
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSulfonated porous organic polymers: strategic design, synthesis, and applications in catalysis, adsorption, and energy-related processes
Ali Reza
Oveisi
 *a,
Saba
Daliran
*a,
Matías
Blanco
bc,
Amarajothi
Dhakshinamoorthy
d,
Unal
Sen
e,
José
Alemán
*bc and
Hermenegildo
García
*f
*a,
Saba
Daliran
*a,
Matías
Blanco
bc,
Amarajothi
Dhakshinamoorthy
d,
Unal
Sen
e,
José
Alemán
*bc and
Hermenegildo
García
*f
aDepartment of Organic Chemistry, Faculty of Chemistry, Lorestan University, Khorramabad 68151-44316, Iran. E-mail: oveisi.a@lu.ac.ir; daliran.s@lu.ac.ir
bOrganic Chemistry Department, Módulo 1, Universidad Autónoma de Madrid, 28049 Madrid, Spain. E-mail: jose.aleman@uam.es
cInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, 28049 Madrid, Spain
dSchool of Chemistry, Madurai Kamaraj University, Madurai, 625021, Tamil Nadu, India
eDepartment of Materials Science and Engineering, Faculty of Engineering, Eskisehir Technical University, Eskisehir 26555, Turkey
fInstituto Universitario de Tecnología Química, (CSIC-UPV), Universitat Politècnica de València, Valencia 46022, Spain. E-mail: hgarcia@qim.upv.es
First published on 19th February 2026
Abstract
Porous organic polymers (POPs) have become an innovative class of tailor-made materials, encompassing a variety of frameworks that range from highly crystalline to fully amorphous structures such as covalent organic frameworks (COFs), covalent triazine frameworks (CTFs), porous aromatic frameworks (PAFs), conjugated microporous polymers (CMPs), polymers of intrinsic microporosity (PIMs), and hyper-cross-linked polymers (HCPs). While their inherent porosity and stability are impressive, the true strength of POPs lies in strategic functionalization. Among the various methods reported, the incorporation of sulfonic acid (–SO3H) groups in these porous scaffolds introduces additional functionality. This review explores a comprehensive overview of sulfonated POPs (SPOPs), where robust frameworks are combined with the strong Brønsted acidity of –SO3H groups. We describe the design and synthesis of SPOPs, highlighting how this functionalization tailors their properties for innovative applications. Moving beyond their well-known role as superior heterogeneous acid catalysts for organic transformations, SPOPs are now emerging as key materials for addressing global challenges. Their remarkable capabilities are evident in environmental applications, including their deployment as high-performance adsorbents for the removal of dyes, antibiotics, and heavy metals from water, as well as functional porous solids for selective gas separation. We also explore their pioneering applications as next-generation proton-conducting membranes for high-performance fuel cells and advanced energy storage systems, offering alternatives to fluorinated membranes. This review delivers both a critical analysis of the current state-of-the-art and a forward-looking perspective on the challenges and opportunities ahead, serving as a roadmap for leveraging the multifunctional properties of SPOPs to advance sustainable chemistry, environmental remediation, and energy technologies.
 Ali Reza Oveisi | Ali Reza Oveisi is an Associate Professor of Organic Chemistry at Lorestan University. After earning his PhD from Bu-Ali Sina University (2015) and conducting research as a visiting scholar in the Hupp and Farha groups at Northwestern University, he founded his independent research group at the University of Zabol in 2017. His work focuses on designing and functionalizing advanced porous materials, including MOFs, COFs, and POPs, for applications in catalysis, photocatalysis, sustainable fuel production, and environmental remediation. His research has been recognized with multiple Scientist Awards, and he currently serves as a Guest Editor for a JoVE Special Issue. |
 Saba Daliran | Saba Daliran holds a BSc from Lorestan University (LU) and an MSc/PhD from Bu-Ali Sina University. She was a visiting scholar with Prof. Hermengildo García at Universitat Politècnica de València and completed INSF- and ISEF-funded postdocs. Now an Assistant Professor at LU, she focuses on (nano)porous materials. Her honors include the Dr Kazemi Ashtiani Award, recognition as a Distinguished Researcher of Lorestan University and Province, and selection among Iran's “Nation's Scientific Elites” (2025). |
 José Alemán (left) and Matías Blanco (right) | José Alemán (left) defended his doctoral thesis in 2006 and was promoted to Full Professor in 2023. He has received numerous prestigious awards, including the Lilly Prize for Best Doctoral Student, the UAM Best Doctoral Thesis Award, the Sigma-Aldrich Award for Young Researchers (RSEQ), the Lilly Young Researcher Award, the José Barluenga-RSEQ Medal, and the Ignacio Rivas-RSEQ Medal. His research focuses on asymmetric catalysis and catalytic materials. He has authored 240 scientific publications, supervised 28 doctoral theses and more than 40 Bachelor's and Master's theses, and led 18 competitively funded research projects. He currently serves as Director of the Advanced Institute of Chemical Sciences at UAM, Vice President of the Organic Chemistry Division of the Royal Spanish Chemical Society, and is a member of the Royal Academy of Exact, Physical and Natural Sciences of Spain. Dr Matías Blanco (right) is Assistant Professor at UAM and contributes his research expertise in the field of catalytic materials. He obtained his degree and PhD from the University of Oviedo and specializes in materials science, combining surface science with the development of heterogeneous catalysts to enhance organic transformations through supramolecular and covalent surface chemistry. His interests in carbon nanomaterials and porous polymers further strengthen the multidisciplinary vision of Alemán's group. |
 Amarajothi Dhakshinamoorthy | Amarajothi Dhakshinamoorthy received his PhD degree in 2009 from Madurai Kamaraj University, Madurai-21, India. Later, he had his postdoctoral stay with Prof. H. Garcia at the Technical University of Valencia for four years. Currently, he is serving as UGC Assistant Professor at the School of Chemistry, Madurai Kamaraj University, India. His research interests include catalytic/photocatalytic applications of MOFs and graphene-related materials and MOFs for energy applications. He is the recipient of the Young Scientist Award 2014 for Chemical Sciences by The Academy of Sciences, India. He has co-authored over 220 international publications, five book chapters, and a patent. He is a Fellow of Royal Society of Chemistry. |
 Unal Sen | Unal Sen earned his BSc in Chemistry from Bilkent University and his PhD in Engineering from Tohoku University. Following postdoctoral research at Northwestern University under Professors Hupp and Farha, he joined Eskisehir Technical University's Department of Materials Science and Engineering. His research centers on MOFs, MOF-derived carbons, and MOF-polymer hybrids for applications in energy storage, gas separation, environmental remediation, and advanced ceramics. His contributions have been recognized with a Japan Society for the Promotion of Science RONPAKU Fellowship. |
 Hermenegildo García | Hermenegildo Garcia is a Full Professor at the Instituto de Tecnología Química of the Technical University of Valencia, Spain. His research focuses on heterogeneous catalysis using porous materials and the photocatalytic production of solar fuels, with over 1000 published articles. His distinctions include the Spanish National Research Award (2021), the Jaume I Prize for Novel Technologies (2016), and being named a Doctor Honoris Causa by the University of Bucharest. He is also the recipient of the 2011 Janssen-Cilag award and was a 2021 Medal Lecturer for the International Association for Advanced Materials. |
1. Introduction
Porous organic polymers (POPs) are a relatively new class of soft materials characterized by scaffolds built predominantly from light elements.1–3 Typically, C, N, O, B, and H integrate the structure of the polytopic monomer subjected to the polymerization for yielding this family of materials, but other elements (e.g., S, P) may also be incorporated.4,5 The evolution of synthetic porous solids began with zeolites and advanced through metal–organic frameworks (MOFs), which combine metal clusters with organic linkers; the foundational work by Kitagawa,6,7 Robson,8 and Yaghi,9 recognized by the Nobel Prize 2025 in Chemistry, started as early as 1989 and was firmly established in the 1990s. The field later expanded into purely organic frameworks, which are the structures of this review focused on. Indeed, POPs are fundamentally categorized by their structural order into amorphous types, which lack long-range molecular arrangement, and crystalline types, which possess a highly ordered repeating structure. The development of POPs has been marked by several key milestones, all based on how the monomers are assembled by covalent bonds in space.10,11 The specific choice of monomer, linkage chemistry, and polymerization conditions dictates the final material's structure, leading to several distinct classes (Fig. 1). Under this general premise, the foundational work on hyper-cross-linked polymers (HCPs) dates back to the 1970s,12 pioneered by Davankov and colleagues,13–15 who created networks with permanent microporosity. The field expanded significantly in the 2000s with the introduction of new material classes. Polymers of intrinsic microporosity (PIMs) were first reported in 2002, emerging from the successful preparation of phthalocyanine-based network polymers (Pc-Network-PIMs).16 A key advancement followed in 2004 with the development of processable PIMs, distinguished by their rigid, contorted backbones that generate intrinsic micropores.17–19 This was followed by the development of first crystalline covalent organic frameworks (COFs) in 2005,3,20,21 which demonstrated the potential for designing ordered, covalently bonded structures with predictable topology.22–25 The subsequent years, beginning in 2007,26 witnessed the concurrent development of conjugated microporous polymers (CMPs)26–29 and further advancements in HCP synthesis.30 Particularly significant subclass, covalent triazine frameworks (CTFs),31 was introduced around 2008 and is defined by nitrogen-rich, robust aromatic triazine linkages.32–36 While traditional CTFs synthesized via irreversible routes are often amorphous, further advanced strategies have yielded crystalline versions.37–39 Historically, due to their π-conjugated skeletons, some amorphous CTFs were grouped under the broader category of CMPs, a class known for combining permanent nanoporosity with optoelectronic properties. The family of porous aromatic frameworks (PAFs) was notably advanced in 2009 with the report of PAF-1,40 a prototype material exhibiting an exceptionally high surface area.41,42 The term “aromatic” in PAFs originates from the abundant benzene rings that form the core building blocks of this and other frameworks in the family.41–44 This progression has yielded a diverse family of materials prized for their high surface areas, tunable porosity, and exceptional stability. | ||
| Fig. 1 Different types of POPs: a) HCP, Reproduced with permission from ref. 45 Copyright 2007 Royal Society of Chemistry; b) PIM, Reproduced with permission from ref. 17 Copyright 2004 Royal Society of Chemistry; c) CMP, Reproduced with permission from ref. 26 Copyright 2007 Wiley-VCH; d) PAF, Reproduced with permission from ref. 43 under a Creative Commons CC BY 4.0 license, 2016 MDPI; e) COF, Reproduced with permission from ref. 46 Copyright 2015 Nature Publishing Group; f) CTF. Reproduced with permission from ref. 35 Copyright 2018 Wiley-VCH. | ||
The main differences among these porous polymers are crystallinity, the type of chemical bonds, and the specific building blocks used, with the choice of reaction directly influencing the resulting polymer (Table 1).
| POP material | Crystallinity | Key characteristics & bonding | Typical reactions/formation |
|---|---|---|---|
| COFs | Crystalline | Designable, ordered structures with high surface area, defined pore size, and reversible synthetic reactions | Reversible reactions include those forming boron-based linkages (e.g., boronate ester, boroxine, borazine), imine-based linkages (e.g., imine, hydrazone, azine, β-ketoenamine), and other dynamic linkages such as squaraine bonds and keto-enol tautomerism |
| HCPs | Amorphous | Inexpensive and scalable | The most common method for irreversible cross-linking is Friedel–Crafts (FC) alkylation |
| CMPs | Amorphous | Extended π-conjugation systems are associated with optoelectronic properties | Irreversible metal-catalyzed coupling reactions, such as Suzuki, Sonogashira, Scholl, and Yamamoto coupling, as well as Buchwald-Hartwig amination, and (less commonly) Schiff base formation |
| PAFs | Amorphous | Exceptional surface area and stability, characterized by rigid aromatic frameworks | Irreversible cross-coupling reactions, including Yamamoto-type Ullmann, Suzuki, and Sonogashira–Hagihara couplings, and nucleophilic substitution reactions |
| CTFs | Mostly amorphous, though crystalline forms are possible | Rich nitrogen content (triazine units), high chemical stability | Nitrile or ketone trimerization utilizing ionothermal synthesis, P2O5 catalysis, and superacid catalysis |
A decade of rapid innovation has significantly advanced the field of porous organic materials. This has led to the reporting of diverse families, known under various general nomenclatures such as POPs,2,47 porous organic frameworks (POFs),48,49 covalent organic polymers (COPs),50–52 porous polymer networks (PPNs),53–55 microporous organic networks (MONs),56 and porous organic networks (PONs).57,58 While nomenclature can vary, POPs are often used as an overarching term for this class of materials, encompassing both crystalline and amorphous frameworks with entirely organic, covalently bonded structures. These materials are typically characterized by high surface areas and permanent porosity, and their applications in gas storage,54,59–61 adsorption/separation,62–71 and catalysis have been extensively investigated.72–83
POPs share several defining characteristics. Their high surface areas stem from deliberate synthetic design,84,85 which creates an interconnected network of monomers and dictates pore size and distribution.86 Synthetic design and conditions may also induce and/or influence the crystallinity degree of the material.87,88 In addition, they present usually semiconducting behavior since most of them contain aromatic patches that allow charge separation and carrier mobility.1,89 Despite being composed of light elements, they often possess good mechanical properties90 and demonstrate notable chemical and thermal stability.91–93 As a whole, the general properties make POPs suitable candidates for advanced applications in (opto)electronics,89,94 gas separation,95 catalysis,2 environmental remediation63 and transformation of renewable feedstocks,96 CO2 capture and fixation,97 bioapplications,98 and energy storage and conversion, among others.62,93,95,99–114 However, the properties of POPs can be further enhanced and tailored for specific applications through post-synthetic functionalization.115 An important functionalization of POPs, as discussed below, is based on the introduction of sulfonic and/or sulfonate sites. By introducing –SO3H groups, and maybe further reactivity with them to yield sulfonates, the properties of the POP material are completely changed due to the incorporation of a highly acidic (or, through proton-metal exchange, a metal-chelating group) in the polymeric structure.116 SPOPs offer compelling advantages over conventional acid systems. Compared to homogeneous strong acids like H2SO4 or p-toluenesulfonic acid (PTSA), which pose challenges in separation, recycling, and corrosion, SPOPs provide comparable acid strength while enabling easy recovery and reuse as heterogeneous solids. This merges high activity with improved process sustainability. Furthermore, SPOPs surpass classical polymeric acid membranes117 (e.g., Nafion®, sulfonated poly(aryl ether)s) by leveraging intrinsic, engineered nanoporosity. Unlike the often random pore structures of traditional materials, the well-defined, tailorable architecture of SPOPs offer superior control over acid-site density, pore environment, and surface chemistry. This precise engineering enhances mass transport, substrate accessibility, and performance in catalysis, separation, and proton conduction. Consequently, SPOPs are uniquely suited for green chemistry challenges. Their combination of strong solid-state acidity, robust frameworks, and tunable porosity makes them ideal catalysts for transforming renewable feedstocks and for critical carbon management processes, including CO2 capture, fixation, and conversion. Additionally, the –SO3H groups facilitate proton–electron conductivity, opening avenues in advanced energy technologies such as fuel cells and batteries. By integrating these functionalities within a designed porous architecture, SPOPs overcome the limitations of traditional solid acids and emerge as flagship materials for sustainable chemical processes and next-generation applications. The development and use of SPOPs have followed a unique evolutionary path since their early beginnings, summarized in Fig. 2 and discussed in the following paragraphs.
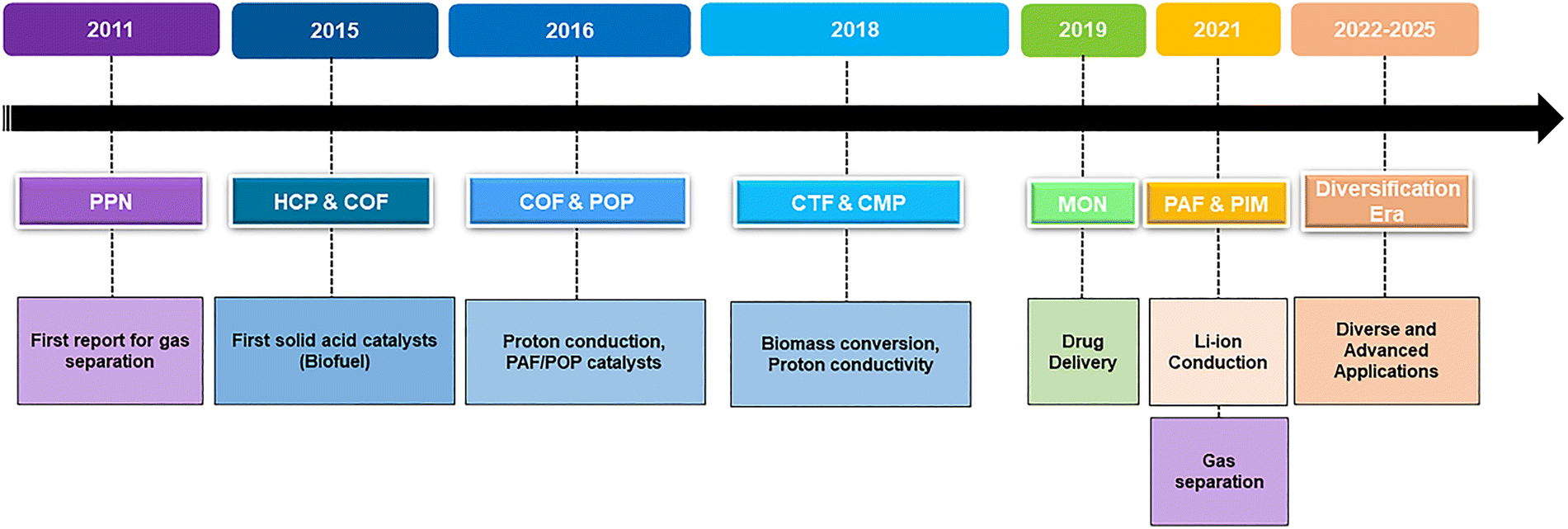 | ||
| Fig. 2 Evolutionary timeline of SPOPs, showing the development of key material families and their expanding applications from 2011 to 2025 toward future advanced applications. | ||
It is important to note that efficient solid acid catalysts synthesized via routes such as the sulfonation of swelling mesoporous polydivinylbenzenes (DVBs),118 pristine polymers often prepared through radical polymerization, were reported prior to this period, highlighting an active parallel research avenue. However, the modern scope focused on diverse synthetic strategies and tailored functionalities gained significant momentum with a key report in 2011, where a PPN was used for selective CO2 adsorption, marking the first exploration of functional POPs in separation science (Fig. 2).53 This key work opened the door for more diversity, and by 2015, HCPs119 and COFs120 were reported as effective solid acid catalysts for biofuel production, thus broadening their applications to catalysis.121 At the same time, early sulfonation techniques started to be systematically studied to improve polymer functionality. The period from 2016 to 2020 witnessed a significant growth in both material families and their applications. In 2016, PAFs122 and related POPs were used as highly effective catalysts.123 In the same year, POPs, including COFs,124 also demonstrated promising proton conductivity, showcasing their increasing versatility in energy-related uses.125 This trend continued with CMPs126 and CTFs127 being studied as efficient proton conductors by 2018–2020. Additionally, the range of applications expanded beyond energy and catalysis, as seen in 2019 when sulfonated MONs were successfully used for drug delivery, highlighting their potential in biomedical and environmental fields.56 Since 2020, the field has entered a maturation phase focused on performance optimization and the development of advanced, diverse applications in catalysis, environmental remediation, and energy technologies.99,116,125,127–133 Major breakthroughs include the engineering of PAFs for rapid Li-ion conduction,134 significant improvements in gas separation properties in sulfonated PIMs,135,136 and ongoing efforts to develop advanced membranes for fuel cells and high-performance proton conductivity.137–139 This ongoing timeline highlights continuous and increasingly rapid research efforts, showing that innovation within the SPOPs family, spanning a diverse range of architectures, for vital roles in separation, catalysis, energy, and environmental science, remains very active and continues to evolve without reaching a limit.
This review focuses on those modified materials, SPOPs: how they are synthesized and/or post-synthetically modified, which new properties arise, and how they govern the application for which the material has been selected. We can anticipate that the defining feature of SPOPs is their strong solid-state acidity. Indeed, the –SO3H is one of the strongest organic acids,140 and in particular, a wide variety of systems typically employ PTSA when a strong organic acid is needed to increase proton concentration and then, trigger the intended phenomenon.141–145 Classically, an acid is a substance that dissociates in solution to donate a proton (H+), while a base accepts this proton (Scheme 1a). The strength of an acid is quantified by its pKa value (the negative logarithm of its dissociation constant). The sulfonic group is a strong acid due to the high electronegativity of the S(VI) center, which is bonded to multiple oxygen atoms, combined with the resonance stabilization of the resulting sulfonate conjugate base (Scheme 1b). Thus, PTSA exhibits a pKa value of −2.8,146 indicating a very strong acid character considering the organic nature of the molecule. When the –SO3H is heterogenized on a platform like POP, each repeating unit in the polymer contains a sulfonic moiety, and therefore, the density of acidic groups per mass of polymer is large (Scheme 1c).147 Therefore, a –SO3H functionalized POP might be considered as a solid acid: an acid insoluble in the reaction medium.148 Since it is insoluble, pKa cannot be determined, and the acid character is only measured with the facility of dissociating the O–H bond and releasing H+ units. Nevertheless, it has to be considered that, when heterogenized, the sulfonic functions are attached to solid support, thus diffusion and surface effects, as well as parallel interactions with other functional groups found in the scaffold of the POP, may occur, which can diminish or enhance the acidic character. Furthermore, the heterogenized sulfonic group can also serve for further post-modification or even metal coordination to generate different but relevant applications apart from acidity. Summing up, the feature of a strong heterogenized acid on the structure of a POP supposes an advantage compared to classic systems. Indeed, SPOPs merge tunable reactivity with the inherent ease of separation of a heterogeneous material, allowing for simple recovery (e.g., via filtration or centrifugation) and thereby enhancing process efficiency and sustainability.
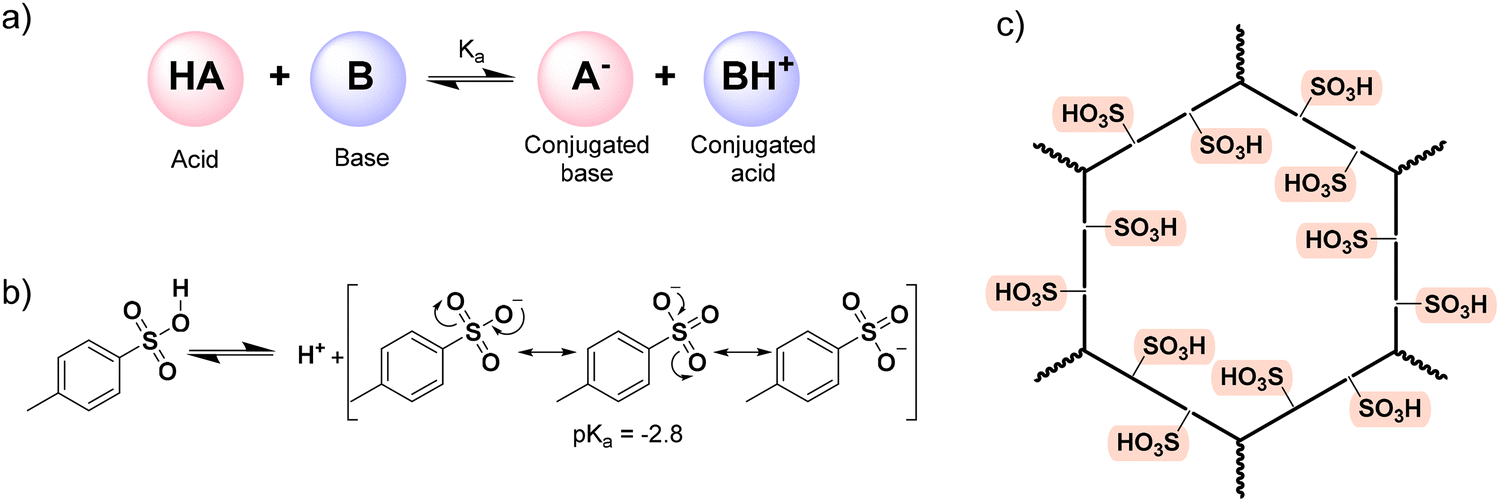 | ||
| Scheme 1 Acidity with POPs. a) general acid–base reaction; b) dissociation equilibrium of PTSA; c) general representation of a sulfonated polymer. | ||
As a whole, this review will discuss the various methods for the design of sulfonic/SPOPs, how the presence of the –SO3H units affects the structure and the properties of the material, including robustness, shape, size, and crystallinity, and then, the most relevant applications in various fields such as catalysis, environmental remediation, gas separation, and energy-related applications (e.g., proton conductivity, fuel cells, and batteries).
2. Sulfonic/sulfonated POPs
2.1. Design and modification strategies of SPOPs
The POPs literature shows that the most prevalent methods for the material synthesis are a collection of reactions, most commonly based on reversible processes, though irreversible and photochemical routes are also employed.149–152 While new reactions are continually emerging, the most widely employed polymerization methods include boronate/boroxime chemistry, the Schiff base condensation (imine, hydrazone, β-ketoenamine, azine, or oxime among others), carbonyl condensation (amidation, Knoevenagel reaction, imide, phenazine and (thio)imidazole ring fusion among others), olefin metathesis, Scholl reaction, FC alkylation, cross coupling methodologies and nitriles trimerization.2,23,42,153–155 This diversity results in a wide gamut of structures with tunable properties.To introduce a –SO3H group, SPOPs have been prepared by two different approaches (Scheme 2a). On the one hand, the –SO3H group can be present in one of the constituting monomers, and the polymerization is performed in its presence without affecting its outcome. As a result, the sulfonic groups of POPs are topologically present by design in each repeating unit. A typical example encompasses the monomer 2,5-diaminobenzenesulfonic acid,120 which can be submitted to Schiff base condensation polymerizations with different electrophiles, but other possibilities are reported too.156 On the other hand, the –SO3H is introduced in a post-synthetic modification (PSM) strategy. Once again, two different reaction pathways are reported. First, the sulfonic group could be introduced using a molecule that directly reacts with the POP core structure, and therefore it is finally fixed to the material network. The most typical strategy employs the aromatic cores of the building blocks to construct the polymer skeleton via FC alkylation, followed by sulfonation with ClSO3H (or SO3 in some cases), especially in HCP modification. However, the number of sulfonic acids in the polymer structure compared to the previous one is not controlled. Indeed, a typical representation is shown in Scheme 2b, where the bond between the aromatics and the sulfonic group is not (or cannot be) precisely determined. The other type of strategies employs pending functional groups present in the “pristine” polymer that can be transformed into the –SO3H. In this case, pending alcohols or amines, including those linking aromatic knots, and even imidazolic units, to be alkylated with sultones, can be employed.132,133,157 As a whole, PSM protocols allow the sulfonation of polymers that cannot host the sulfonic group in the monomers, thus expanding the number of systems able to present a sulfonic/sulfonated moiety.
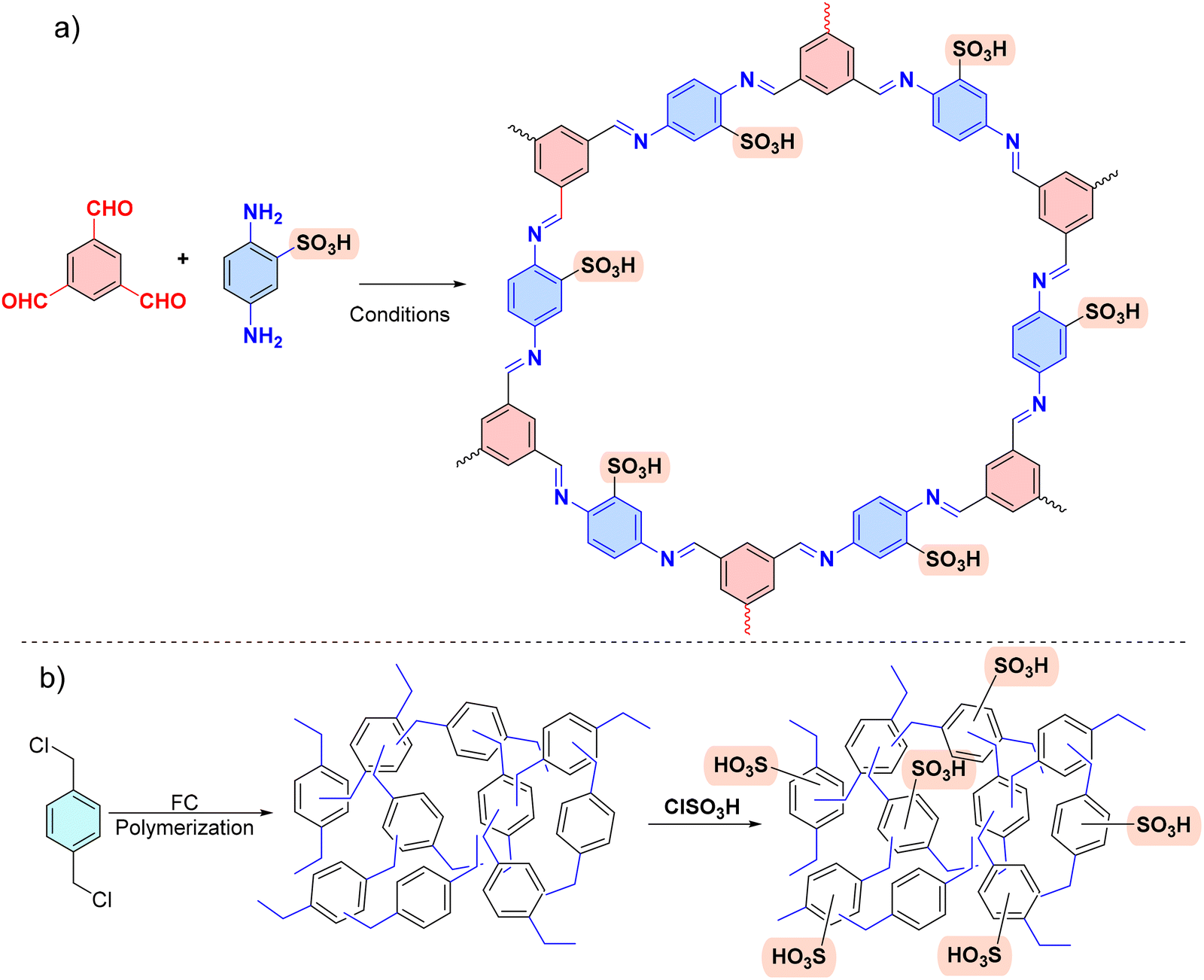 | ||
| Scheme 2 Different approaches for the synthesis of SPOPs. a) Monomer containing the sulfonic group; b) PSM strategy on HCPs based on FC alkylation. | ||
2.2. Effects of the sulfonic group on the POP properties
Studies on SPOPs typically include a control sample without sulfonation for comparison. Both types of samples have been characterized in the same way and have been tested in the same application, usually highlighting the advantages and disadvantages of the functionalization with –SO3H. Therefore, a straightforward comparison can be made. In general, the routine characterization of both “pristine” and sulfonated sample informs about the presence of the sulfonic group by a series of techniques like Fourier-Transformed infrared (FT-IR) spectroscopy (stretching vibrations of the sulfonic group bonds at ∼1200, ∼1000 and ∼625 cm−1),158 thermogravimetric analysis (TGA) with slightly higher weight loses as a result of the presence of the sulfonic moiety,159 X-ray photoelectron spectroscopy (XPS) with the detection and analysis of S 2p XPS core level region (similar results are obtained by energy dispersive X-ray spectroscopy (EDX), which can be done in combination with a microscopic technique),160 by wettability tests because the sulfone is more polar161 and increases contact angle, and by electrochemical tests related to the presence of surface acids, such as electrochemical impedance spectroscopy. Very frequently, the amount of –SO3H available for the required application is determined by either elemental analysis or titration measurements.162 Indeed, HCl and NaOH with adequate indicators in the presence of the POP are employed to determine the acidity of the material.163When the presence of the sulfonic functionality is confirmed, other tests have to be carried out to ensure that the POP structure is preserved after sulfonation. Thus, the assembly of the material is analyzed by solid-state nuclear magnetic resonance (SS-NMR) to check on the aromatic core signals.138 Generally, the sulfonation does not strongly modify the skeleton of the POP, and a similar set of signals is frequently achieved. In addition, the crystal structure is usually maintained after the sulfonation. Indeed, powder X-ray diffraction (PXRD) patterns tended to be almost identical,158 although some loss of crystallinity can be reported in the shape of less intense and broader peaks since the treatment somehow may affect POP structure and ordering.164 Finally, the textural properties are checked too, and generally, slightly smaller surface areas with less gas adsorption are obtained as a result of the sulfonation,165 undoubtedly assigned to the presence of bulky chemical groups in the pore that reduce the porosity of the function material, although in some cases the opposite trend has been observed.161
The final check usually done is how the sulfonation has affected the shape, particle size, and morphology of the samples, and this task is often carried out by microscopic analysis. While the aspect of the materials depends on the polymerization conditions, and very different configurations might be obtained as a function of the reaction conditions and monomers structures, some trends are observed in the literature. In general, the shape and morphology were nicely maintained. For example, the rod-like structure on the material presented by Yang et al. had the same aspect both at the scanning and transmission electron microscopes (SEM and TEM).161 Amorphous laminar structures consistent in shape and lateral dimensions were achieved by Wolska and collaborators, even considering that they yielded HCPs with an increased degree of sulfonation using ClSO3H.166 However, very strong functionalization treatments may result in the partial collapse of the structure, as resembled by the microscopic analysis reported by Dong and coworkers.167 Therefore, the sulfonation must be performed with some caution, because not the highest sulfonic content would lead to the best material, as also shown in the application sections.
