamics of Excited States, ed. L. González and R. Lindh, John Wiley & Sons Ltd, 2020, pp. 413–433 Search PubMed.
- B. G. Levine, J. D. Coe, A. M. Virshup and T. J. Martínez, Chem. Phys., 2008, 347, 3–16 CrossRef CAS.
- M. Ben-Nun and T. J. Martínez, Chem. Phys. Lett., 1998, 298, 57–65 CrossRef CAS.
- A. Kirrander and M. Vacher, in Quantum Chemistry and Dynamics of Excited States, ed. L. González and R. Lindh, John Wiley & Sons Ltd, 2020, pp. 469–497 Search PubMed.
- J. C. Tully, Faraday Discuss, 1998, 110, 407–419 RSC.
- J. C. Tully, J. Chem. Phys., 2012, 137, 22A301 CrossRef PubMed.
- J. C. Tully, Catal. Lett., 1991, 9, 205–217 CrossRef CAS.
- L. González and R. Lindh, in. Quantum chemistry and dynamics of excited states: methods and applications, John Wiley & Sons, 2020 Search PubMed.
- S. Mai, B. Bachmair, L. Gagliardi, H. G. Gallmetzer, L. Grünewald, M. R. Hennefarth, N. M. Høyer, F. A. Korsaye, S. Mausenberger, M. Oppel, T. Piteša, S. Polonius, E. S. Gil, Y. Shu, N. K. Singer, M. X. Tiefenbacher, D. G. Truhlar, D. Vórós, L. Zhang and L. González, SHARC4.0: Surface Hopping Including Arbitrary Couplings — Program Package for Non-Adiabatic Dynamics, https://sharc-md.org/, 2025.
- S. Mai, P. Marquetand and L. González, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1370 Search PubMed.
- M. Barbatti, G. Granucci, M. Ruckenbauer, F. Plasser, J. Pittner, M. Persico and H. Lischka, NEWTON-X: a package for Newtonian dynamics close to the crossing seam, version 1.2, https://www.netwonx.org, 2011.
- M. Barbatti, M. Ruckenbauer, F. Plasser, J. Pittner, G. Granucci, M. Persico and H. Lischka, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 26–33 CAS.
- J. Li, P. Reiser, B. R. Boswell, A. Eberhard, N. Z. Burns, P. Friederich and S. A. Lopez, Chem. Sci., 2021, 12, 5302–5314 RSC.
- J. Li, R. Stein, D. M. Adrion and S. A. Lopez, J. Am. Chem. Soc., 2021, 143, 20166–20175 CrossRef CAS PubMed.
- L. Zhang, S. V. Pios, M. Martyka, F. Ge, Y.-F. Hou, Y. Chen, L. Chen, J. Jankowska, M. Barbatti and P. O. Dral, J. Chem. Theory Comput., 2024, 20, 5043–5057 CrossRef CAS PubMed.
- L. Du and Z. Lan, J. Chem. Theory Comput., 2015, 11, 1360–1374 CrossRef CAS PubMed.
- M. Barbatti, M. Bondanza, R. Crespo-Otero, B. Demoulin, P. O. Dral, G. Granucci, F. Kossoski, H. Lischka, B. Mennucci, S. Mukherjee, M. Pederzoli, M. Persico, M. Pinheiro Jr, J. Pittner, F. Plasser, E. Sangiogo Gil and L. Stojanovic, J. Chem. Theory Comput., 2022, 18, 6851–6865 CrossRef CAS PubMed.
- M. Pinheiro, M. de Oliveira Bispo, R. S. Mattos, M. T. Do Casal, B. C. Garain, J. M. Toldo, S. Mukherjee and M. Barbatti, Digital Discovery, 2025 Search PubMed.
- Y. Zhu, J. Peng, C. Xu and Z. Lan, J. Phys. Chem. Lett., 2024, 15, 9601–9619 CrossRef CAS PubMed.
- F. Häse, I. F. Galván, A. Aspuru-Guzik, R. Lindh and M. Vacher, Chem. Sci., 2019, 10, 2298–2307 RSC.
- Y. Zhu, J. Peng, X. Kang, C. Xu and Z. Lan, Phys. Chem. Chem. Phys., 2022, 24, 24362–24382 RSC.
- A. M. Virshup, J. Chen and T. J. Martínez, J. Chem. Phys., 2012, 137, 22A519 CrossRef PubMed.
- G. W. Richings and S. Habershon, Molecules, 2021, 26, 1–31 CrossRef PubMed.
- J. Kára, K. Acheson and A. Kirrander, J. Chem. Theory Comput., 2025, 21(12), 5789–5802 CrossRef PubMed.
- X. Li, D. Hu, Y. Xie and Z. Lan, J. Chem. Phys., 2018, 149, 244104 CrossRef PubMed.
- M. A. Kochman, New J. Chem., 2024, 48, 14327–14335 RSC.
- K. Acheson and A. Kirrander, J. Chem. Theory Comput., 2023, 19, 6126–6138 CrossRef CAS PubMed.
- M. Brehm, M. Thomas, S. Gehrke and B. Kirchner, J. Chem. Phys., 2020, 152, 164105 CrossRef CAS PubMed.