3. Catalysis
The strategic incorporation of –SO3H groups into POPs creates a versatile class of solid Brønsted acids. SPOPs transcend the limitations of conventional solid acids (e.g., Amberlyst-15, Nafion®) and homogeneous analogues (e.g., H2SO4, PTSA) by combining four key design principles: (i) high and tunable acid-site density anchored in a robust framework, (ii) engineered nanoporosity for enhanced mass transport and substrate preorganization, (iii) synergistic multifunctionality by co-incorporating other active sites, and (iv) tailored surface chemistry (e.g., hydrophobicity) to optimize the reaction microenvironment. This section will cover those relevant examples where SPOPs are employed as heterogeneous catalysts in different chemical reactions, the main characteristics and catalytic parameters are summarized in Table 2.| POP | BBs/monomers | Functionalization | BET surface area (m2 g−1)/pore size (nm) | Application | Performance metric | Acid density (mmol g−1) | Stability assessment | Scale up (yes/no) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| HCP | 1,1′-dichloroxylene (DCX) or 4,4′-bis(chloromethyl)-1,1′-biphenyl (BCMP) | FC alkylation with ClSO3H | 1335–1900/0.85–2.2 | FC alkylation of anisole and benzyl alcohol and Beckmann rearrangement of cyclohexanone oxime | 100% conv. and 100% sel. | 0.65–2.41 | TGA | No | 168 |
| HCP | 2,2′,4,4′,6,6′-Hexafluoro-3,3′-divinyl-1,1′-biphenyl | FC alkylation with oleum | 340/0.3 | FC acylation of benzoyl chloride and anisole | 95.3% conv. and >99.5% sel. | 3.56 | TGA, N2 adsorption, | Yes, flow chemistry | 169 |
| Double decker type Silsesquioxane | Phenyltrimethoxysilane | FC alkylation with ClSO3H | 9–590/1.52 | Epoxide opening | 100% conv. and 100% sel. | 0.783–1.844 | TGA | No | 170 |
| HCP | Divinyl benzene (DVB) | FC alkylation with ClSO3H | 816–1271/30–70 | Acetal formation | 99.5% conv. and 96% sel. | 2.5–3.0 | No | No | 171 |
| HCP | DCX, 2,2′,6,6′-tetramethylbiphenyl (MeM) and 2,2′,6,6′-tetramethoxybiphenyl (OMeM) | FC alkylation with ClSO3H | 766–1277/1.4–4.8 | Synthesis of diphenylbenzopyrans/Naphthopyrans and triazoles | Large scope, yields >90% | 4.81% (Elemental analysis) | TGA | No | 172 |
| HCP-BBA-0.5 | Benzene and benzylamine | PSM | 604/<2, 2.2 and 33 | Oil shale upgrading (Catalytic cracking) | Shale oil yield: 41.85%, | 2.69 | — | — | 173 |
| PDVB-SO3H-IS | DVB and sodium p-styrene sulfonate (SPSS/SBS) | De novo | 650/ average 11 | BisphenolA synthesis | Conv.: ∼30% | 1.58 | Reusability test | To 120 mmol | 174 |
| Sel.: >90% | |||||||||
| SHCPs (SHCP-x = 1–10) | Bis(chloromethyl) biphenyl | One-pot | 540–1060 (micro-macropores)/- | Cyclohexyl acetate hydrolysis | Conv.: up to 75% | 0.03–3.76 | Reusability test, EA, XPS, TGA | — | 154 |
| Sel.: >99% | |||||||||
| P(QP-SBA)-3 | SPSS/SBS and phosphonium-based monomer (PQ) | De novo | 230 (micro-macropores)/- | Cyclohexyl acetate hydrolysis | Conv.: 93% | 3.6 | Reusability test | — | 129 |
| Sel.: 95% | |||||||||
| Yield: 89% | |||||||||
| COF | 1,3,5-triformyl phloroglucinol (TFPG) and 2,5-diamino benzenesulfonic acid (DABSA) | De novo | 600/- | CO2 capture solvent regeneration | Rate of up to 3.74 CO2 mmol min−1: increase of 733% | 0.185 | TGA | No | 175 |
| [DBUH]2Cys@COF-3 | 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and L-cysteine (Cys) ionic liquid (IL) into TpPa-COF-SO3H 1,3,5-triformylphloroglucinol (TP), 2,5-diaminobenzenesulfonic acid (Pa-SO3H) | Impregnation | 160/1.65–1.90, and 3.10 | CO2 cycloaddition | Yield: up to 96% | — | Reusability test, FT-IR, PXRD, SEM | — | 176 |
| Co(II)@TFPPy-PyTTA-COF | Tetrakis(4-formylphenyl) pyrene (TFPPy) and pyrene-1,3,6,8-tetrayl tetraaniline (PyTTA) | PSM | 600/1.2 | CO2 cycloaddition | Conv.: 70–99% | — | Reusability test, FT-IR, PXRD | — | 177 |
| Yield: 65–99% | |||||||||
| p2NPh-OSO3H HCP | 2-naphthol (NPh-OH) | PSM | 180/∼0.8 | Using acetic anhydride to acylating phenols, alcohols, thiols, amines, and aldehydes | Yield: 75–98% | 1.14 | Reusability test | — | 178 |
| PAF-30-SO3H | Tetrakis(p-bromophenyl)methane and 4,4′-biphenyldiboronic acid | PSM | 174/2.8 | Alkylation of guaiacol | Conv.: ∼70% (yields: 34% mono and 35% dialkylate) | 2.4 | — | — | 179 |
| PAF-20-SO3H | Tetrakis(p-bromophenyl)methane and 1,4-benzenediboronic acid | PSM | 354/2.3 | Alkylation of guaiacol | Conv.: 33% (yield: 23% C-isopropyl guaiacol) | 2.0 | — | — | 179 |
| Pt-PAF-30-SO3H | Tetrakis(p-bromophenyl)methane and 4,4′-biphenyldiboronic acid | PSM | 193 (PAF-30-SO3H)/- | Biofuel hydrodeoxygenation | e.g: from guaiacol: methoxycyclohexanol (32%), methoxycyclohexane (32%) and cyclohexane (27%) | 1.4 | — | — | 131 |
| COF | 1-Vinylimidazole (VIM), 2,5-dimethoxybenzene-1,4-dicarboxaldehyde (DMTPA), and 1,3,5-tri(4-aminophenyl)benzene (TAPB). | Imidazole alkylation with sultone | 504/2.5 | Biginelli reaction | Large scope, yields >90% | 3.75% (Elemental analysis) | TGA, organic solvents, acids and reductants | Yes, aerogel-based cup-like reactor 0.3 mol | 132 |
| COF | 1,4-terephthaldehyde and meso-tetrakis(4-aminophenyl)porphyrin | Amine reduction and alkylation with sultone | 60/2–4 | Dihydro-2-oxopyrrol four component synthesis | Yields > 85% | 1.70 | TGA and recovered catalyst analysis | No | 180 |
| Cashew nut shell liquid (CNSL)-formaldehyde resin | CSNL and formaldehyde | FC alkylation with ClSO3H | 8/- | Bis(indolyl)methane and Di[bis(indolyl)methane] synthesis | Large scope, >80% yield | 7.96–9.56% S (elemental analysis) | TGA | No | 181 |
| HCP | Bisphenol | FC alkylation with ClSO3H | 452/0.55 | Phosphonate synthesis | Large scope, yields > 90% | — | TGA | No | 182 |
| COF | Tri(4-formyl phenoxy) cyanurate (TFPC) and 1,4-diaminebenzene | FC alkylation with ClSO3H | 12/0.12 | Triazolo quinazolinones and 4H-pyrimido benzothiazoles synthesis | Large scope, yields 60–90% | 30% weight loss (TGA) | TGA | No | 183 |
| COF | 1,3,5-triformyl phloroglucinol (TFPG) and DABSA | De novo | 251/- | 3-component hydroxy carbamate synthesis | Yields 60–90% | 11% (EDS) | No | No | 184 |
| CTF | Melamine | FC alkylation with ClSO3H | — | 5-hydroxymethylfurfural (5-HMF) production | Temperature–time–catalyst ratio optimization | 1.3 | TGA | No | 185 |
| SBA-15 decorated CTF | Cyanuric chloride, melamine | Amine alkylation with sultone | 157/2.1 | 5-HMF production | Production from maltose | 5.5% weight loss of sulfonic groups (TGA) | TGA | No | 157 |
| HCP | Vinyl benzyl chloride, DVB | FC alkylation with ClSO3H | 550–693/1 | 5-HMF production | Similar or better activity than PTSA | 3.35–4.14 | No | No | 186 |
| COF | TFPG and DABSA | De novo | 159/1.4 | 5-HMF production | One pot synthesis of diformylfurfural from fructose | 3.15 | TGA | No | 120 |
| HCP | 1,3,5-triphenylbenzene and 1,2-dichloroethane | H2SO4 treated | 234/3.91 | 5-HMF production | Solvent selection | 2.73 | TGA, recovered catalyst analysis | No | 187 |
| CTF | 1,3-dicyanobenzene, 2,6-pyridinedicarbonitrile, 1,4-dicyanobenzene or 4,4′-biphenyldicarbonitrile | Gas phase sulfonation with SO3 | 600–2400/- | Hydrolysis of cellobiose | 86% conv. 90% sel. | 0.38–0.61 | TGA | No | 188 |
| COF | Triaminophenylbenzene (TAPB) and 2,6-dimethoxyterephthaldehyde (DMTP) | Ionic liquid encapsulation | 1266–2231/2.2–2.7 | Dehydrative formation of isosorbide from sorbitol | 100% conv. and >91% select. | 0.33–1.23 | Basic treatment | No | 189 |
| PDVB | DVB and sodium p-styrenesulfonate hydrate (SPSS) | Ion exchange with dilute sulfuric acid | 243–1008/4.3–9.49 | Dehydrative formation of isosorbide from sorbitol | 80% yield and 80% select. | 0.29–3.73 | Acid treatment, TGA | No | 190 |
| Hypercrosslinked hollow spherical polymers | BHN (2,6-bis(hydroxymethyl)naphthalene) and BHB (1,4-bis(hydroxymethyl)benzene) | FC alkylation with ClSO3H | 323–365/2.74 –6.31 | FAME formation | Esterification of real samples with yield 64–92.3% | 0.45–3.13 | TGA | No | 191 |
| HCP | DCX, naphthalene | FC alkylation with ClSO3H | 486–816/- | Tetrahydropyranylation of alcohols and transesterification of waste oil with methanol | 90% yield of free fatty acids in 4 h | 2.7–3.2 | TGA | No | 192 |
| HCP | 4,4′-bis(chloromethyl)-1,1′-biphenyl, ClSO3H | De novo | 645–1093/- | Esterification of hexanoic acid with benzyl alcohol | 95% conv. in 22 h run | 1.26–4.44 | TGA | No | 193 |
| HCP | Carbazole and α,α′-dibromo-p-xylene | FC alkylation with ClSO3H | 346/- | Biofuel synthesis from free fatty acids | Esterification with real samples in yields >90% | 3.7 | TGA | No | 119 |
| HCP | Phenol or bisphenol A | FC alkylation with ClSO3H | 210–324/7 | Biofuel synthesis from free fatty acids | Esterification with real samples in yields 30–90% | 0.85–1.60 | TGA | No | 163 |
| HCP | 1,3,5-triphenylbenzene and diphenylphosphate | FC alkylation with ClSO3H | 847/0.62–2.7 | Esterification of levulinic acid | 5 examples with C1–C5 alcohols in high conversion and selectivity | 1.24 | Stability after reuse | No | 194 |
| PDVB | DVB | FC alkylation with ClSO3H | 280–587/3.6–40.1 | Esterifications and acylations | Conversion 47–88% with high selectivities >97.5% | 2.13–3.72 | TGA | No | 118 |
| COF | 1,3,5-triformyl phloroglucinol, DABSA | Functionalized with 1-methyl-3-(3-sulfopropyl)-1H-imidazol-3-ium hydrosulfate | 103–264/7.4–8 | Biodiesel formation | 91% conversion of the model soybean oil into FAMEs | Hammet function = 1.45–1.6 | TGA and reused material analysis | No | 195 |
| HCP | 1,3,5-tri(4-vinylphenyl)-benzene and 1,4-divinylbenzene | FC alkylation with ClSO3H | 478–738/4.2–8.7 | Biodiesel formation: esterification of oleic acid with methanol | Yields > 95% with TOF: 44–100 mmol g−1 h−1 | 3.1–3.6 | TGA | No | 116 |
| PAF | 1,3,5-tris(4-Bromophenyl) benzene, benzene-1,4-diboronic acid and 1,3,5-tris(4-bromophneyl)amine | Treatment with oleum | 210–270 | Esterification of acetic acid with n-butanol | Conversion >70%, superior to benchmark Amberlist 15 | 5 | TGA | No | 122 |
| Hollow microporous organic polymer | Tetrakis(4-ethynylphenyl) methane and 1,4-diiodobenzene | FC alkylation with ClSO3H | 232/<2 | Polyketone synthesis | ∼5 g of polymer per 1.5 mg of catalyst without fouling the reactor | 6.91% S (Elemental analysis) | NMR | Yes | 121 |
| Silica - microporous organic polymer | Tetrakis(4-ethynylphenyl) methane and 1,4-diiodobenzene | FC alkylation with ClSO3H | 41–90/1–3 | Terpolymerization of CO, ethylene, and propylene | 17.2 kg of polymer per gram without fouling the reactor | 0.28–0.47 | DSC | Yes | 196 |
| PDVB | DVB and 4-vinylbenzenesulfonic acid sodium salt | De novo monomer truncation | 228–542/1–10 | Polypropylene synthesis | 36.4 kg of polymer per gram with high stereoregularity | 1.43 | TGA - DSC | Yes | 197 |
| PDVB | DVB and 4-vinylbenzenesulfonic acid sodium salt or sodium allylsulfonate | De novo monomer truncation | 385–471/- | Polyethylene synthesis | 7 g of polymer per g of catalyst per h with ultrahigh molecular weight | — | No | No | 198 |
| COF | 4,4′-biacetyl-2-hydroxylphenyl | –OH functionalization with sultone | 362/2 | Cascade synthesis of benzimidazoles, benzodiazepines and reductive amination of carbonyls | Large scope, yields > 90% | 2.87 | TGA | No | 199 |
| HCP | Tetraphenylethylene and formaldehyde dimethylacetal | FC alkylation with ClSO3H | 995/1–4 | Synthesis of symmetrical triarylmethanes | Large scope, different nucleophiles, yields: 30 – 95% | 11.11 | TGA and reused material characterization | No | 200 |
| PDVB | DVB and 4-styrenesulfonic acid sodium salt | De novo monomer truncation | 250–535/4.5–15.7 | Deacetalization−Knoevenagel condensation | 100% conversion 99% yield of the cascade product | 0.58–3.5 | TGA | No | 162 |
| CTF | Cyanuric chloride and p-terphenyl | FC alkylation with ClSO3H | 223/0.6–5.5 | One pot hydrogenation-esterification | 90% yield to cascade product in 8 h, reusable material | 2.99 | No | No | 201 |
| COF | 1,3,5-triformyl phloroglucinol, DABSA | Functionalized with (1R,2R)-1,2-diaminocyclohexane | 45/1.5 | Asymmetric Michael addition | Enantiomeric excess: 84 – 96% | 1.65 | NMR | No | 202 |
3.1. General solid acid behavior in catalysis
The synthesis of POPs where the constituting monomers possess –SO3H yields materials in which the topological pores are intrinsically decorated with sulfonic moieties. Therefore, the number of available active sites is remarkably high, and such materials are generally regarded in the literature as exhibiting strong acidic behavior. On the other hand, PSMs over the aromatic networks (such as the sulfonation with ClSO3H) or using ad hoc functional reactive points also cover the material with –SO3H groups.135,153,187 These properties have been harvested for different purposes, for example, the construction of membranes for fuel cells,139 as proton-conductive materials,124 for adsorption of dyes,159 or in electrochemical devices, such as desalination, batteries, and supercapacitors.203,204In catalysis with organic molecules, SPOPs have found application as catalysts in reactions where an acid is needed. Their appeal lies in high surface areas, robust stability, and tunable functionality through monomer selection and/or PSMs like sulfonation. This is a common advantage across POPs. Indeed, this versatility, combined with the cost-effectiveness of subclasses like HCPs, makes this class of materials highly suitable for a diverse range of acid-catalyzed reactions. The intimate link between porosity, acid-site density, and spatial confinement frequently leads to catalytic performances that surpass conventional solid acids (e.g., Amberlyst-15, Nafion®) and even rival homogeneous analogues (e.g., H2SO4, PTSA) in certain reactions. The foundational work by Li et al. on sulfonated HCPs demonstrated that a balance between acid density and preserved microporosity were key factors to achieve complete conversion and selectivity in FC alkylation and the Beckmann rearrangement (Fig. 3).168 The material achieved a turnover number (TON) of 43 and demonstrated excellent recyclability, benchmarking favorably against both homogeneous H2SO4 and solid acid references (Amberlyst-15, SBA-15-S) (see Table 2, entry 1). This performance directly highlights the dual advantage of the SPOP as a robust, porous heterogeneous acid. The pore structure connected to the acid moieties was concluded to be responsible for the observed performance as plausible explanation of the observed behavior. In a related study, Sun et al. developed a strong acidic SPOP (H0 ≤ −12.4) by sulfonating a fluorinated POP, where the electron-withdrawing backbone enhanced –SO3H acidity.169 This material showed exceptional activity in FC acylation of benzyl alcohol (see Table 2, entry 2). Under continuous flow conditions in a fixed-bed reactor, it maintained stable performance for over 150 min, achieving a steady product yield of approximately 97%. In sharp contrast, commercial solid acids such as Amberlyst-15 and Nafion® NR50 deactivated rapidly, falling below 5% conversion after 180 min. By 300 min, FPOP-SO3H had produced 1.83-, 91.3-, and 131.7-fold more product than POP-SO3H, Amberlyst-15, and Nafion® NR50, respectively. This marked stability is attributed to a synergistic combination of hierarchical porosity and fluorine incorporation. The interconnected pore structure facilitates efficient mass transport, limiting pore blockage and diffusional limitations. Furthermore, as observed in systems like Nafion®, the electron-withdrawing fluorine atoms enhance both the thermal stability and the acid strength of the –SO3H groups. Together, these features, strong acidity, stabilized active sites, and optimized transport pathways, establish FPOP–SO3H as a leading porous solid–acid catalyst for this transformation, rivaling state-of-the-art benchmarks. This is a premier example of how the inherently rigid, non-swelling architecture of SPOPs overcome a major limitation of polymeric resin catalysts, enabling practical application in continuous processes. Later, Zhao and coworkers were able to open styrene oxide under acid conditions using a double-decker silsesquioxane-based polymer synthesized via a Scholl coupling reaction, which was sulfonated after synthesis in similar strategies than previous polymers.170 The reaction was completed with perfect activity (full conversion and selectivity in 30 min at room temperature, including a flow chemistry scaling set-up), and the good results were attributed to the high loading of sulfonic acid moieties although the activity was reduced to 50% conversion after the 4th cycle, likely due to the leaching of –SO3H groups into the alcohol medium. Notably, its performance surpassed that of the benchmark solid acid catalyst MIL-DABCO-SO3H.205 The acid character of this family of materials was employed as a catalyst to construct an acetal from glycols and acetylacetonates using a sulfone-decorated polydivinylbenzene-based POP with yields above 95% in just 2 h of reaction.171 This reaction is typically carried out with PTSA in homogeneous phase, therefore the advantage of an easy separation catalyst that increases the overall reaction efficiency is highly beneficial. Yevad et al. prepared highly porous HCPs that were sulfonated in a second step using as monomers a mixture of DCX, 2,2′,6,6′-tetramethylbiphenyl (MeM) and 2,2′,6,6′-tetramethoxybiphenyl (OMeM) (Table 2).172 These materials were able to catalyze a different condensation reaction that, in this case, yielded diphenylbenzopirans in numbers higher than 90% at room temperature after 2 h. Moreover, they also prepared triazoles from nitroalkenes and azide excellent yields (exceeding 90%) at room temperature within 1 h. In summary, material synthesis and further modification seem to be valid strategies for the development of efficient catalysts in organic transformations. As a whole, different types of polymeric structures may be suitable to be employed in organic reactions, and the control on the functionalization results in solid acids that present advantages in important organic reactions. However, the choice of one platform over another remains an unresolved question, and current mechanistic understanding is limited by the lack of precise characterizations, especially regarding the nature of active sites and in-operando measurements. Indeed, vague and simplistic homogeneous mimics are commonly found. These aspects demand further and deeper investigation that integrates theory and unconventional characterization.
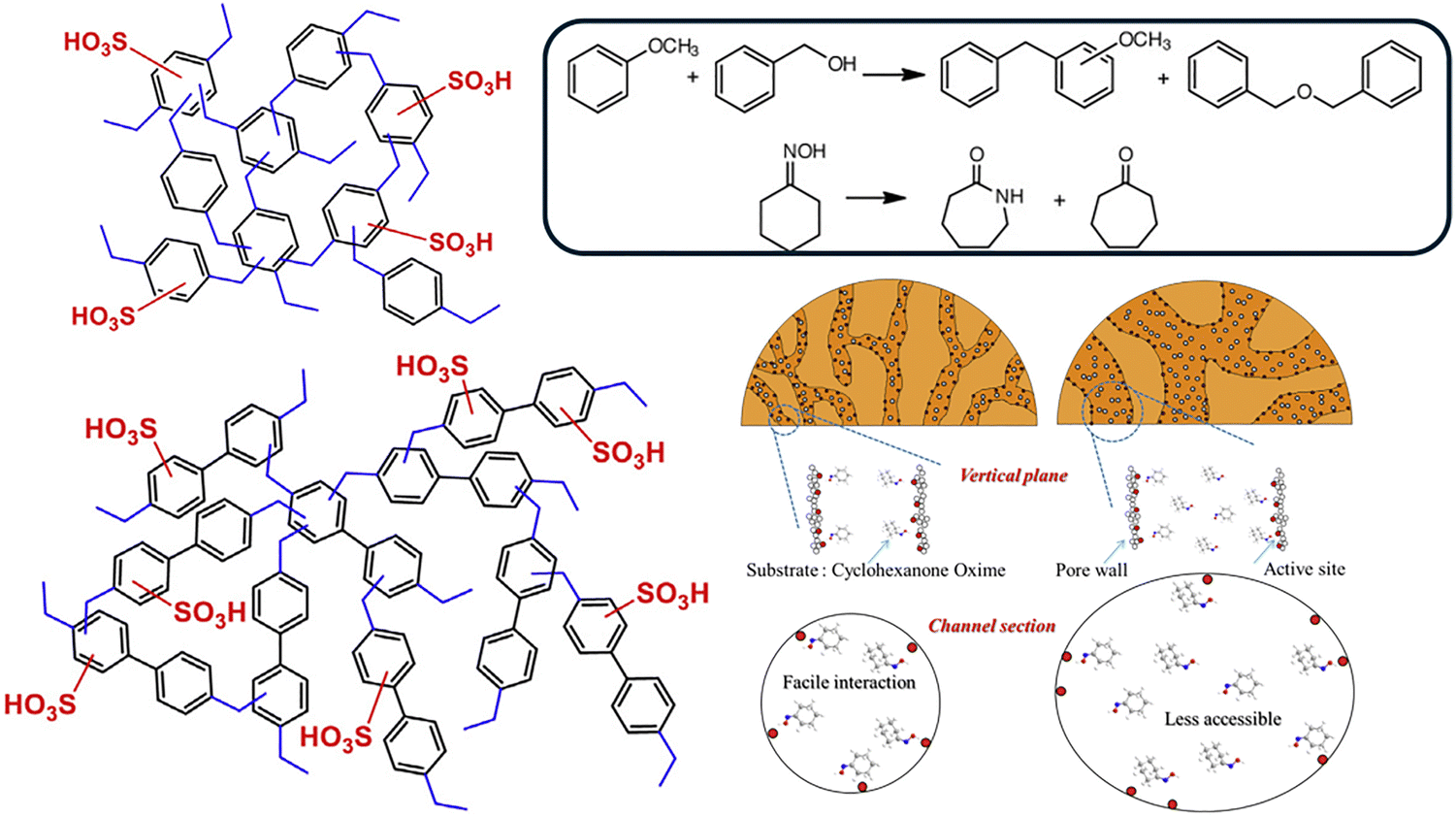 | ||
| Fig. 3 HCPs with activity in FC alkylation and Beckmann rearrangement, and a plausible explanation. Reproduced with permission from ref. 168 Copyright 2015 Elsevier. | ||
A key advantage of SPOPs is their tunable multifunctionality, which is critical for upgrading complex, real-world feedstocks. Wang et al. engineered a bifunctional acid–base HCP (HCP-BBA-S-x) for the in situ catalytic pyrolysis of oil shale (Fig. 4a).173 This system exemplifies the structure–performance trade-offs inherent to SPOP design and their superiority over single-function catalysts. The study revealed a non-linear relationship between acid-site density and performance. While increased sulfonation (variable “x”) raised acid density from ∼0.9 to ∼3.0 mmol g−1, excessive functionalization (x > 0.5) caused pore blockage, collapsing the BET surface area from ∼1050 m2 g−1 to ∼350 m2 g−1 and diminishing activity. The optimal catalyst, HCP-BBA-S-0.5, balanced a moderate acid density (∼2.7 mmol g−1) with preserved hierarchical porosity (avg. pore size ∼4.5 nm), achieving a 19% increase in shale oil yield (∼42%) over the non-catalytic process. Mechanistically, the proximity of acidic (–SO3H) and basic (–NH2) sites within the rigid framework enabled synergistic pathways: acid sites cracked heavy hydrocarbons, while adjacent base sites facilitated deoxygenation and isomerization. This cooperation directly enhanced fuel quality, reducing oxygen content by 55% (to 1.64%) and increasing the H/C ratio to 1.80. This case provides a critical design principle for SPOPs in demanding applications, an optimal balance between acid-site density, pore integrity, and site cooperation is more crucial than maximizing any single property. This tunable, multifunctional design surpasses the capabilities of conventional single-site solid acids.
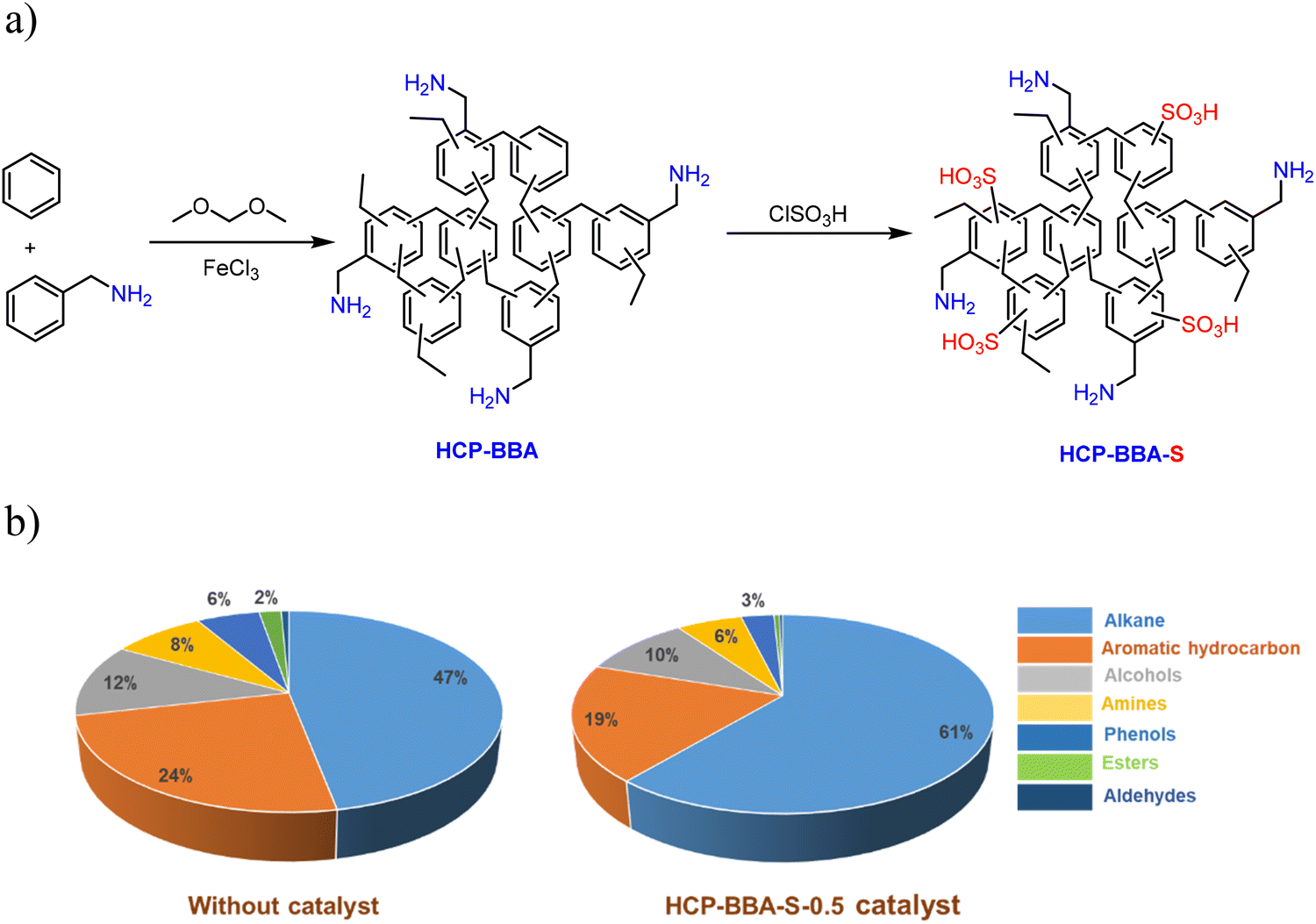 | ||
| Fig. 4 Catalyst synthesis and its performance: a) preparation steps for the HCP-BBA-S-x catalysts; b) comparative composition of shale oil produced with and without the HCP-BBA-S-0.5 catalyst. Adapted from ref. 173. | ||
In a complementary study focusing on controlled acid-site incorporation, Wei and co-workers demonstrated an alternative synthetic approach via in situ sulfonation during polymerization. Their catalyst, PDVB-SO3H-IS, was prepared by copolymerizing sodium p-styrenesulfonate (also referred to in the literature as sodium vinylbenzenesulfonate or SPSS) with DVB, followed by proton exchange, yielding a mesoporous polydivinylbenzene-based solid acid with an acid density of 1.58 mmol H+/g.174 This material exhibited excellent performance in the acid-catalyzed condensation of phenol and acetone to produce bisphenol A, achieving ∼38% phenol conversion (near the theoretical limit) and >90% selectivity at 90 °C (Scheme 3). Its robust reusability over five cycles was attributed to the covalent anchoring of –SO3H groups within the cross-linked polymeric matrix, preventing leaching.
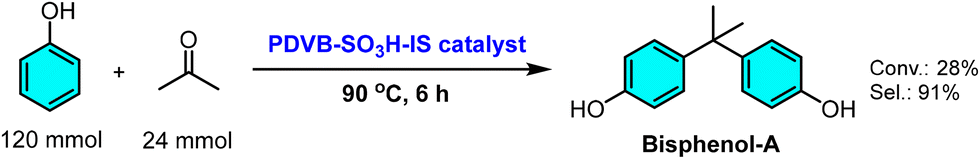 | ||
| Scheme 3 Phenol conversion and bisphenol-A selectivity achieved using PDVB-SO3H-IS as a catalyst. Adapted from ref. 174. | ||
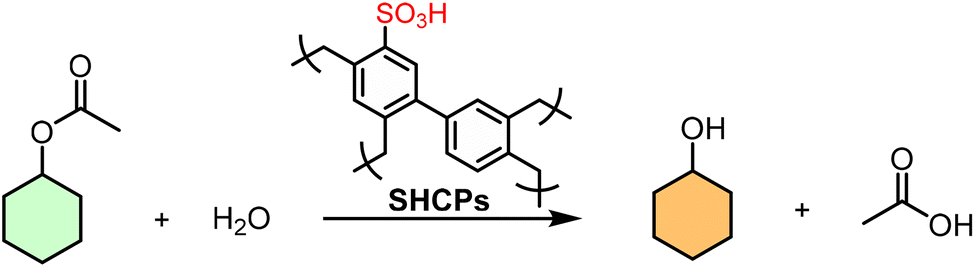
A pivotal study by Woodward et al. redefined the synthesis of SHCPs through a one-pot, metal-free methodology using ClSO3H as both a FC catalyst and sulfonating agent.154 This integrated approach condensed a conventionally multi-step, multi-day process into under 24 h, eliminating separate metal catalysts and significantly reducing reagent use. By simply varying the ClSO3H-to-monomer molar ratio, a series of SHCPs (SHCP-1 to SHCP-10) with finely tuned acid densities (up to ∼3.8 mmol g−1) could be prepared. They found that SHCP-5, with an optimal acid density of 2.20 mmol g−1 and a preserved BET surface area of 890 m2 g−1, achieved ∼75% conversion in hydrolyzing cyclohexyl acetate, surpassing both Amberlyst-15 and homogeneous H2SO4. This study underscores that maximum –SO3H loading can compromise porosity; optimal performance arises from a synergistic interplay of both properties (Table 3).
|
|
|||
|---|---|---|---|
| Catalyst | –SO3H content (mmol g−1) | BET surface area (m2 g−1) | Conversiona (mol%) (with selectivity > 99%) |
| a Calculated by GC-MS. | |||
| SHCP-1 | 0.03 | ∼540 | 0 |
| SHCP-2 | 0.44 | ∼900 | ∼31 |
| SHCP-3 | 1.15 | ∼1060 | ∼68 |
| SHCP-4 | 1.72 | ∼950 | ∼72 |
| SHCP-5 | 2.20 | ∼890 | ∼75 |
| SHCP-6 | 2.82 | ∼805 | ∼72 |
| SHCP-7 | 3.24 | ∼725 | ∼64 |
| SHCP-8 | 3.40 | ∼705 | ∼68 |
| SHCP-9 | 3.60 | ∼680 | ∼65 |
| SHCP-10 | 3.76 | ∼690 | ∼59 |
The strategic integration of amphiphilicity into SPOPs presents a powerful approach to overcome mass-transfer limitations in aqueous-phase catalysis. In a study, an amphiphilic sulfonated HCP (SHCP) catalyst series, denoted P(QP-SBA)-n (with n being the SBS-to-QP molar ratio), was synthesized via solvothermal copolymerization of a phosphonium-based surfactant monomer (QP) and vinyl benzenesulfonate (SBS, previously referred to as SPSS), followed by acidification.129 The design rationale centered on creating a polymer matrix with intrinsic swelling behavior and dual hydrophilic–hydrophobic character. The optimal catalyst, P(QP-SBA)-3, proved highly effective for hydrolyzing cyclohexyl acetate (93% conversion, 95% selectivity, 89% yield), outperforming conventional acids (H2SO4, PhSO3H) and benchmark solid acids like Amberlyst-15, [HSO3bmim]HSO4 IL,206 and high-surface-area carbon solid acid.207 Critical analysis of the structure-performance relationship reveals that its superiority is not merely a function of high acid density (∼0.36 mmol g−1) but stems from a synergistic trifecta: (i) favorable BET surface area (228 m2 g−1) enabling accessible sites, (ii) excellent surface amphiphilicity (evidenced by near-zero contact angles for both water and organic phases) ensuring effective dispersion and interfacial contact, and (iii) a swollen gel-like phase that enriches hydrophobic substrates within the polymer network, effectively mimicking phase-transfer catalysis. This holistic design enabled efficient catalysis even under static conditions, highlighting reduced energy input for mixing. The catalyst maintained robust performance over four cycles, with minor deactivation attributed primarily to physical handling losses, underscoring the structural stability imparted by the framework.
Moving beyond traditional acid catalysis, the functionality of –SO3H groups can be leveraged to modulate the electronic structure and coordination environments within sophisticated frameworks for advanced applications. A seminal example is the work by Li et al., who strategically incorporated –SO3H groups into the organic struts during the synthesis of a β-ketoenamine COF.175 This design served a dual purpose: first, the –SO3H groups induced a morphological transformation from nanorods to interwoven nanowires, significantly increasing the specific surface area from 320 to 600 m2 g−1 and creating a more favorable environment for metal anchoring. Second, and more critically, these electron-withdrawing groups acted as electronic modulators for the subsequent immobilization of single-atom cobalt sites. Through comprehensive characterization including XPS, X-ray absorption fine structure (XAFS), high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and density functional theory (DFT) calculations, the study elucidated that the sulfonic functionality cooperatively tuned the local Co–N–C coordination (Fig. 5).
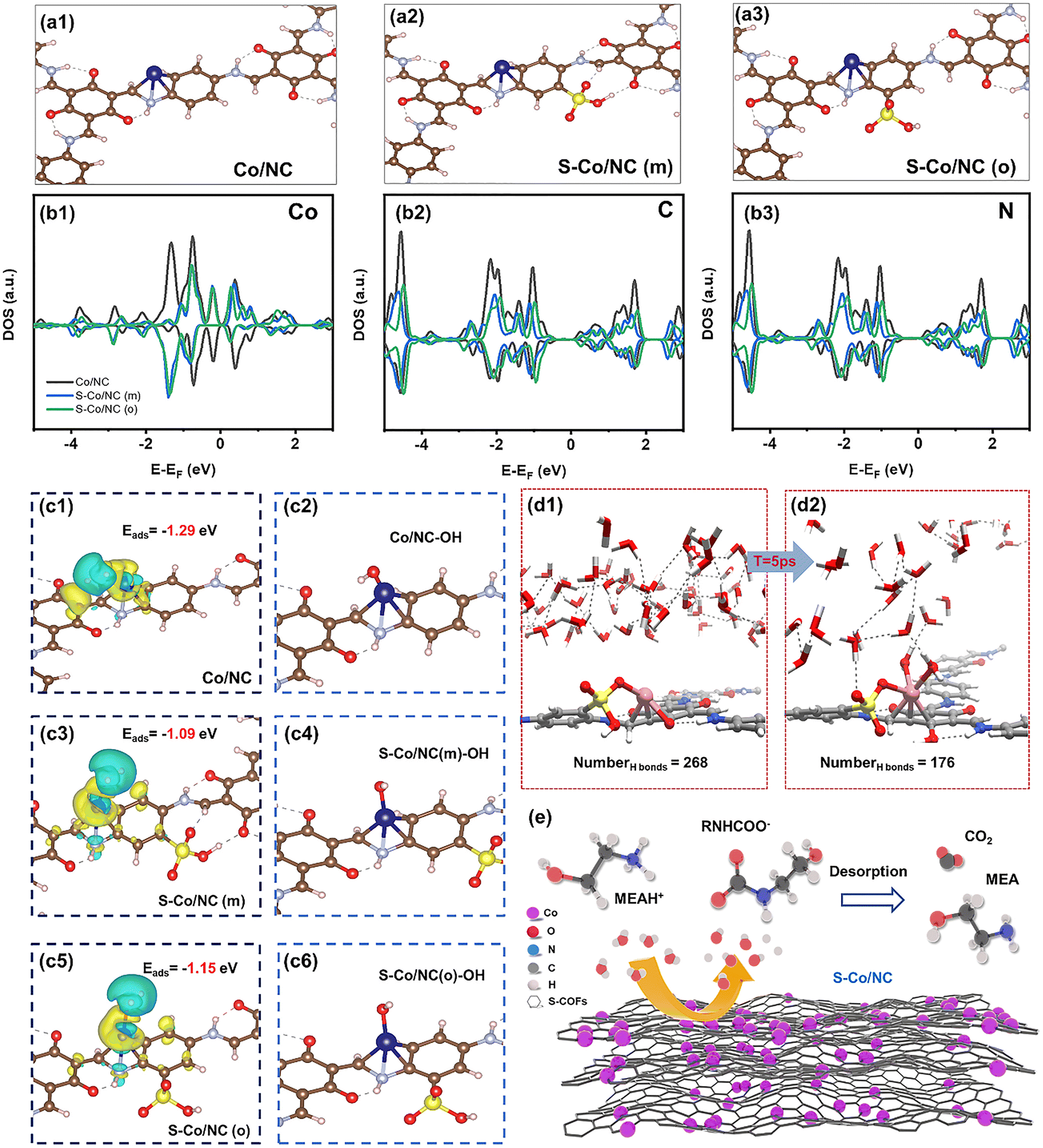 | ||
| Fig. 5 Structural and electronic characterization of COF catalysts and the catalytic mechanism for carbamate decomposition. a) Atomic structure of the Co active sites in (a1) Co/NC and (a2) S-Co/NC, with their corresponding reaction pathways; b) Electronic structure analysis showing the density of states for (b1) Co, (b2) C, and (b3) N atoms in different catalysts; c) Charge redistribution upon H2O adsorption (blue/yellow) and optimized catalyst structures with surface –OH groups; d) Ab initio molecular dynamics simulation of H2O interaction with the S–Co/NC surface; e) Proposed catalytic cycle for carbamate decomposition over the S–Co/NC catalyst. Monoethanolamine (MEA) was used as the benchmark CO2 capture solvent. Reproduced with permission from ref. 175 Copyright 2024 Elsevier. | ||
This modulation enhanced the Lewis acidity of the Co center while the proximal sulfonic groups provided Brønsted acid sites, thereby facilitating proton transfer. Consequently, the engineered solid acid catalyst, S–Co/NC, demonstrated exceptional performance for the energy-intensive solvent regeneration step in amine-based CO2 capture. By accelerating carbamate decomposition via synergistic Lewis and Brønsted acid catalysis, S–Co/NC increased the reaction rate by over 700% relative to the non-catalytic baseline. This enhancement facilitated effective CO2 desorption at a reduced temperature, which corresponded to an approximately 29% decrease in the energy required for solvent regeneration. This study transcends conventional use of –SO3H as merely a Brønsted acid site; it showcases its pivotal role as a morphological director and electronic modulator in constructing advanced single-atom catalysts, opening new avenues for SPOPs in addressing critical energy and environmental challenges.
Moving beyond simple impregnation, a sophisticated host–guest design strategy leveraging acid–base interactions was employed to construct a highly selective heterogeneous catalyst. Xue et al. ingeniously immobilized an alkaline amino acid ionic liquid (AAIL), [DBUH]2Cys (synthesized from 1,8-diazabicyclo[5.4.0]undec-7-ene and L-cysteine), within the precisely defined nanochannels of a –SO3H-functionalized COF (TpPa-SO3H).176 The resulting composites, [DBUH]2Cys@COF-X (X = 1–4; 0.1–0.4 mmol IL), were applied to CO2 cycloaddition reaction (Fig. 6). This design achieved two key outcomes: (1) it created confined bifunctional active sites within the pores, and (2) it engineered the pore environment. While IL incorporation reduced the BET surface area, it strategically increased pore size and volume, creating defined mesopores (∼3.0 nm) in the optimal catalyst, [DBUH]2Cys@COF-3. This engineered nanostructure translated directly to exceptional and selective performance. The catalyst achieved a 96% yield for the epichlorohydrin conversion under remarkably mild, solvent-free conditions (1 atm CO2, 100 °C). More importantly, it exhibited pronounced size-selective catalysis: small epoxides (e.g., epichlorohydrin, glycidol) were converted near-quantitatively (95–96%), while bulky substrate (e.g., styrene oxide) was sterically excluded, yielding only trace amounts. This transforms the composite into a molecular gatekeeper, a rare property for solid acids. Consequently, this system outperforms many reported IL/COF hybrids,208–211 which often require higher pressure, loading, or co-catalysts. DFT calculations revealed the synergistic mechanism enabled by confinement: the [DBUH]+ cation activates the epoxide via H-bonding, while the proximal cysteine carboxylate (–COO−) activates CO2 and attacks the epoxide, cooperatively lowering the energy barrier. While the catalyst was recyclable over three cycles, activity loss in the fourth cycle highlighted an ongoing challenge with IL leaching stability. Nevertheless, this work exemplifies how precise spatial organization of functional groups within SPOPs can unlock unique selectivity and cooperative catalysis under mild conditions.
 | ||
| Fig. 6 Proposed catalytic mechanism for the synergistic cycloaddition of CO2 with epoxides using [DBUH]2Cys@COF-3 as a catalyst. Reproduced with permission from ref. 176 Copyright 2024 Elsevier. | ||
An ionic, heterogeneous 2D COF, denoted as Co(II)@TFPPy-PyTTA-COF, was synthesized via a multi-step PSM of the imine-linked TFPPy-PyTTA-COF (where TFPPy = tetrakis(4-formylphenyl)pyrene and PyTTA = pyrene-1,3,6,8-tetrayl tetraaniline) (Scheme 4).177 The PSM process involved: (i) reduction of the imine (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) linkages to amine (C–N) groups, (ii) ring-opening alkylation of these amines with 1,3-propane sultone to install alkyl sulfonate groups, and (iii) metalation with cobalt(II) acetate to anchor Co(II) ions via ionic coordination to the sulfonate moieties, yielding Co(II)@TFPPy-PyTTA-COF. This linkage-focused PSM is significant because it bypasses incompatibility between functional groups and harsh solvothermal synthesis conditions, enabling precise installation of active sites within pre-formed, stable pores. The resulting ionic, bifunctional pore environment featuring Lewis acidic Co2+ centers and nucleophilic sulfonate anions proved highly effective for the converting CO2 and epoxides into cyclic carbonates. Under the optimized reaction parameters (100 °C, 3.0 MPa CO2), the catalyst achieved high conversions (70–99%) and yields (65–99%) for a range of epoxides, matching the activity of the homogeneous Co(OAc)2. More importantly, it exhibited heterogeneity and stability, retaining its crystallinity and activity over five catalytic cycles without metal leaching. This work establishes linkage engineering via PSM as a powerful strategy for integrating cooperative acid–base sites into robust porous scaffolds, creating high-performance, reusable catalysts for efficient CO2 utilization.
N) linkages to amine (C–N) groups, (ii) ring-opening alkylation of these amines with 1,3-propane sultone to install alkyl sulfonate groups, and (iii) metalation with cobalt(II) acetate to anchor Co(II) ions via ionic coordination to the sulfonate moieties, yielding Co(II)@TFPPy-PyTTA-COF. This linkage-focused PSM is significant because it bypasses incompatibility between functional groups and harsh solvothermal synthesis conditions, enabling precise installation of active sites within pre-formed, stable pores. The resulting ionic, bifunctional pore environment featuring Lewis acidic Co2+ centers and nucleophilic sulfonate anions proved highly effective for the converting CO2 and epoxides into cyclic carbonates. Under the optimized reaction parameters (100 °C, 3.0 MPa CO2), the catalyst achieved high conversions (70–99%) and yields (65–99%) for a range of epoxides, matching the activity of the homogeneous Co(OAc)2. More importantly, it exhibited heterogeneity and stability, retaining its crystallinity and activity over five catalytic cycles without metal leaching. This work establishes linkage engineering via PSM as a powerful strategy for integrating cooperative acid–base sites into robust porous scaffolds, creating high-performance, reusable catalysts for efficient CO2 utilization.
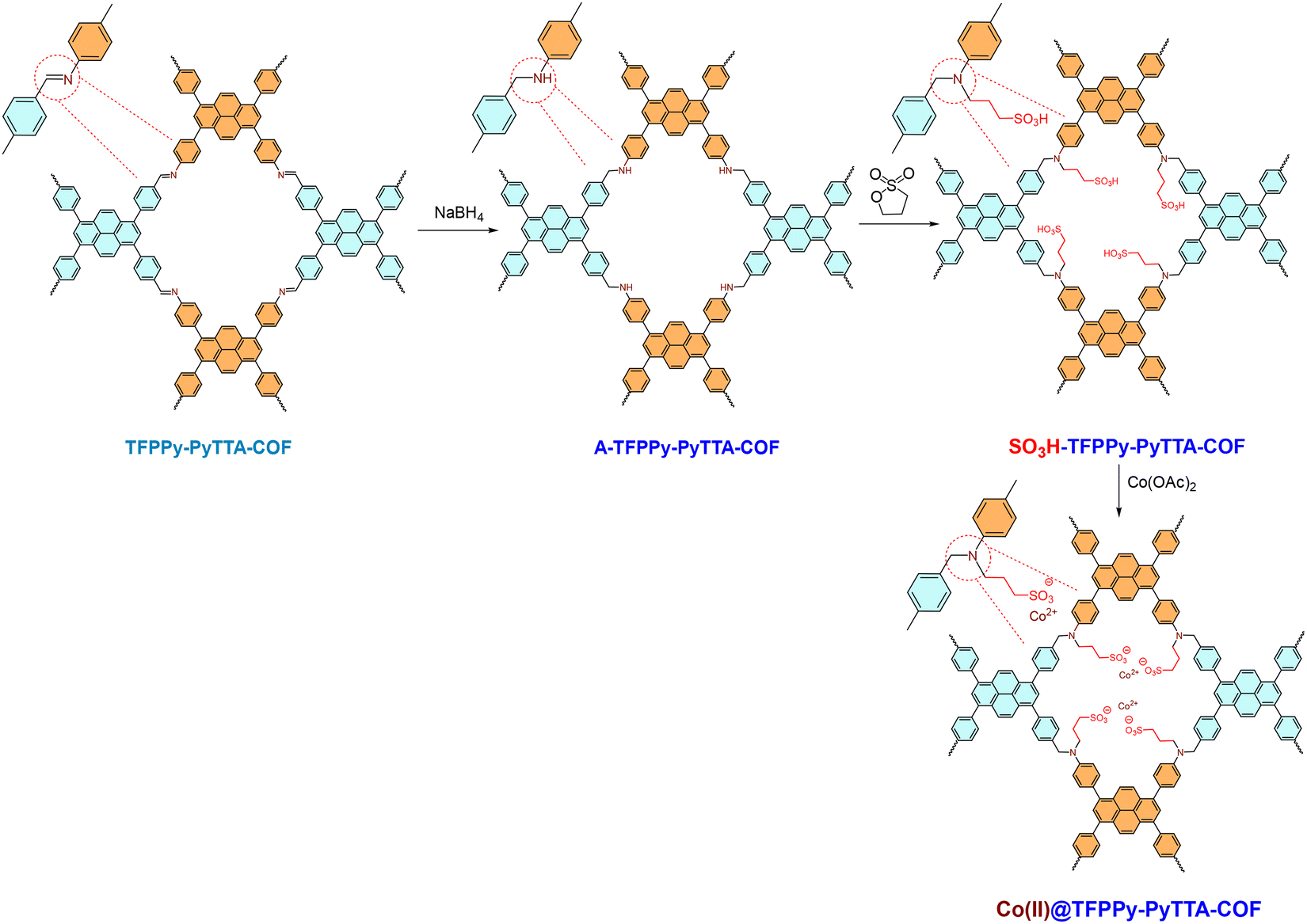 | ||
| Scheme 4 Schematic illustration of the PSM process for synthesizing Co(II)@TFPPy-PyTTA-COF. Adapted from ref. 177. | ||
Kim and colleagues178 synthesized a 2-naphthol-based HCP (p2NPh-OH) via FC alkylation using dimethoxymethane as the cross-linker (Scheme 5). Subsequent functionalization with ClSO3H yielded the sulfonated solid acid catalyst, p2NPh-OSO3H. This recoverable catalyst exhibited excellent performance for the protection and acylation of diverse substrates including phenols, alcohols, thiols, amines, and aldehydes using acetic anhydride under solvent-free conditions at room temperature. In the acylation of benzyl alcohol, p2NPh-OSO3H demonstrated superior catalytic activity compared to both polydopamine-derived sulfamic acid-functionalized magnetic nanoparticles (Fe3O4@PDA-SO3H),212 noting its tedious synthesis, and Ph3P(OAc)2.213 Notably, the catalyst exhibited no significant loss in activity after ten cycles, confirming its high stability and reusability.
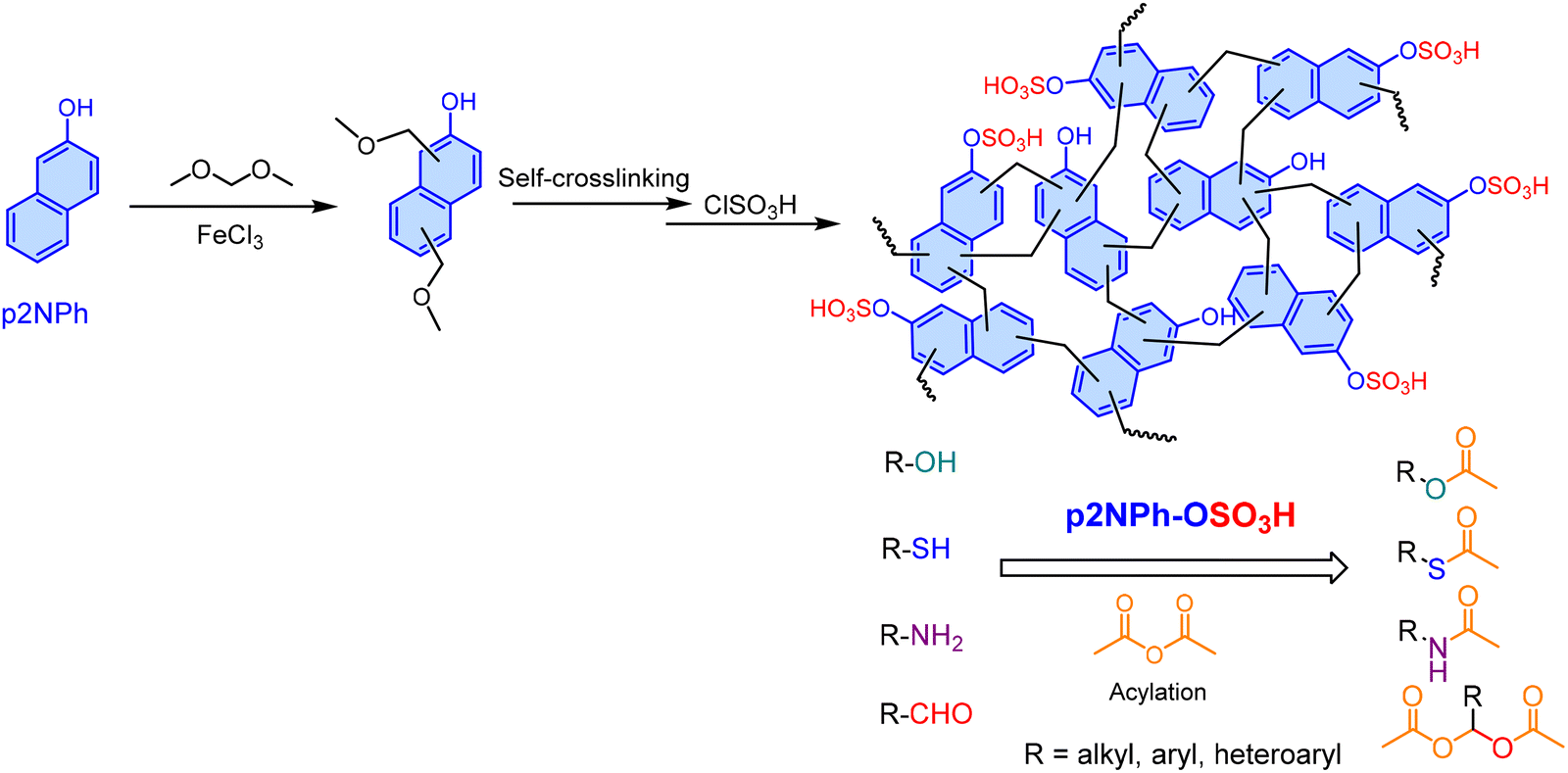 | ||
| Scheme 5 Synthetic route to sulfonated poly(2-naphthol) and its use as a catalyst for the acylation of various functional groups with acetic anhydride. Adapted from ref. 178. | ||
The well-defined nanoporosity of SPOPs acts as a nanoreactor, imposing spatial constraints that can dramatically influence reaction pathways and selectivity, moving SPOPs beyond being mere acid reservoirs to becoming precision tools for selective synthesis. This is exemplified by the alkylation of guaiacol over two sulfonated PAF, PAF-20-SO3H and PAF-30-SO3H.179 Their synthesis via Suzuki cross-coupling and post-sulfonation created a deliberate structural contrast: PAF-20-SO3H had higher surface area but smaller pores (∼2.3 nm), while PAF-30-SO3H featured larger pores (∼2.8 nm) that facilitated a higher –SO3H loading (2.34 vs. 1.88 mmol g−1) (Scheme 6). This variance directly dictated catalytic selectivity. With isopropanol, the larger-pore PAF-30-SO3H achieved higher conversion (∼70% vs. ∼30% in 6 h) and yielded both mono- and dialkylates. In contrast, the more confined pores of PAF-20-SO3H favored monoalkylation, suggesting steric control over product distribution. The challenge of reactant competition was evident with bulkier cyclohexanol, where its dehydration product (cyclohexene) blocked sites. However, process optimization using a non-polar solvent and limiting alcohol concentration enabled PAF-30-SO3H to achieve ∼60% guaiacol conversion to valuable cyclohexyl derivatives in 2 h. This case demonstrates that for SPOPs in reaction systems with competing chemical pathways, achieving high performance requires a dual-design approach: engineering the pore nanoconfinement to control selectivity and simultaneously optimizing the bulk reaction environment to manage these pathways.
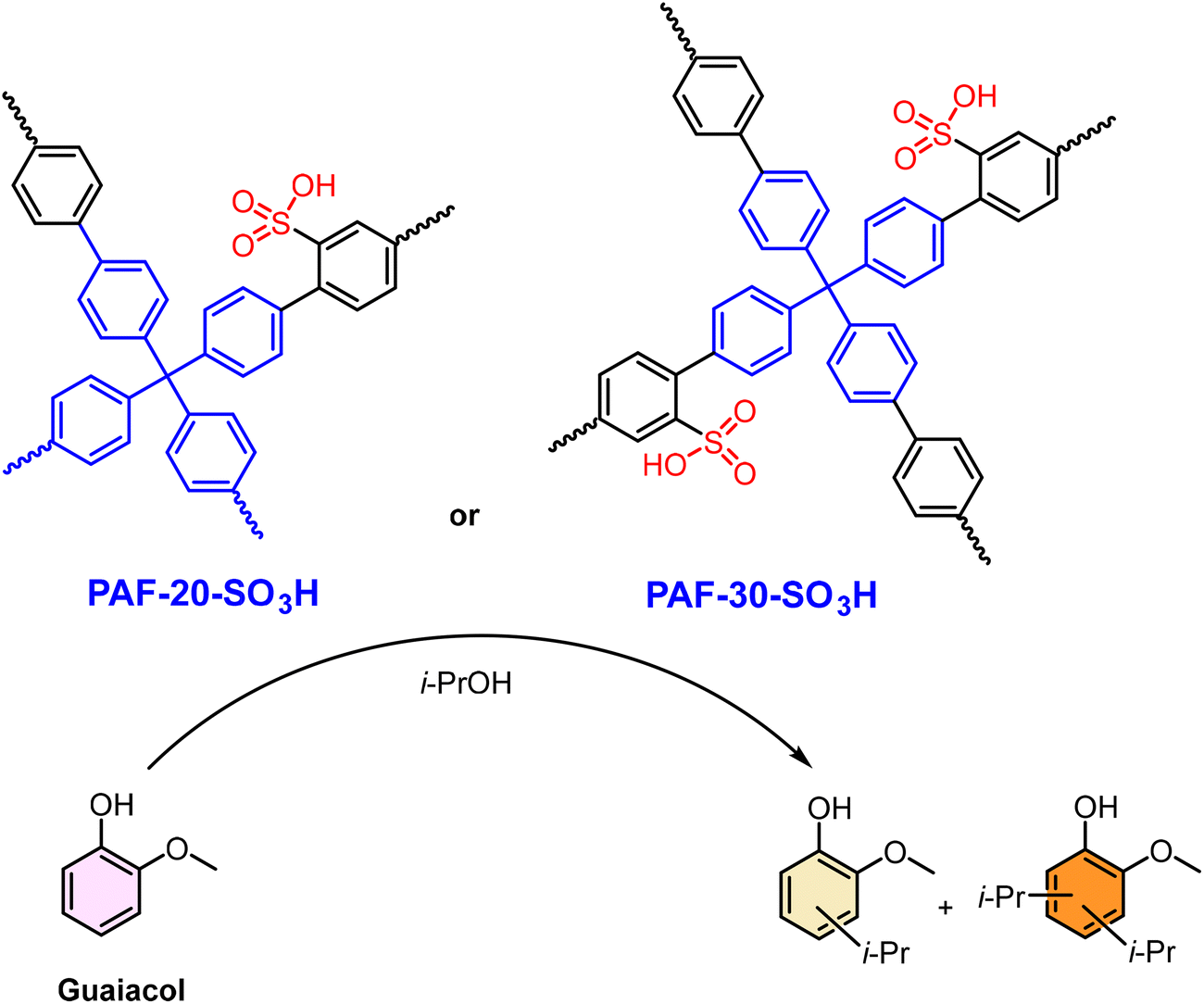 | ||
| Scheme 6 Reaction scheme for the alkylation of guaiacol with isopropanol catalyzed by PAF-20-SO3H and PAF-30-SO3H, showing the main products. Adapted from ref. 179. | ||
In related work, Kalinina et al. directly compared a platinum catalyst supported on a non-acidic PAF (Pt-PAF-30) with its sulfonated analog (Pt-PAF-30-SO3H) for the hydrodeoxygenation (HDO) of bio-derived molecules (guaiacol, veratrole, catechol) (Scheme 7).131 The contrast in catalytic pathways was stark. Pt/PAF-30 primarily hydrogenated aromatic rings (e.g., converting guaiacol to methoxycyclohexanol), with deoxygenation limited to partially hydrogenated intermediates. Critically, it could not perform demethoxylation on saturated rings. In contrast, Pt-PAF-30-SO3H efficiently catalyzed both dehydroxylation and demethoxylation of saturated species, dramatically boosting the yield of fully deoxygenated cyclohexane to 65–95%. This underscores that proximal acid sites are essential for cleaving robust C(alkyl)–O bonds (e.g., in methoxycyclohexane), a step often rate-limiting in HDO. The synergy likely involves acid-catalyzed dehydration or methanol elimination from protonated intermediates, a pathway inaccessible to the metal site alone. This work exemplifies how integrating –SO3H functionality transforms a simple hydrogenation catalyst into a highly effective deoxygenation system, a design principle leveraged in subsequent work for broader lignin model substrates.214
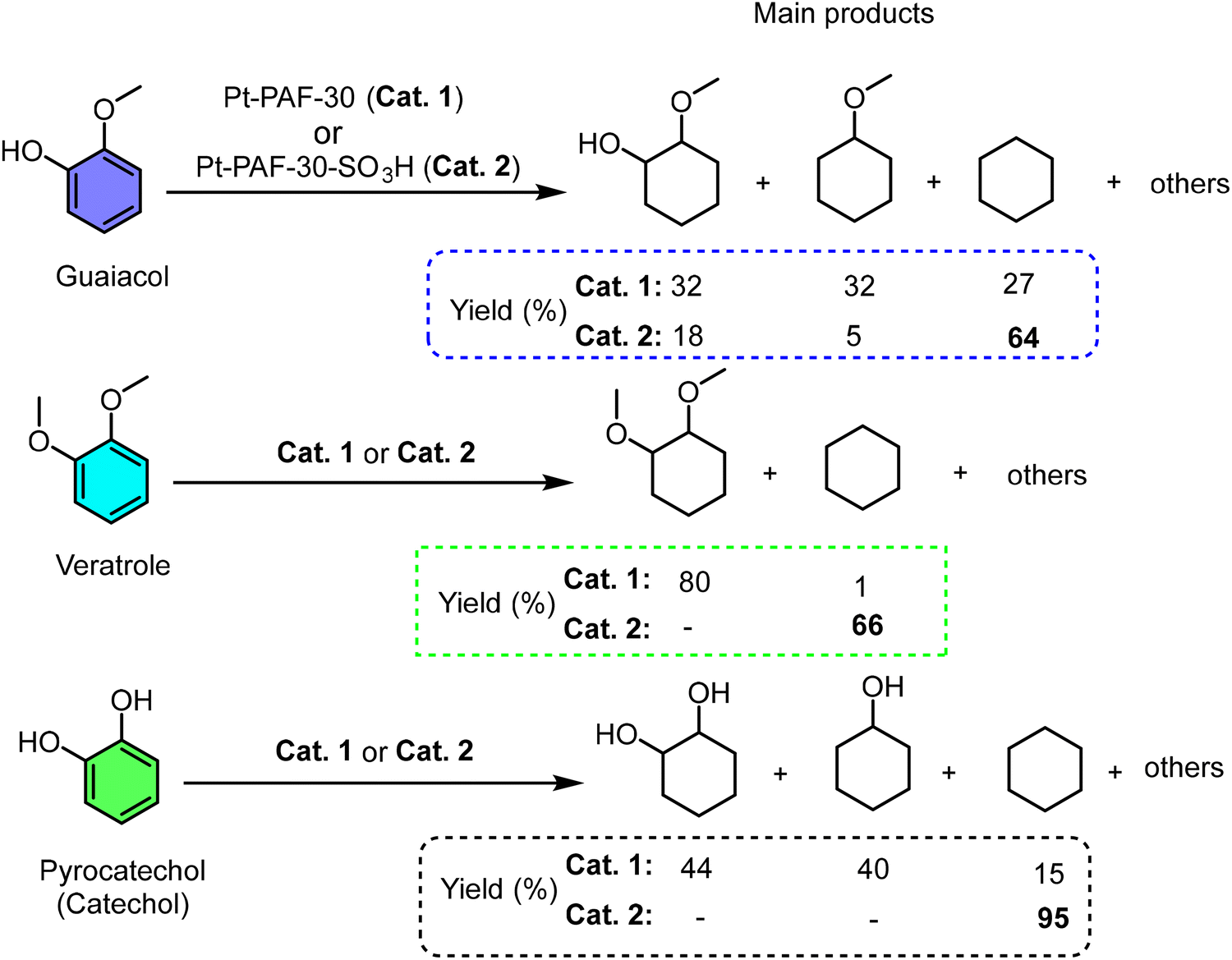 | ||
| Scheme 7 PAF-SO3H catalyzed hydrogenation-deoxygenation of lignin-derived model compounds. Adapted from ref. 131. | ||
3.2. Heterocycle and pharmacologically relevant compounds
The synthesis of N- and O-heterocycles, crucial pharmacophores, often requires precise acid catalysis and efficient product separation.215–218 SPOPs offer a unique platform for these transformations by combining strong and accessible Brønsted acid sites with a tunable hydrophobic/hydrophilic pore environment, which can influence substrate preorganization and selectivity. In the previous section, Yevad et al., have discussed the synthesis of diphenylbenzopirans with good activity.172 This work represents a good example of the power of SPOPs for the synthesis of relevant heterocyclic compounds (Scheme 8). In this sense, Yao and coworkers presented a COF created in a multicomponent Povarov reaction among polytopic aldehydes and amines with vinyl-imidazole.132 Hence, they made a porous material connected by strong quinoline linkages compared to the standard and labile imine bonds to increase robustness. In addition, the pending imidazole allowed an easy PSM to introduce the –SO3H by a straightforward alkylation with 1,3-propanesultone. As a result, a –SO3H-ionic liquid COF was yielded. The combination of both moieties was concluded responsible for the activity in the catalytic Biginelli hydropyrimidone formation reaction studied: yields above 90% with excellent recyclability and even a scale-up attempt at 0.3 mmol level were reported. Authors took advantage of heterogeneous catalyst properties in terms of activity, robustness and straightforward separation to outperform typical homogeneous catalysts (e.g. PTSA) and facing a greener effort in the Biginelli reaction. A similar synthetic route was followed by our group.180 A porphyrin-iminic COF was reduced and furtherly alkylated with 1,3-propanesultone. This facile –SO3H functionalization underscores the synthetic versatility of the polymeric platform, enabling the efficient synthesis of highly substituted dihydro-2-oxypyrroles in high yield (>90%) within 60 min. A comparative study with typical catalysts for that transformation was included, and the resulting polymer presented better results than homogeneous (e.g. I2)219 and heterogeneous systems (e.g. Fe3O4@nanocarbon) catalysts.220 This enhanced performance is attributed to a synergistic effect arising from two key features: the strong Brønsted acidity of the anchored –SO3H groups and the porous structure of the polymer framework. Kotkar and Tilve presented an acidic resin from cashew nutshell liquor, a biomass waste product, for synthesizing bis(indolyl)methanes from xylochemicals.181 This work addresses a significant gap, as such products are rarely synthesized using catalysts derived from biomass, let alone SPOPs from this source. To create the catalyst, they performed a stepwise oxidation of the liquor followed by sulfonation with ClSO3H, yielding a low-cost polymer randomly decorated with carboxylic and –SO3H groups. Although the material had a non-uniform acid site distribution, its high total acidity (∼2.8 mmol H+/g) was proved highly effective. In the proposed mechanism, this acidity efficiently activates the aldehyde substrate for nucleophilic attack by indole. The SPOP's merits were demonstrated quantitatively: it achieved excellent yields (>90%) under mild conditions, outperforming conventional acid catalysts like silica-supported sulfuric acid. Crucially, the biomass-derived SPOP catalyst could be reused for multiple cycles with minimal activity loss, highlighting its robustness and potential economic advantage. This case underscores a key strength of SPOPs: their synthesis can be tailored using sustainable, low-cost feedstocks to create competitive, reusable solid acids, offering a simpler and safer alternative to traditional benchmark systems. N-Containing bisphosphonates could be efficiently obtained using a –SO3H functionalized HCP, according to Reddy et al.182 The decorated polymer was synthesized using FC polymerization of bisphenol crosslinked with dimethoxymethane, followed by the typical post-synthetic functionalization of HCPs consisting in the sulfonation. As a result, it produced a catalyst active in the three-component reaction of 2-(tert-butyl)aniline, triethyl orthoformate, and diethyl phosphite, affording the target product under solvent-free conditions at 50 °C in only 1 h, with excellent recyclability. Authors proposed a catalytic mechanism involving the presence of surface sulfonate groups as generic proton donors over suitable substrates as main reasons for their results, in what they considered the first example of HCP catalyzing that reaction. The easy synthesis, where the –SO3H resembles present and accessible for the construction of solid acids may be the key for achieving this active material. In summary, a common mechanistic theme across these studies is the proposed role of sulfonate groups in activating reaction substrates via hydrogen bonding. However, the contribution of polymer backbone rigidity (COF vs. HCP) to stabilizing the resulting charged intermediates remains an area requiring deeper investigation, potentially through DFT studies and/or advanced spectroscopic/electrochemical techniques. | ||
| Scheme 8 Relevant heterocyclic molecules prepared by sulfonated polymers. | ||
Zolfigol and coworkers showed that the incorporation of magnetic nanoparticles in the COF facilitated catalyst recovery without compromising –SO3H site accessibility, leading to superior performance in triazoloquinazolinone synthesis. This exemplifies the design principle of adding separable functionality orthogonal to the catalytic site.183 Using only 20 mg of material, the target product was obtained with superior performance compared to benchmark catalysts for the same reaction (10 min, 80 °C, neat reaction, 93% yield). In this case, the motivation for having magnetic easy-to-separate material with acid sites becomes crucial, representing a very good example of efficient combination of the material properties. Mondal and coworkers employed an imine COF material where the sulfone was present in one of the monomers submitted to the polymerization to study the synthesis of alkylhydroxy carbamates using amines, epoxides and CO2 in just 8 h of reaction.184 This example shows a topological design and introduction of the –SO3H, which may be present in all the material pores, in contrast with the other examples discussed in this section. As a result of that intrinsic acidity, a variety of products could be obtained. In addition, the material was robust enough to present good cyclability over five consecutive reaction runs, although pore blockage was observed after the 6th cycle, that hinders the full potential of this type of topologically designed COFs.