- M. Brehm and B. Kirchner, J. Chem. Inf. Model., 2011, 51, 2007–2023 CrossRef CAS PubMed.
- N. Michaud-Agrawal, E. J. Denning, T. B. Woolf and O. Beckstein, J. Comput. Chem., 2011, 32, 2319–2327 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graph., 1996, 14, 33–38 CrossRef CAS PubMed.
- R. Curth, T. Röhrkasten, C. Müller and J. Westermayr, SHNITSEL - Surface Hopping Nested Instances Training Set for Excited-state Learning, 2025, doi: DOI:10.5281/zenodo.15482819.
- R. Curth, T. E. Röhrkasten, C. Müller and J. Westermayr, Sci. Data, 2025, 12, 1300 CrossRef CAS PubMed.
- K. Höllring, T. Röhrkasten and C. Müller, SHNITSEL - Surface Hopping Nested Instances Training Set for Excited-state Learning, 2026, doi: DOI:10.5281/zenodo.18436276.
- S. Hoyer and J. Hamman, J. Open Res. Softw., 2017, 5, 1–10 Search PubMed.
- P. Virtanen, R. Gommers, T. E. Oliphant, M. Haberland, T. Reddy, D. Cournapeau, E. Burovski, P. Peterson, W. Weckesser, J. Bright, S. J. van der Walt, M. Brett, J. Wilson, K. J. Millman, N. Mayorov, A. R. J. Nelson, E. Jones, R. Kern, E. Larson, C. J. Carey, İ. Polat, Y. Feng, E. W. Moore, J. VanderPlas, D. Laxalde, J. Perktold, R. Cimrman, I. Henriksen, E. A. Quintero, C. R. Harris, A. M. Archibald, A. H. Ribeiro, F. Pedregosa and P. van Mulbregt, Nat. Methods, 2020, 17, 261–272 CrossRef CAS PubMed.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss and V. Dubourg, et al. , J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed.
- S. Mausenberger, C. Müller, A. Tkatchenko, P. Marquetand, L. González and J. Westermayr, Chem. Sci., 2024, 15, 15880–15890 RSC.
- K. Hoellring, T. Roehrkasten, C. Müller and C. Group, CompPhotoChem/shnitsel-tools: v2026.01.2, 2026, doi: DOI:10.5281/zenodo.19135766.
- NetCDF, version 4.8.1. Unidata, 2021, DOI:10.5065/D6H70CW6.
- A. H. Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. B. Jensen, J. Kermode, J. R. Kitchin, E. L. Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. B. Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. Schiøtz, O. Schütt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, J. Phys.: Condens. Matter, 2017, 29, 273002 CrossRef PubMed.
- S. R. Bahn and K. W. Jacobsen, Comput. Sci. Eng., 2002, 4, 56–66 CrossRef CAS.
- J. Westermayr, M. Gastegger, M. F. S. J. Menger, S. Mai, L. González and P. Marquetand, Chem. Sci., 2019, 10, 8100–8107 RSC.
- R. Barrett, C. Ortner and J. Westermayr, Transferable Machine Learning Potential X-MACE for Excited States using Integrated DeepSets, arXiv, 2025, preprint arXiv:2502.12870, DOI:10.48550/arxiv.org/abs/2502.12870.
- V. Delmas, A. N. Nardi, I. C. D. Merritt, A. Ferté, I. F. Galván and M. Vacher, J. Chem. Theory Comput., 2025, 21, 6611–6621 CrossRef CAS PubMed.
- T. Tsutsumi, Y. Ono and T. Taketsugu, J. Chem. Theory Comput., 2022, 18, 7483–7495 CrossRef CAS PubMed.
- S. Sen, R. K. Kar, V. A. Borin and I. Schapiro, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2022, 12, e1562 CrossRef CAS.
- I. C. D. Merritt, D. Jacquemin and M. Vacher, Phys. Chem. Chem. Phys., 2021, 23, 19155–19165 RSC.
- I. Polyak, L. Hutton, R. Crespo-Otero, M. Barbatti and P. J. Knowles, J. Chem. Theory Comput., 2019, 15, 3929–3940 CrossRef CAS PubMed.
- SHNITSEL-tools, 2026, https://github.com/CompPhotoChem/shnitsel-tools.
- D. Casanova and A. I. Krylov, Phys. Chem. Chem. Phys., 2020, 22, 4326–4342 RSC.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- D. Rappoport and F. Furche, J. Chem. Phys., 2010, 133, 134105 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- F. Neese, WIREs Comput. Mol. Sci., 2022, 12, e1606 CrossRef.
- T. K. Allison, H. Tao, W. J. Glover, T. W. Wright, A. M. Stooke, C. Khurmi, J. van Tilborg, Y. Liu, R. W. Falcone, T. J. Martínez and A. Belkacem, J. Chem. Phys., 2012, 136, 124317 CrossRef CAS PubMed.
- S. Gómez, E. Spinlove and G. Worth, Phys. Chem. Chem. Phys., 2024, 26, 1829–1844 RSC.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2026 |