As demonstrated, sulfonated polymers can be used to construct relevant and complex architectures, and they can be achieved with different materials, cores and functionalization methods, although topological sulfonated materials presented pore blockage problems. Nevertheless, the acidity and accessibility of active sites in all materials decorated with –SO3H groups, combined with the facile recovery, was beneficial for the reaction and allowed the outperforming of benchmark and/or homogeneous catalysts. A major future challenge lies in quantitatively correlating acid strength distribution within SPOP pores with reaction kinetics and selectivity, moving beyond yield-based comparisons. Thus, a direct relationship between the different material types will shed light on these questions too.
3.3. Biomass valorization
Sulfonated polymers are also pivotal as acid catalysts in biomass valorization. Their strong acidic character makes them particularly well-suited for reactions involved in biomass valorization, a process that adds value to plant matter and organic residues. Chemically (Fig. 7), this often entails acid-catalyzed dehydration of glycosidic polymers such as cellulose, lignin, or biosugars, leading to the formation of furfural derivatives, which serve as valuable precursors for plastics and textiles, where the specific surface acidity and heterogeneous character of SPOPs may play a crucial role. Nevertheless, other processes where biomolecules are transformed into commodities by acid catalysts are also considered under this definition. This section covers the role of SPOPs as acidic platforms for performing this important task framed in the greener effort towards finding a sustainable alternative for petrochemicals in the obtaining of highly functionalized useful chemicals.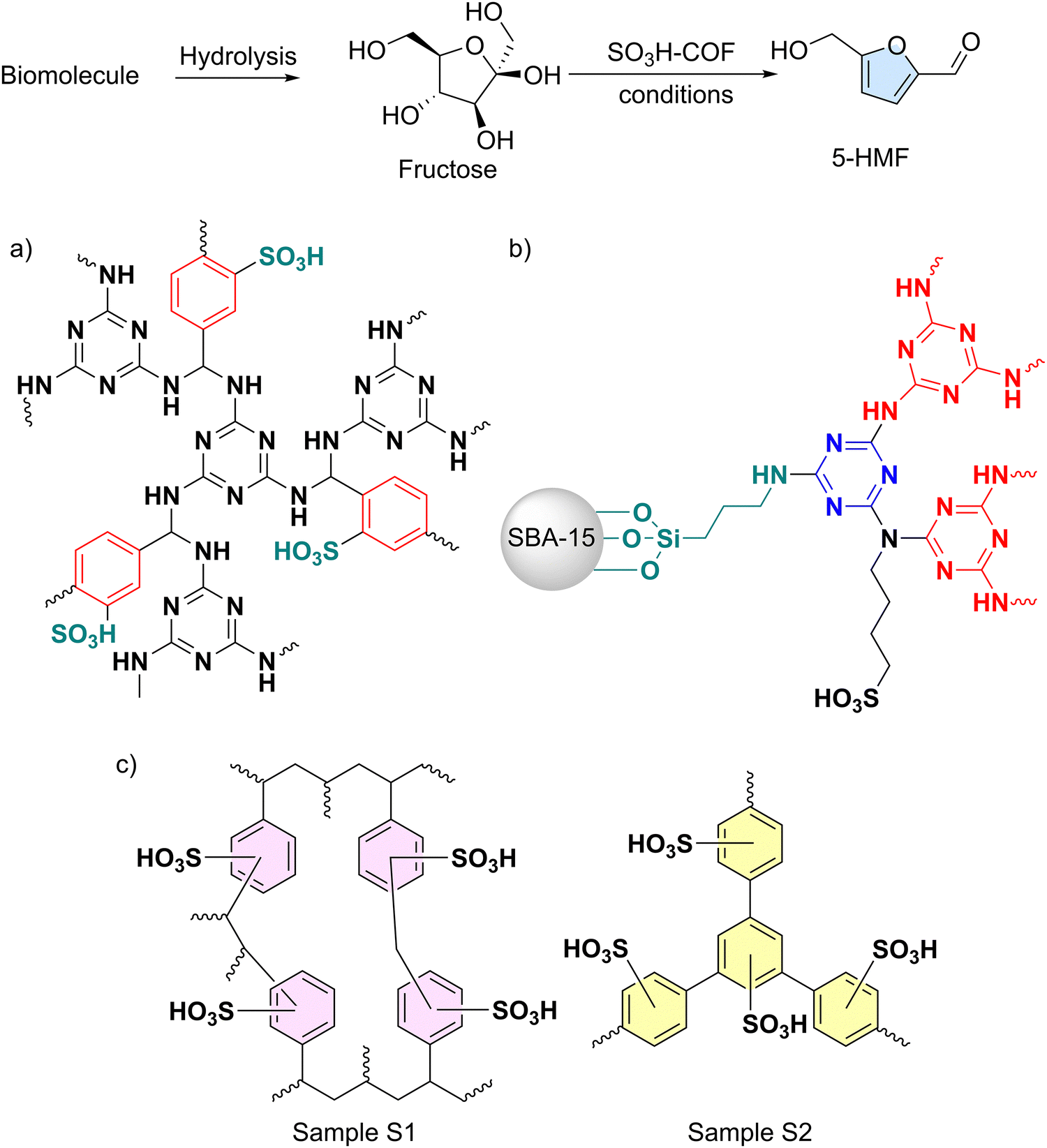 | ||
| Fig. 7 Fructose conversion to 5-HMF catalyzed by a) CTFs functionalized with the –SO3H on the aromatic core; adapted from ref. 185, b) on the linking N atom; adapted from ref. 157, c) comparison between a HCP (sample S1) and a PAF (sample S2). Adapted from ref. 186. | ||
Indeed, when sulfonated polymers are considered, 5-hydroxymethylfurfural (5-HMF) is one of the most fashionable products (Fig. 7).221 Very recently, the work by Darvishi and co-workers demonstrates that a CTF functionalized with groups in a post-synthetic reaction is able to dehydrate fructose to 5-HMF with high efficiency (Fig. 7a).185 CTF are indeed robust platforms that can host high loadings of sulfonation. Thus, they achieved 97% yield in this reaction (110 °C, 40 min, DMSO as solvent) using a CTF with 1.33 mmol of sulfonic acid per gram of material, with good recyclability due to the heterogeneous character and without leaching of active species, while the pristine material showed negligible activity. Babaei et al. achieved similar results with SBA-15 silica particles coated with a CTF layer that was finally functionalized on the amine that links the triazine rings instead of using the aromatic core (Fig. 7b).157 In this study, the production of 5-HMF was first modeled using fructose, followed by the use of larger biomolecules such as sucrose, maltose, and glucose to provide insight into potential real-world applications. It seems that the employment of the robust core of CTFs can be beneficial for this particular acid-mediated dehydration. In addition, the accessibility and richness of aromatic cores allow an easy material functionalization with sulfonic acids prompted to trigger reactivity.
Nevertheless, other materials can be employed too. In another work, Du et al. compared the catalytic activity of a HCP functionalized on the aromatic core with –SO3H (sample S1) with a PAF functionalized in identical manner (sample S2) in the production of 5-HMF from fructose (Fig. 7c).186 They observed that both materials matched the activity of PTSA in a reaction performed under microwave irradiation at 140 °C in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture dioxane:water, and the activity was superior to Amberlist-15 catalyst. They concluded that the large and accessible surface area of S1 and S2 samples (550–693 m2 g−1) was responsible for the observed phenomena. An iminic COF also showed promising activity in the synthesis of 5-HMF.120 The selected monomers contained the sulfonic moiety, and the material could obtain the target molecule in 1 h in DMSO with good conversion and selectivity. In addition, the acidic and reactive character of this sulfonated COF allowed authors to reach diformylfurane in moderate performance. Ye and coworkers studied the dehydration of fructose using a functionalized HCP in different solvents.187 They obtained the best conversion of fructose and selectivity towards 5-HMF using DMSO (100% and 85%, respectively) among DMF, THF or water, but it deactivated quickly. Thus, they switched to a mixture of dioxane-water, and their study pointed out that a small amount of water in the mixture (2.5% v/v) could depress the oligomerization of 5-HMF and enhance the stability of the material. Common trends are therefore observed, indicating that a range of SPOP architectures can achieve high activity (e.g., yields up to 97% under optimal conditions) in 5-HMF synthesis, often matching or surpassing homogeneous acids (e.g., PTSA) and benchmark heterogeneous systems like Amberlyst-15.157,185,186 However, the specific advantages of each material class remain obscured by a lack of direct, standardized comparison. For instance, while high surface area is often cited, the comparative studies by Du et al. do not isolate this effect from other variables like acid strength or hydrophobicity.186 Consequently, selecting the most suitable SPOP for a given application remains challenging. Future work must prioritize systematic, side-by-side evaluations under identical conditions and employ advanced techniques (e.g., DFT analysis, in situ spectroscopy) to establish clear structure–activity relationships that link specific material properties such as pore hierarchy, acid site density and accessibility, and framework hydrophobicity to catalytic performance and stability.187
:1 mixture dioxane:water, and the activity was superior to Amberlist-15 catalyst. They concluded that the large and accessible surface area of S1 and S2 samples (550–693 m2 g−1) was responsible for the observed phenomena. An iminic COF also showed promising activity in the synthesis of 5-HMF.120 The selected monomers contained the sulfonic moiety, and the material could obtain the target molecule in 1 h in DMSO with good conversion and selectivity. In addition, the acidic and reactive character of this sulfonated COF allowed authors to reach diformylfurane in moderate performance. Ye and coworkers studied the dehydration of fructose using a functionalized HCP in different solvents.187 They obtained the best conversion of fructose and selectivity towards 5-HMF using DMSO (100% and 85%, respectively) among DMF, THF or water, but it deactivated quickly. Thus, they switched to a mixture of dioxane-water, and their study pointed out that a small amount of water in the mixture (2.5% v/v) could depress the oligomerization of 5-HMF and enhance the stability of the material. Common trends are therefore observed, indicating that a range of SPOP architectures can achieve high activity (e.g., yields up to 97% under optimal conditions) in 5-HMF synthesis, often matching or surpassing homogeneous acids (e.g., PTSA) and benchmark heterogeneous systems like Amberlyst-15.157,185,186 However, the specific advantages of each material class remain obscured by a lack of direct, standardized comparison. For instance, while high surface area is often cited, the comparative studies by Du et al. do not isolate this effect from other variables like acid strength or hydrophobicity.186 Consequently, selecting the most suitable SPOP for a given application remains challenging. Future work must prioritize systematic, side-by-side evaluations under identical conditions and employ advanced techniques (e.g., DFT analysis, in situ spectroscopy) to establish clear structure–activity relationships that link specific material properties such as pore hierarchy, acid site density and accessibility, and framework hydrophobicity to catalytic performance and stability.187
Conversely, other biomolecules may be valorized too using SPOPs. For instance, Artz et al. employed sulfonated CTFs as catalysts in the hydrolysis of cellobiose to glucose.188 The versatility and monomer engineering for CTF synthesis was exploited in this example. Indeed, they screened different materials using monomers of varied complexity, i.e., 1,3-dicyanobenzene, 1,4-diazobenzene, 2,6-dicyanopyridine and 4,4′-dicyanobiphenyl. The catalysts could achieve the selective synthesis of glucose in just 4 h of reaction, yields up to 86% at a very good substrate-to-catalyst ratio of 10. Their most active material used 1,3-dicyanobenzene, even though some sulfur leaching was observed during operation due to the catalytic hydrothermal conditions. On the other hand, confined Brønsted acidic ILs based on 1-methyl-3-(3-sulfopropyl)-1H-imidazol-3-ium hydrosulfate in the cavities of an iminic COF presented very good activity in the dehydrative formation of isosorbide from sorbitol.189 Even though no covalent bonding between polymer and active site was created, the material resulted in the best catalyst under study, outperforming benchmark and homogeneous related catalyst, with a yield of isosorbide as high as 97% and good recyclability. H-bonding between the salt and the polymer, which possess the adequate scaffold to launch that interactions and the suitable morphology upon design of that COF were concluded to be the mechanistically consequence of the robustness observed together with the contribution to the stabilization of the active site. In contrast, Yuan and coworkers synthesized a series of polydivinylbenzene-based solid acid catalysts (PDSF-x) with tunable acid strength by co-grafting –SO3H and strongly electron-withdrawing trifluoromethanesulfonic acid (–SO2CF3) groups onto a PDVB framework.190 The variable “x” in PDSF-x denotes the amount of CF3SO3H used in the functionalization. They investigated the effect of increasing acid strength on the solvent-free dehydration of sorbitol to isosorbide. Interestingly, while the total sulfur content (and thus acid site loading) increased from 1.75 to 3.73 mmol g−1, the catalytic activity decreased, with initial turnover frequencies (TOFs) dropping from 231 h−1 to around 200 h−1. The authors attributed this decline to pore blockage from excessive grafting, which reduced the specific surface area and made active sites less accessible. Consequently, the catalyst with moderate functionalization, PDSF-0.05, delivered the optimal performance, complete sorbitol conversion, an 80% yield of isosorbide at 140 °C, and excellent recyclability.
In summary, the performance of these catalysts is intrinsically governed by their material properties. A critical insight from comparing these works is that for SPOPs, the highest degree of functionalization (e.g., sulfonation) does not automatically guarantee the highest catalytic activity or operational stability. Optimal performance arises from a balanced interplay of acidity, porosity, and structural robustness. To advance this field, systematic studies are needed to decouple these factors, correlating specific linker functionalities, pore architectures, and acid site densities with activity and durability. Furthermore, expanding the valorization of diverse organic residues into SPOPs represents a promising pathway for synthesizing a broader range of high-value green chemicals.
3.4. Biofuel synthesis: esterification and transesterification
Biofuel is a fuel produced from a short span of time from biomass.222 The definition brings together different categories of chemicals, ranging from biologically produced methane and short-chain alcohols such as methanol, ethanol, propanol, and butanol (in their various isomers) to long-chain compounds commonly referred to as biodiesel. Indeed, biodiesel is commonly produced from oils and fats by transesterification reactions to convert into fatty acid methyl esters (FAMEs).223 Since the formation of esters, including FAMEs, is a carbonyl reaction typically catalyzed by acids, SPOPs are excellent candidates to carry out this reaction thanks to their intrinsic acidity. In addition, the advantages of specific and precisely located –SO3H groups within the polymer network, tunable design and heterogenous characteristics towards separation after reaction make them appealing candidates for an important challenge as the production of fuels avoiding petroleum practices.In this regard, SHCPs have been deeply studied, maybe due to their easy synthesis and facile functionalization. For instance, hollow nanosphere polymer particles were used by Song et al. to catalyze biodiesel formation. They synthesized spheric HCP particles ranging 500 nm in diameter that were sulfonated after synthesis with ClSO3H, and they tested the methanolysis (esterification with MeOH) of both lauric acid and coconut oil.191 Good results were achieved (yields higher than 85%) with all samples under study compared to the non-sulfonated material due to the acidity provided by the sulfonic groups as mechanistic reason. Guo and Liang prepared a hyper crosslinked xylene-based polymer doped with naphthalene to, according to authors design, provide more sulfonation sites.192 They achieved packed spheres configuration that were active in the tetrahydropyranylation of alcohols and in the transesterification of waste oil with methanol, achieving over 90% yield of free fatty acids in their reaction mixture after 4 h. The doped agent resulted beneficial for the functionalization-catalytic activity purposes. The materials presented excellent recyclability too thanks to the inherent robustness of the HCP structure. The tunability that presents HCPs was exploited by Schweng and coworkers (Fig. 8a).193 Indeed, they synthesized four different materials varying the density of –SO3H groups and tested them in the model esterification of hexanoic acid with benzyl alcohol. They discovered that the higher the acidity the higher the catalytic activity, in contrast with the 5-HMF production discussed above. Indeed, a maximum was reached of 95 ± 4% after 22 h for the most acidic material. However, this high level of acidity led to uncontrolled selectivity compared to the other less acidic materials, where the target product was obtained, although with less yield, in high selectivity. Therefore, in stark concordance with the previous data, a balance must be met to develop efficient catalysts. In a similar approach, Bhunia et al. presented a supermicroporous HCP thanks to monomer design (FC polymerization of carbazole with α,α′-dibromoxylene) that was post-synthetically sulfonated.119 The material efficiently catalyzed the esterification with methanol of different fatty acids, such as oleic, palmitic and stearic among others, with enhanced performance compared to the non-sulfonated polymer. Biodiesel was also produced by a SHCP based on polyphenols (Fig. 8b).163 The samples produced were tested in the esterification and transesterification of various long-chain fatty acids and fatty oils as soy or coconut oil. Authors discovered that catalytic activity boosted up with increasing acid functionalization, and a significant degree of sulfonation was required, being the most acidic the most active material, although selectivity and the effect of phenolic groups were not addressed. Furthermore, the materials presented good recyclability without leaching of acid sites. Summing up, SHCPs are outstanding candidates for catalyzing esterification reactions. Their suitability stems from straightforward synthesis, ease of functionalization via sulfonation, and the ability to precisely control the acid site density. They account accessible sulfonic groups that serve as heterogeneous acids prompted to catalyze the esterification mechanism, and the hydrophobicity of the material benefits substrate diffusion compared to homogeneous systems. Benchmarking against state-of-the-art catalysts confirms that SPOPs often deliver superior activity under significantly milder conditions, underscoring their transformative potential when judiciously designed. However, more insights into the actual mechanisms, including theory and experiment, are needed as in the precedent examples.
 | ||
| Fig. 8 Different esterification or transesterification reactions catalyzed by HCPs. a) HCP for the esterification of heptanoic acid with benzyl alcohol; b) polyphenol-based HCP for the synthesis of biodiesel; c) phosphate-linking polymer for the esterification of levulinic acid. Adapted from ref. 163, 193 and 194. | ||
Other acid groups could also be employed to complement the sulfonic group acidity, such as the sulfonated porous polymer presented by Kim et al. that also contained a linking phosphate (Fig. 8c).194 This material catalyzed the esterification of levulinic acid with aliphatic alcohols (from methanol to pentanol) with good recyclability and enhanced catalytic character compared to the non-sulfonated material, thus indicating the low acidity of the phosphate and the need of stronger acid sites. However, the role of the phosphate in the activity might require further research by advanced techniques to assess the viability of this strategy towards real applications. Finally, esterification reactions including acetic acid with cyclohexanol or fatty acids (hexanoic, lauric) with ethanol were efficiently catalyzed by a sulfonated mesoporous polydivinylbenzene, prepared via PSM using ClSO3H.118 Therefore, other structures rather than HCP are compatible with the biofuel pool. Indeed, sulfonated COFs were also employed as catalysts in biodiesel formation. Jia et al. presented an iminic COF whose monomers contained the sulfonic group.195 The acidity of the material was increased introducing in the pores the IL 1-methyl-3-(3-sulfopropyl)-1H-imidazol-3-ium hydrosulfate. This material presented a 91% conversion of the model soybean oil into FAMEs, which is far better than the 5% conversion achieved by the pristine material, and it also worked on real systems with good reusability. Surprisingly, the iminic COF resisted the acid mediated reaction, in a sign of inherent robustness that validates this kind of material for acid-mediated transformations. Many acid-catalyzed reactions involve hydrophobic organic reactants and suffer in aqueous or polar environments due to catalyst deactivation or poor substrate affinity. SPOPs offer a unique solution through inherent or engineered hydrophobicity of their organic backbones, which creates a favorable microenvironment for organic transformations. Munyentwali et al. highlighted this in their study of a rigid, hydrophobic POP for oleic acid esterification.116 They concluded that strong surface hydrophobicity was key to outperforming hydrophilic benchmarks like Nafion® NR50 and Amberlyst-15. The hydrophobic pores favored the partitioning of fatty acid reactants, leading to higher local concentrations at the –SO3H sites. This intrinsic property of many POPs provides a built-in advantage over hydrophilic solid acids in non-polar reaction media. On their work, Goesten et al. showed that a sulfonated PAF maintained comparable or superior initial activity to Amberlyst-15 in acetic acid esterification with n-butanol, but with minimal deactivation over multiple cycles.122 This contrasts with Amberlyst-15, which suffers from swelling and acid site leaching in polar media, highlighting the stability advantage of rigid SPOP frameworks.
In summary, the acid character of the sulfonated polymers allows the efficient esterification reaction to be performed to obtain biodiesel and related compound. However, characterization of the active sites in operando rather than invoking the acidic character as general mechanism may be key for definitively assessing the true nature of the active sites, synergies, structure–properties relationships and the actual benefits for employing SPOPs in this type of chemistry. The activity values are clear, but more is needed to unseat classic systems.
3.5. Polymerization
Innovative and more sustainable polymerization reactions or processes, that demonstrate enhanced activity, efficiency, control and selectivity continues to be key for the synthesis of important added value materials with specific chemistry and application.224,225 In this way, several synthetic processes in polymer technology are still catalyzed or mediated by PTSA.226 This strong acid activated the monomer (or the initiator) generating the active species that sustains polymer growth. Thus, sulfone-decorated polymers might have room for development in the search for more efficient technologies, where the heterogeneous character might have advantages such as more efficient reagent diffusion during polymerization to avoid reactor fouling and easy separation after reaction, combined with localized solid acid nature.In this regard, Park et al. used a sulfonated microporous polymer as a catalyst for polyketone synthesis from CO and ethylene (Fig. 9a).121 The porous catalyst produced ∼5 g of polymer per 1.5 mg of catalyst without fouling the reactor, a stark contrast to PTSA homogeneous system. The confined pores likely control monomer access and polymer chain growth, demonstrating how SPOP architecture can solve practical engineering challenges in polymer production. Similar catalytic system was presented by Kang et al.196 They coated another microporous organic polymer in silica particles and then, samples were sulfonated. The resulting material was employed as seed in the catalytic terpolymerization of CO, ethylene, and propylene. Under optimal conditions, they obtained 17.2 kg of polymer per gram without fouling, and the resulting polymers exhibited a narrower molecular weight distribution than those produced by homogeneous systems. In both cases, the SPOP synthetic method resulted in materials with the adequate porosity to yield the target polymers without pore blockage. Thus, templated sulfonated microporous organic polymers are excellent hybrid catalysts where monomers can easily find the active and diffuse after reaction because the sulfonation seemed to both host the metal complex and enhance the dispersion in the reaction medium, facilitating the polymerization. In a different approach without the hard template method, the stepwise protocol based on synthesis and sulfonation was also applied to yield a material by FC polymerization.147 Therefore, self-synthesized SPOPs are also valid candidates to catalyze polymerizations too. This POP was tested in the dimerization of isopentane. Indeed, the alkylation and/or dimerization of alkanes is an important reaction since it provides products that can be directly introduced in the gasoline pool. This reaction is typically carried out both academic and industrially under acidic conditions, and that SPOP carried it out with good results: 24 h of reaction at 140 °C afforded the best results in terms of conversion, yield and selectivity (80%, 75% and 95%, respectively), with better activity than acidic zeolites compared in the study. The localized acidity in the SPOP may be responsible for the observed activity. Finally, the group of Wang presented two works dealing with the Ziegler–Natta process227 using CH3MgCl and TiCl4 as precursors to be added over the sulfonated polymer.197,198 The reaction was classically performed over alumina substrates, but the SPOP may present some advantages. With this material, authors achieved the catalytic synthesis of ultrahigh molecular weight polyethylene, as well as polypropylene (Fig. 9b),197 which exhibited both high stereocontrol and a broad polydispersity. Both works highlight the chemical environment of Ti and Mg sites due to the –SO3H groups present in the material (even coordinating the Ziegler–Natta centers), which resulted beneficial for the catalytic purposes. In contrast, conventional systems did not present this chemical ambient, thus highlighting the excellent performance of that SPOPs in industrially important reactions as Ziegler–Natta polymerizations.
 | ||
| Fig. 9 Sulfonated porous polymers as platform to sustain the polymerization of a) polyketones. Reproduced with permission from ref. 121 Copyright 2016 American Chemical Society and b) polypropylene with the Ziegler–Natta catalyst. Reproduced with permission from ref. 197 Copyright 2022 Elsevier. | ||
However, few studies on SPOPs to sustain polymerizations have been carried out, in particular lacking more types of POPs. More investigations, both at synthesis and mechanism levels and experimental and theoretical approaches are required to introduce these soft materials into a realm dominated by doped inorganic systems, where milder synthesis and the intrinsic tunability of the SPOPs can compete for a more efficient polymer synthesis.
3.6. Cascade reactions
The acidic nature of porous frameworks can be employed to trigger cascade or tandem processes.228,229 Indeed, this section devoted to catalysis with sulfonated polymers has discussed how the solid acid character can catalyze different reactions, such as condensation, dehydrations, esterifications and polymerizations among others. Thus, a proper reaction design can exploit this character and include an acid-mediated step in a more complex overall reaction. On the other hand, the acid nature can be coupled with other active sites to trigger complex catalytic processes. Merging all the required parameters into a solid polymer, versatile enough to allow the synthesis upon design, and combined with the inherent robustness and easy separation after reaction would be quite beneficial for the development of advanced heterogeneous catalysts. For cascade reactions, the confined environment of SPOPs pores can be essential for facilitating intermediate transfer. In their study, Gong and colleagues reported presented a sulfonated COF able to catalyze different reactions.199 They obtained their catalyst by acid-mediated trimerization from a diketone containing a –OH group, and then the decoration was carried out by alkylation with 1,3-propanesultone (Fig. 10a). COFs demonstrate outstanding flexibility to allow this synthetic design and polymerization conditions. This material was able to catalyze the synthesis of benzimidazoles, benzodiazepines and one-pot reductive aminations, reactions that require an acid activation that was efficiently performed by the chosen SPOP. For the first process, the sulfonic material catalyzed the imine formation, which let the substrate ready for an intramolecular cyclization; thus, a broad scope of benzimidazoles with good activity (yields above 90% in 1 h reaction) was presented. The authors proposed that the periodic porous architecture of the COF does more than simply immobilize –SO3H groups; it enforces proximity between reactants and catalytic sites, creating a local environment where reactive intermediates are stabilized and their intermolecular diffusion is minimized. This overcomes the fundamental kinetic challenges of diffusion-controlled reactions and allows for the efficient, sequential execution of multiple catalytic steps, a cooperative synergy difficult to replicate with a physical mixture of homogeneous catalysts, which often suffer from incompatibility and low local concentrations of intermediates. A similar strategy was designed for the benzodiazepine synthesis, and another broad scope was studied with very good catalytic activity. Finally, in the presence of reductant that did not affect the structure of the catalyst thanks to the intrinsic COF robustness, an imine and an aldehyde were condensed and reduced in a one-pot process. However, the proposed mechanism just stays in a simple acid activation of the substrates. In a different work, Kalita et al. synthesized a –SO3H functionalized tetraphenylethylene-based HCP featuring a well-developed porous network and accessible –SO3H sites.200 The yielded SHCP was tested in the ambitious synthesis of symmetrical triarylmethanes via a dual C–C bond cleavage. Good activity in terms of high yields (33–94%) was reported. This reaction is typically performed by strong and aggressive Lewis acids as FeCl3, but authors could outperform this system with their polymer. The scope of method was also explored with different substrates varying their characteristics. In addition, recovery of the catalyst by filtration allowed five consecutive reuse cycles without observing decrement in the activity, overcoming industrial limitations in terms of separation and recovery that the homogeneous catalyst cannot present. The material prepared by Sun and colleagues enabled two successive acid–base mediated reactions.162 Indeed, they performed the polymerization of DVB in the presence of a monotopic sulfonate, which was further modified with an amine and acidified to provide a bifunctional catalyst. Thus, the resulting POP was acidic enough to break an acetal and basic enough to condensate the resulting aldehyde with a malonate derivative (Fig. 10b). Only this bifunctional material accomplished the cascade process, in contrast to pristine, sulfonated polymer or homogeneous catalysts that lacked the required active sites. Therefore, the aromatic core of the selected SPOP was reactive enough to support the intended functionalizations that led with the observed catalytic activity. Corma et al. demonstrated that the bifunctional PAF catalyst PPAF–SO3H–NH2 achieved 100% yield in a one-pot cascade reaction involving acetal hydrolysis followed by Knoevenagel condensation, far surpassing mixtures of its monofunctional counterparts or commercial resins. This result confirms that the proximity and cooperative action of acid and base sites within a single, mesoporous framework are essential for high-efficiency cascade catalysis.229 A transformative frontier for SPOPs is the integration of –SO3H sites with other functional groups (e.g., metals, basic sites) within a single framework to enable cooperative catalysis. This creates bifunctional or multifunctional catalysts capable of driving sophisticated tandem processes. A prime example is the work by Raza et al., who fabricated a Pd nanoparticle-loaded sulfonated CTF.201 This material catalyzed a one-pot reduction-esterification cascade, converting 4-nitrobenzoic acid to benzocaine (Fig. 10c). Control experiments proved both functionalities were essential: Pd nanoparticles reduced the nitro group, while the proximal –SO3H sites esterified the resulting acid. The capacity of SPOP frameworks to spatially organize disparate active sites such as proximal –SO3H groups and metal nanoparticles, enables efficient telescoped syntheses in a one-pot operation, circumventing the need for intermediate separation and re-catalysis. Current literature predominantly shows two-step cascades, where the designed acidity and hydrolytic stability of the polymer are critical. While this demonstrates the successful integration of acid activation with other functions (e.g., redox metals, basic sites), the field must now advance toward longer, more complex catalytic sequences. Crucially, a deeper mechanistic understanding, elucidating how substrates interact with the confined pore environment and co-localized sites, is essential to rationally design SPOPs that fully unleash their potential for precise multi-step catalysis. | ||
| Fig. 10 Cascade processes catalyzed by –SO3H decorated POPs. a) Synthesis of benzimidazoles, benzodiazepines and reductive amination by a sulfonated COF. Reproduced with permission from ref. 160 Copyright 2022 Elsevier; b) acetalization and Knoevenagel condensation using modified HCP. Reproduced with permission from ref. 197 Copyright 2019 Royal Society of Chemistry; c) reduction and esterification with a Pd-sulfonated CTF. Reproduced with permission from ref. 162, 199 and 201 Copyright 2021 Elsevier. | ||
Asymmetric catalysis is an emerging field in the realm of organic materials,230–232 and it was also attempted using –SO3H functionalized organic polymers. Indeed, Pang et al. constructed a β-ketoenamine COF with –SO3H groups that were further functionalized with a chiral diamine to create a chiral sulfonamide catalyst.202 This material catalyzed an asymmetric Michael addition of acetone to β-nitrostyrene with high efficiency of ∼80% and enantiomeric excess (ee) of 90%. For a series of β-nitroolefins, enantioselectivities ranged from 84% to 96%. The proposed mechanism involves a synergy where the chiral center guides enantioselection while the free –SO3H group activates the substrate via acid–base interaction (Fig. 11). Although current systems may face stability limitations, this work pioneers the use of SPOPs as modular platforms for heterogeneous enantioselective acid catalysis.
 | ||
| Fig. 11 Enantioselective Michael addition with a proposed mechanism using sulfonated-functionalized β-ketoenamine porous polymer (R = H or organocatalyst). Adapted from ref. 202. | ||
4. Environmental remediation
The incorporation of –SO3H groups into POPs represents a powerful strategy to engineer high-performance adsorbents for environmental remediation. This approach leverages a synergistic interplay between the intrinsic properties of the porous scaffold and the introduced functional motifs. The inherent high surface area and permanent porosity of POPs facilitate mass transfer and physical entrapment of pollutants via pore-filling. Concurrently, sulfonation serves a dual purpose: (i) it drastically improves the hydrophilicity and aqueous dispersibility of otherwise often hydrophobic frameworks, ensuring optimal contact with waterborne contaminants, and (ii) it decorates the pore walls with a high density of anionic sulfonate (–SO3−) groups, enabling strong, specific electrostatic interactions with cationic organic pollutants (e.g., dyes, antibiotics) and facilitates ion–exchange processes, wherein protons from the –SO3H groups are exchanged for cationic metal ions. Furthermore, the aromatic backbones prevalent in many POPs contribute through π–π stacking interactions with pollutant molecules, while hydrogen bonding can also be a contributing factor. The culmination of these complementary mechanisms including electrostatic attraction, ion exchange, π–π interactions, hydrogen bonding, and pore filling often results in exceptional adsorption performances, characterized by remarkably fast kinetics and capacities that can exceed 1000 mg g−1. However, a critical challenge in designing such materials lies in balancing the degree of sulfonation with the preservation of porosity, as the grafting of bulky –SO3H groups can lead to pore blocking and a significant reduction in surface area, a common trade-off that impacts overall adsorption performance. Table 4 summarizes the use of SPOPs for environmental remediation including dye adsorption, antibiotic/drugs removal, metal ion adsorption, and gas separation.| POP | BBs/monomers | Functionalization | BET surface area (m2 g−1)/Pore size (nm) | Application | Adsorption capacity and performance | Acid density (mmol g−1) | Stability assessment | Scale up (yes/no) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| SHCTPE | HCTPE | FC polymerization | 860/1.734 | Adsorption of organic dyes in aqueous solution | 99% removal after 5 s. 1410 mg g−1 for MB and 1096 mg g−1 for RhB | NR | Five cycles | No | 233 |
| TC-COP-SO3H | Triphenylmethane and cyanuric chloride | FC reaction | Langmuir surface area of 50.15 | Adsorption of cationic dyes | 897.5 mg g−1 for basic blue 41 and 862.5 mg g−1 for basic red 46 | NR | Three cycles and TGA | No | 234 |
| PAF-215 | DVB | Conventional PSM | 580/1.19 | Adsorption of cationic dyes and antibiotics | 1075 mg g−1 for RhB, 1020 mg g−1 for MB, 826 mg g−1 for TC and 1134 mg g−1 for CIP | 13.2 wt% | Five cycles | No | 235 |
| Sulfone-grafted CMPs (SPTP and CPTP) | 1,3,6,8-Tetrabromopyrene | Sulfone grafted PSM | SPTP - 290.3/- | Adsorption of cationic dyes | 1650 mg g−1 and 97% removal efficiency of MB (100 mg L−1) in 25 min | NR | N2 adsorption and desorption studies, TGA | No | 130 |
| CPTP - 391.3/- | |||||||||
| TpStb-SO3Na | Tp and Stb-SO3H | Condensation reaction | 15.4/2.65 | Adsorption of organic pollutants from wastewater | For CV - 1861 mg g−1, MB - 1078 mg g−1, MG - 5857 mg g−1, and JGB - 1339 mg g−1 | NR | PXRD, FT-IR, N2 adsorption and desorption studies, TGA | No | 156 |
| HCP | Benzene and FDA | FC reaction | 855/0.51 | Dye separation | m-HCP-5 - CR from aqueous media 465 mg g−1 | NR | N2 Sorption isotherm | No | 236 |
| H−S−CMPs | 1,3,5-Triethynylbenzene and 2,7-dibromocarbazole | Songashira-Hagihara cross-coupling | 105.79/- | Adsorption of antibiotics and cationic dyes from wastewater | RhB - 206.2 mg g−1, MB - 324.7 mg g−1, CIP - 222.2 mg g−1, norfloxacin (NOR)- 216.9 mg g−1 | — | Five cycles, | No | 237 |
| BaPy–SO3H | Resorcinarene-based BaPy | Suzuki–Miyaura coupling reaction | 3.07/0.10 | Adsorption of organic pollutants | 1850 mg g−1 for RhB, 2295 mg g−1 for MB, 1503 mg g−1 for CV, and 1304 m g−1 | — | Five cycles, FT-IR, XPS, SEM | — | 238 |
| TH-SMONs | Tetra(4-ethynylphenyl)methane with 1,4-diiodobenzene | Sonagashira coupling | 43/0.26 | Drug delivery | — | 6.14 wt% | SEM, Elemental mapping, N2 Isotherm curves, PXRD, IR | — | 56 |
| SHCPs | Triphenylmethane | FC reaction and PSM | 939/3.7–3.9 | Adsorption of antibiotic pollutants from water | 427.5 mg g−1 of CIP | 3.82 wt% | Ten cycles, TGA, FT-IR | — | 239 |
| TP-PPOPs-SO3H | TPP & triptycene | Conventional method | 368.44/1.48 | Adsorption of MB and CIP | MB - 1283.33 mg g−1 and CIP - 485.39 mg g−1 | — | Eight cycles, SEM, FT-IR | — | 240 |
| Am-COF-SO3H | Im-COF-SO3H | 1-ethyl-3-methylimidazolium tetrafluoroborate (Emim BF4) mediated oxidation | 8.79/1.48 | Adsorption of fluoroquinoline antibiotics in water | Enrofloxacin (ENR) - 614 mg g−1, enoxacin (ENO) - 615 mg g−1, NOR - 579 mg g−1 | — | Six cylces, FT-IR, XPS, PXRD | — | 241 |
| SHCP | 4,4′-bis(chloromethyl)-1,1′-biphenyl (BCMB) | One pot chemical synthesis | 704/- | Removal of antibiotic pollutants | CIP - 757.7 mg g−1 | 12.4 wt% | Seven cycles, ATR-IR | — | 166 |
| TFPB-DABDA iCOFs | 1,3,5-tris(p-formylphenyl)benzene (TFPB) and 2,5-diaminobenzene-1,4-disulfonic acid (DABDA) | Schiff base reaction | 39.80/3.769 | Adsorption and separation of lanthanide compounds | The maximum adsorption capacity of early lanthanides can reach up to 0.1554 mmol g−1 | — | — | — | 242 |
| SHCP-P-X X = 0.5, 1.5, 3.0, 4.0 | 4,4′-Bis(chloromethyl)biphenyl (BCMBP) | Chemical synthesis | 1044.6/– | Recover rare earth elements from tailing wastewater | 106.78 mg g−1 (La), 111.99 mg g−1 (Eu) and 126.27 mg g−1 (Lu) | 3.27 mmol g−1 | Ten cycles, IR, BET | Yes | 243 |
| TAS-COF | 2,4,6-triformylphloroglucinol (Tp) and 3,7-diaminodibenzo[b,d]thiophene (DAS) | Schiff base condensation | — | Colorimetric detection of UO22+ | LOD = 0.07 µmol L−1 | — | SEM, EDS mapping, FT-IR, PXRD | — | 244 |
| TFPOTDB-SO3H | DABSA and 2,4,6-tris-(4-formylphenoxy)-1,3,5-triazine | Solvothermal reaction | 190.73/1.55 | Adsorption of As(III) from water | 344.8 mg g−1 | — | Four cycles, SEM, TEM, PXRD, N2 Sorption isotherm | — | 245 |
| SHCP | Fluorene-9-bisphenol (BHPF) | FC reaction | 427.6/0.235 | Adsorption of rubidium and cesium | For Ru+ - 121.3 and Cs+ - 133.9 mg g−1 | — | Six cycles, SEM, FT-IR, PXRD, N2 Sorption isotherm, TGA | — | 246 |
| Composite aerogel | Sulfonated CMP (sCMP) and carboxymethyl cellulose (CMC) | physical doping and freeze-drying | 16.737/0.028425 | Adsorption of Pb2+ and MB. | Pb2+ - 94.93 and MB - 294.84 mg g−1 | — | FT-IR, XPS, TGA, PXRD, SEM, EDS mapping | — | 247 |
| PIMs | Hexaphenylbenzene (HPB), triphenylbenzene (TPB), spirobisfluorene (SBF), triptycene (TRIP) | FC Polymerization | 1585/- | CO2 adsorption and gas separation of N2 and CH4 from CO2 | 298 mg g−1 | — | FT-IR, SEM, N2 adsorption isotherms | — | 136 |
| TpPa-SO3Cu0.5 | Triformylphloroglucinol (Tp), DABSA (Pa-SO3H) | Mechanical grinding synthetic method | 85.98/1.3 | Adsorption of NH3 nitrogen from water | 30.45 mg N g−1 | — | Five cycles, PXRD, XPS, FT-IR, TGA | — | 248 |
| 1TCS@PDMS10 | Sulphonated TCS | Post-oxidation and post-sulfonation | 72.5/– | NH3 capture | Up to 1.41 mmol g−1 | — | ATR-IR, PXRD, SEM, Elemental analysis | — | 249 |
| Sulfonated polyimide (SPI) | ODA, BTDA | Polymerization | — | Methane storage as methane hydrate | Storage capacity 119 V/V, | — | Six cycles, FT-IR, SEM | — | 250 |
| PPN-6-SO3Li | PPN | Sulfonation and lithiation | 1186/– | CO2 adsorption | adsorption selectivity for CO2 over N2 (155 and 414 for PPN-6-SO3H and PPN-6-SO3Li, respectively) | — | N2 Sorption isotherm, SEM, Elemental analysis | — | 53 |
| SHCP-1 | DCX/BDM/BCMBP | FC alkylation and PSM | 940.33/3.96 | Capture of NH3 from exhaust gases | 11.54 mmol g−1 | 3.38 wt% | Ten cycles, N2 Sorption isotherm, SEM, Elemental analysis, IR, XPS | Yes | 251 |
| SPIM-1 | 5,5′,6,6′-Tetrahydroxy-3,3,3′,3′-tetramethyl-1,1′-spirobisindane (TTSBI) and 1,4-dicyanotetrafluorobenzene (DCTB) | Sulfonation using SO3 solution | 642/– | Gas separation | Selectivity of H2/N2 and O2/N2 were 125 and 8.43 coupled with H2 and O2 permeability of 1077 and 73.4 Barrer resp | 4.77 wt% | FT-IR, XPS, SEM/EDS mapping, | — | 135 |
4.1. Dye adsorption
Incorporating –SO3H groups into POPs is a highly effective method for enhancing the adsorption of dyes (Scheme 9), with a pronounced effect on cationic dyes. | ||
| Scheme 9 Representative synthetic dye molecules to assess the adsorption capacities of SPOPs, including methyl orange (MO), methylene blue (MB), basic blue 41 (BB41), rhodamine B (RhB), malachite green (MG), crystal violet (CV), paraquat, diquat, basic red 46 (BR46), congo red (CR), and janus green B (JGB). | ||
This approach activates multiple complementary mechanisms. The innate high surface area and porous structure of POPs provide a foundation for physical entrapment through pore filling. Sulfonation, on the other hand, introduces a strong electrostatic force that captures cationic dyes. Additionally, the resulting anionic frameworks demonstrate increased hydrophilicity, which improves their dispersion in water and allows better access for dyes to the internal pore surfaces. The combination of this electrostatic attraction, the inherent π–π stacking ability of the aromatic polymer backbone, and the high porosity creates a synergy that leads to remarkably fast adsorption kinetics and capacities. In many cases, these performance metrics surpass those of unfunctionalized POPs and numerous benchmark adsorbents. A notable example is the synthesis of a sulfonate-grafted tetraphenylethylene-based HCP (SHCTPE) via molecular knitting and post-sulfonation (Fig. 12).233 While the sulfonation step reduced the BET surface area from 1561 m2 g−1 (precursor HCTPE) to 860 m2 g−1, it conferred superhydrophilicity and a negatively charged skeleton, leading to exceptional, ultrafast adsorption kinetics for MB and RhB with >99% removal in just 5 seconds and pseudo-second-order rate constants of 58.7 and 83.4 g mg−1 min−1 for MB and RhB, respectively. Furthermore, the adsorption capacities achieved for MB and RhB were 1410 mg g−1 and 1096 mg g−1, respectively. SHCTPE also retained its performance over five cycles, demonstrating excellent stability. This serves as a promising example of a superhydrophilic HCP for the efficient adsorption of organic dyes in aqueous solutions. This underscores that for rapid aqueous-phase adsorption, enhanced hydrophilicity and electrostatic driving force can outweigh the importance of ultra-high surface area.
 | ||
| Fig. 12 Synthesis of HCTPE and SHCTPE solids. Adapted from ref. 233. | ||
In a subsequent study, a sulfonated COP (TC-COP-SO3H) (Fig. 13) derived from triphenylmethane and cyanuric chloride experienced a more drastic loss in porosity (Langmuir surface area dropped from 137 to 50 m2 g−1 upon sulfonation), yet still achieved enhanced capacities for BB-41 and −46 compared to its non-sulfonated counterpart.234 This improved performance of TC-COP-SO3H is due to the high density of adsorptive sites, which facilitate favorable electrostatic interactions with cationic dye molecules, compensating for its significantly reduced porosity.
 | ||
| Fig. 13 Preparation process of TC-COP-SO3H. Adapted from ref. 234. | ||
A significant methodological advance to circumvent the porosity-loss dilemma was demonstrated by Yang et al.,235 who emphasized the critical role of the polymer's swelling state during modification. By employing a one-pot strategy where the PAF precursor remained in a fully swollen state before sulfonation, they synthesized PAF-215. This material achieved an impressive balance of a high sulfur content (13.2 wt%), indicating dense functionalization, and a retained BET surface area of 580 m2 g−1. Consequently, PAF-215 exhibited top-tier adsorption capacities for RhB (1075 mg g−1), MB (1020 mg g−1), and antibiotics including tetracycline (TC, 826 mg g−1) and ciprofloxacin (CIP, 1134 mg g−1), showcasing that preserving pore accessibility during sulfonation is key to maximizing overall adsorbent performance. This exceptional adsorption performance is attributed to its hydrophilicity, high density of –SO3H groups, substantial specific surface area, and hierarchical pore structure.
Similarly, for inherently hydrophobic CMPs, sulfonation is essential to impart water dispersibility. To overcome these challenges, sulfonate-grafted CMPs were prepared with high dispersity in water (Fig. 14). The sulfonate-grafted pyrene-based CMPs, for instance, achieved a high MB capacity of 1650 mg g−1, primarily attributed to the combined effects of electrostatic attraction and π–π stacking within their ordered porous structure.
 | ||
| Fig. 14 The synthetic route of sulfonated-CMP. Adapted from ref. 130. | ||
In a recent contribution, an anionic sulfonate-grafted COF (TpStb-SO3Na) was synthesized from Tp (Tp: 2,4,6-triformylphloroglucinol) and Stb-SO3H (Stb-SO3H: 4,4′-diaminostilbene-2,2′-disulfonic acid) through the Schiff-base condensation.156 The framework features densely aligned sulfonate groups within its periodic one-dimensional channels (Fig. 15), which impart a strong negative surface charge and enable ultrahigh adsorption capacities for cationic dyes.
 | ||
| Fig. 15 Schematic of the TpStb-SO3Na structure in an AA stacking motif. Reproduced with permission from ref. 156 Copyright 2023 Elsevier. | ||
Remarkably, TpStb-SO3Na achieved record-high adsorption values for CV (1861 mg g−1), MG (5857 mg g−1), and JGB (1339 mg g−1), surpassing all previously reported COFs, MOFs, and benchmark adsorbents.252–255 It also exhibited rapid kinetics (∼99% uptake within 10 min) and high capacity for MB (1078 mg g−1) at room temperature. The exceptional performance is attributed to the synergy of electrostatic attraction between the anionic sulfonate sites and cationic dye molecules, coupled with π–π interactions between the conjugated COF skeleton and the aromatic dye structures. Furthermore, TpStb-SO3Na demonstrated excellent selectivity for cationic dyes over anionic ones and could be regenerated and reused for five cycles without significant loss of adsorption capacity, underscoring its potential for practical wastewater remediation.
In a complementary study, HCP membranes (m-HCPs) were fabricated via a versatile interfacial polymerization (IP) approach using benzene as the monomer (Fig. 16).236 The resulting m-HCP-5 membrane exhibited a high BET surface area of ∼760 m2 g−1 with a micropore-dominant structure. In dye-removal tests, m-HCP-5 showed an adsorption capacity of ∼460 mg g−1 for CR, while filtration experiments revealed a high-water flux of 55 L m−2 h−1 bar−1 and anionic dye rejections exceeding 93%. The separation performance stems from the combined effects of size sieving (micropores ∼0.5–1.2 nm) and electrostatic repulsion provided by the negatively charged sulfonate groups introduced during the interfacial polymerization process. This work highlights the potential of engineered porous organic membranes for simultaneous adsorption and charge-selective separation in advanced water-treatment applications.
 | ||
| Fig. 16 Schematic diagram of m-HCPs fabrication with benzene as the monomer and formaldehyde dimethyl acetal (FDA). Reproduced with permission from ref. 236 Copyright 2024 Nature Publishing Group. | ||
In another precedent, a CMP possessing a hollow sphere was fabricated by using a template, SiO2 nanoparticles and 1,3,5-triethynylbenzene and 2,7-dibromocarbazole as building blocks.237 Subsequently, hydrophilic CMPs were obtained by a sulfonation modification (H-S-CMPs) with better dispersibility in water. The performance of H-S-CMPs was investigated in the adsorption of dyes and antibiotics. The maximum adsorption capacities using H-S-CMPs were 206.2, 324.7, 222.2, and 216.9 mg g−1 for RhB, MB, CIP, and norfloxacin, respectively. In addition, the adsorption mechanism of H-S-CMPs was due to the synergism between hydrogen bonding, electrostatic forces, π–π stacking and pore filling. The adsorption capacity of H-S-CMPs remained stable after 5 cycles, and the removal rate of dyes exceeded 70%. A resorcinarene-based POP (BaPy) was designed and subsequently sulfonated to produce BaPy–SO3H (Fig. 17).238 BaPy–SO3H demonstrates exceptionally rapid adsorption rate, achieving equilibrium within just 1 min for a variety of organic pollutants (dyes, herbicides, and antibiotics) due to the establishment of electrostatic interactions, π–π interactions, and host–guest effects. BaPy–SO3H showed the apparent rate constants of 31.24, 31.27, and 30.63 g mg−1 min−1 for CV, MG, and paraquat, respectively, which are the highest performance among reported adsorbents to the best of our knowledge.256–261 Additionally, the BaPy–SO3H exhibited impressive maximum adsorption capacities of 1850, 2295, 1503 and 1304 mg g−1 for RhB, MB, CV and trimethoprim, respectively.
 | ||
| Fig. 17 Synthetic procedure of BaPy–SO3H. Adapted from ref. 238. | ||
4.2. Antibiotic/pharmaceutical adsorption
Sulfonate groups are essential for the adsorption of antibiotic molecules, many of which have cationic functional groups under ambient conditions. The electrostatic attraction between the anionic sulfonate sites and the cationic parts of antibiotics like fluoroquinolones is a key mechanism for their effective removal from water, often resulting in high adsorption capacities and selectivity even in complex water conditions.In one earlier report, a sulfonated MON (SMON) with triple- (TH-SMON) and double-shelled (DH-SMON) hollow spheres were synthesized, and their drug delivery performance was evaluated against that of single-shelled hollow sulfonated MON spheres (H-SMON) (Fig. 18).56 The study revealed that both drug loading capacity and release efficiency generally increased with the number of shells. Using RhB as a model molecule, the loading capacity was achieved for H-SMON, DH-SMON and TH-SMON with 0.7 ± 0.3 wt%, 6.8 ± 0.2 wt% and 7.3 ± 0.2 wt%, which correspond to the loading efficiency of 61 ± 3%, 73 ± 3%, and 78 ± 2%, respectively. Similarly, the cumulative release of RhB in PBS at pH 7.4 after 7 days at 37 °C increased as a function of thickness, from 41% to 60% from H-SMON to TH-SMON, respectively. The observed performance trend can be attributed to an expansion of the intershell space, enabling more facile infiltration of the aqueous electrolyte. All materials demonstrated excellent and stable dispersibility in aqueous media.
 | ||
| Fig. 18 Schematic outlining the synthesis of H-/DH-/TH-SMONs. Reproduced with permission from ref. 56 Copyright 2019 American Chemical Society. | ||
In one of the recent studies, experimental studies have shown that the adsorption capacity of HCPs is increased as the loading of –SO3H groups increases in the polymer while inversely proportional to the cross-linking density of the parent.239 Furthermore, chemisorption was found to be the main adsorption mechanism for SHCPs in removing pollutants via ionic interactions. Interestingly, SHCP1 showed around four-fold enhanced adsorption capacity for CIP compared to Norit (the commercial activated carbon) and Amberlyst-15. In another report, a 3D hyper-crosslinked porphyrin based POP (TP-PPOPs) was prepared using tetraphenylporphyrin and triptycene as monomers and the resulting solid was reacted with ClSO3H to obtain TP-PPOPs-SO3H, a sulfonated material with enhanced electronegativity and hydrophilicity (Fig. 19).240 The performance of TP-PPOPs-SO3H was studied in the adsorption of MB and CIP, achieving 1283.33 mg g−1 and 485.39 mg g−1 for MB and CIP, respectively. This enhanced performance was due to several factors that include strong electrostatic interactions and high surface area with extended π-conjugated structure.
 | ||
| Fig. 19 Synthesis of a) TP-PPOPs and b) TP-PPOPs-SO3H. Reproduced with permission from ref. 240 Copyright 2024 Elsevier. | ||
A –SO3H-decorated COF (ImCOF-SO3H; Im stands for imine-linked) was prepared and later, converted to a COF (AmCOF-SO3H; Am stands for amide-linked) through the reaction with Emim BF4 (Fig. 20)241 and its performance was studied in the adsorption of enrofloxacin (ENR), enoxacin (ENO), and norfloxacin (NOR). The adsorption capacities were 614 mg g−1, 615 mg g−1, and 579 mg g−1, for ENR, ENO and NOR, respectively, representing a 2.5-fold enhancement over ImCOF-SO3H. This enhanced activity of AmCOF-SO3H is due to the stronger electrostatic interactions with –SO3H groups and the facile formation of hydrogen bonding with the amide groups. These interactions are further complemented by π–π stacking and favorable pore-size matching effects. Notably, AmCOF-SO3H showed 90% removal of fluoroquinolone antibiotics, thus demonstrating the practical application of this solid in wastewater treatment.
 | ||
| Fig. 20 Synthesis of AmCOF-SO3H catalyst. Reproduced with permission from ref. 241 Copyright 2025 Elsevier. | ||
SHCPs were synthesized and their performance was tested for the adsorptive removal of various antibiotic pollutants under environmentally relevant conditions.166 The SHCPs were capable of highly efficient removal of antibiotic pollutants at relatively high (30 mg L−1) and low (50 µg L−1) initial concentrations, both from a simple as well as complex water matrices. Interestingly, the rate of CIP removal and the adsorption capacity observed for SHCP was ∼758 mg g−1 which is approximately two-fold than that of both post-synthetically sulfonated polymers (∼477 mg g−1) and Amberlyst-15 (∼439 mg g−1). These comparisons clearly indicate the beneficial effects of the highly functionalized SHCP as a potential adsorbent for the selective removal of organic pollutants from aqueous media.
4.3. Metal ion capture and recovery
To enable the selective capture and recovery of metal ions, –SO3H groups are integrated into POPs, creating powerful chelating platforms. This functionality is particularly transformative for the challenging separation of lanthanides, whose nearly identical chemical properties render conventional separation inefficient. SPOPs overcome this by exhibiting unique and tunable ion-selectivity, often based on ionic radii. Lanthanides are a group of important elements that have been widely used in many fields.262 Their separation is crucial for technical applications, but it remains a troublesome task due to their subtly different properties. In one of the recent studies, a nanoporous sulfonic ionic COFs (iCOFs) was obtained by reacting 1,3,5-tris(p-formylphenyl)benzene (TFPB) and 2,5-diaminobenzene-1,4-disulfonic acid (DABDA) to obtain TFPB-DABDA iCOFs and its performance was assessed in the selective adsorption separation of lanthanide elements (Scheme 10).242 The experimental results indicate that the adsorption capacity of TFPB-DABDA iCOFs is highly sensitive to the ionic radius of the lanthanides. The adsorption capacity for light rare earth elements (LREEs: La3+, Ce3+, Pr3+, Nd3+, Sm3+, Eu3+, and Gd3+) gradually increased, while for the trend for heavy rare earth elements (HREEs: Tb3+, Dy3+, Ho3+, Er3+, Tm3+, and Lu3+) was different, initially increasing at lower concentrations and then decreasing significantly with further concentration increases. Furthermore, TFPB-DABDA iCOFs exhibited the maximum adsorption capacity of 0.1554 mmol g−1 for early lanthanides, while for late lanthanides it is negligible. The separation mechanism was due to the uniform distribution of –SO3H groups attached within the ordered channels of the COFs. | ||
| Scheme 10 Synthesis of sulfonated TFPB-DABDA iCOFs. Adapted from ref. 242. | ||
A –SO3H-functionalized HCPs (SHCPs) was synthesized with 3.27 mmol g−1 density of –SO3H having 1044.6 m2 g−1 specific surface area with uniform micropores, and superhydrophilicity.243 These inherent properties enabled fast adsorption kinetics (30 min) and high adsorption capacities for REEs (106.78 mg g−1 for La, 111.99 mg g−1 for Eu, and 126.27 mg g−1 for Lu). Furthermore, the material showed extremely high REE3+/Al3+ selectivity in a sulfate medium (SF(La/Al) = 6929, SF(Eu/Al) = 4810, SF(Lu/Al) = 3003), surpassing most existing REE adsorbents. When applied to wastewater, SHCP adsorbed 87% of the total REE3+, while the adsorption rate for Al3+ was zero. These results imply that the high selectivity stems from the difference in binding stability between REE3+ and Al3+ complexes with sulfate ions, demonstrating that –SO3H groups offer a promising approach for REE recovery from sulphate-rich aqueous solutions. Beyond REEs, SPOPs have also been applied to the detection and removal of other critical and toxic species. The Schiff base condensation of 2,4,6-triformylphloroglucinol (Tp) and 3,7-diaminodibenzo[b,d]thiophene (DAS) resulted in an imine-based COF (TAS-COF).244 The benzothiophene sulfone unit in TAS-COF endows oxidase-like activity upon exposure to visible light irradiation by generating superoxide radicals. Thus, under visible light irradiation, TAS-COF photocatalyzes the oxidation of colorless 3,3′,5,5′-tetramethylbenzidine (TMB) to blue oxTMB. The subsequent coordination of uranyl ions (UO22+) with oxTMB quenches this blue color, providing the basis for a facile colorimetric sensor. Using this strategy, TAS-COF achieved a detection limit of 0.07 µmol L−1 for UO22+. A sulfonated COF, designated TFPOTDB-SO3H, was synthesized by reacting DABSA and 2,4,6-tris-(4-formylphenoxy)-1,3,5-triazine, and its efficacy was tested for the adsorption of As(III) (Fig. 21).245 The material achieved a 97.1% removal rate of As(III) within 10 min, corresponding to a high adsorption uptake of 345 mg g−1 at pH 8 and room temperature. Furthermore, the influence of other competing anions was studied and observed a reduction of As(III) uptake by around 1–7%. Moreover, the adsorbent exhibited excellent reusability, retaining 89% of its initial uptake capacity after four successive adsorption–desorption cycles.
 | ||
| Fig. 21 Synthesis of TFPOTDB-SO3H solid catalyst. Reproduced with permission from ref. 245 Copyright 2024 Elsevier. | ||
SPOPs also show versatility in adsorbing alkali metals. A –SO3H-functionalized (SHCP) was synthesized through FC reaction and the performance of SHCP was studied in the adsorption of Rb+ and Cs+.246 The adsorption efficiencies for Rb+ and Cs+ using SHCP was 94.7% and 95.9% at 30 min. Interestingly, SHCP showed a good adsorption selectivity for Rb+ and Cs+ ions in the presence of several competitive ions. The physical doping and freeze-drying of sulfonated CMP (SCMP) with carboxymethyl cellulose (CMC) resulted in a composite aerogel (CMC/SCMP) and its activity was tested in the adsorption of Pb2+ and MB.247 The adsorption capacity was up to 94.93 and 294.84 mg g−1, respectively for Pb2+ and MB. The characterization and experimental data indicate that this activity is attributed to chelation, electrostatic attraction, H-bonding and π–π interaction.
4.4. Gas separation
In gas separation applications, the integration of –SO3H groups endows porous polymers with high selectivity for gases like CO2 and NH3. These strongly acidic, polar sites chemisorb basic gas molecules, significantly boosting their uptake over non-polar species such as N2 and CH4. This is evidenced by dramatically increased isosteric heats of adsorption and exceptional selectivity, with reported CO2/N2 values exceeding 400 for some sulfonated networks. Furthermore, when integrated into membranes, sulfonate groups induce tighter polymer chain packing, which critically enhances diffusion selectivity. This synergistic effect between improved sorption selectivity (from strong gas-functional group interactions) and enhanced diffusion selectivity (from modified polymer morphology) leads to a superior overall separation performance that often surpasses the upper bounds of current trade-off curves for key gas pairs like CO2/N2 and O2/N2. For instance, a study on HCPs with PIMs demonstrated how functionalizing the polymer backbone with polar groups like –NO2, –NH2, and –HSO3 tunes their selectivity for CO2 over N2 and CH4.136 Although the introduction of these functional groups generally reduced the overall porosity, this was more than compensated for by a significant improvement in CO2 affinity. The sulfonated polymers achieved the highest CO2 uptake capacity of 298 mg g−1 (6.77 mmol g−1). In terms of selectivity, the aminated (–NH2) polymers performed best, reaching values of 26.5 for CO2/N2 and up to 8.6 for CO2/CH4. The superior performance of these functionalized PIMs is attributed to a synergistic contribution from the residual porosity, the specific nature of the functional group, and an optimal isosteric heat of adsorption.A COF (TpPa-SO3H) was prepared via a modified mechanical grinding synthetic method and further functionalized with metal sites by post-loading to obtain TpPa-SO3Cu0.5.248 Due to the presence of abundantly accessible active sites and well-ordered COF channels, TpPaSO3Cu0.5 demonstrated superior adsorption kinetics and a high capacity of 30.45 mg N g−1, outperforming most reported adsorbents (<0.001–0.994 g mg−1 min−1 and 0–25 mg N g−1). Besides, TpPa-SO3Cu0.5 showed a selectivity coefficient 328 times higher than to TpPa-SO3H in real water (10 mg N L−1, pH = 10). This solid exhibited a high NH3 recycling ratio (∼95%) during five adsorption-regeneration cycles. In a separate approach, a hyper-crosslinked POP was sequentially oxidized and sulfonated in a post-synthetic treatment to generate a porous solid decorated with readily accessible carboxylic and –SO3H groups.249 Interestingly, the polymer was coated with poly(dimethylsiloxane) and the resulting solid exhibited a 40-times increase (from 0.04 to 1.41 mmol g−1) compared to non-modified polymer towards a low-pressure NH3 adsorption; key merits of this approach include easy preparation, cost-effectiveness, and scalability for mass production. In another study, sulfonated polyimide (SPI) foams with tunable surface wettability were developed for methane hydrate formation.250 The research demonstrated that an SPI foam with a balanced hydrophobic–hydrophilic surface significantly enhanced formation kinetics. Under static conditions, the optimized foam achieved an extremely short induction period and a rapid hydrate formation rate of 2.3 V V−1 min−1, along with a high storage capacity of 119 V V−1, all without the use of chemical additives. An early investigation reported that sulfonated polymeric networks (PPN-6-SO3H and its Li+ salt, PPN-6-SO3Li) exhibited a significant enhancement in both the isosteric heat of CO2 adsorption and the overall CO2 uptake capacity.53 Ideal adsorption solution theory calculations using single-component-isotherm data and a 15/85 CO2/N2 ratio at 295 K and 1 bar, revealed exceptionally high selectivity for CO2 over N2 (155 and 414 for PPN-6-SO3H and PPN-6-SO3Li, respectively). Three SHCPs with distinct structural features were synthesized and their performance was evaluated in NH3 adsorption.251 Among the various solids screened, SHCP-1 showed superior NH3 uptake of 11.54 mmol g−1 at 30 °C and 1 bar. Interestingly, NH3 adsorption capacity was more than 10 mmol g−1 even after ten regeneration cycles, thus illustrating its stability. This superior performance of this solid is attributed to the presence of well-developed micro/mesoporous structure consisting of abundant adsorption sites which favors the chemisorption between –SO3H groups and –NH3. In another study, a PIM-1 membrane was modified through straightforward sulfonation with a sulfur trioxide to afford sulfonated PIM-1 (SPIM-1) membranes (Scheme 11).135 Characterization data revealed the attachment and uniform distribution of –SO3H groups along the polymer backbone. Further, SPIM-1–6 (6 stands for PIM-1 was sulfonated for 6 min) showed the highest gas separation performance, with selectivity values approaching or exceeding the latest trade-off limits for O2/N2, CO2/N2, H2/N2, and CO2/CH4 pairs. The superior activity can be attributed to the SO3H group, inducing compact polymer chain packing, which remarkably enhances diffusion selectivity. After 60 days of aging, the SPIM-1–6 membrane exhibited even higher selectivity (H2/N2 = 125, O2/N2 = 8.43) alongside high gas permeabilities (H2 = 1077 Barrer, O2 = 73.4 Barrer).
 | ||
| Scheme 11 Synthesis of a) PIM-1; b) the SO3 sulfonation solution and c) SPIM-1 membranes, where x and y vary with sulfonation time. Adapted from ref. 135. | ||
5. Energy-related applications
5.1. Proton conductivity
Proton-conducting materials are central to electrochemical energy technologies because they govern the efficiency, durability, and cost of fuel cells, sensors, and chemical reactors.263,264 Among them, polymer electrolyte membrane fuel cells (PEMFCs) are particularly attractive for portable, automotive, and stationary applications. In such devices, the polymer electrolyte membrane is widely regarded as the “heart” of the system, since its properties dictate key performance metrics including power density, operational lifetime, and system complexity. To operate reliably, a PEM must simultaneously provide high proton conductivity, negligible electronic conductivity, minimal fuel and oxidant crossover, and robust thermal, chemical, and mechanical stability, while remaining cost-effective and processable at scale.265,266 These stringent and often competing requirements have driven intensive research not only into dense fluorinated and hydrocarbon polymers, but increasingly into architecturally tunable porous frameworks that allow decoupling and rational optimization of ion transport, mechanical integrity, and chemical robustness.267,268Historically, organic proton-conducting polymers have dominated the PEM field. Nafion®, a perfluorosulfonic acid (PFSA) membrane developed by DuPont, has set the technological benchmark owing to its high proton conductivity under humidified conditions, well-developed morphology, and proven long-term operation in devices. Together with Nafion®, high-temperature polybenzimidazole (PBI) membranes and sulfonated hydrocarbon polymers such as sulfonated poly(ether ether ketone) (sPEEK) represent the state-of-the-art in commercial and pre-commercial systems.269,270 Despite these successes, however, conventional polymeric membranes exhibit persistent limitations. Nafion® suffers from a sharp loss of conductivity at elevated temperatures and reduced relative humidity, complex and energy-intensive processing, and concerns related to fluorinated waste and cost. Likewise, phosphoric-acid-doped PBI and sulfonated polyaryl membranes often display trade-offs between proton conductivity, oxidative and mechanical stability, and dimensional control, as highlighted in recent benchmark studies.271,272 Collectively, these issues indicate that even optimized PFSA and hydrocarbon membranes do not fully satisfy the simultaneous requirements of high conductivity, durability, and sustainability, motivating the exploration of alternative architectures such as sulfonated POPs.
To address the persistent trade-offs inherent to conventional polymer electrolytes, research has increasingly turned to composite and hybrid membrane architectures that introduce auxiliary phases capable of reorganizing local proton-transport environments. Representative approaches include acid–base complexes such as PBI-H3PO4,273 heterocycle-assisted proton conductors,274 and nanostructured Nafion®-CNT hybrids,275 as well as inorganic–organic systems such as CsHSO4-SiO2 and CsH2PO4-SiO2.276 While these strategies demonstrate that hydrogen-bond organization, water retention, and phase stability can be modulated through secondary components, their impact remains fundamentally constrained by the limited architectural freedom of dense polymer matrices, which restricts long-range transport pathways and hinder deliberate positioning of acid sites. Consequently, improvements tend to be incremental rather than transformative, reinforcing the need for frameworks in which porosity, acidity, and transport channels can be co-engineered from the molecular level. Architecturally tunable porous materials particularly SPOPs and SCOFs267 directly address this gap by enabling decoupled control over proton-donating groups, pore topology, and conduction mechanisms, thereby offering a fundamentally different design space than conventional composite membranes.
A clear understanding of proton-transport mechanisms is essential for designing advanced electrolytes, particularly in systems where acidity, pore topology, and local chemical environments can be engineered. Proton motion in polymeric and porous materials typically arises from a combination of Grotthuss-type structural diffusion and vehicular transport by protonated species. The former operates through rapid hydrogen-bond reorganization and is generally associated with lower activation energies (<0.4 eV), while the latter involves the diffusion of hydronium or other protonated carriers and dominates when mobility is restricted or hydration is limited.277 In architecturally tunable porous frameworks such as SPOPs and SCOFs, these mechanisms are strongly influenced by the spatial distribution of –SO3H groups, pore confinement, and the degree of structural ordering (Fig. 22). Periodically aligned acid sites and narrow channels can promote directional hopping, whereas disordered or highly hydrated domains favor mixed pathways. Such transitions are typically reflected in activation-energy trends obtained from impedance spectroscopy.278,279 Importantly, the ability to decouple pore geometry, acid-site placement, and hydration environments in porous frameworks, an option not accessible in dense polymers, creates a distinct design platform for tailoring proton-conduction pathways across a broad range of operating conditions.
 | ||
| Fig. 22 Conceptual schematic of proton transport in dense polymer membranes versus architecturally tunable porous frameworks, highlighting hydration-dependent tortuous pathways and ordered pore-confined proton conduction, respectively. | ||
Beyond classical polymers and composite membranes, crystalline and semi-crystalline porous materials have emerged as a powerful platform for advancing proton-conducting electrolytes. Among these, COFs have attracted particular attention due to their long-range structural order, well-defined pore channels, and precise chemical tunability. Constructed from lightweight elements linked through strong covalent bonds, COFs offer an unprecedented level of control over pore size, topology, and functional-group placement, enabling direct correlation between framework architecture and proton-transport behavior. In contrast to amorphous polymer systems, the periodicity of COFs allows systematic investigation of how channel confinement, acid-site alignment, and hydrogen-bond organization govern conduction mechanisms. Early studies demonstrated that COFs functionalized with acidic groups can sustain continuous proton-hopping pathways within ordered nanochannels, providing model systems to decouple structural effects from compositional complexity. Although their absolute conductivities initially lagged behind benchmark polymer membranes, these materials established critical structure-mechanism relationships that now underpin the rational design of high-performance SCOFs and related porous organic frameworks.280
Parallel to COFs, other POPs have emerged as a complementary and highly versatile class of proton-conducting materials. Unlike crystalline COFs, POPs are typically amorphous or semi-ordered, yet they offer exceptional chemical and thermal stability, broad synthetic flexibility, and scalable preparation routes. Their inherently disordered nature allows dense incorporation of functional groups without the strict topological constraints imposed by crystallinity. As a result, POPs provide a unique platform in which high surface area, tunable pore environments, and chemical robustness can be combined to tailor proton-transport behavior. Importantly, the absence of long-range order does not preclude efficient proton conduction; instead, localized hydrogen-bond networks formed within interconnected micropores can sustain effective transport pathways when appropriately functionalized. Compared with COFs, POPs therefore offer greater tolerance toward high acid densities and structural heterogeneity, making them particularly attractive for aggressive functionalization strategies such as sulfonation. These features position POPs as a practical bridge between model crystalline frameworks and application-oriented polymer electrolytes, especially in contexts where processability, durability, and chemical stability are as critical as absolute conductivity.281
Among the functionalization strategies explored for porous organic frameworks, sulfonation has emerged as the most effective route to impart strong Brønsted acidity and enable efficient proton conduction (Table 5). The introduction of –SO3H groups provides abundant proton-donating sites while simultaneously promoting the formation of extended hydrogen-bond networks that favor Grotthuss-type transport. In POPs, sulfonation can be achieved either through PSM of preformed backbones or via the direct incorporation of sulfonated monomers during polymerization. These two approaches offer fundamentally different levels of control over acid-site distribution, framework integrity, and pore accessibility. Post-sulfonation enables high acid densities and broad applicability across diverse POP chemistries but may introduce structural heterogeneity or partial pore blockage if not carefully controlled. In contrast, in-monomer sulfonation allows more uniform placement of –SO3H groups and preserves pore connectivity, albeit often at the expense of synthetic complexity. Crucially, studies across multiple POP families consistently demonstrate that proton conductivity is governed not only by the total acid content but also by the spatial organization of sulfonic sites relative to pore architecture and hydration domains. This decoupling of acidity, porosity, and framework stability represents a key advantage of SPOPs over dense polymer electrolytes and underpins their emerging role as tunable proton-conducting membranes.
| POP | BBs/monomers | Functionalization | BET surface area (m2 g−1)/pore size (nm) | Application | Performance metric | Acid density (mmol g−1) | Stability assessment | Scale up (yes/no) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| *The first and second values indicate measurements before and after sulfonation, respectively (if given). | |||||||||
| NUS-10(R) | 1,3,5-triformylphloroglucinol (TFP), DABDA | Mechanosynthesis (LAG) → Recrystallization (de novo) | –, 69/0.84 | 3.96 × 10−2/0.21 eV | 25 °C, 97% RH | 5.20 mmol g−1 (IEC) | Time-dependent conductivity (15 days, stable) | — | 124 |
| MPOPS | Cyanuric chloride and DABSA | Extended condensation polymerization (de novo) | 114/1.27 | 3.07 × 10−2/0.407 | 80 °C, humid conditions | 1.836 mmol g−1 (IEC) | Stability test (I vs t, >16000 s), MEA test |
— | 125 |
| SBO-CMP | 1,4-Phenylenediboronic acid, 4,4′-biphenyldiboronic acid, tetrabromobisolefin | Suzuki–Miyaura coupling | 440, 40/2 | 1.29 × 10−2/0.32 | 70 °C, 100% RH | 27.6 wt% | — | 126 | |
| Phloroglucinol-based POP | Phloroglucinol and terephthaldehyde | Microwave-assisted de novo synthesis | 992, 29/— | 7.72 × 10−2/0.34 | 80 °C, 90% RH | — | Time-dependent conductivity | — | 282 |
| TaPOP-SO3H | Tetrakis(4-azidophenyl)methane, 1,3,5-triethynyl-benzene, propargyl sulfonate | In situ sulfonation (click) | 629.5, 63.8/1.45 | 0.92 × 10−2/0.25 | 80 °C, 98% RH | — | Long-life reusability test (>20 h) | — | 283 |
| S-POP-BP-TPOT | 1,4-Bis(chloromethyl)benzene (BP), 2,4,6-triphenoxy-1,3,5-triazine (TPOT) | FC alkylation followed by postsulfonation | 395, 93/0.62, 0.27 Pore volume cm3 g−1 | 1.5 × 10−2/0.19 | 25 °C, 95% RH | 30 wt%/3.6 mmol g−1 (IEC) | Time-dependent proton conductivity, Fenton's reagent test, fuel-cell test | — | 127 |
| S-POP-TPM | Tetraphenylmethane (TPM), formaldehyde dimethyl acetal (FDA) | FC alkylation followed by postsulfonation | 1312, 720/0.54 cm3 g−1 pore volume | 0.1 × 10−1/0.23 | 80 °C, 95% RH | 36 wt% | Time-dependent proton conduction (6 h) | — | 284 |
| 2-MeL40-S | Biphenyldicarboxaldehyde, 5-methylresorcinol (MeL), phloroglucinol (PG) | Microwave-assisted condensation followed by postsulfonation | Decreased (after MeL/sulfonation)/— | 2.39 × 10−1/0.085 | 80 °C, 95% RH | IEC 5.85 mmol g−1, DoS 116.8% for 1-MeL40-S | Cyclic stability, time-dependent proton conductivity | — | 285 |
| SPAF | Tetrakis(4-bromophenyl)methane (BrTPM), Benzene-1,4-diboronic acid (BDB) | Suzuki coupling followed by gas-phase sulfonation (PSM) | 1692, 124/∼1 | 1.4/0.36 (35 kJ mol−1) | 80 °C, 100% RH | 5.67 mequiv g−1 (IEC) | NMR, IR, Titration (Long-term stability), ESEM | — | 286 |
| SMPAF-303 | PAF-303 (BrTPM + BDB), Formaldehyde, HCl/Na2SO3 | PSM (Chloromethylation → Sulfonation) | 1317(Ar), 743(Ar)/0.681, 0.337 pore volume cm3 g−1 | 0.1/0.23 (22.6 kJ mol−1) | 77 °C, 100% RH | 1.73 mequiv g−1 (IEC) | High thermal stability (500 K) | — | 287 |
| 50 wt% SPEEK@Ox-BD-COF-SO3H | Tri(4-aminophenyl)benzene (TAPB), benzo[d][1,3]dioxole-4,7-dicarbaldehyde, SPEEK | Sequential PSM (oxidation, sulfonation) followed by infiltration | 1580, 21/3.2 | 6.22 × 10−3/0.199 | 90 °C, 98% RH | COF: 6.2 wt% SO3H | Repeated conductivity tests (48-h run) | — | 135 |
| PyTTA-DHTA-COF-SO3H | 4,4′,4″,4‴-(pyrene-1,3,6,8-tetrayl)tetraaniline (PyTTA), 2,5-dihydroxyterephthalaldehyde (DHTA) | Solvothermal condensation followed by PSM | 2013, 493/2.1, 1.7 | 2.0 × 10−2/0.38 | 70 °C, 100% RH | 19.5 wt% | Time-dependent proton conduction (8 h, stable) | — | 283 |
| S-COF-2 | 2,4,6-Trimethoxybenzene-1,3,5-tricarbaldehyde, DABDA | De novo synthesis (solvothermal) | 351, 139/1.30, 1.26 | 1.5 × 10−2/0.17 | 25 °C, 95% RH | 15.88 wt%/40.2% content | Time-dependent proton conduction (24 h, stable) | — | 137 |
| H3PO4@COF-SO3H | 1,3,5-tris(p-formylphenyl)benzene, DABDA | Solvothermal synthesis, mechanochemical impregnation (PSM) | 53.51, 1.74/3.51 | 3.57 × 10−2/0.14 eV | Ambient conditions | — | Proton battery test, cycling stability (1000 cycles) | — | 288 |
| SPEEK/COF-EN_3 | Hexahydroxybenzene, tetrafluoroterephtalonitrile | Solvothermal synthesis | —/— | 2.17× 10−1/0.19 eV | 80 °C, 90% RH | Speek 77% | Cell performance | — | 289 |
| COF-Fx-SO3 | 2,4,6-tris(4-formylphenyl) pyrroline, DABSA | Solvothermal synthesis | 233/1.7 | 1.93 × 10−2/0.24 eV | 140 °C, anhydrous conditions | — | Time-dependent conductivity | — | 290 |
| 7.7 × 10−3/— | 90 °C, 97% RH | ||||||||
| PA@TpDMAP/Pa-SO3H | DABSA, dimethylaminopyridine, triformylphloroglucinol | Solvothermal followed by PSM | 247, 987/2.21 | 1.56× 10−2/0.19 eV | 140 °C, anhydrous conditions | Speek 83% | AC electro-chemical impedance spectroscopy | — | 291 |
| SPEEK/HPW@COF | 1,3,5-Triformylphloroglucinol, 3,5-diamino-1,2,4-triazole | Solvothermal synthesis of triazole COF into PSM | 164, 92/1.44 1.32 | 2.80 × 10−1/0.25 eV | 75 °C, 100% RH | 1.75 mmol g−1 (IEC) | Time-dependent conductivity | — | 128 |
| SPEEK/TpPaeSO3He5 | 1,3,5-triformylphloroglucinol, DABSA | RT solvothermal | —/1.5 nm | 3.46 × 10−1/0.27 eV | 80 °C, 100% RH | 2.34 mmol g−1 IEC | Single fuel cell performance | — | 292 |
| SPAF/SPEEK | 1,5-Cyclooctadiene, 2,2′-bipyridyl, Ni(COD)2 | PSM sulfonation | 3360, 608/– | 5.88 × 10−2/0.13 eV | RT in pure water | SPEEK sulfonation 70.6% | Fuel cell durability test | — | 293 |
| PA-PBI/SCOF | 1,3,5-Triformylphloroglucinol, DABSA | In situ synthesized in PBI matrix. | —/— | 5.42 × 10−1/4.46 kJ mol−1 (∼0.046 eV) | 80 °C, 100% RH | — | PA retention stability | — | 294 |
| MIP-COF/SPBI | Melamine, isophthalaldehyde | Solvothermal synthesis | 437.18/pore volume 0.55 cm3 g−1 | 1.08 × 10−1/— | 80 °C, 100% RH | 2.69/3.70 meq g−1 | Fuel cell testing, oxidative stability (93.14%) | — | 295 |
| Calculated/Measured IEC values | |||||||||
| SBCNF/TpBd-2SO3H | 4,4′-Diamino-3,3′-biphenyldisulfonic acid, 1,3,5-triformylphloroglucinol | Interfacial synthesis | —/— | 1.48 × 10−1/0.32 | 80 °C, 100% RH | 3.27 mmol g−1 IEC | Time-dependent conductivity | — | 296 |
| NUS-9/Nafion® | DABSA, 1,3,5-benzenetricar-boxaldehyde | Interfacial synthesis | 95.8/3 | 2.1 × 10−1/<0.4 | 95 °C, 100% RH | 3.67 wt% S content | Fuel cell testing | — | 297 |
| 0.9–1.02 mmol g−1 IEC | |||||||||
| BD(SO3H)2/PTFE | 2,4,6-Triformylphloroglucinol, 4,4′-diamino-3,3′-biphenyldisulfonic acid | Solvothermal synthesis followed by casting onto porous PTFE | —/1.78 | 2.49× 10−1/— | 80 °C, 100% RH | 29.2 wt% | OCV test period under a galvanostat | — | 298 |
Looking forward, SPOPs and SCOFs offer a highly promising platform for next-generation proton-conducting membranes, owing to their modular architectures and the ability to systematically tune chemical composition, pore environment, and functional-group density. These attributes enable fine control over proton-transport mechanisms that are difficult to achieve in conventional dense polymers. At the same time, key challenges remain, including achieving high conductivity under anhydrous or low-humidity conditions, ensuring long-term operational stability, and developing scalable synthesis routes. Addressing these issues is essential for translating laboratory-scale performance into practical PEMFC technologies. In the following sections, we therefore highlight representative advances in SPOPs, SCOFs, and their hybrid membranes with conventional polymers, with particular emphasis on synthetic strategies, structure–property relationships, conductivity benchmarks, and mechanistic design principles relevant to proton transport.
The development of proton-conducting POPs has predominantly relied on post-sulfonation as a versatile and chemically robust strategy to introduce strong Brønsted acid sites into stable porous backbones. Early studies on phenolic and aromatic POPs demonstrated that carefully designed frameworks can withstand harsh sulfonation conditions while preserving structural integrity, thereby enabling exceptionally high densities of covalently anchored –SO3H groups within accessible pore environments. Importantly, these systems revealed that proton conductivity is not dictated solely by acid loading; instead, the continuity of hydrogen-bond networks formed within interconnected pores plays a decisive role in facilitating efficient Grotthuss-type transport under humidified conditions.282 This principle was further validated in CMPs, where rigid π-conjugated backbones provided mechanically resilient platforms for post-sulfonation. Despite partial reductions in surface area after functionalization, SCMPs retained effective proton-transport pathways and exhibited activation energies consistent with dominant proton hopping, underscoring that pore connectivity and acid-site distribution can outweigh absolute porosity.126 More recently, click-derived triazole-based POPs have introduced an additional level of mechanistic sophistication by combining heterocyclic nitrogen sites as intrinsic proton relays with –SO3H groups as proton sources. The cooperative interaction between triazole units and –SO3H functionalities promotes dense hydrogen-bond networks and enhanced water affinity, leading to conductivity enhancements of several orders of magnitude compared with pristine polymers (Fig. 23).283 Together, these representative post-sulfonated POP systems establish critical design principles grounded in framework robustness, preservation of accessible pore networks, and cooperative acid–base interactions that continue to guide the rational development of high-performance SPOP proton-conducting membranes.
 | ||
| Fig. 23 Synthetic route for TaPOP and TaPOP-SO3H structures. Adapted from ref. 283. | ||
Among post-sulfonated POP families, triazine-rich networks are particularly instructive because their electron-deficient nodes simultaneously facilitate electrophilic sulfonation and enhance hydrophilicity and water retention, two factors that are critical for sustaining continuous hydrogen-bond networks. In sulfonated triazine POPs prepared via FC polymerization, dense and uniformly distributed –SO3H groups lead to proton conductivities approaching 10−2 S cm−1 at room temperature and high relative humidity, accompanied by low activation energies (∼0.19 eV), indicative of a dominant Grotthuss-type transport mechanism (Fig. 24a).127 Importantly, comparative studies reveal that triazine-containing frameworks consistently outperform non-heteroatom analogues at comparable sulfonation levels, underscoring the cooperative role of backbone polarity and acid density. Beyond amorphous systems, crystalline sulfonated POPs incorporating triazine motifs demonstrate that periodic alignment of –SO3H groups within ordered micropores further lowers transport barriers and enhances humidity tolerance, reaching conductivities above 10−2 S cm−1 under both hydrous and near-anhydrous conditions (Fig. 24b).125 These studies establish that proton transport in sulfonated triazine POPs is governed not solely by acid content, but by a synergistic interplay between framework electronics, pore architecture, and water-mediated hydrogen-bond connectivity, providing a transferable design rationale for next-generation polymer electrolytes.
 | ||
| Fig. 24 a) Design strategy and synthetic pathway for triazine-derived POPs and S-POPs. ref. 127 Copyright 2020 American Chemical Society; b) depiction of the formation of MPOPS-1. Reproduced with permission from ref. 125 Copyright 2020 American Chemical Society. | ||
Rather than indicating that higher sulfonation degrees alone guarantee superior performance, recent studies on post-sulfonated POPs collectively reveal that proton conduction is an emergent property of framework-level organization. Effective SPOPs operate in a regime where chemical functionality, pore continuity, and local polarity are co-optimized, allowing proton transport pathways to persist even under conditions where dense polymer membranes fail. In this context, post-sulfonation should be viewed not merely as a functionalization step, but as a structural perturbation that reshapes hydration domains and transport topology within the framework. Architectures that tolerate this perturbation without collapsing pore accessibility or disrupting inter-pore connectivity consistently exhibit lower activation barriers and improved humidity tolerance. Importantly, these insights reposition post-sulfonated POPs as a distinct materials class in which conductivity can be tuned independently from mechanical reinforcement and chemical stability a level of decoupling that remains inaccessible in conventional polymer electrolytes. This shift from composition-driven to architecture-driven optimization defines the principal conceptual advance offered by post-sulfonated POPs for proton-conducting membrane design.
Beyond framework selection and sulfonation chemistry, recent advances have increasingly targeted synthetic strategies that explicitly couple acid strength with mesoscale transport organization. Rather than relying on incremental increases in sulfonation degree, these approaches aim to synchronize proton mobility, hydration stability, and long-range pathway continuity within porous architectures. A representative strategy employed hierarchically porous SPOPs with high densities of covalently tethered –SO3H groups, achieving proton conductivities close to 10−1 S cm−1 at 80 °C and 95% RH while preserving mechanical and chemical integrity during prolonged operation. Notably, this work demonstrated that sustained high conductivity arises from continuous, interconnected proton-transport pathways enabled by hierarchical pore organization, rather than from acid loading alone.284 Building on this structural paradigm, a subsequent study introduced electronic modulation of –SO3H sites through the incorporation of electron-withdrawing substituents into the POP backbone. This electronic tuning enhanced intrinsic Brønsted acidity and stabilized hydrogen-bond networks, resulting in record conductivities of 0.15–0.239 S cm−1 and exceptionally low activation energies (0.085–0.096 eV) at 80 °C and 90% RH.285 Together, these studies establish that high-performance proton conduction in SPOPs emerges from the synergistic optimization of pore connectivity and acid-site electronics, providing a clear mechanistic design framework that transcends conventional structure–property trade-offs in polymer electrolytes (Fig. 25).
 | ||
| Fig. 25 Hierarchically connected hydrated pores combined with electronically tuned, covalently tethered –SO3H sites enable continuous Grotthuss-type proton transport, giving rise to high proton conductivity and low activation energy in SPOPs. | ||
Alongside these developments, PAFs have emerged as a closely related but conceptually distinct subclass of POPs, in which rigid, highly connected aromatic skeletons enable exceptionally high densities of –SO3H sites without catastrophic loss of structural integrity. In contrast to more flexible polymeric backbones, the intrinsic rigidity of PAFs suppresses excessive segmental motion and dimensional swelling, allowing proton transport to be governed predominantly by pore-confined hydrogen-bond networks rather than bulk polymer relaxation. Early sulfonated aromatic frameworks demonstrated that densely packed –SO3H groups directly anchored to rigid aromatic cores can sustain ultrafast proton conduction approaching 1 S cm−1 under humidified conditions, a performance attributed to minimized activation barriers and continuous acid-lined micropores (Fig. 26).286 More recent advances introduced methylene-linked –SO3H side chains into microporous aromatic frameworks, decoupling acid-site stability from the aromatic backbone and significantly enhancing thermal robustness. In these systems, side-chain engineering preserves strong Brønsted acidity while mitigating the thermal fragility associated with directly sulfonated aromatic rings, enabling conductivities spanning from 10−7 to 10−1 S cm−1 depending on hydration level. Importantly, these studies reveal that proton transport in SPAFs is highly sensitive to local polarity modulation within the pore environment: bottleneck effects dominate at low relative humidity due to electrolyte clustering, whereas fully hydrated pores eliminate transport barriers and enable near-bulk proton mobility.287 PAF-based systems highlight a critical design principle distinct from both COFs and conventional POPs, that rigid aromatic connectivity combined with controlled acid-site tethering can simultaneously maximize conductivity, thermal stability, and confinement, positioning SPAFs as a key bridge between amorphous SPOPs and crystalline framework electrolytes.
 | ||
| Fig. 26 Synthesis of SPAF-2x from tetrakis(4-bromophenyl)methane (BrTBPM) and 1,4-diboronic acid (BDB) via PAF-2 formation and subsequent sulfonation (x = degree of sulfonation). Reproduced with permission from ref. 286 Copyright 2023 American Chemical Society. | ||
While SPOPs and SPAFs clearly demonstrate that high acid density and pore confinement can sustain efficient proton transport even in disordered architectures, their amorphous nature inevitably limits precise control over transport pathways and acid-site periodicity. In such systems, proton conduction is largely governed by local hydrogen-bond networks and mesoscale connectivity, which complicates the decoupling of intrinsic structure–property relationships from statistical disorder. To overcome these limitations and to gain molecular-level insight into proton-transport mechanisms, increasing attention has shifted toward COFs. Owing to their crystallinity, long-range order, and modular synthetic design, COFs enable the periodic alignment of acidic functionalities within well-defined nanochannels, providing an unparalleled platform to systematically interrogate the roles of pore geometry, acid-site spacing, and hydrogen-bond organization in proton conduction. As a result, COFs have emerged not only as promising proton-conducting materials in their own right, but also as model systems that complement amorphous SPOPs by enabling fundamental structure–transport correlations that are otherwise difficult to access in disordered frameworks.
A decisive milestone in this context was reported by Peng and co-workers in 2016, who introduced a mechanoassisted strategy to synthesize intrinsically SCOFs with densely and periodically decorated –SO3H groups along crystalline nanochannels. The resulting frameworks (NUS-9 and NUS-10) achieved proton conductivities up to 3.96 × 10−2 S cm−1 under humidified conditions, accompanied by activation energies of approximately 0.20 eV, consistent with a dominant Grotthuss-type transport mechanism (Fig. 27).124 Beyond their electrochemical performance, this work was transformative in two key respects: first, it demonstrated that strong Brønsted acid sites can be permanently integrated into COF backbones without sacrificing crystallinity or structural integrity; second, it showed that such crystalline proton conductors can be processed into PVDF-based mixed-matrix membranes with long-term operational stability. By simultaneously establishing structure-defined proton transport and membrane-level processability, this study laid the conceptual and practical foundation for scalable, functionalized COFs as next-generation proton-conducting materials.
 | ||
| Fig. 27 Synthetic scheme of sulfonated COFs NUS-9(G) and NUS-10(G) via liquid assisted grinding at room temperature. Adapted from ref. 124. | ||
Building on the mechanoassisted SCOF paradigm, studies reported in 2021 marked a decisive shift toward structurally precise proton conductors by refining how –SO3H groups are introduced and stabilized within crystalline frameworks. In particular, work on intrinsically sulfonated two-dimensional COFs demonstrated that high densities of covalently anchored –SO3H groups can be incorporated while fully preserving long-range order, enabling proton transport to be governed by periodic channel architecture rather than local disorder. These materials exhibited proton conductivities approaching 10−2 S cm−1 at high relative humidity, with low activation energies (∼0.17–0.20 eV), providing compelling evidence for dominant Grotthuss-type hopping along ordered one-dimensional nanochannels.158 Complementary post-synthetic sulfonation studies further clarified the delicate balance between linker electronics and steric accessibility, showing that crystallinity retention is essential for sustaining continuous hydrogen-bond networks and minimizing transport bottlenecks.133 In parallel, bottom-up approaches in which –SO3H groups were anchored directly into COF backbones eliminated acid leaching and ensured uniform proton-source distribution across channels, resulting in binder-free electrolytes with enhanced durability (Fig. 28).137 These studies transformed SCOFs from conceptual proton conductors into structurally controlled model systems, where proton conductivity can be quantitatively correlated with acid-site periodicity, pore confinement, and framework order.
 | ||
| Fig. 28 Synthesis of S-COF-1 and S-COF-2 solid catalysts. Reproduced with permission from ref. 137 Copyright 2022 Wiley-VCH. | ||
The application scope of proton-conducting COFs has recently expanded beyond classical fuel-cell membranes toward emerging proton-based energy-storage devices, where ordered ion transport and stability under demanding conditions are critical. In a notable 2025 study, a SCOF loaded with phosphoric acid was shown to function as a solid electrolyte with superprotonic conductivity of 1.06 × 10−1 S cm−1 at 423 K under anhydrous conditions. The combination of periodically aligned acidic sites and confined phosphoric acid enabled fast proton transport while suppressing acid volatilization, allowing stable operation within an electrochemical window of 2.4 V. Integration of this electrolyte into rechargeable proton batteries with vanadium hexacyanoferrate cathodes delivered a specific capacity of 101.8 mAh g−1 and retained 80% of the initial performance after 1000 cycles, underscoring the viability of COFs for solid-state proton batteries.288 Complementing this device-oriented approach, a parallel 2025 study demonstrated that nitrile- and ether-rich COFs can also act as active nanofillers in sulfonated poly(ether ether ketone), where their polar channels improve water management, mechanical integrity, and proton transport. The resulting composite membranes achieved conductivities up to 217 mS cm−1 at 80 °C and 90% RH and delivered a 2.3-fold increase in fuel-cell power density, highlighting the dual functionality of COFs as both structural and proton-conducting components.289 Structural innovations reported in 2025 further advanced COF-based proton conductors by directly addressing long-term durability and operation under harsh conditions. One representative strategy involved the development of perfluoroalkyl- and –SO3H-functionalized COFs that deliberately mimic key architectural features of Nafion®, combining hydrophobic stabilization with densely packed Brønsted acid sites. The introduction of fluorinated segments created water-resistant pore environments that suppressed acid leaching while markedly enhancing thermal and chemical stability, with frameworks remaining intact up to 350 °C and under strongly acidic conditions. Although moderate conductivities were observed under humidified operation (7.7 × 10−3 S cm−1 at 90 °C and 97% RH), phosphoric acid doping enabled efficient anhydrous proton transport, reaching 1.93 × 10−2 S cm−1 at 140 °C (Fig. 29).139 Building on this durability-driven design, a complementary 2025 study introduced dual-acid-tailored COFs incorporating both sulfonic and phosphonic acid groups within a single framework. The cooperative interaction between these acidic functionalities generated dense hydrogen-bond networks that dramatically lowered activation energies (0.12–0.15 eV) and enabled ultrafast proton conduction up to 0.243 S cm−1 at 80 °C and 95% RH.291 Together, these studies highlight how chemical synergy and pore-level engineering can reconcile high conductivity with long-term operational stability, paving the way toward practical next-generation proton exchange membranes.
 | ||
| Fig. 29 Bottom-up strategy for the synthesis of COF-C2/Fx-SO3 (x = 6, 8). Reproduced with permission from ref. 139 Copyright 2025 American Chemical Society. | ||
The incorporation of crystalline and porous frameworks into sulfonated polyether ether ketone (SPEEK) has recently emerged as a promising strategy to overcome the conductivity-stability trade-off inherent to conventional polymer electrolytes. One representative example is the use of triazole-functionalized COFs capable of immobilizing phosphotungstic acid (HPW). Fan et al. demonstrated that blending HPW-loaded triazole COFs into SPEEK could simultaneously enhance proton conduction and stability. The triazole moieties provided anchoring sites that prevented HPW leaching while also facilitating hydrogen-bonded proton transport. The resulting hybrid membranes achieved conductivities above 10−2 S cm−1 under humid conditions, clearly outperforming pristine SPEEK. This approach preserved the intrinsic acidity of HPW while mitigating its solubility, thereby combining the high conductivity of inorganic acids with the processability of polymer electrolytes.128
In a different design, Yin and co-workers incorporated two-dimensional SCOF nanosheets into the SPEEK matrix. Unlike guest-loaded systems, here the –SO3H groups were covalently anchored to the COF network, establishing a robust and ordered network of Brønsted acid sites. The nanosheet morphology promoted homogeneous dispersion within the polymer phase, suppressed phase separation, and created continuous pathways for proton migration. These hybrid membranes achieved conductivity as high as ∼0.35 S cm−1 at 80 °C and full relative humidity, nearly twice that of pure SPEEK, while also displaying superior mechanical integrity and oxidative stability. This study highlighted the advantages of introducing nanoscale crystalline order and fixed sulfonic functionalities into polymer systems, offering a pathway to high-performance membranes with improved durability.292
More recently, Xu et al. reported the development of SPAFs blended into SPEEK. In contrast to crystalline COFs, SPAFs provide rigid but amorphous aromatic networks with high –SO3H density. Their incorporation restricted excessive swelling of the polymer while simultaneously enhancing water retention and proton conduction. The composite membranes maintained conductivity above 10−2 S cm−1 over a wide humidity range and demonstrated improved dimensional and mechanical stability compared with pristine SPEEK. Furthermore, long-term durability tests indicated reduced performance degradation, underscoring the effectiveness of SPAFs in addressing the limitations of conventional sulfonated polymers (Fig. 30).293
 | ||
| Fig. 30 Scheme for the preparation of SPAF/SPEEK hybrid membrane. Reproduced with permission from ref. 293 Copyright 2025 Elsevier. | ||
Polybenzimidazole (PBI) has long been recognized as a robust high-temperature polymer for fuel cell membranes, particularly when doped with phosphoric acid, yet its relatively low proton conductivity and acid leaching have motivated the development of hybrid strategies. A recent approach involves the in situ growth of a sulfonated ionic COF (SCOF) directly within the PBI matrix, generating a dense distribution of –SO3H groups and ordered transport channels while forming acid–base ionic crosslinks with the imidazole sites of the polymer. This design effectively immobilizes phosphoric acid, enhances mechanical strength, and reduces proton transport barriers, with reported conductivities of ∼542 mS cm−1 at 80 °C under full humidity and stable performance in both electrochemical hydrogen compressor and single-cell tests.294 Another study integrated melamine-derived COFs into sulfonated PBI (SPBI) to form nanocomposites where interfacial hydrogen bonding and higher ion exchange capacity (IEC) promoted continuous hydrophilic domains. At an optimized 15 wt% COF loading, the membranes exhibited conductivities around 0.108 S cm−1 at 80 °C, improved tensile strength (∼23 MPa), controlled water uptake, and delivered a peak power density of ∼394 mW cm−2 at 80 °C and full humidity, outperforming Nafion® 117 in cell tests, with oxidative stability retained at ∼91–93%.295 These results demonstrate that COF integration can significantly enhance the conductivity and durability of PBI-based membranes.
Recent efforts have also explored the integration of COFs with unconventional polymeric and biopolymer substrates, broadening the design space of hybrid proton-conducting membranes. One representative example is the combination of TpBd-2SO3H COF nanosheets with sulfonated bacterial cellulose nanofibers (SBCNFs). The strong hydrogen bonding between COFs and cellulose fibers produced dense proton-conducting networks and significantly reinforced the mechanical strength of the resulting composites. The optimized SBCNF/TpBd-2SO3H-0.5 membrane reached a conductivity of 145.7 mS cm−1 at 80 °C and 100% RH, while maintaining tensile strength of nearly 88 MPa, much higher than pristine cellulose films. Importantly, the hybrid exhibited controlled swelling and improved dimensional stability, highlighting how biopolymer-COF systems can deliver both environmental compatibility and high performance for sustainable PEM development.296 A different design strategy involved the incorporation of ultrathin SCOF nanosheets into a Nafion® matrix. The two-dimensional morphology and dense –SO3H groups of SCOFs enabled homogeneous dispersion within Nafion®, enhancing both phase separation and water retention. The optimum composite, containing 0.5 wt% SCOF loading, delivered a proton conductivity of 0.207 S cm−1 at 95 °C and 100% RH, surpassing pristine Nafion®. Correspondingly, single-cell PEMFC tests exhibited a maximum power density of 1.024 W cm−2, representing an ∼80% improvement over unmodified Nafion®. The hybrid also maintained mechanical integrity and oxidative stability, demonstrating the ability of SCOF nanosheets to address Nafion's® long-standing weaknesses under harsh operating conditions.297
Most recently, a novel hybrid strategy was demonstrated by confining SCOFs within porous polytetrafluoroethylene (PTFE) substrates to produce highly flexible composite membranes. Unlike conventional filler-polymer systems, the in situ growth of SCOFs in PTFE pores eliminated interfacial voids and ensured strong anchoring of the frameworks. The resulting membranes combined the hydrophobic stability of PTFE with the high acidity of SCOFs, achieving outstanding performance. The optimized SCOF/PTFE hybrid exhibited 0.249 S cm−1 conductivity at 80 °C and 95% RH and a peak fuel cell power density of 1195 mW cm−2, exceeding the benchmark Nafion® 117 membrane. Mechanical testing confirmed tensile strengths above 30 MPa and excellent thermal stability up to 350 °C, while durability studies showed minimal performance decay over extended operation. This dual-phase approach thus provides a scalable pathway to membranes with both exceptional proton conduction and robust long-term stability.298
5.2. Battery
COFs and POPs represent a molecularly precise platform for designing solid-state ionic conductors, directly addressing critical limitations in modern batteries. In lithium-ion batteries (LIBs), they enable safe, single-ion conduction by engineering anion-immobilized networks or nanoconfined ionic channels. Simultaneously, their ordered nanopores in vanadium redox flow batteries (VRFBs) solve the fundamental trade-off between high proton conductivity and low vanadium crossover. This section examines how strategic material design at the molecular level through fixed-anion architectures and tailored ionic nanoenvironments is advancing the performance of next-generation battery electrolytes and membranes. The promise of COF/POP-derived single-ion conductors for LIBs is illustrated by two complementary strategies that engineer Li+ transport at the molecular level. An anionic PAF converted by chlorosulfonation and lithiation (Li-SPAF1) immobilizes the counter-anion on a rigid 3D diamond-type network, creating continuous hopping sites that raise the Li+ transference number without sacrificing conductivity. Pressed Li-SPAF1 pellets deliver 0.102 mS cm−1 at 25 °C with Ea ≈ 0.21 eV, while DFT/NEB analyses attribute the performance to organized Li+ migration along sulfonate-lined micropores (calculated barrier ∼0.33 eV), and Li‖Li cells exhibit low interfacial resistance-evidence of favorable solid|Li contact.134 A complementary route boosts ion dissociation and pathway accessibility by post-modifying a pyridine COF to a zwitterionic pyridinium-sulfobetaine that self-exfoliates into nanosheets. The resulting PS-CON hosts Li+ in strongly solvating ionic environments and shortens diffusion lengths, yielding 2.19 × 10−4 S cm−1 at 25 °C (two orders higher than the parent COF), t_Li+ = 0.46, a widened electrochemical window to 4.05 V, and ultra-stable Li|Li cycling (>1200 h, <48 mV overpotential), all consistent with robust interfacial kinetics and suppressed concentration polarization.299 Together, the fixed-anion 3D SPAF architecture and the zwitterionic, self-exfoliated 2D COF nanosheets delineate a clear design space for organic solid electrolytes: anchor anionic sites to direct Li+ hopping, and engineer ionic backbones/nanosheet morphologies to enhance dissociation and lower transport barriers. These approaches are compatible with scalable processing while closing the conductivity gap to inorganic SSEs.The challenge of simultaneously achieving high proton conductivity and low vanadium ion permeability in VRFBs has spurred the development of COF-based membrane designs. In 2022, sulfonated COF nanosheets were incorporated into a SPEEK matrix, where the highly ordered nanochannels facilitated efficient proton conduction while creating tortuous pathways that hindered VO2+ diffusion. The resulting hybrid membranes exhibited reduced crossover, improved coulombic efficiency, and stable long-term cycling compared to pristine polymer electrolytes.164 A significant advance followed in 2023, when Angewandte Chemie reported the fabrication of self-standing SCOF membranes using macromolecular mediation. These thin crystalline films delivered well-defined proton-conducting channels with low activation energies, alongside high permselectivity in aqueous electrolytes, achieving energy efficiencies that surpassed those of conventional Nafion® membranes.300 In parallel, a study in the Journal of Applied Polymer Science developed hybrid membranes by immobilizing an imidazolium IL within TpPa-SO3H COFs and dispersing the IL@COF filler into a sulfonated polymer matrix. This approach stabilized the IL, enhanced proton conductivity, suppressed vanadium ion crossover, and improved the mechanical properties of the membrane, resulting in higher energy efficiencies during VRFB operation.301
COF-based membranes have been shown to significantly improve the trade-off between proton conductivity and vanadium ion selectivity in VRFBs. Incorporation of sulfonated frameworks into polymeric matrices generated hydrophilic domains enriched with Brønsted acid sites, which facilitated fast proton hopping while restricting VO2+ migration. The membranes displayed enhanced water retention and dimensional stability, leading to higher coulombic and energy efficiencies during prolonged cycling tests.100 Structural engineering through hollow-fiber COFs further established directional nanochannels with intrinsic robustness, enabling energy efficiencies above 85% under elevated current densities and confirming the ability of ordered crystalline frameworks to sustain conductivity without compromising selectivity (Fig. 31).302 A complementary approach involved blending SCOFs with ionomeric backbones to create homogeneous ionic domains, resulting in membranes that preserved high conductivity, suppressed vanadium diffusion, and maintained stable performance over extended charge–discharge cycles.290
 | ||
| Fig. 31 Preparation, morphology, and structure of the hollow fiber HF-SCOF. a) Schematic preparation process; morphology of PAN@SCOF fiber of b) surface and c) cross-sectional SEM images, d) TEM and EDS images; morphology of HF-SCOF fiber of e) surface and f) cross-sectional SEM images, g) TEM and EDS images. Reproduced with permission from ref. 302 Copyright 2025 Wiley. | ||
6. Core challenges and key research directions
The impressive scope of applications for SPOPs outlined in this review underscores their significant potential. However, a critical assessment reveals that the field is still maturing from a discovery phase toward predictive design and practical implementation. Several interconnected core challenges must be systematically addressed to advance the field beyond proof-of-concept demonstrations.(1) A predominant limitation is the frequent lack of molecular-level understanding. While high acid density is often correlated with performance, the precise nature of the active site, its local chemical environment, effective acidity (pKa distribution), spatial accessibility, and dynamic interaction with confined reactants remains poorly characterized. Mechanistic explanations often default to vague homogeneous analogies or generic references to “confinement effects”, without quantitative validation. Future work must prioritize the integration of advanced in situ and operando characterization techniques (e.g., solid-state NMR, X-ray absorption spectroscopy, vibrational spectroscopy) with computational modeling (DFT, molecular dynamics) to map acid-site strength, reactant diffusion barriers, and transition-state stabilization within engineered pores. Establishing predictive, quantitative models requires moving beyond reporting bulk properties (BET surface area, total acid density) to defining atomic-scale descriptors of active sites.
(2) The field suffers from a scarcity of direct, standardized comparisons between different SPOP classes (e.g., crystalline COF vs. amorphous HCP) or functionalization strategies (de novo vs. PSM) under identical application conditions. Consequently, fundamental questions about the advantages of long-range order, specific linkage chemistry, or pore topology for a given function remain unanswered. Optimal performance parameters (acid density vs. porosity preservation) are often found empirically. Systematic, side-by-side benchmarking studies are urgently needed. Research should focus on controlled experiments that isolate key variables such as acid density in isostructural frameworks or pore size with identical functionality to decouple their effects. This will establish true, causal structure–property relationships and enable rational material selection for targeted applications.
(3) For translation from laboratory to technology, long-term stability under real-world conditions is paramount but under-reported. The chemical stability of –SO3H groups and the hydrolytic/mechanical robustness of the framework in aqueous, thermal, or flowing reactant environments require rigorous assessment. Furthermore, the scalability of synthesis, especially for crystalline SPOPs, and their processability into application-ready forms (dense, defect-free membranes; structured monoliths; coated fabrics) present significant practical barriers. Research must adopt standardized stability testing protocols that mimic intended operational environments (e.g., accelerated stress tests for fuel cells). Parallel efforts should develop greener, scalable synthetic routes and innovative processing or composite strategies (e.g., mixed-matrix membranes, aerogels) to transform lab-scale powders into functional, engineered devices.
(4) Many studies showcase SPOPs in single-step reactions or the adsorption of model compounds. The next frontier requires moving from monofunctional materials to systems with deliberately integrated, cooperative functionalities. The deliberate design of multivariate frameworks, where –SO3H sites are combined with orthogonal functional groups (e.g., metal complexes, basic sites, redox-active moieties, or photosensitizers), is crucial. This will enable sophisticated applications such as acid–base or redox-acid tandem catalysis, stimuli-responsive capture/release, and dual ion-electron conduction. Achieving this requires sophisticated linker design, controlled multi-step PSM, and a deeper understanding of site–site interactions within the confined pore.
Addressing these core challenges will transform SPOP research from an exploratory, largely descriptive endeavor into a predictive, engineering-focused discipline. The following section outlines the specific opportunities and application horizons that become accessible once these foundational issues are resolved.
7. Outlook and future perspectives
The critical analysis above defines a clear roadmap for the field. By confronting the challenges of mechanistic understanding, benchmarking, stability, and complexity, researchers can unlock the next tier of opportunities for SPOPs. Their development has been propelled by the evolution of porous materials, from Nobel Prize-recognized MOFs to purely organic, covalently bonded frameworks. POPs, spanning amorphous HCPs to crystalline COFs, offer exceptional porosity, high surface areas, and outstanding stability. Yet, these intrinsic properties are merely the foundation. Strategic PSM, particularly sulfonation, tailors these materials to meet demanding application-specific needs, establishing a powerful structure–property–performance relationship where designed modifications yield synergistic effects and enhanced efficiency. This review has chronicled the transformative impact of creating SPOPs. By merging the robust, tunable porosity of POPs with the strong Brønsted acidity of –SO3H groups, SPOPs emerge as multifunctional platforms that surpass non-functionalized POPs and conventional solid acids like Amberlyst-15 or Nafion®. They are more than simple heterogeneous analogues of molecular acids like PTSA; their advanced functionality stems from the synergistic combination of nano-confinement, engineered porosity, and a high density of accessible acidic sites. A critical, unifying insight is the nonlinear role of acid loading, where a “more is better” approach often reduces performance due to pore blocking and hindered mass transport. Optimal performance results from a precise synergy of three key design elements: (i) High and accessible acidity: the density and strength of –SO3H sites available to reactants; (ii) Engineered nanoconfinement and porosity: pore architecture that governs mass transport, creates confined nanoreactor environments, and imparts selectivity; and (iii) Tailored surface chemistry and multifunctionality: the ability to incorporate co-functional groups (e.g., –NH2, metal complexes) to enable cooperative mechanisms or cascade processes. This design principle empowers SPOPs across three critical technological domains: heterogeneous catalysis, where they bridge homogeneous acid potency with heterogeneous practicality for biodiesel production, biomass conversion, and CO2 utilization; advanced environmental remediation and separation, where they act as highly efficient adsorbents for pollutants and selective captors for gases and critical resources; and next-generation electrochemical energy technologies, where they decouple ion conductivity from stability in fuel cells, redox flow batteries, and solid-state batteries. To realize this potential, future research should focus on several key avenues that directly address the core challenges:• Multifunctional system design: integrating –SO3H sites with metal complexes, basic sites, or photoresponsive units within a single framework for tandem catalysis and smart adsorbents.
• Stability and processability: rigorously testing long-term hydrolytic and mechanical stability under harsh conditions and developing scalable processing routes to create membranes, monoliths, and coated fabrics.
• Application in emerging energy technologies: leveraging innate proton conductivity to replace fluorinated membranes (e.g., Nafion®) in fuel cells and flow batteries, and exploring roles as solid electrolytes.
• Advanced environmental remediation: deploying SPOPs for the selective removal of emerging contaminants like pharmaceuticals and PFAS from complex water streams.
• Defect engineering: introducing controlled defects to modulate porosity, enhance active-site accessibility, and create new functionalities.
• Complex and multivariate architectures: constructing hierarchical heterostructures (e.g., POP-on-POP, POP-on-MOF) to unlock synergistic effects in catalysis, separation, sensing, and energy storage.
In conclusion, SPOPs is becoming an emerging class of functional porous materials with considerable potential in many areas. They provide a flexible and powerful platform that goes beyond the traditional concept of solid acid. By improving synthetic control, deepening fundamental understanding, and actively exploring new application areas, SPOPs are ready to move from laboratory to commercial products becoming essential materials that support solutions for sustainable chemistry, environmental protection, and energy technology. Their future is not just promising; it is necessary given the urgency of developing renewable energy technologies and replacing fluorinated polymers.
Conflicts of interest
There are no conflicts to declare.Data availability
The present review has not generated new scientific data. Any issue regarding the present submission will be addressed by the corresponding authors.Acknowledgements
This work was supported by MICINN (PID2024-155520NB-I00, PID2022-138470NB-I00, PID2021-122299NB-100, TED2021-129999B-C32, TED2021-130470B-100), VIRMAT projects in response to COVID-19 financed by the ERDF-REACT-EU resources, MSCD-Doctoral Network 2023 – “Photocatalytic Industrial Applications” (PIA) (Reference: 101168878), “Comunidad de Madrid” for European Structural Funds (S2018/NMT-4367), Laboratorios inteligentes para la ciencia del futuro: descubrimiento de materiales avanzados para fotosíntesis artificial (FOTOART5.0-CM) (Reference: TEC2024/TEC-308) and proyectos sinérgicos I + D (Y2020/NMT-6469). The authors also gratefully acknowledge the support of Lorestan University (LU).References
- T. Zhang, G. Xing, W. Chen and L. Chen, Mater. Chem. Front., 2020, 4, 332–353 RSC.
- P. Kaur, J. T. Hupp and S. T. Nguyen, ACS Catal., 2011, 1, 819–835 CrossRef CAS.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef.
- T. Zhang, V. G. Gregoriou, N. Gasparini and C. L. Chochos, Chem. Soc. Rev., 2022, 51, 4465–4483 RSC.
- A. K. Sahoo, D. Sarkar, A. Kumar and J. N. Moorthy, Chem. – Eur. J., 2025, 31, e01267 CrossRef CAS.
- S. Kitagawa, R. Kitaura and S.-I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS.
- M. Kondo, T. Yoshitomi, H. Matsuzaka, S. Kitagawa and K. Seki, Angew. Chem., Int. Ed. Engl., 1997, 36, 1725–1727 CrossRef CAS.
- B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1989, 111, 5962–5964 CrossRef CAS.
- O. M. Yaghi, G. Li and H. Li, Nature, 1995, 378, 703–706 CrossRef CAS.
- X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC.
- A. R. Oveisi, K. Zhang, A. Khorramabadi-zad, O. K. Farha and J. T. Hupp, Sci. Rep., 2015, 5, 10621 CrossRef CAS.
- J. Huang and S. R. Turner, Polym. Rev., 2018, 58, 1–41 CrossRef CAS.
- V. Davankov, S. Rogozhin and M. Tsyurupa, Mezhdunar. Kniga, 1973, 15, 463–465 CAS.
- V. A. Davankov and M. P. Tsyurupa, React. Polym., 1990, 13, 27–42 CrossRef CAS.
- M. P. Tsyurupa, Z. K. Blinnikova, N. A. Proskurina, A. V. Pastukhov, L. A. Pavlova and V. A. Davankov, Nanotechnol. Russ., 2009, 4, 665–675 CrossRef.
- N. B. McKeown, S. Makhseed and P. M. Budd, Chem. Commun., 2002, 2780–2781 RSC.
- P. M. Budd, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib and C. E. Tattershall, Chem. Commun., 2004, 230–231 RSC.
- N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675–683 RSC.
- N. B. McKeown, Polymer, 2020, 202, 122736 CrossRef CAS.
- A. López-Magano, S. Daliran, A. R. Oveisi, R. Mas-Ballesté, A. Dhakshinamoorthy, J. Alemán, H. Garcia and R. Luque, Adv. Mater., 2023, 35, 2209475 CrossRef PubMed.
- P. J. Waller, F. Gándara and O. M. Yaghi, Acc. Chem. Res., 2015, 48, 3053–3063 CrossRef CAS PubMed.
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- K. T. Tan, S. Ghosh, Z. Wang, F. Wen, D. Rodríguez-San-Miguel, J. Feng, N. Huang, W. Wang, F. Zamora, X. Feng, A. Thomas and D. Jiang, Nat. Rev. Methods Primers, 2023, 3, 1 CrossRef CAS.
- R. P. Bisbey and W. R. Dichtel, ACS Cent. Sci., 2017, 3, 533–543 CrossRef CAS PubMed.
- B. Liu, Y. Wu, L. Wang, H.-L. Jiang and Q. Li, Nat. Chem., 2026, 18, 64–172 CrossRef.
- J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 Search PubMed.
- Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC.
- J.-S. M. Lee and A. I. Cooper, Chem. Rev., 2020, 120, 2171–2214 CrossRef CAS PubMed.
- J. Kim, C. M. Moisanu, C. N. Gannett, A. Halder, J. J. Fuentes-Rivera, S. H. Majer, K. M. Lancaster, A. C. Forse, H. D. Abruña and P. J. Milner, Chem. Mater., 2021, 33, 8334–8342 CrossRef CAS.
- L. Tan and B. Tan, Chem. Soc. Rev., 2017, 46, 3322–3356 RSC.
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS PubMed.
- C. Krishnaraj, H. S. Jena, K. Leus and P. Van Der Voort, Green Chem., 2020, 22, 1038–1071 RSC.
- L. Liao, M. Li, Y. Yin, J. Chen, Q. Zhong, R. Du, S. Liu, Y. He, W. Fu and F. Zeng, ACS Omega, 2023, 8, 4527–4542 CrossRef CAS PubMed.
- B. Darsanj, E. Heydari-Bafrooei and M. Dinari, Energy Fuels, 2025, 39, 4450–4457 CrossRef CAS.
- S.-Y. Yu, J. Mahmood, H.-J. Noh, J.-M. Seo, S.-M. Jung, S.-H. Shin, Y.-K. Im, I.-Y. Jeon and J.-B. Baek, Angew. Chem., Int. Ed., 2018, 57, 8438–8442 CrossRef CAS PubMed.
- M. Liu, L. Guo, S. Jin and B. Tan, J. Mater. Chem. A, 2019, 7, 5153–5172 RSC.
- J. Tan, S. Namuangruk, W. Kong, N. Kungwan, J. Guo and C. Wang, Angew. Chem., Int. Ed., 2016, 55, 13979–13984 CrossRef CAS PubMed.
- B. Zhang, M. Wei, H. Mao, X. Pei, S. A. Alshmimri, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 12715–12719 CrossRef CAS PubMed.
- Y. Zhai, G. Liu, F. Jin, Y. Zhang, X. Gong, Z. Miao, J. Li, M. Zhang, Y. Cui, L. Zhang, Y. Liu, H. Zhang, Y. Zhao and Y. Zeng, Angew. Chem., Int. Ed., 2019, 58, 17679–17683 CrossRef CAS PubMed.
- T. Ben, H. Ren, S. Ma, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng, J. M. Simmons, S. Qiu and G. Zhu, Angew. Chem., Int. Ed., 2009, 48, 9457–9460 Search PubMed.
- T. Ben and S. Qiu, CrystEngComm, 2013, 15, 17–26 RSC.
- Y. Tian and G. Zhu, Chem. Rev., 2020, 120, 8934–8986 CrossRef CAS PubMed.
- E. Karakhanov, Y. Kardasheva, L. Kulikov, A. Maximov, A. Zolotukhina, M. Vinnikova and A. Ivanov, Catalysts, 2016, 6, 122 CrossRef.
- Y. Yuan, F. Sun, H. Ren, X. Jing, W. Wang, H. Ma, H. Zhao and G. Zhu, J. Mater. Chem., 2011, 21, 13498–13502 RSC.
- J. Germain, J. M. J. Fréchet and F. Svec, J. Mater. Chem., 2007, 17, 4989–4997 RSC.
- H. Xu, J. Gao and D. Jiang, Nat. Chem., 2015, 7, 905–912 CrossRef CAS.
- N. Naz, M. H. Manzoor, S. M. G. Naqvi, U. Ehsan, M. Aslam and F. Verpoort, Appl. Mater. Today, 2024, 38, 102198 Search PubMed.
- S. Zhang, Q. Yang, C. Wang, X. Luo, J. Kim, Z. Wang and Y. Yamauchi, Adv. Sci., 2018, 5, 1801116 CrossRef PubMed.
- J. Perego, S. Bracco, A. Comotti, D. Piga, I. Bassanetti and P. Sozzani, Angew. Chem., Int. Ed., 2021, 60, 6117–6123 CrossRef CAS PubMed.
- Z. Xiang, X. Zhou, C. Zhou, S. Zhong, X. He, C. Qin and D. Cao, J. Mater. Chem., 2012, 22, 22663–22669 RSC.
- N. A. Babujohn, A. Eluri and V. P. Nabeela, Chem. Eng. J., 2023, 464, 142459 CrossRef CAS.
- L. Yuan, N. Zheng, T. Yang, A. Li, Y. Yuan, J. Hua, L. Li and C. Zhou, J. Taiwan Inst. Chem. Eng., 2023, 144, 104733 CrossRef CAS.
- W. Lu, D. Yuan, J. Sculley, D. Zhao, R. Krishna and H.-C. Zhou, J. Am. Chem. Soc., 2011, 133, 18126–18129 CrossRef CAS PubMed.
- W. Lu, D. Yuan, D. Zhao, C. I. Schilling, O. Plietzsch, T. Muller, S. Bräse, J. Guenther, J. Blümel, R. Krishna, Z. Li and H.-C. Zhou, Chem. Mater., 2010, 22, 5964–5972 CrossRef CAS.
- X. Yang, L. Zou and H.-C. Zhou, Polymer, 2017, 126, 303–307 Search PubMed.
- J. Y. Jang, T. M. D. Le, J. H. Ko, Y.-J. Ko, S. M. Lee, H. J. Kim, J. H. Jeong, T. Thambi, D. S. Lee and S. U. Son, Chem. Mater., 2019, 31, 300–304 CrossRef CAS.
- H. J. Jeon, J. H. Choi, Y. Lee, K. M. Choi, J. H. Park and J. K. Kang, Adv. Energy Mater., 2012, 2, 225–228 CrossRef CAS.
- H. S. Choi, H. J. Jeon, J. H. Choi, G.-H. Lee and J. K. Kang, Nanoscale, 2015, 7, 18923–18927 RSC.
- A. O. Mousa, M. G. Mohamed, C.-H. Chuang and S.-W. Kuo, Polymers, 2023, 15, 1891 CrossRef CAS PubMed.
- B. Yu, Y. Tao, X. Yao, Y. Jin, S. Liu, T. Xu, H. Wang, H. Wu, W. Zhou, X. Zhou, X. Ding, X. Wang, X. Xiao, Y.-B. Zhang and J. Jiang, J. Am. Chem. Soc., 2024, 146, 28932–28940 CrossRef CAS PubMed.
- L. Zhu, Y. Cao, T. Xu, H. Yang, L. Wang, L. Dai, F. Pan, C. Chen and C. Si, Energy Environ. Sci., 2025, 18, 5675–5739 Search PubMed.
- K. S. Song, P. W. Fritz and A. Coskun, Chem. Soc. Rev., 2022, 51, 9831–9852 RSC.
- S. Fajal, S. Dutta and S. K. Ghosh, Mater. Horiz., 2023, 10, 4083–4138 RSC.
- B. Lopez-Iglesias, F. Suárez-García, C. Aguilar-Lugo, A. González Ortega, C. Bartolomé, J. M. Martínez-Ilarduya, J. G. de la Campa, Á. E. Lozano and C. Álvarez, ACS Appl. Mater. Interfaces, 2018, 10, 26195–26205 CrossRef CAS.
- J.-X. Hu, H. Shang, J.-G. Wang, L. Luo, Q. Xiao, Y.-J. Zhong and W.-D. Zhu, Ind. Eng. Chem. Res., 2014, 53, 11828–11837 Search PubMed.
- M. Gomez-Suarez, Y. Chen and J. Zhang, Polym. Chem., 2023, 14, 4000–4032 RSC.
- Q. Li, Y. Wu, X. Tan, B. Ruan, Y. Yu, X. Chen, B. Zhao and L. Huang, ACS Appl. Polym. Mater., 2025, 7, 15429–15442 Search PubMed.
- M. C. Nilavu, H. Aggarwal and N. Rajesh, ACS Appl. Eng. Mater., 2025, 3, 2007–2019 Search PubMed.
- A. Hassan and N. Das, ACS Appl. Eng. Mater., 2024, 2, 56–66 Search PubMed.
- H. Wang, Y. Zhai, Y. Li, Y. Cao, B. Shi, R. Li, Z. Zhu, H. Jiang, Z. Guo, M. Wang, L. Chen, Y. Liu, K.-G. Zhou, F. Pan and Z. Jiang, Nat. Commun., 2022, 13, 7123 CrossRef CAS PubMed.
- T. Ashirov, J. Lim, A. Robles, T. Puangsamlee, P. W. Fritz, A. Crochet, X. Wang, C. Hewson, P. Iacomi, O. Š. Miljanić and A. Coskun, Angew. Chem., Int. Ed., 2025, 64, e202423809 CrossRef CAS PubMed.
- M. Debruyne, V. Van Speybroeck, P. Van Der Voort and C. V. Stevens, Green Chem., 2021, 23, 7361–7434 Search PubMed.
- W. Qu, J. Hu, H. Xu, C. Chen, Q. Sun, J. Chen and X. Jia, Catal. Sci. Technol., 2025, 15, 7004–7008 RSC.
- M. C. Borrallo-Aniceto, M. Pintado-Sierra, A. Valverde-González, U. Díaz, F. Sánchez, E. M. Maya and M. Iglesias, Green Chem., 2024, 26, 1975–1983 RSC.
- M. Debruyne, N. Raeymackers, H. Vrielinck, S. Radhakrishnan, E. Breynaert, M. Delaey, A. Laemont, K. Leus, J. Everaert, H. Rijckaert, D. Poelman, R. Morent, N. De Geyter, P. Van Der Voort, V. Van Speybroeck, C. V. Stevens and T. S. A. Heugebaert, ChemCatChem, 2024, 16, e202301205 CrossRef CAS.
- A. Karn, F. Sama, V. V. Chernyshev and J. N. Moorthy, ACS Appl. Polym. Mater., 2025, 7, 9120–9130 Search PubMed.
- H. Li, F. Pan, C. Qin, T. Wang and K.-J. Chen, Adv. Energy Mater., 2023, 13, 2301378 CrossRef CAS.
- R. K. Totten, Y.-S. Kim, M. H. Weston, O. K. Farha, J. T. Hupp and S. T. Nguyen, J. Am. Chem. Soc., 2013, 135, 11720–11723 CrossRef CAS PubMed.
- J. Mondal, Q. T. Trinh, A. Jana, W. K. H. Ng, P. Borah, H. Hirao and Y. Zhao, ACS Appl. Mater. Interfaces, 2016, 8, 15307–15319 CrossRef CAS.
- J. H. Ko, N. Kang, N. Park, H.-W. Shin, S. Kang, S. M. Lee, H. J. Kim, T. K. Ahn and S. U. Son, ACS Macro Lett., 2015, 4, 669–672 CrossRef CAS PubMed.
- C. Sarkar, C. Biswas, R. Paul and J. Mondal, Chem. Commun., 2025, 61, 12283–12300 Search PubMed.
- M. A. Shirani, M. Afshari, I. Romero-Muñiz, A. E. Platero-Prats, M. Dinari and F. Zamora, J. Mater. Chem. A, 2025, 13, 19172–19192 Search PubMed.
- J. Guo and D. Jiang, ACS Cent. Sci., 2020, 6, 869–879 CrossRef CAS PubMed.
- Q. Yu, Y.-Z. Cheng, Z. Li, D.-H. Yang, Q. Bo Meng and B.-H. Han, Chem. Eng. J., 2022, 442, 136275 Search PubMed.
- A. R. Oveisi, S. Heydarani, S. Daliran, E. Sanchooli, M. Khajeh, J. Albero, C. Molinet-Chinaglia and H. Garcia, Adv. Funct. Mater., 2025, 35, e09095 CrossRef CAS.
- Y. Zhu and W. Zhang, Chem. Sci., 2014, 5, 4957–4961 Search PubMed.
- A. Giri, Y. Khakre, G. Shreeraj, T. K. Dutta, S. Kundu and A. Patra, J. Mater. Chem. A, 2022, 10, 17077–17121 RSC.
- S. Oudi, A. R. Oveisi, S. Daliran, M. Khajeh, A. Dhakshinamoorthy and H. García, Nanomaterials, 2023, 13, 558 Search PubMed.
- H. Bildirir, V. G. Gregoriou, A. Avgeropoulos, U. Scherf and C. L. Chochos, Mater. Horiz., 2017, 4, 546–556 RSC.
- Y. Feng, H. Zhao, J. Liu, Z. Song, W. Che, K. Ma and Y. Wang, Constr. Build. Mater., 2023, 409, 134042 CrossRef CAS.
- M. G. Mohamed, A. F. M. El-Mahdy, M. G. Kotp and S.-W. Kuo, Mater. Adv., 2022, 3, 707–733 RSC.
- P. Zhao, L. Tian, Y. Guo, B. Lv, X. Mao, T. Li, J. Cui, J. Guo and B. Yang, Eur. Polym. J., 2022, 162, 110918 CrossRef CAS.
- Q. Sun, B. Aguila, Y. Song and S. Ma, Acc. Chem. Res., 2020, 53, 812–821 Search PubMed.
- A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef CAS.
- W. Liu, Y. Yang, L. Guo, J. Di, C. Hon Lau, M. V. Bermeshev and L. Shao, Chem. Eng. J., 2024, 498, 155569 CrossRef CAS.
- A. Modak, A. Ghosh, A. R. Mankar, A. Pandey, M. Selvaraj, K. K. Pant, B. Chowdhury and A. Bhaumik, ACS Sustainable Chem. Eng., 2021, 9, 12431–12460 Search PubMed.
- J. Ozdemir, I. Mosleh, M. Abolhassani, L. F. Greenlee, R. R. Beitle and M. H. Beyzavi, Front. Energy Res., 2019, 7, 77 CrossRef.
- Y. Zhu, P. Xu, X. Zhang and D. Wu, Chem. Soc. Rev., 2022, 51, 1377–1414 Search PubMed.
- X. H. Xiong, Y. Tao, Z. W. Yu, L. X. Yang, L. J. Sun, Y. L. Fan and F. Luo, Chem. Eng. J., 2020, 384, 123240 Search PubMed.
- B. Pang, R. Du, W. Chen, F. Cui, N. Wang, H. Zhao, G. Xie, L. Tiantian, G. He and X. Wu, Energy Storage Mater., 2024, 67, 103293 Search PubMed.
- Y. Huang, L. Zhi, C. Cheng, Q. Wei, S. Sun and C. Zhao, Colloids Surf., B, 2025, 253, 114716 Search PubMed.
- T.-X. Wang, H.-P. Liang, D. A. Anito, X. Ding and B.-H. Han, J. Mater. Chem. A, 2020, 8, 7003–7034 Search PubMed.
- X. Liu, C.-F. Liu, S. Xu, T. Cheng, S. Wang, W.-Y. Lai and W. Huang, Chem. Soc. Rev., 2022, 51, 3181–3225 RSC.
- N. Singh, S. Son, J. An, I. Kim, M. Choi, N. Kong, W. Tao and J. S. Kim, Chem. Soc. Rev., 2021, 50, 12883–12896 Search PubMed.
- D. Luo, M. Li, Q. Ma, G. Wen, H. Dou, B. Ren, Y. Liu, X. Wang, L. Shui and Z. Chen, Chem. Soc. Rev., 2022, 51, 2917–2938 Search PubMed.
- S. Wang, H. Li, H. Huang, X. Cao, X. Chen and D. Cao, Chem. Soc. Rev., 2022, 51, 2031–2080 RSC.
- K. Miller, J. M. Gayle, S. Roy, M. H. Abdellah, R. Hardian, L. Cseri, P. G. Demingos, H. R. Nadella, F. Lee, M. Tripathi, S. Gupta, G. Guo, S. Bhattacharyya, X. Wang, A. B. Dalton, A. Garg, C. V. Singh, R. Vajtai, G. Szekely and P. Ajayan, Small, 2024, 20, 2401269 Search PubMed.
- M. G. Rabbani, R. K. Sasse, S. Behera, P. Jena, J. Liu, P. K. Thallapally, T. Islamoglu, M. K. Shehab, M. M. Kaid, O. K. Farha and H. M. El-Kaderi, Langmuir, 2024, 40, 8024–8034 Search PubMed.
- H. Li, Z. Zhou, T. Ma, K. Wang, H. Zhang, A. H. Alawadhi and O. M. Yaghi, J. Am. Chem. Soc., 2024, 146, 35486–35492 Search PubMed.
- F. Gandomkar, M. Khajeh, A. R. Oveisi, P. Karimi and H. García, Polymer, 2025, 316, 127858 Search PubMed.
- A. H. Zare, M. Khajeh, A. R. Oveisi, S. Daliran and M. Ghaffari-Moghaddam, J. Polym. Environ., 2024, 32, 5771–5782 CrossRef CAS.
- D. Karimi, M. Khajeh, A. R. Oveisi, M. Bohlooli, A. Khatibi, R. S. Neyband and R. Luque, Environ. Pollut., 2023, 334, 122109 Search PubMed.
- N. Nouruzi, M. Dinari, B. Gholipour, M. Afshari and S. Rostamnia, ACS Appl. Nano Mater., 2022, 5, 6241–6248 CrossRef CAS.
- H. Ali, Y. Orooji, A. Y. A. Alzahrani, M. H. Al Mughram, A. M. Abu-Dief, I. Omar, A. Hayat, D. Yue and Y. Xu, Coord. Chem. Rev., 2025, 543, 216884 Search PubMed.
- J. H. Kim, D. W. Kang, H. Yun, M. Kang, N. Singh, J. S. Kim and C. S. Hong, Chem. Soc. Rev., 2022, 51, 43–56 Search PubMed.
- A. Munyentwali, C. Li, H. Li and Q. Yang, Chem. – Asian J., 2021, 16, 2041–2047 Search PubMed.
- M. B. Karimi, F. Mohammadi and K. Hooshyari, Int. J. Hydrogen Energy, 2019, 44, 28919–28938 Search PubMed.
- F. Liu, X. Meng, Y. Zhang, L. Ren, F. Nawaz and F.-S. Xiao, J. Catal., 2010, 271, 52–58 Search PubMed.
- S. Bhunia, B. Banerjee and A. Bhaumik, Chem. Commun., 2015, 51, 5020–5023 RSC.
- Y. Peng, Z. Hu, Y. Gao, D. Yuan, Z. Kang, Y. Qian, N. Yan and D. Zhao, ChemSusChem, 2015, 8, 3208–3212 CrossRef CAS PubMed.
- N. Park, Y. N. Lim, S. Y. Kang, S. M. Lee, H. J. Kim, Y.-J. Ko, B. Y. Lee, H.-Y. Jang and S. U. Son, ACS Macro Lett., 2016, 5, 1322–1326 Search PubMed.
- M. G. Goesten, À. Szécsényi, M. F. de Lange, A. V. Bavykina, K. B. S. S. Gupta, F. Kapteijn and J. Gascon, ChemCatChem, 2016, 8, 961–967 CrossRef CAS.
- R. Gomes, P. Bhanja and A. Bhaumik, J. Mol. Catal. A: Chem., 2016, 411, 110–116 CrossRef CAS.
- Y. Peng, G. Xu, Z. Hu, Y. Cheng, C. Chi, D. Yuan, H. Cheng and D. Zhao, ACS Appl. Mater. Interfaces, 2016, 8, 18505–18512 CrossRef CAS PubMed.
- P. Bhanja, A. Palui, S. Chatterjee, Y. V. Kaneti, J. Na, Y. Sugahara, A. Bhaumik and Y. Yamauchi, ACS Sustainable Chem. Eng., 2020, 8, 2423–2432 Search PubMed.
- S.-J. Yang, X. Ding and B.-H. Han, Langmuir, 2018, 34, 7640–7646 Search PubMed.
- Z. Li, Z. Liu, H. Li, M. Hasan, A. Suwansoontorn, G. Du, D. Wang, Y. Zhang and Y. Nagao, ACS Appl. Polym. Mater., 2020, 2, 3267–3273 Search PubMed.
- C. Fan, H. Wu, Y. Li, B. Shi, X. He, M. Qiu, X. Mao and Z. Jiang, Solid State Ion., 2020, 349, 115316 Search PubMed.
- Y. Lei, M. Zhang, G. Leng, C. Ding and Y. Ni, Microporous Mesoporous Mater., 2020, 299, 110110 Search PubMed.
- W. Zhao, Y. Jiao, R. Gao, L. Wu, S. Cheng, Q. Zhuang, A. Xie and W. Dong, Chem. Eng. J., 2020, 391, 123591 CrossRef CAS.
- M. A. Kalinina, L. A. Kulikov, K. A. Cherednichenko, A. L. Maximov and E. A. Karakhanov, Pet. Chem., 2021, 61, 1061–1070 Search PubMed.
- B.-J. Yao, W.-X. Wu, L.-G. Ding and Y.-B. Dong, J. Org. Chem., 2021, 86, 3024–3032 CrossRef CAS PubMed.
- Y. Zhang, C. Li, Z. Liu, Y. Yao, M. M. Hasan, Q. Liu, J. Wan, Z. Li, H. Li and Y. Nagao, CrystEngComm, 2021, 23, 6234–6238 Search PubMed.
- S. Lee, J. Jeong, J. Moon, M. Kim, K. V. L. Amarasinghe, K. Yoon Chung, H.-D. Lim and D. Whang, Chem. Eng. J., 2021, 424, 130527 CrossRef CAS.
- H. Dong, Z. Zhu, K. Li, Q. Li, W. Ji, B. He, J. Li and X. Ma, J. Membr. Sci., 2021, 635, 119440 CrossRef CAS.
- H. Zhou, C. Rayer, A. R. Antonangelo, N. Hawkins and M. Carta, ACS Appl. Mater. Interfaces, 2022, 14, 20997–21006 CrossRef CAS PubMed.
- L. Zhai, Y. Yao, B. Ma, M. M. Hasan, Y. Han, L. Mi, Y. Nagao and Z. Li, Macromol. Rapid Commun., 2022, 43, 2100590 Search PubMed.
- Z. Shao, X. Xue, K. Gao, J. Chen, L. Zhai, T. Wen, S. Xiong, H. Hou and L. Mi, J. Mater. Chem. A, 2023, 11, 3446–3453 Search PubMed.
- Y. Du, Y. Yang, Z. Shan, X. Xu, H. Xu, B. He, Y. Liu, X. Zhang, B. Xu and G. Zhang, ACS Appl. Nano Mater., 2025, 8, 6747–6754 CrossRef CAS.
- P. S. Tully, in Kirk-Othmer Encyclopedia of Chemical Technology, Sulfonic Acids, Wiley, New York, 2000 DOI:10.1002/0471238961.1921120620211212.a01.
- B. Bita, Curr. Org. Chem., 2011, 15, 3091–3097 Search PubMed.
- S. Pal, D. Das and S. Bhunia, Org. Biomol. Chem., 2024, 22, 1527–1579 RSC.
- J. C. Wu, B. H. Wang, D. L. Zhang, G. F. Song, J. T. Yuan and B. F. Liu, J. Chem. Technol. Biotechnol., 2001, 76, 619–623 Search PubMed.
- M. P. D. Mahindaratne and K. Wimalasena, J. Org. Chem., 1998, 63, 2858–2866 Search PubMed.
- S. Kang, G. Zhang, X. Yang, H. Yin, X. Fu, J. Liao, J. Tu, X. Huang, F. G. F. Qin and Y. Xu, Energy Fuels, 2017, 31, 2847–2854 CrossRef CAS.
- J. P. Guthrie, Can. J. Chem., 1978, 56, 2342–2354 CrossRef CAS.
- L. Jin, Z. Ren, C. Yang, J. Li, Y. Shan and P. Liu, Appl. Catal., A, 2025, 698, 120236 Search PubMed.
- A. Corma, Curr. Opin. Solid State Mater. Sci., 1997, 2, 63–75 Search PubMed.
- G. De Crescenzo, B. de Santos, M. J. Capitán, J. Álvarez, S. Cabrera, A. Fraile, M. Blanco and J. Alemán, J. Mater. Chem. A, 2025, 13, 23928–23938 RSC.
- C.-J. Wu, X.-Y. Li, T.-R. Li, M.-Z. Shao, L.-J. Niu, X.-F. Lu, J.-L. Kan, Y. Geng and Y.-B. Dong, J. Am. Chem. Soc., 2022, 144, 18750–18755 CrossRef CAS PubMed.
- C.-J. Wu, M.-Z. Shao, J.-L. Kan, W.-J. Liang, T.-R. Li, L.-J. Niu, H.-J. Wang, Y. Geng and Y.-B. Dong, ACS Mater. Lett., 2024, 6, 5016–5022 CrossRef CAS.
- G.-B. Wang, Y.-J. Wang, J.-L. Kan, K.-H. Xie, H.-P. Xu, F. Zhao, M.-C. Wang, Y. Geng and Y.-B. Dong, J. Am. Chem. Soc., 2023, 145, 4951–4956 Search PubMed.
- S. Daliran, M. Blanco, A. Dhakshinamoorthy, A. R. Oveisi, J. Alemán and H. García, Adv. Funct. Mater., 2024, 34, 2312912 Search PubMed.
- A. Blocher, F. Mayer, P. Schweng, T. M. Tikovits, N. Yousefi and R. T. Woodward, Mater. Adv., 2022, 3, 6335–6342 Search PubMed.
- S. Ge, K. Wei, W. Peng, R. Huang, E. Akinlabi, H. Xia, M. W. Shahzad, X. Zhang, B. B. Xu and J. Jiang, Chem. Soc. Rev., 2024, 53, 11259–11302 Search PubMed.
- R. Li, X. Tang, J. Wu, K. Zhang, Q. Zhang, J. Wang, J. Zheng, S. Zheng, J. Fan, W. Zhang, X. Li and S. Cai, Chem. Eng. J., 2023, 464, 142706 CrossRef CAS.
- Z. Babaei, A. N. Chermahini, M. Dinari, M. Saraji and A. Shahvar, Sustainable Energy Fuels, 2019, 3, 1024–1032 Search PubMed.
- Z. Yang, P. Chen, W. Hao, Z. Xie, Y. Feng, G. Xing and L. Chen, Chem. – Eur. J., 2021, 27, 3817–3822 Search PubMed.
- J. Liu, H. Zhu, Z. Qiang, Q. Zhao, C. Que, C. Liu, M. Li, J. Cao, J. Wang and X. Yang, Sep. Purif. Technol., 2025, 364, 132466 CrossRef CAS.
- W. Meng, Q. Xue, J. Zhu and K. Zhang, npj Clean Water, 2024, 7, 23 CrossRef CAS.
- Y. Yang, D. Wang, Q. Liu, Y. Zou, M. Wang, L. Li, Y. Lan, Y. Xiao, J. Hu, H. Zhang, M. Hu, K. Zhang, J. Wu and D. Gao, Sep. Purif. Technol., 2025, 354, 129280 Search PubMed.
- Z. Sun, X. Yang, X. Huang, M. Zhang, G. Bian, Y. Qi, X. Yang and W. Zhang, New J. Chem., 2019, 43, 16676–16684 RSC.
- R. M. N. Kalla, M.-R. Kim and I. Kim, Ind. Eng. Chem. Res., 2018, 57, 11583–11591 CrossRef CAS.
- J. Li, F. Xu, Y. Chen, Y. Han and B. Lin, ACS Appl. Energy Mater., 2022, 5, 15856–15863 CrossRef CAS.
- Y. He, X. Lin, J. Chen and H. Zhan, J. Membr. Sci., 2021, 635, 119476 CrossRef CAS.
- J. Wolska, M. Frankowski, J. Jenczyk and L. Wolski, Sep. Purif. Technol., 2024, 343, 127147 Search PubMed.
- K. Dong, J. Zhang, W. Luo, L. Su and Z. Huang, Chem. Eng. J., 2018, 334, 1055–1064 Search PubMed.
- J. Li, X. Wang, G. Chen, D. Li, Y. Zhou, X. Yang and J. Wang, Appl. Catal., B, 2015, 176–177, 718–730 CrossRef CAS.
- Q. Sun, K. Hu, K. Leng, X. Yi, B. Aguila, Y. Sun, A. Zheng, X. Meng, S. Ma and F.-S. Xiao, J. Mater. Chem. A, 2018, 6, 18712–18719 RSC.
- X. Zhao, Q. Wang, R. Kunthom and H. Liu, ACS Appl. Mater. Interfaces, 2023, 15, 6657–6665 Search PubMed.
- B.-J. Chen, J.-P. Wu, Rukhsana, F.-Y. Xiang, Y.-M. Shen and X.-Z. Liang, Kinet. Catal., 2024, 65, 513–520 Search PubMed.
- C. Yadav, V. K. Maka, S. Payra and J. N. Moorthy, ACS Appl. Polym. Mater., 2020, 2, 3084–3093 Search PubMed.
- X. Wang, P. Ma, C. Yu, J. Bian and X. Meng, Appl. Catal., A, 2025, 700, 120298 Search PubMed.
- N. Wei, Q. Wang, L. Guo, D. Wang, T. Chen and G. Wang, ChemistrySelect, 2024, 9, e202304065 Search PubMed.
- Y. Li, L. Xing, Z. Chen, G. Zhan, B. Yuan, Y. Peng, L. Wang and J. Li, Appl. Catal., B, 2024, 358, 124350 CrossRef CAS.
- Q. Xue, L. Cheng, Q. Qu, L. Yang, C. Fang, H. Li, J. Ding, H. Wan and G. Guan, J. Environ. Chem. Eng., 2024, 12, 113718 CrossRef CAS.
- Q. Yan, H. Xu, X. Jing, H. Hu, S. Wang, C. Zeng and Y. Gao, RSC Adv., 2020, 10, 17396–17403 RSC.
- R. M. N. Kalla, S. S. Reddy and I. Kim, Catal. Lett., 2019, 149, 2696–2705 Search PubMed.
- L. A. Kulikov, M. A. Bazhenova, Y. S. Bolnykh, D. A. Makeeva, M. V. Terenina, Y. S. Kardasheva, A. L. Maximov and E. A. Karakhanov, Pet. Chem., 2022, 62, 1195–1203 CrossRef CAS.
- S. Daliran, S. F. Hosseini, A. R. Oveisi, E. Sanchooli, E. Rodríguez-Castellón and D. Ballesteros-Plata, BMC Chem., 2025, 19, 215 CrossRef CAS PubMed.
- G. D. Kotkar and S. G. Tilve, ACS Omega, 2024, 9, 47543–47556 Search PubMed.
- S. S. Reddy, R. M. N. Kalla, A. Varyambath and I. Kim, Catal. Commun., 2019, 126, 15–20 CrossRef CAS.
- Z. Alishahi, M. Torabi, M. A. Zolfigol and M. Yarie, J. Solid State Chem., 2023, 324, 124119 CrossRef CAS.
- T. Mondal, J. Seth, S. Sarkar and S. M. Islam, New J. Chem., 2023, 47, 8885–8893 RSC.
- S. Darvishi, S. Sadjadi and A. Rezvanian, J. Phys. Chem. Solids, 2025, 199, 112555 CrossRef CAS.
- M. Du, A. M. Agrawal, S. Chakraborty, S. J. Garibay, R. Limvorapitux, B. Choi, S. T. Madrahimov and S. T. Nguyen, ACS Sustainable Chem. Eng., 2019, 7, 8126–8135 Search PubMed.
- B. Ye, W. Zhang, R. Zhou, Y. Jiang, Z. Zhong and Z. Hou, New J. Chem., 2022, 46, 6756–6764 RSC.
- J. Artz, I. Delidovich, M. Pilaski, J. Niemeier, B. M. Kübber, K. Rahimi and R. Palkovits, RSC Adv., 2018, 8, 22392–22401 RSC.
- Y.-R. Du, B.-H. Xu, J.-S. Pan, Y.-W. Wu, X.-M. Peng, Y.-F. Wang and S.-J. Zhang, Green Chem., 2019, 21, 4792–4799 Search PubMed.
- D. Yuan, L. Li, F. Li, Y. Wang, F. Wang, N. Zhao and F. Xiao, ChemSusChem, 2019, 12, 4986–4995 Search PubMed.
- W. Song, Y. Zhang, A. Varyambath, J. S. Kim and I. Kim, Green Chem., 2020, 22, 3572–3583 RSC.
- Z. Guo and X. Liang, Res. Chem. Intermed., 2024, 50, 705–722 Search PubMed.
- P. Schweng, D. Guerin-Faucheur, F. Kleitz and R. T. Woodward, Chem. Commun., 2024, 60, 14395–14398 RSC.
- J. Kim, S. Ravi, K. Kim, Y. Choi, H.-H. Park and Y.-S. Bae, ACS Appl. Mater. Interfaces, 2023, 15, 48485–48494 Search PubMed.
- X. Jia, W. Xie and H. Li, React. Chem. Eng., 2025, 10, 1932–1948 RSC.
- S. Y. Kang, Y. N. Lim, Y.-J. Cheong, S. M. Lee, H. J. Kim, Y.-J. Ko, B. Y. Lee, H.-Y. Jang and S. U. Son, Ind. Eng. Chem. Res., 2017, 56, 10235–10241 Search PubMed.
- W. Kang, C. Zhang, G. Li, P. Zhang and Y. Li, Microporous Mesoporous Mater., 2022, 343, 112151 Search PubMed.
- W. Kang, X. Wang, Y. Ren, P. Zhang, A. Huang and G. Li, RSC Adv., 2024, 14, 34578–34585 Search PubMed.
- K. Gong, C. Li, D. Zhang, H. Lu, Y. Wang, H. Li and H. Zhang, Mol. Catal., 2022, 519, 112139 Search PubMed.
- G. Kalita, N. Deka, D. Paul, L. Thapa, G. K. Dutta and P. N. Chatterjee, Synlett, 2020, 304–308 Search PubMed.
- A. A. Raza, S. Ravi, S. S. Tajudeen and A. K. I. Sheriff, J. Solid State Chem., 2021, 302, 122417 CrossRef CAS.
- Y. Pang, B. Wang, Y. Kang, J. Li, X. Gu, G. Kang, X. Yan, Y. Li and L. Chen, Chem. Eng. Sci., 2022, 260, 117933 CrossRef CAS.
- D. Jiang, X. Xu, Y. Bando, S. M. Alshehri, M. Eguchi, T. Asahi and Y. Yamauchi, Bull. Chem. Soc. Jpn., 2024, 97, uoae074 CrossRef CAS.
- L. Cui, H. Yuan, B. Yang, Q. Zhang, H. Zhang and J. Li, Desalination, 2025, 612, 118971 CrossRef CAS.
- S.-S. Mortazavi, A. Abbasi and M. Masteri-Farahani, Colloids Surf., A, 2020, 599, 124703 CrossRef CAS.
- F. Yang, W. Xue, D. Zhang, F. Li and Y. Wang, React. Kinet., Mech. Catal., 2016, 117, 329–339 CrossRef CAS.
- W. Xue, L. Sun, F. Yang, Z. Wang and F. Li, Materials, 2016, 9, 833 CrossRef PubMed.
- B. Hoque, M. Y. Khan, A. Hanif, M. Abdelnaby, A. Helal, A. Khan, M. Usman and Q. A. Drmosh, J. Environ. Chem. Eng., 2024, 12, 112695 CrossRef CAS.
- D. Ma, Y. Song, H. Zhao, C. Yu, Y. Zhang, C. Li and K. Liu, ACS Sustainable Chem. Eng., 2023, 11, 6183–6190 CrossRef CAS.
- Y.-R. Du, X. Yang, Y.-F. Wang, P.-X. Guan, R. Wang and B.-H. Xu, Mol. Catal., 2022, 520, 112164 CAS.
- M. Yin, L. Wang and S. Tang, ACS Appl. Mater. Interfaces, 2022, 14, 55674–55685 CrossRef CAS PubMed.
- H. Veisi, S. Taheri and S. Hemmati, Green Chem., 2016, 18, 6337–6348 RSC.
- N. Iranpoor, H. Firouzabadi and E. E. Davan, Tetrahedron Lett., 2013, 54, 1813–1816 CrossRef CAS.
- L. A. Kulikov, M. A. Bazhenova, I. S. Bolnykh, A. L. Maximov and E. A. Karakhanov, Catal. Lett., 2024, 154, 6106–6122 CrossRef CAS.
- F. Doraghi, F. Mohaghegh, O. H. Qareaghaj, B. Larijani and M. Mahdavi, RSC Adv., 2023, 13, 13947–13970 RSC.
- J. Chun, S. Padmanaban and Y. Lee, J. Am. Chem. Soc., 2025, 147, 16642–16652 CrossRef CAS PubMed.
- A. R. Oveisi, A. Khorramabadi-zad and S. Daliran, RSC Adv., 2016, 6, 1136–1142 RSC.
- R. Ghorbani-Vaghei, A. Davood, S. Daliran and A. R. Oveisi, RSC Adv., 2016, 6, 29182–29189 RSC.
- A. T. Khan, A. Ghosh and M. Musawwer Khan, Tetrahedron Lett., 2012, 53, 2622–2626 CrossRef CAS.
- N. Salehi and B. B. Fatameh Mirjalili, RSC Adv., 2017, 7, 30303–30309 RSC.
- M. Masteri-Farahani, M. Ghahremani and M. Niakan, Energy Fuels, 2025, 39, 15225–15241 CrossRef CAS.
- A. Priya, Y. Hu, J. Mou, C. Du, K. Wilson, R. Luque and C. S. K. Lin, in Handbook of Biofuels Production, ed. R. Luque, C. S. K. Lin, K. Wilson and C. Du, Woodhead Publishing, 3rd edn, 2023, ch. 1, pp. 3–24 DOI:10.1016/B978-0-323-91193-1.00002-0.
- H. Fukuda, A. Kondo and H. Noda, J. Biosci. Bioeng., 2001, 92, 405–416 CrossRef CAS PubMed.
- M. Hong, J. Chen and E. Y. X. Chen, Chem. Rev., 2018, 118, 10551–10616 CrossRef CAS PubMed.
- X. Wang, D. Wu, X. Mu, W. Kang, G. Li, A. Huang and Y. Xie, Polymers, 2023, 15, 555 CrossRef CAS PubMed.
- L.-p Yue, F.-x Kong, Y. Wang, G.-d Sun and J.-f Chen, J. Membr. Sci., 2023, 685, 121877 CrossRef CAS.
- S. Borsap, M. Charernsuk, K. Tangjituabun and J. Tantirungrotechai, ChemCatChem, 2025, 17, e202500342 CrossRef CAS.
- D. Azarifar, R. Ghorbani-Vaghei, S. Daliran and A. R. Oveisi, ChemCatChem, 2017, 9, 1992–2000 CrossRef CAS.
- E. Merino, E. Verde-Sesto, E. M. Maya, M. Iglesias, F. Sánchez and A. Corma, Chem. Mater., 2013, 25, 981–988 CrossRef CAS.
- Q. Liu, Z. Li, J. Sun, Y. Lan, J. Hu, Y. Xiao, F.-Q. Yang and D. Gao, J. Sep. Sci., 2025, 48, e70101 CrossRef CAS PubMed.
- L. Ma, M. M. Wanderley and W. Lin, ACS Catal., 2011, 1, 691–697 CrossRef CAS.
- K. N. Parida, C. Yadav, S. Jindal, P. Tamuly, Y. D. Vankar and J. N. Moorthy, Chem. – Eur. J., 2025, 31, e202404525 CrossRef CAS PubMed.
- X. Luo, S. Zhou, S. Zhou, D. Wang, Y. Zhang, X. Hao, G. Liu, P. Liu and P. Gu, ACS Appl. Polym. Mater., 2024, 6, 10615–10624 CrossRef CAS.
- N. Taheri and M. Dinari, Appl. Surf. Sci., 2023, 18, 100543 CrossRef.
- L. Yang, Z. Zhan, L. Zhao, C. Zhang, S. Wang, W. Hu and G. Zhu, Chem. Sci., 2025, 16, 775–783 RSC.
- Q. Li, S. Li, C. Yu, Z. Zhan, G. Cheng, B. Tan and S. Ren, Commun. Mater., 2024, 5, 255 CrossRef CAS.
- Y. Guo, Q.-M. Hasi, S. Hu, S. Jiang, J. Long, C. Xiao, Y. Zhang and L. Chen, Langmuir, 2024, 40, 23382–23397 CrossRef CAS PubMed.
- Y. Zhang, X. Hu, K. Jiang, Z. Fan, D. Wang, P. Gu, S. Zhou and Z. Li, Polymer, 2024, 313, 127738 CrossRef CAS.
- J. Wolska, M. Muńko, H. El Siblani, I. Telegeiev, M. Frankowski, A. Szwajca, J. Walkowiak-Kulikowska, M. El-Roz and L. Wolski, J. Environ. Chem. Eng., 2023, 11, 110429 CrossRef CAS.
- Y. Zhu, R. Ding, S. Chen, X. Qu, Y. Yang and X. Zhang, Sep. Purif. Technol., 2024, 333, 125857 Search PubMed.
- M. Bi, Y. Ma, J. Chai, Y. Su and Y. Xia, Sep. Purif. Technol., 2025, 375, 133810 CrossRef CAS.
- B. Li, J. Chen, J. Xiao, L. Zhao and H. Qiu, ACS Appl. Nano Mater., 2023, 6, 2498–2506 CrossRef CAS.
- S. Liang, Q. Meng, Z. Liu, H. Cai, H. Liu, M. Rong and L. Yang, Chem. Eng. J., 2024, 499, 155850 CrossRef CAS.
- Y. Xu, J. Wei and X. Chen, Chemosensors, 2022, 10, 248 CrossRef CAS.
- M. Khosravani, M. Dehghani Ghanatghestani, F. Moeinpour and H. Parvaresh, Heliyon, 2024, 10, e25423 CrossRef CAS PubMed.
- Y. Wang, L. Zhang, S. Wang, H. Feng, F. Pang, H. Li, B. Liu, X. Zang, S. Zhang and Z. Wang, J. Water Process Eng., 2024, 68, 106321 CrossRef.
- Y. Guo, Q.-M. Hasi, J. Yu, Y. Guo, L. Song, S. Wu, X. Luo and L. Chen, Sep. Purif. Technol., 2023, 324, 124518 CrossRef CAS.
- W. Jin, J. Liu, N. Huang, Z. Wang, Y. Zhang, Y. Peng, C. Gong, Y. S. Ok and Z. Xu, ACS ES&T Eng, 2023, 3, 1511–1520 Search PubMed.
- D. W. Kang, M. Kang, M. Moon, H. Kim, S. Eom, J. H. Choe, W. R. Lee and C. S. Hong, Chem. Sci., 2018, 9, 6871–6877 RSC.
- Z. Xuan, X. Sun, X. Zhang, G. Zhang, F. Wang and D. Liu, Chem. Eng. J., 2025, 515, 163958 CrossRef CAS.
- K. Zong, K. Jiang, X. Bao and D. Deng, J. Environ. Chem. Eng., 2025, 13, 116082 Search PubMed.
- S.-Q. Deng, Y.-L. Miao, Y.-L. Tan, H.-N. Fang, Y.-T. Li, X.-J. Mo, S.-L. Cai, J. Fan, W.-G. Zhang and S.-R. Zheng, Inorg. Chem., 2019, 58, 13979–13987 CrossRef CAS PubMed.
- Q. Liao, C. Ke, X. Huang, G. Zhang, Q. Zhang, Z. Zhang, Y. Zhang, Y. Liu, F. Ning and K. Xi, J. Mater. Chem. A, 2019, 7, 18959–18970 RSC.
- J. Abdi, M. Vossoughi, N. M. Mahmoodi and I. Alemzadeh, Chem. Eng. J., 2017, 326, 1145–1158 CrossRef CAS.
- N. A. Khan, H. J. An, D. K. Yoo and S. H. Jhung, J. Hazard. Mater., 2018, 360, 163–171 Search PubMed.
- X. Song, J. Lu, M. Liu, L. Tang, L. Sun, R. Jiang and L. Zhang, Chem. Eng. J., 2024, 481, 148714 CrossRef CAS.
- J. Missau, D. A. Bertuol and E. H. Tanabe, Appl. Clay Sci., 2021, 214, 106297 Search PubMed.
- D. Shetty, S. Boutros, T. Skorjanc, B. Garai, Z. Asfari, J. Raya and A. Trabolsi, J. Mater. Chem. A, 2020, 8, 13942–13945 RSC.
- S. Chanpee, N. Kaewtrakulchai, N. Khemasiri, A. Eiad-ua and P. Assawasaengrat, Molecules, 2022, 27, 5309 CrossRef CAS PubMed.
- Y. Zhang, Y. Zhang, X. Luo, Q. Gao, Y. Liu, X. Wang, S. Zhou, D. Wang, P. Gu and Z. Li, Sep. Purif. Technol., 2023, 327, 124799 CrossRef CAS.
- W. Li, Z. Xie, S. Xue, H. Ye, M. Liu, W. Shi and Y. Liu, Sep. Purif. Technol., 2021, 276, 119435 Search PubMed.
- J. A. Cotruvo Jr., ACS Cent. Sci., 2019, 5, 1496–1506 CrossRef PubMed.
- A. Javed, P. Palafox Gonzalez and V. Thangadurai, ACS Appl. Mater. Interfaces, 2023, 15, 29674–29699 CrossRef CAS PubMed.
- Y. Nagao, ChemElectroChem, 2024, 11, e202300846 Search PubMed.
- R. Sahoo, B. Pramanik and M. C. Das, Adv. Energy Mater., 2025, 15, 2501491 CrossRef CAS.
- D. E. Matkin, I. A. Starostina, M. B. Hanif and D. A. Medvedev, J. Mater. Chem. A, 2024, 12, 25696–25714 RSC.
- V. Joseph and A. Nagai, RSC Adv., 2023, 13, 30401–30419 RSC.
- J. Si, S. Zhang, S. Zhao, J. Li, X. Feng and B. Wang, Energy Mater. Adv., 2025, 6, 0334 CrossRef CAS.
- U. Sen, H. Usta, O. Acar, M. Citir, A. Canlier, A. Bozkurt and A. Ata, Macromol. Chem. Phys., 2015, 216, 106–112 CrossRef CAS.
- B. Maria Mahimai, G. Sivasubramanian, K. Sekar, D. Kannaiyan and P. Deivanayagam, Mater. Adv., 2022, 3, 6085–6095 RSC.
- M. Arun, S. Giddey, P. Joseph and D. S. Dhawale, J. Mater. Chem. A, 2025, 13, 11236–11263 RSC.
- A. Zucconi, J. Hack, R. Stocker, T. A. M. Suter, A. J. E. Rettie and D. J. L. Brett, J. Mater. Chem. A, 2024, 12, 8014–8064 RSC.
- Q. Li, J. O. Jensen, R. F. Savinell and N. J. Bjerrum, Prog. Polym. Sci., 2009, 34, 449–477 CrossRef CAS.
- U. Sen, S. Ünügür Çelik, A. Ata and A. Bozkurt, Int. J. Hydrogen Energy, 2008, 33, 2808–2815 CrossRef CAS.
- C.-M. Chang, H.-Y. Li, J.-Y. Lai and Y.-L. Liu, RSC Adv., 2013, 3, 12895–12904 Search PubMed.
- S. M. Haile, D. A. Boysen, C. R. I. Chisholm and R. B. Merle, Nature, 2001, 410, 910–913 CrossRef CAS PubMed.
- N. Agmon, Chem. Phys. Lett., 1995, 244, 456–462 CrossRef CAS.
- H. Xu, S. Tao and D. Jiang, Nat. Mater., 2016, 15, 722–726 CrossRef CAS PubMed.
- T. Ogawa, K. Kamiguchi, T. Tamaki, H. Imai and T. Yamaguchi, Anal. Chem., 2014, 86, 9362–9366 CrossRef CAS PubMed.
- Y. Lu, S. Zhou, C. Zhu, J. Zhou and X. Feng, Chem. – Eur. J., 2025, 31, e202501116 CrossRef CAS PubMed.
- D. W. Kang, M. Kang and C. S. Hong, J. Mater. Chem. A, 2020, 8, 7474–7494 RSC.
- D. W. Kang, K. S. Lim, K. J. Lee, J. H. Lee, W. R. Lee, J. H. Song, K. H. Yeom, J. Y. Kim and C. S. Hong, Angew. Chem., Int. Ed., 2016, 55, 16123–16126 CrossRef CAS PubMed.
- D. Zhang, Y. Gao, T.-X. Luan, K. Cheng, C. Li and P.-Z. Li, Microporous Mesoporous Mater., 2021, 325, 111348 CrossRef CAS.
- Z. Li, Y. Yao, D. Wang, M. M. Hasan, A. Suwansoontorn, H. Li, G. Du, Z. Liu and Y. Nagao, Mater. Chem. Front., 2020, 4, 2339–2345 RSC.
- S. Y. Kim, M. Kang, D. W. Kang, H. Kim, J. H. Choe, H. Yun and C. S. Hong, Angew. Chem., Int. Ed., 2023, 62, e202214301 CrossRef CAS PubMed.
- S. F. Winterstein, A. F. Privalov, C. Greve, R. Siegel, B. Pötzschner, M. Bettermann, L. Adolph, J. Timm, R. Marschall, E. A. Rössler, E. M. Herzig, M. Vogel and J. Senker, J. Am. Chem. Soc., 2023, 145, 27563–27575 CrossRef CAS PubMed.
- S. F. Winterstein, M. Bettermann, J. Timm, R. Marschall and J. Senker, Molecules, 2024, 29, 1669 CrossRef PubMed.
- B. Tang, S.-T. Liu, X.-R. Ma, F.-J. Zhao, G.-Q. Zhang, X.-S. Gao, J. Yang, H.-B. Luo and X.-M. Ren, ACS Appl. Mater. Interfaces, 2025, 17, 42004–42010 CrossRef CAS PubMed.
- H. Kim, I. Song, J. Kim, H. Seo, N. Park, H. J. Choi, W. Yang, Y.-E. Sung, W. B. Lee and J.-C. Lee, ACS Appl. Mater. Interfaces, 2025, 17, 23155–23164 CrossRef CAS PubMed.
- Afzal, Y. Ren, S. Wang, H. Ma, S. Yuan, Q. Zhao, M. B. Wadud, X. Liang, Q. Pan, G. He and Z. Jiang, J. Membr. Sci., 2025, 722, 123863 CrossRef CAS.
- V. Joseph, K. Maegawa, M. Wlazło, M. J. Potrzebowski, K. Łyczko and A. Nagai, Chem. Mater., 2025, 37, 2561–2568 CrossRef CAS.
- Z. Yin, H. Geng, P. Yang, B. Shi, C. Fan, Q. Peng, H. Wu and Z. Jiang, Int. J. Hydrogen Energy, 2021, 46, 26550–26559 CrossRef CAS.
- T. Xu, R. Yu, H. Bian and N. Zhang, Inorg. Chem. Commun., 2025, 176, 114348 CrossRef CAS.
- L. Zhang, Z. Gao, Y. Kong, N. Xing, X. Pang, Z. Liu, Z. Yao, S. Zhu, H. Wu and Z. Jiang, J. Membr. Sci., 2024, 708, 123088 CrossRef CAS.
- S. Moorthy, R. Sudhakaran, P. Dayalan and P. Deivanayagam, J. Polym. Sci., 2025, 63, 4153–4163 CrossRef CAS.
- B. He, Y. Lin, Q. Zhou and S. Tang, Int. J. Hydrogen Energy, 2024, 69, 391–400 Search PubMed.
- D. Zhang, X. Li, L. Yang, L. Qian, G. Song, J. Guo and C. He, Int. J. Hydrogen Energy, 2025, 148, 150089 CrossRef CAS.
- H. Zhao, B. Pang, F. Cui, W. Chen, G. Xie, X. Wu, R. Du, C. Liu and G. He, J. Membr. Sci., 2025, 715, 123479 CrossRef CAS.
- C.-X. Liu, S. Hwang, Y. Lee, Y. H. Ko, S. S. Park and E. Lee, ACS Appl. Mater. Interfaces, 2024, 16, 48203–48210 CrossRef CAS PubMed.
- Y. Wu, Y. Wang, D. Zhang, F. Xu, L. Dai, K. Qu, H. Cao, Y. Xia, S. Li, K. Huang and Z. Xu, Angew. Chem., Int. Ed., 2023, 62, e202313571 CrossRef CAS PubMed.
- X. Meng, Q. Peng, J. Wen, K. Song, L. Peng, T. Wu, C. Cong, H. Ye and Q. Zhou, J. Appl. Polym. Sci., 2023, 140, e53802 CrossRef CAS.
- B. Pang, G. Xie, H. Zhao, F. Cui, C. Liu, W. Chen, X. Yan, X. Wu and G. He, Adv. Energy Mater., 2025, 15, 2500523 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |