
Symmetry breaking of single-atom catalysts in heterogeneous electrocatalysis: reactivity and configuration
Bin Wu†
 a,
Zuohuan Chen†b,
Yifan Yeb,
Justin Zhu Yeow Seowac,
Daniel Mandlerd,
Adrian Fishere,
Dingsheng Wangf,
Shaojun Guog and
Zhichuan J. Xu*a
a,
Zuohuan Chen†b,
Yifan Yeb,
Justin Zhu Yeow Seowac,
Daniel Mandlerd,
Adrian Fishere,
Dingsheng Wangf,
Shaojun Guog and
Zhichuan J. Xu*a
aSchool of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, Singapore, 639798, Singapore. E-mail: xuzc@ntu.edu.sg
bNational Synchrotron Radiation Laboratory, University of Science and Technology of China, Hefei, 230029, China
cNTI-NTU Corporate Laboratory, Nanyang Technological University, 50 Nanyang Avenue, Singapore, 639798, Singapore
dInstitute of Chemistry, The Hebrew University of Jerusalem, Jerusalem, 9190401, Israel
eDepartment of Chemical Engineering and Biotechnology, University of Cambridge, Phillipa Fawcett Drive, Cambridge, CB3 0AS, UK
fDepartment of Chemistry, Tsinghua University, Beijing, 100084, China
gSchool of Materials Science and Engineering, Peking University, Beijing, 100871, China
First published on 21st November 2025
Abstract
Single-atom catalysts (SACs) have emerged as transformative materials in heterogeneous electrocatalysis, yet their conventional symmetric coordination environments often yield suboptimal catalytic efficacy. This review systematically examines the deliberate disruption of local symmetry as a powerful design strategy to precisely tailor the electronic properties of SACs. We categorize and analyze atomic-level modulation approaches, including strain-induced lattice distortion, defect-engineered coordination tailoring, and curvature-derived interfacial fields, demonstrating how these strategies effectively break the intrinsic symmetry of motifs such as M–N4. Our analysis reveals that such symmetry breaking redistributes electron density around the metal center, lifts orbital degeneracy, and optimizes the d-band center, leading to enhanced intermediate adsorption, accelerated reaction kinetics, and broken scaling relationships. Furthermore, these asymmetrically configured SACs exhibit improved stability through strengthened metal–support interactions. While significant progress has been made, we conclude that future efforts must address the challenges of atomic-level precision, stability under operation, and scalable synthesis to fully realize the potential of symmetry-broken SACs across various electrocatalytic applications, thereby establishing a new paradigm for the rational design of advanced electrocatalytic materials.
Key learning points1. Symmetry breaking as a modulation principle of SACs: intentional disruption of local atomic symmetry in SACs enables precise modulation of electronic structures and reactivity beyond the constraints of conventional symmetric M–N4 coordination environments.2. Electronic structure tuning and reactivity control of SCAs: asymmetrization in single-atom configuration not only redistribute charge density but lift orbital degeneracy, optimizing adsorption energetics and lowering activation barriers in key electrocatalytic reactions. 3. Mechanistic implications of symmetry modulation of SACs: symmetry breaking decouples linear scaling relationships among intermediates, allowing independent control of activity, stability and selectivity, and offering new pathways to overcome traditional catalytic trade-offs among activity, stability and selectivity of SACs. 4. Multifaceted symmetry-breaking strategies for SACs: diverse modulation routes, including strain engineering, defect-induced coordination distortion, curvature-induced electronic field effects, enable precise symmetry control and reactivity tuning at the atomic scale. 5. Dynamic evolution of symmetry of SACs: symmetry breaking in SACs under operating conditions is sometimes not static but dynamically evolves under electrochemical environments and therefore, understanding these reversible symmetry perturbations through in situ and operando techniques is essential for correlating structural distortion with catalytic function and stability. |
1. Introduction
Amid escalating fossil fuel depletion and intensifying global environmental challenges, the pursuit of alternative, clean energy systems has emerged as a critical scientific and technological priority.1 Electrochemical energy technologies, such as fuel cells, water electrolyzers, and the electroreduction of carbon dioxide and nitrogen, are central to this transition, offering promising pathways for efficient energy conversion and storage.2 Their efficacy hinges on the performance of electrocatalysts in facilitating key reaction processes, which significantly influences overall device efficiency.3 Advancing the design of highly efficient electrocatalysts is thus essential for propelling sustainable energy innovations.4 Electrocatalysts are broadly categorized as heterogeneous or homogeneous based on their phase relative to the reactants.5,6 Heterogeneous catalysts, which include elemental metals or supported nanoparticles, are characterized by high durability under extreme conditions and ease of recovery, making them extensively employed in industrial applications.5–13 While traditional heterogeneous catalysis often utilizes metal nanoparticles (NPs), their limited atom utilization efficiency, where many atoms remain buried and inactive, has prompted the emergence of single-atom catalysts (SACs).6,14–16 SACs feature atomically dispersed metal species on engineered supports, offering maximum atom utilization and unprecedented control over active site structure and reactivity.17 As illustrated in Fig. 1, a rigorous investigation into the distinctions between SACs and other catalytic architectures is essential. SACs, achieving the theoretical maximum of metal atom dispersion, are noted for their superior atom economy and well-defined, uniform active sites, drawing comparisons to homogeneous catalysts and positioning them as bridges between homogeneous and heterogeneous catalysis, especially in applications requiring high specificity.6,18,19 | ||
| Fig. 1 Structural features of metal catalysts in the forms of NPs and SAs supported on the host, exhibiting active sites with distinct properties. | ||
The most extensively studied structural motif in SACs is the M–N4 configuration, where a metal center is coordinated by four nitrogen atoms in a planar geometry with local D4h symmetry.20–23 This configuration confers distinct electronic properties that improve electrocatalytic kinetics, but its electronically balanced and symmetric nature limits the fine modulation of metal reactivity, leading to suboptimal adsorption energies for reaction intermediates.24–27 Consequently, symmetry-breaking strategies are increasingly employed in SAC design to enhance performance.27 Symmetry breaking, the loss of intrinsic symmetrical order due to alterations in atomic positioning, is a significant concept in modulation engineering for electrocatalysts.28–33 Intentional distortion of symmetric frameworks modifies the electronic structure and spatial configuration of catalytic centers, often creating anisotropic surface features that enrich reactant concentration and enhance catalytic turnover.34,35 In SACs, where single metal atoms are anchored by coordination atoms or supports, the coordination environment directly influences performance.34,36 Symmetry breaking regulates the electronic properties of SACs, altering electronic configuration and reactant adsorption behavior, which lowers the reaction activation energy, boosts activity, and enhances selectivity and stability.37–39 It also optimizes electron migration pathways, strengthens metal–support bonding to prevent agglomeration, and improves overall catalyst stability.40–42 Current symmetry-breaking strategies for SACs are classified into three major categories: strain-induced, defect-induced, and curvature effects-induced symmetry breaking, all recognized as efficient for improving electrocatalyst performance.
In SAC systems, applying lattice strain can induce subtle modifications in metal–ligand bonds at the angstrom scale, disrupting geometric symmetry and perturbing the electronic configuration of the active site.43–45 Tensile strain typically downshifts the metal d-band center, while compressive strain upshifts it, providing a strategy for fine-tuning electronic structure and enhancing catalytic activity and selectivity.46–48 The coordination environment surrounding the metal center is equally critical, governing catalytic properties such as activity, selectivity, and stability.49 The electronic configuration and oxidation state of the metal are strongly influenced by the first coordination shell, where variations in the identity, coordination number, and positioning of bonded atoms modulate the metal's electronic structure.50,51 Tailoring the local coordination environment, such as by introducing asymmetry, modulates the polarity of metal–ligand interactions and redistributes charge density, optimizing adsorption behavior and reaction kinetics.52–54 Furthermore, the inherent curvature of the carbon matrix support can induce symmetry breaking.55 Unlike a planar structure, surface curvature generates an interfacial electric field and local strain, potentially enhancing catalytic activity.56–59 For instance, tip-like FeN4 sites on spherical carbon surfaces create a strong local electric field that accumulates ORR-related species and accelerates ORR kinetics, demonstrating how high-curvature induces compressive strain, optimizing the electronic structure and reducing the reaction energy barrier.59 Despite these advances, the fundamental mechanisms by which symmetry breaking enhances electrocatalytic performance remain inadequately understood, particularly the detailed mechanistic insights into how symmetry perturbations influence electrochemical behavior. Therefore, rational design of symmetry-breaking strategies, coupled with comprehensive mechanistic elucidation and a balance between disrupting local symmetry and maintaining structural stability, is essential. Atomic-scale symmetry breaking has emerged as a powerful and versatile strategy for precisely tailoring the electronic and geometric structures of SACs, thereby significantly enhancing their electrocatalytic performance.19 This review systematically examines how deliberate disruption of local symmetry, achieved through strain engineering, defect-induced coordination modulation, and curvature effects, enables fine-tuning of the metal centers’ electronic properties, optimizes the adsorption of key intermediates, reduces activation barriers, and breaks conventional scaling relationships. By comprehensively analyzing recent advances in these symmetry-breaking strategies, we highlight their critical roles in improving catalytic activity, selectivity, and stability across a range of electrocatalytic reactions. Finally, we outline future research directions and persistent challenges, such as achieving atomic-level precision in symmetry control, ensuring structural durability under operational conditions, and scaling up the synthesis of symmetry-broken SACs. This work serves as a timely and insightful resource for researchers seeking to move beyond conventional symmetric M–N4 motifs, providing a foundational understanding and a forward-looking perspective on how atomic-scale asymmetry can be deliberately engineered to overcome persistent activity–selectivity trade-offs and material stability challenges, thereby accelerating the development of next-generation SACs for sustainable energy conversion and storage technologies.39
2. Fundamentals of symmetry breaking in SACs
The concept of symmetry breaking was initially introduced in the field of condensed matter physics in the early 1960s.60 Since then, it has become a foundational principle within the research framework of materials science.60 Symmetry breaking in SACs has emerged as a critical design principle for advancing electrocatalysis, offering a fundamentally distinct strategy for tuning the local environment of catalytically active sites.21,39 SACs are characterized by isolated metal atoms stabilized on suitable supports through coordination with surrounding atoms, often forming well-defined metal–ligand motifs.21,39 While SACs inherently provide maximized atomic utilization and uniform active sites, their performance is strongly governed by the geometric and electronic characteristics of the active center.61 In this context, symmetry breaking refers to the intentional disruption of the ideal, often highly symmetric, local coordination geometry or electronic structure around the metal atom.21,37 This perturbation leads to anisotropic charge distributions, modified orbital interactions, and reconfigured metal–ligand bonding, thereby enabling fine-tuning of catalytic properties beyond the limitations of conventional, symmetric active sites.39The conceptual foundation of symmetry breaking in SACs lies in the modulation of the spatial and electronic configuration of the active site.21 In a perfectly symmetric coordination environment (e.g., M–N4 in square-planar geometry), the metal center typically exhibits spatially balanced orbital degeneracy and a uniform distribution of electron density, which may not always be optimal for mediating complex electrochemical reactions that require precise intermediate binding.39 Introducing asymmetry alters the local crystal field and splits degenerate d-orbitals, resulting in the redistribution of electronic density.62 This shift in electronic structure can be harnessed to optimize interactions with adsorbates and to lower activation energy barriers for specific steps in electrocatalytic processes.38 Importantly, symmetry breaking does not merely introduce disorder, rather it imposes a controlled heterogeneity at the atomic level from symmetry that can be tailored to elicit desirable catalytic properties.39
Symmetry breaking in SACs can be accomplished through various strategies including coordination asymmetry, axial ligand modification, curvature effects of support and application of external strain.62–65 For instance, asymmetric coordination, where the metal atom is coordinated by non-equivalent ligands such as nitrogen (N), phosphorus (P), or sulfur (S) in varying configurations, can create local electron density gradients, enabling site-specific reactivity.66 Similarly, introducing axial ligands of metal centers or modifying the support surface can induce out-of-plane distortions that disrupt spatial symmetry and further modulate electronic structure.63 Another effective approach is strain engineering, wherein compressive or tensile strain imposed on the host matrix can deform bond angles and lengths at the angstrom scale, leading to significant changes in the metal–ligand interaction and the overall charge distribution of the catalyst.54,65 Besides, incorporation of support with high surface curvature can effectively introduce intrinsic micro-strain and regulate the interfacial electric field surrounding the catalytic surface.67 This modulation exerts a profound impact on the d-orbital characteristics and overall electronic structure of the anchored metal atoms, in contrast to SACs stabilized on flat, low-curvature surfaces.67 The induced internal strain and electric field effects synergistically alter the local electronic environment, thereby influencing catalytic reactivity and stability at the atomic scale. From a mechanistic perspective, symmetry breaking introduces site-specific heterogeneity which posits a universal volcano-type relationship between activity and binding energy deviates from the classical Sabatier principle.68 In SACs with broken symmetry, the presence of localized electronic domains and spatially confined orbitals leads to non-uniform reactivity patterns, challenging the conventional scaling relationships that often constrain catalyst optimization.68 This opens new avenues for designing SACs with independently tunable adsorption energies for different intermediates, thus overcoming activity–selectivity or activity–stability trade-offs. While activity, selectivity, and stability are interrelated, symmetry breaking provides a multi-dimensional tuning strategy that can decouple their governing factors. This allows for targeted optimization that is both thermodynamically and kinetically feasible, as evidenced by the numerous previous experimental and theoretical reports.39 Furthermore, the structural distortion associated with symmetry breaking can influence the dynamic behavior of the active site under reaction conditions, enabling adaptive reconfiguration and self-optimization of the catalyst surface.39
In summary, symmetry breaking is a fundamental and versatile strategy in tailoring active sites in SACs for enhanced electrocatalytic performance. Disrupting local coordination symmetry alters the electronic and structural environment, enabling optimized interactions with intermediates, reduced kinetic barriers, and improved catalytic activity. Specifically, its key effects are outlined as follows (Fig. 2):
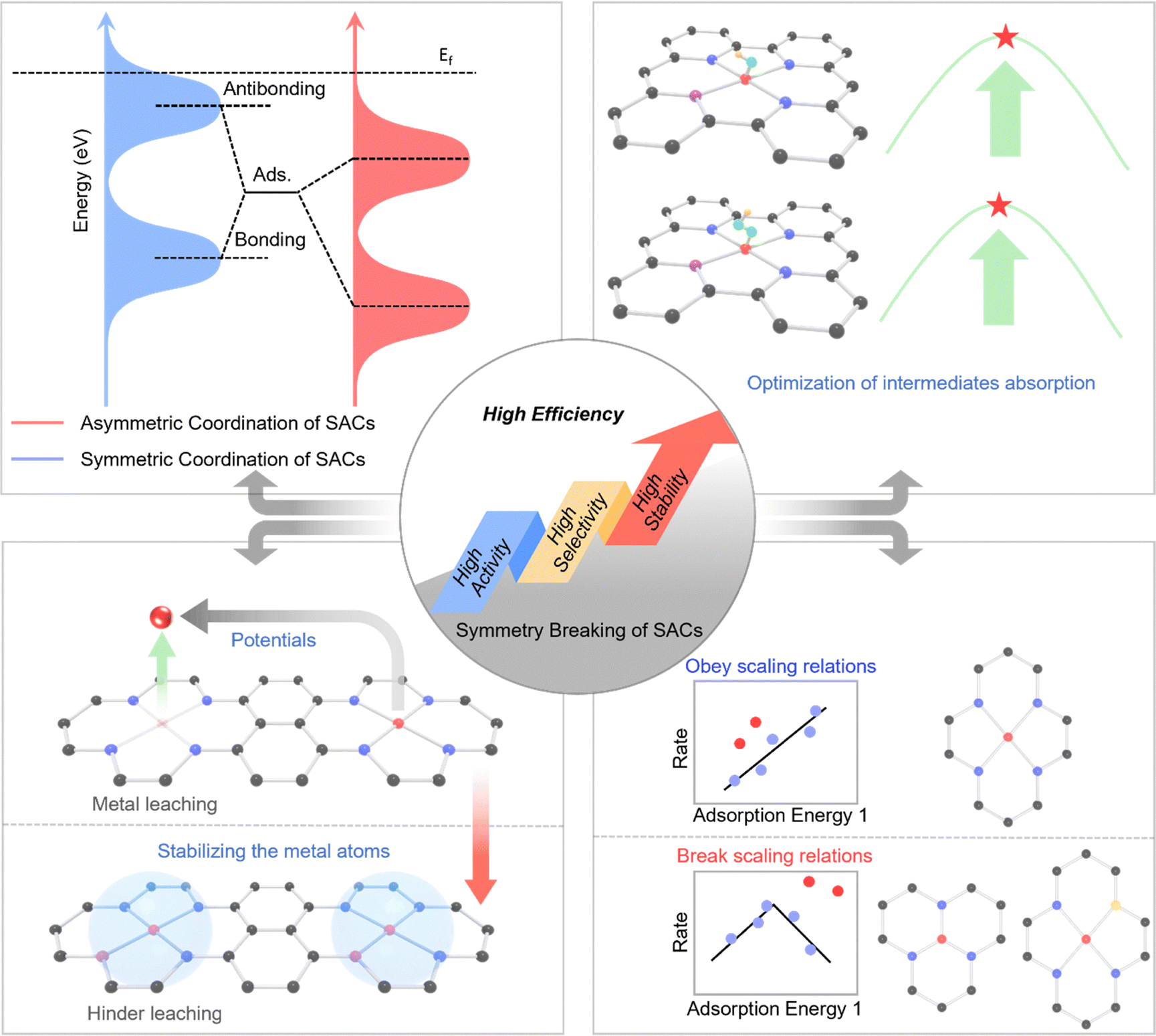 | ||
| Fig. 2 The key effects of symmetry breaking on electrocatalysis in SACs. | ||
2.1 Tuning the electronic structure
Symmetry breaking alters the local crystal field around the metal atom, leading to the lifting of orbital degeneracy and the redistribution of electronic density.39 This adjustment can shift the d-band center of the active site, thereby modulating the interaction strength between the active site and reaction intermediates. Symmetry breaking lifts orbital degeneracy, redistributes electron density, and shifts the d-band center, collectively enabling fine-tuning of adsorption energetics. This aligns with the d-band center theory and provides a mechanistic basis for the enhanced catalytic performance of symmetry-broken SACs. As a result, adsorption energies can be optimized to facilitate faster reaction kinetics and lower activation barriers.2.2 Enhancing adsorption and intermediate stabilization
Introducing asymmetry into the coordination environment creates local charge polarization, which enhances the binding specificity toward particular intermediates.36,53 This is crucial for reactions such as the oxygen reduction reaction (ORR), hydrogen evolution reaction (HER), oxygen evolution reaction (OER) and carbon dioxide reduction reaction (CO2RR), where selective adsorption and stabilization of key intermediates determine the overall catalytic efficiency and product distribution.2.3 Breaking scaling relationships
In conventional catalytic systems, adsorption energies of different intermediates are often linearly correlated, imposing intrinsic limitations on performance optimization.69 Symmetry breaking allows for decoupling these relationships by creating heterogeneous active sites with independently tunable binding affinities, enabling a more favorable reaction pathway and higher catalytic turnover rates.68 The decoupling of linear scaling relationships via symmetry breaking fundamentally originates from the creation of anisotropic active sites where different reaction intermediates interact with distinct orbital environments and local charge distributions, thereby breaking the energetic coupling that exists in symmetric sites where all adsorption configurations are geometrically and electronically equivalent. Specifically, in a symmetric M–N4 site, intermediates like OOH and OH typically bind through similar metal–oxygen interactions, leading to linearly correlated adsorption energies, whereas in an asymmetric site such as M–N3C1 or axially-coordinated M–N5, the spatial and electronic asymmetry allows OOH to engage with both the metal center and adjacent heteroatoms through a multi-center adsorption mode, while OH may bind predominantly to the metal, thus enabling these energies to be tuned independently through localized ligand and strain effects.39 This concept is not merely theoretical but is increasingly experimentally verifiable through a multi-faceted approach. The operando X-ray absorption spectroscopy (XAS) can detect the structural asymmetry and track potential-dependent bond length changes that correlate with intermediate stabilization, while isotopic kinetic experiments and potential-dependent selectivity measurements can reveal altered reaction pathways indicative of decoupled adsorption energetics. In summary, symmetry breaking in SACs provides a physically grounded and experimentally verifiable strategy to overcome scaling relationships by introducing atomic-level heterogeneity that enables independent optimization of intermediate binding energies.2.4 Improving stability and durability
Asymmetric coordination structures can lead to stronger metal–support interactions and more robust anchoring of single atoms, reducing the likelihood of metal leaching during prolonged electrochemical operation.70 This stabilization is essential for maintaining high activity and extending catalyst lifetime in practical applications. These modulation strategies related to symmetry breaking in SACs offer a versatile platform for precise modulation of active sites by breaking the symmetry of SACs configuration, providing an efficient pathway for the rational design of next-generation electrocatalysts.71Overall, the atomic-level symmetry breaking in SACs is fundamentally distinct from the surface heterogeneity or random defects in conventional catalysts, primarily in its precision, uniformity, and electronic tunability. While traditional heterogeneity, such as step edges, kinks, or vacancies in nanoparticles, arises from stochastic structural imperfections that create a spectrum of active sites with a broad distribution of catalytic behaviors, symmetry breaking in SACs is a deliberate and atomically precise engineering strategy. It involves the controlled disruption of a well-defined, initially symmetric coordination site such as M–N4 into a designed asymmetric configuration (e.g., M–N3, M–N2S2, or axially-coordinated M–N5). This targeted perturbation is not a random defect but a tailored modification that uniformly lifts the orbital degeneracy of the metal center, leading to predictable shifts in the d-band center and a systematic redistribution of electron density. Consequently, symmetry breaking enables the fine-tuning of adsorption energetics for specific intermediates across all active sites in a uniform manner, thereby breaking scaling relationships and optimizing selectivity, a level of control unattainable with the heterogeneous mix of sites in conventional catalysts. Furthermore, this atomic-level design intentionally enhances stability through strengthened metal–support interactions, moving beyond the incidental and often unstable defect sites in traditional systems to create a generation of electrocatalysts where activity, selectivity, and durability can be rationally and independently engineered.
3. Strain-induced symmetry breaking
Strain-induced symmetry breaking can directly tune the electronic structure of SACs without introducing foreign atoms, thereby optimizing the local electronic configuration of active sites, enhancing intrinsic activity, and achieving excellent catalytic performance.72 Specifically, the introduction of strain can modulate the electronic states at the Fermi level of SACs, prompting electron reconfiguration at the active sites, altering electron transfer properties, and inducing metallic or semiconducting behavior in SACs. This, in turn, affects orbital hybridization and binding between single-atom sites and intermediates, leading to variations in catalytic activity.73 In this context, strain is usually quantified by the ratio of surface to bulk interatomic distances in catalysts.74 When the interatomic distance on the surface is larger than that in the bulk material, tensile strain is produced; whereas when the interatomic distance on the surface is smaller than that in the bulk material, compressive strain is generated. These differences in interatomic distance alter the coordination environment of the active sites, thereby affecting the performance of the catalyst. For SACs, strain can be adjusted by stretching or bending the substrate, doping with heteroatoms,75 introducing defects,76 and changing the lattice parameters of the substrate.77As shown in Fig. 3, in SACs with ideal planar M–N4 configurations, symmetry breaking of the coordination environment can be effectively achieved through in-plane strain engineering. When linear strain-inducing factors are introduced, the M–N4 site will generate anisotropic biaxial strain parallel to the plane direction of the carbon matrix. To balance this strain, the carbon matrix will correspondingly produce strain in the opposite direction in the direction perpendicular to the plane of the carbon matrix.78 In M–N–C type SACs, the active metal centers exhibit discrete energy levels, which differ from the continuous energy bands of metal catalysts.79 Under compressive strain, the contraction of metal–ligand bonds lead to an upshift in the d-orbitals of the active metal centers. This compression enhances the repulsive interactions between the ligand electrons and the d-orbitals, raising their energy levels and thereby boosting the reactivity of the metal centers. Conversely, tensile strain has the opposite effect, lowering the d-orbital energy levels in SACs and thus diminishing the reactivity of metal centers. In this chapter, we will explore how tensile strain, compressive strain, and the coexistence of both strains in SACs lead to the symmetry breaking of single-atom metal coordination and subsequently affect the activity of catalysts.
 | ||
| Fig. 3 Schematic of strain-induced symmetry breaking. When compressive or tensile strain is applied in a direction, the symmetry of the M–N4 site is disrupted, resulting in corresponding tensile or compressive strain in a direction perpendicular to the applied strain to accommodate the distortion. | ||
3.1. Tensile strain strategy
During electrochemical catalysis, the active sites may undergo unique changes in terms of configuration and electronic states, especially under tensile strain conditions. Miao et al. successfully modified the Fe–N coordination number and bond length by adjusting the binding strength between metal ions (Fe) and polymers, achieving tensile strain adjustment of Fe–N coordination (Fig. 4a).80 Using polyacrylic acid (PAA) homopolymers and poly(acrylic acid-maleic acid) (P(AA–MA)) copolymers with different ratios of acrylic acid–maleic acid (AA/MA) as chelating agents, it was found that the P(AA–MA)–Fe–N catalyst has a higher Fe3+–polymer binding constant, longer Fe–N4 bond length than PAA–Fe–N, and almost no low-coordination Fe–N2/N3 structure, while PAA–Fe–N contains about 15% of low-coordination Fe–N2/N3 structure (Fig. 4b). The optimized P(AA–MA)–Fe–N catalyst has longer Fe–N bond lengths and unique Fe–N4/C groups, which enable the catalyst to exhibit excellent ORR stability under actual operating conditions of proton exchange membrane fuel cells at room temperature (Fig. 4c). Through density functional theory (DFT) calculations, the difference between adsorption energy (Ead) of Fe atoms on the support and bulk cohesive energy (Ecoh) was studied for each configuration. It was found that Fe–N2/C and Fe–N3/C have positive Ead–Ecoh values, indicating that they are more unstable in terms of metal aggregation, while Fe–N4/C with tensile strain is the most stable in preventing metal loss and aggregation. | ||
| Fig. 4 (a) Schematic representation of P(AA–MA)–Fe–N and PAA–Fe–N obtained from different precursors, (b) k3-weighted Fourier transformed extended X-ray absorption fine structure (FT-EXAFS) spectra from Fe K-edge absorption spectra of P(AA–MA)–Fe–N, PAA–Fe–N, and Fe foil samples and (c) current density retention of P(AA–MA)–Fe–N and PAA–Fe–N catalysts at 0.55 V in a PEMFC. (Reproduced with permission from ref. 80, copyright 2021, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim). (d) Schematic representation of FePc–CNTs directly bonded to carbon nanotubes and FePc–Py–CNTs bonded to carbon nanotubes via amino pyridine bonds. Blue, purple, red, and green spheres represent nitrogen, iron, oxygen, and carbon atoms respectively. (Reproduced with permission from ref. 81, copyright 2021, The Royal Society of Chemistry). (e) The relationship between the onset potentials of the two ORR pathways on Ni–N4/C and the degree of tensile strain, and (f) occupancy in each of the d orbitals of Ni–N4/C under different tensile strain rates. (Reproduced with permission from ref. 82, copyright 2024, American Chemical Society). | ||
Loyola et al. combined Fe phthalocyanine (FePc) with carbon nanotubes (CNTs) modified with pyridine (Py) groups, using pyridine groups as axial ligands to form a five-coordinate structure at the Fe center (Fig. 4d).81 This change in coordination can be considered as a structural strain adjustment. Compared with traditional four coordinated FePc–CNT, FePc–Py–CNT has an increased Fe–N bond length, indicating the effect of tensile strain. At pH 13, the onset potential and half-wave potential of FePc–Py–CNT is less negative compared to commercial Pt/C (20%, Vulcan), exhibiting excellent ORR activity. Theoretical calculations have shown that this improvement may be related to changes in Fe–N bond length and coordination environment caused by tensile strain, which may optimize the adsorption and activation process of O2 molecules at the Fe center.
Although studies have explored how strain improves the catalytic activity of M–Nx/C catalysts, the underlying mechanism is still unclear. To this end, Zhao and his collaborators constructed Ni–N4/C models with different degrees of stretching by adjusting the lattice constant on the carbon skeleton, and conducted in-depth research on the effect of tensile strain on the ORR activity and selectivity.82 In 4-electron ORR pathway, the ORR activity shows a monotonically increasing trend with the increase of tensile strain. For 2-electron ORR pathway, the onset potential exhibits a volcanic curve, reaching a maximum value of 0.691 V at a tensile strain of 6% (Fig. 4e). Tensile strain causes the d-band center of Ni–N4/C to shift towards the Fermi level, thereby enhancing the ability to adsorb *OOH and ultimately increasing the activity of 4e− ORR. Meanwhile, it also found that the spin state of Ni–N4/C catalyst can change with 9 different tensile strains. Specifically, as the tensile strain increases, the spin state of Ni–N4/C can transition from a low spin state to a high spin state, especially at 6% tensile strain, which corresponds to the peak of 2e− ORR activity (Fig. 4f). This indicates that tensile strain plays an important role in enhancing the selectivity and activity of ORR by adjusting the catalytic electronic structure.
In addition, the introduction of S and N heteroatoms into Mo isolated single atoms (Mo ISA) loaded on two-dimensional (2D) carbon materials modifies Mo coordination environment.75 Owing to the introduction of larger S atoms into Mo-ISA/SxN1C (where SxN1C refers to S and N co-doped graphene), the hydrogen adsorption free energy of Mo-ISA/SxN1C is significantly reduced, demonstrating good stability and ultra-high HER activity. The presence of S atoms induces a tensile strain in the vicinity of S and Mo atoms, significantly disrupting the local symmetry surrounding Mo atoms. This strain effect leads to an accumulation of negative charges on neighboring C atoms, facilitating enhanced charge transfer from Mo to H atoms and culminating in a redistributed charge landscape. For Co SACs supported on γ-graphyne for CO2RR, Liu et al. utilized DFT calculation and constrained ab initio molecular dynamics (AIMD) simulations to demonstrate that tensile strain weakens the interactions between Co atoms and the reaction intermediates COOH and H.83 However, the adsorption strength of COOH decreases much more slowly than that of H, which significantly enhances the selectivity of the CO2RR.
3.2. Compressive strain strategy
In CO2RR, symmetry breaking of M–N4 SACs induced by compressive strain is particularly advantageous in increasing its selectivity against the competing HER. Li et al. initiated the contraction of graphitized carbon lattice by changing the thermal activation temperature, thereby introducing compressive strain into Ni–N–C catalysts.77 As the activation temperature increased from 400 °C to 900 °C, the length of the Ni–N bond shortened from 1.85 Å to 1.62 Å, indicating that the Ni–N bond was gradually affected by compressive strain (Fig. 5a). This thermal activation process also caused the graphene layer to transition from a flat state to a twisted and wrinkled configuration, and these structural changes in the graphene layer further influenced the strain state of the Ni–N bond. The DFT calculation results revealed that under 2% compressive strain, the CO2RR limiting potential (UL(CO2RR)) of NiN4 sites decreases from −1.50 V to −1.40 V, indicating that compressive strain sites have higher CO2RR activity (Fig. 5b). Moreover, the CO2RR selectivity (UL(CO2RR) − UL(HER)) of NiN4 sites under compressive strain was significantly higher than that of strain-free sites, indicating that compressive strain can effectively suppress HER and improve CO2RR selectivity. The compressive strain induces uneven changes in the Ni–N bond lengths, which effectively disrupted the symmetry of the Ni–N coordination environment. This symmetry breaking altered the distribution and orbital overlap of electron clouds. As the compressive strain increased further, some N coordination atoms evaporated, forming NiN3 sites. This transition not only modified the local coordination environment of Ni atoms but also potentially introduced new active sites, thereby influencing the electronic structure of Ni atoms. In fact, for 2D carbon materials, the thermal activation process can introduce compressive strain into SACs due to the negative thermal expansion coefficient of the carbon layer. This property causes the carbon layer to contract at elevated temperatures, leading to a nonplanar structure. Similarly, Li and colleagues successfully introduced compressive strain into Fe–N4 sites by adjusting the thermal activation temperature.84 It has been observed that during the thermal activation, the off-plane ripples of the carbon layer triggered local contraction, thereby changing the structure of the Fe–N4 sites, generating compressive strain, and forming highly active Fe–N4 sites with the optimal coordination number and bond length at 700 °C (Fig. 5c). The induced compressive strain effectively modulates the electronic structure, causing a positive shift in the 3d orbitals of Fe and enhancing charge transfer from Fe to the adjacent nitrogen atoms. These modifications facilitate oxygen adsorption and O–O bond cleavage, thereby promoting the ORR (Fig. 5d and e). It is worth noting that excessive compressive strain can lead to further deformation of the Fe–N4 coordination. Such an unstable structure becomes thermodynamically unfavorable for the ORR. Therefore, although compressive strain can improve the activity of Fe–N4 sites to some extent, the magnitude of the strain must be precisely controlled to avoid excessive structural deformation and a decline in activity. This underscores the importance of balancing the magnitude of strain in catalyst design to achieve optimal catalytic performance. | ||
| Fig. 5 (a) Symmetry breaking of Ni–N4 catalysts via variation in temperature during the thermal activation step of the synthesis process and (b) atomic structures of Ni–N4 sites, relaxed and with −2% compressive strain, and the limiting potential of CO2RR at Ni–N4 sites at different degrees of compressive strain. (Reproduced with permission from ref. 71, copyright 2022, The Royal Society of Chemistry). (c) R space of Fe K-edge absorption spectra of different catalyst samples, (d) geometric structures and charge distributions of Fe–N4 sites under complete relaxation and 2% compressive strain, and (e) free energy evolution of fully relaxed Fe–N4 sites and Fe–N4 sites with 2% compressive strain during ORR. (Reproduced with permission from ref. 78, copyright 2019, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim). (f) The process of encapsulating FeCo nanoparticles with Fe–N–C SAC coating, where FeCo nanoparticles apply compressive strain on the SAC. (Reproduced with permission from ref. 79, copyright 2022, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim). (g) Change in onset potential of nitrogen reduction reaction (NRR) when strain is applied from −3% to 3% in Mn–N3C1 and Mn–N1C3. (Reproduced with permission from ref. 80, copyright 2021, American Chemical Society). | ||
Compared to isolated Fe–N–C catalysts, incorporating nanoparticles with lattice mismatch into the catalysts can also induce compressive strain in Fe–N–C. The {110} facet of FeCo nanoparticle can induce interlayer compressive strain in the surrounding graphene Fe–N–C layer due to lattice mismatch (Fig. 5f).85 The Fe–N bond length on the graphene sublayer is shortened, and the geometric symmetry of Fe–N4 coordination is disrupted. The interlayer compressive strain also breaks the electronic symmetry of Fe–N4 sites, shifts the overlap between Fe d and O p orbitals upward, weakens the interaction between Fe–N4 sites and *OH intermediates, and affects the adsorption energy and free energy changes of the reaction.
Li et al. used 2D Mn SACs as model catalysts and utilized DFT to investigate the impact of coordination bond strain on two monolayer single-atom catalysts, Mn–N3C1 and Mn–N1C3, which are identified for efficient nitrogen reduction reaction (NRR) catalysis.86 Despite the distinct coordination environments of these catalysts, under tensile strain, both of them show similar trends in the change of free energy as a function of strain for the adsorption of N2 and the formation of *NNH during the first protonation (Fig. 5g). Specifically, only compressive strain (negative strain) can reduce the d-band center energy of Mn sites, enhancing the N2 adsorption ability of Mn center and promoting further activation of N2. This process not only reduces the overpotential of NRR, but also significantly improves the overall performance of the catalyst.
3.3. Synergistic strategy
Many of the improvements in catalytic activity in SACs are derived from synergistic effects of multiple symmetry breaking strategies. For example, synergistic effects in dual-atom sites can effectively modulate the atomic-scale strain environment at Fe single-atom active centers, which can improve their HER or ORR performance.75,84,85 Compared to isolated Fe single-atom sites, dual-atom configurations (e.g., Fe–Mn, Fe–Ru, and Fe–Cu) induce distinctly different strain states, with computed strain values of −2.6%, +2.1%, and +3.1% respectively (Fig. 6a and b).87 This indicates that the introduced second metal atom is a key factor in modulating the local strain environment at the Fe site. Remarkably, in the Fe–Ru dual-atom system, the introduction of moderate tensile strain causes a downward shift in the d-band center of Fe atoms (Fig. 6c). This enhances electron occupancy in antibonding orbitals, thereby lowering the key energy barrier in the acidic ORR process. Benefiting from this strain modulation mechanism, the FeRu–N–C catalyst exhibits superior catalytic activity for acidic ORR (Fig. 6d and e). This symmetry breaking, induced by the dual-atom configuration, fundamentally alters the electronic structure of the Fe active center, optimizing its interaction with reaction intermediates and ultimately accelerating the ORR reaction kinetics. Concurrently, the synergistic interaction between Fe and Ru atoms, combined with the altered strain environment, collectively enhances the structural stability of the Fe sites under electrocatalytic reaction conditions. | ||
| Fig. 6 (a) Schematic diagram featuring atomic strain modulation of iron sites through introduction of a second metal atom, (b) the relationship between atomic strain, d-band center, and Gibbs energy barrier of ORR in Fe–M dual-atom systems, (c) schematics of the positions of the d-band relative to Fermi level, Ef in Fe and Fe–Ru, here εd represents atomic strain which quantitatively describes the distortion of geometry structure, (d) linear sweep voltammogram of FeRu–N–C in O2-saturated 0.1 M HClO4 and (e) onset potential and half wave potential of FeRu–N–C compared with Fe–N–C, as well as the kinetic current density at 0.80 V and 0.85 V. (Reproduced with permission from ref. 81, copyright 2024, Elsevier). (f) The relationship between lattice strain in different directions and distance between active sites (dsite) in Fe–N4 and (g) volcanic diagram of OER overpotential (ηOER) and dsite in Fe–N4. (Reproduced with permission from ref. 72, copyright 2023, American Chemical Society). | ||
Besides introduction of a second metal atom, a synergistic effect can also manifest between active site density and site geometry. The density and geometric arrangement of active sites together determine the magnitude and direction of the strain, which in turn impacts the catalytic performance. More specifically, altering the distance between Fe–N4 active sites (dsite, which is negatively correlated with Fe site density) introduces anisotropic strain in the in-plane direction of the graphene substrate, including tensile strain along the x-direction and compressive strain along the y-direction.78 The degree of strain is closely associated with the active site density (Fig. 6f). When the density of active sites is relatively low, the strain is primarily governed by the local effects of individual active sites, resulting in relatively small strain energy and lattice distortion. The volcanic dependence of OER activity on dsite indicates that when the density of active sites increases to a certain extent (Fig. 6g), the interactions between multiple active sites become significant, leading to a rapid increase in strain energy and lattice distortion. This interaction optimizes the electronic structure of the active site, thereby enhancing catalytic activity. These findings indicate that different steps in multi-step reactions may respond differently to strain. Therefore, the rational design of SACs that incorporate the coexistence of tensile and compressive strains can more effectively enhance overall catalytic activity.
Overall, the distinct influences of tensile and compressive strain on catalytic pathways can be summarized by their opposing effects on the electronic structure of the metal center and the subsequent modulation of intermediate adsorption. As discussed above, compressive strain typically contracts metal–ligand bonds, upshifting the d-band center and strengthening the adsorption of key intermediates. This is particularly beneficial for reactions like the ORR, where optimized binding of OOH and facilitated O–O bond cleavage can enhance the 4e− pathway activity, as demonstrated in strained Fe–N4 and Ni–N4 sites.77,84 Conversely, compressive strain's strong adsorption of H can effectively suppress the competing HER, thereby improving the selectivity for CO2RR to CO, as seen in Ni–N–C catalysts.77 In contrast, tensile strain elongates metal–ligand bonds, generally leading to a downshift of the d-band center and a weakening of adsorbate binding. While this might seem detrimental, it can be strategically employed to optimize specific pathways. For instance, in the ORR, moderate tensile strain can fine-tune the OOH adsorption energy to a more optimal value, boosting the kinetics of both 2e− and 4e− pathways, as computationally predicted for Ni–N4/C.82 More profoundly, in CO2RR, the differential response of intermediates to tensile strain is key. The studies on Co SACs and Ni–N4 sites show that while the adsorption of both COOH and H is weakened under tensile strain, the adsorption strength of H decreases more significantly.82,83 This selective destabilization of H adsorption over COOH directly enhances the CO2RR selectivity against HER. Therefore, the strategic application of tensile or compressive strain offers a powerful lever to not only enhance intrinsic activity but also to steer reaction selectivity by differentially stabilizing or destabilizing critical reaction intermediates in complex multi-pathway electrocatalytic processes.
4. Defect-induced symmetry breaking of SACs
In addition to strain-induced symmetry breaking, defect-induced symmetry breaking endow the asymmetric configuration of SACs (M–N4) by the modulation of intrinsic coordination and geometry structure, and both the coordination change of active sites by introduction of heteroatoms or the construction of geometric asymmetry can also provide more active sites and promote the catalytic reaction.66,88On one hand, coordination-breaking strategies are frequently derived from simplified theoretical models, such as static or dynamic coordination disruption, which may not comprehensively represent the dynamic and complex nature of real catalytic environments.50 On the other hand, geometric symmetry breaking primarily disrupts the saturated structures of SACs, leading to structural distortions that enhance electron transfer efficiency, stabilize reaction intermediates, and lower activation energy barriers, thereby promoting faster catalytic reaction kinetics.89,90 Therefore, the defect-induced symmetry breaking strategy of a M–N4 SAC can lead to the formation of four typical structural configurations of SACs as shown in Fig. 7: unsaturated low coordination M–Nx (x = 1, 2, 3), planar heteroatom coordination M–NxWy (W = other light-element heteroatoms, x + y = 4), axial heteroatom coordination M–N5 or M–N4Wx (W = other light-element heteroatoms, x = 1, 2) and bimetallic coordination M1M2–N4. Overall, by breaking the local symmetry of M–N4 coordination, defect-induced symmetry breaking allows for effective tuning of electrocatalytic performance of SACs, enabling them to exhibit superior activity and selectivity in various catalytic reactions.39,62,91
 | ||
| Fig. 7 SACs in representative configurations featuring various coordination environments, including regular M–N4 structure, low coordination structures, axial heteroatom coordination structures, planar heteroatom coordination structures, and dual-metal coordination structures. | ||
4.1. Low-coordination structures
Engineering coordination unsaturation has emerged as an effective strategy to tailor the electronic environment of SACs by decreasing the number of nitrogen ligand atoms surrounding the metal center.92,93 This approach converts the fully coordinated M–N4 structure into lower coordination states such as M–N1, M–N2, or M–N3, introducing an asymmetric ligand field. Such asymmetry perturbs the local electronic distribution and facilitates enhanced interaction with adsorbed intermediates, thereby accelerating catalytic turnover.94 The induced unsaturation not only alters the electronic density at the metal center but also promotes effective charge transfer, particularly relevant for transition metals with partially filled 3d orbitals.95,96 This change in coordination environment, often achieved through synthetic control or defect engineering, highlights the critical influence of local ligand structure on catalytic reactivity.50 Currently, studies on rational modulation of M–Nx sites continue to gain momentum, driven by efforts to optimize structure–activity relationships in heterogeneous electrocatalysis.The M–N3 coordination motif features a metal center bonded to three nitrogen atoms, resulting in a partially unsaturated structure relative to the more saturated M–N4 configuration.97,98 Such asymmetry enables finer tuning of the metal's electronic properties, facilitating enhanced interaction with reaction intermediates.97,98 Dong et al. developed symmetry-disrupted CuN3 single-atom catalysts (PSB-CuN3 SACs) via microwave-assisted synthesis, demonstrating significantly improved CO2RR performance relative to conventional Cu–N4 counterparts.38 By intentionally breaking the local D4h symmetry, the formation of planar CuN3 and CuN2 sites exhibiting lower C2v symmetry was achieved as shown in Fig. 8a and b. This geometric distortion promotes unusual dsp orbital hybridization and induces notable charge redistribution at the active centers (Fig. 8c). Their investigation further revealed a marked shift in the electronic structure: Cu atoms in the CuN3 sites exhibited an elevated d-band center energy compared to CuN4 configurations, while a subsequent reduction in the d-band center was observed for CuN2 coordination, highlighting the symmetry-dependent modulation of electronic states. Additionally, Yuan et al. investigated SACs incorporating Ni–N3 coordination environments, characterized by a lower nitrogen coordination number relative to the conventional Ni–N4 configuration (Fig. 8d and g).99 In this unsaturated structure, electron density, indicated by the cyan-shaded region, is more prominently localized around the Ni center, implying a higher accumulation of negative charge. This electronic enrichment enhances the binding affinity toward CO2 molecules, thereby improving the efficiency of their electrochemical reduction to CO as shown in Fig. 8h. Compared to other transition metals, Zn–NC systems have been less explored due to the full occupancy of 3d10 orbital of Zn, which limits electron transfer and catalytic activity.100 Li et al. addressed this limitation by designing a N-coordinated Zn SACs with unsaturated Zn–N3 sites.101 These low-valence sites effectively stabilize the COOH* intermediate, significantly lowering the energy barrier for CO2 reduction. As a result, the catalyst achieves high CO selectivity (up to 95%) and large current densities (up to 1 A cm−2) in flow cell configurations. Meanwhile, the M–N2 coordination environment, characterized by two nitrogen atoms bonded to a central metal site, strikes a desirable equilibrium between maintaining coordination integrity and maximizing catalytic reactivity.102 Gong et al. fabricated Ni single-atom catalysts (NiSA–Nx–C) on N-doped carbon using a dual protection strategy, enabling precise control of the Ni–N coordination number from 4 to 2 by adjusting pyrolysis temperature.103 This tuning optimized the local atomic environment for enhanced CO2RR activity. CO2RR proceeds via a two-electron, two-proton transfer pathway, where the rate-determining step involves COOH* formation. Gibbs free energy analyses revealed that NiSA–N2C exhibited the lowest ΔG (1.42 eV) for COOH* formation compared to NiSA–N3C (1.45 eV) and NiSA–N4C (1.73 eV), highlighting Ni–N2 as the most active site for efficient CO generation. Besides that, Li et al. developed a SAC featuring Mn–N2C2 coordination, achieving an impressive half-wave potential (E1/2) of 0.915 V in alkaline media, indicating high ORR activity.104In situ XAS and DFT studies identified Mn2+–N2C2 as the active site and Mn4+–N2C2 as the main reaction site. This work highlights how Mn–support interactions reduce intermediate reaction free energy, thereby boosting the performance of oxygen-involving electrocatalysis.
 | ||
| Fig. 8 (a) and (b) The electronic structure was characterized through differential charge density analysis and projected density of states (PDOS) calculations for the Cu active site and its coordinated oxygen atom within the *OCHO intermediate adsorbed on the CuN3C3 substrate. The atomic color scheme is as follows: H (white), C (grey), N (blue), O (red), and Cu (cyan). The Fermi energy level is demarcated by a black dashed line. (c) Illustrative schematics depicting band energy shifts and orbital hybridization in a planar CuN4 moiety possessing D4h local symmetry and a defective CuN3 moiety with lower C2v local symmetry. (Reproduced with permission from ref. 33, copyright 2023, Springer Nature). Ni K-edge fitting curves in R-space for the (d) Ni–N3–C and (e) Ni–N4–C structures. Insets depict the corresponding nickel-centered coordination environments, with Ni (green), N (blue), and C (brown) atoms, (f) and (g) wavelet transformation results of Ni–N3–C and Ni–N4–C, respectively and (h) a continuous 25 h CO2 electrolysis experiment of Ni–N3–C at −0.79 V (vs. RHE). (Reproduced with permission from ref. 93, copyright 2023, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim). | ||
4.2. Planar heteroatom coordination structures
Introducing light-element heteroatoms such as boron (B), S, P, or oxygen (O) into the coordination environment of SACs has emerged as a promising approach to modulate local symmetry and enhance catalytic functionality.105 By substituting nitrogen atoms in conventional M–N4 frameworks, these heteroatoms generate mixed coordination motifs denoted as M–NxWy (where W represents heteroatoms and x + y = 4). Such heteroatom-replacing coordination induces substantial alterations in the geometric and electronic configuration of the metal center, thereby tailoring the binding strength of reaction intermediates and improving overall catalytic activity and adaptability across different reactions.106 The influence of heteroatoms like S, P, and O on charge density redistribution and reactivity can be quantitatively rationalized through the interplay of their electronegativity and orbital size, which collectively dictate the polarity of the metal–ligand bond and the degree of orbital hybridization. A heteroatom with lower electronegativity than nitrogen such as S or P reduces the electron-withdrawing effect from the metal center, leading to a higher electron density on the metal atom, which can be quantified by Bader charge analysis and observed as a negative chemical shift in X-ray photoelectron spectroscopy (XPS). Conversely, a more electronegative atom like O intensifies electron withdrawal. Simultaneously, the larger atomic radius and more diffuse orbitals of S and P (utilizing 3p orbitals versus N's 2p) enhance orbital overlap with the metal d-orbitals, particularly the dz2 and dxz/dyz orbitals, leading to a broader and more covalently hybridized density of states near the Fermi level. This modified hybridization, combined with the charge transfer, directly shifts the d-band center position, a key quantitative descriptor for reactivity. For instance, the incorporation of less electronegative S in a Cu–S1N3 motif increases electron density on Cu while the strong 3p–3d overlap optimizes the d-band center, collectively weakening the over-strong *OH binding and enhancing ORR kinetics. Therefore, the strategic selection of a heteroatom allows for the fine-tuning of the metal site's electronic structure along two coordinated axes, in other words, electronegativity governing the ionic character and charge density, and orbital size governing the covalent interaction strength, together enabling a predictable optimization of adsorption energetics for target reactions.S atoms, owing to their lower electronegativity, can effectively modulate the electronic environment of metal centers in electrocatalysts, thereby contributing to improved ORR performance.91 Bai et al. introduced a dual-doping strategy using nitrogen and sulfur to tune the coordination environment of Mn sites for enhanced ORR activity as shown in Fig. 9a.107 Structural analyses confirmed an optimized Mn–N2S2 configuration in the Mn–N/S–C catalyst, where S-induced asymmetry significantly improved performance, yielding an E1/2 of 0.91 V. This catalyst enabled zinc–air batteries with a high power density of 193 mW cm−2 and robust output stability. In anion exchange membrane fuel cells, Mn–N/S–C outperformed its undoped counterpart (Mn–N4) with a 1.53-fold increase in peak power density. The DFT calculations revealed that the asymmetric Mn–N2S2 site, especially in ortho-position, plays a key role in boosting catalytic activity (Fig. 9b). Meanwhile, Shang et al. designed a hierarchically porous carbon-supported Cu single-atom catalyst with an asymmetric coordination environment for efficient ORR.108 Extended X-ray absorption fine structure (EXAFS) analysis of XAS revealed the coexistence of Cu–N and Cu–S bonds, forming a Cu–S1N3 structure with bond lengths of 1.98 Å and 2.32 Å, respectively (Fig. 9c). The introduction of S ligand significantly boosted ORR performance, achieving an E1/2 of 0.918 V and excellent stability in alkaline media. The asymmetric configuration also led to a lower reaction free energy, indicating more favorable adsorption and desorption of key intermediates.
 | ||
| Fig. 9 (a) Synthesis routes of Mn–S–C, Mn–N–C, and Mn–N/S–C and (b) the free energy diagram for the ORR pathways on the Mn–N4G, Mn–N3SG, ortho-Mn–N2S2G, para-Mn–N2S2G, and Mn–NS3G catalysts is presented, alongside the optimized structures of the ortho-Mn–N2S2G moiety and three key reaction intermediates. (Reproduced with permission from ref. 101, copyright 2023, Elsevier). (c) Schematic atomic interface model of S–Cu–ISA/SNC and FT-EXAFS fitting curves of S–Cu–ISA/SNC at Cu K-edge. (Reproduced with permission from ref. 102, copyright 2020, Springer Nature). (d) Synthesis procedure of Fe–NCP. (Reproduced with permission from ref. 105, copyright 2023, American Chemical Society). | ||
Despite P and N being in the same group of the periodic table, they exhibit notable differences in electron spin density, atomic size, and electronegativity.109 The incorporation of P as a coordinating atom significantly improves the ORR activity by effectively tuning the electronic structure of the active site.110 Roh et al. enhanced the ORR activity of Fe–N–C catalysts by phosphine gas treatment, which modified the coordination environment of the Fe centers (Fig. 9d).111 This process partially substituted Fe–N bonds with Fe–P bonds, forming more stable FeN3P active sites and improving Fe adsorption properties. Fe K-edge X-ray absorption near-edge structure (XANES) spectra exhibited a reduced pre-edge peak in Fe–NCP compared to Fe–NC, indicating lower symmetry around the Fe center. Additionally, the EXAFS spectrum of Fe–NCP revealed a shoulder peak attributed to Fe–P coordination, confirming the formation of FeN3P moieties. The d-band center of Fe–NCP also shifted downward, facilitating *OH desorption and thereby enhancing ORR performance. Analogously, Xue et al. developed a P and N co-doped carbon-supported Fe single-atom catalyst (Fe–SA/PNC) via a noncontact chemical vapor deposition method.112 This catalyst, with densely dispersed Fe atoms coordinated to N and P in a hierarchical carbon matrix in the form of FeN2P2 motif of SACs, shows excellent ORR activity (E1/2 = 0.92 V in alkaline media) and remarkable pH-universal stability. In zinc–air batteries, Fe–SA/PNC outperforms commercial Pt/C, delivering a peak power density of 260 mW cm−2 and a current density of 433 mA cm−2, compared to 159 mW cm−2 and 272 mA cm−2 for Pt/C-based cells.
Moreover, O doping can also introduce structural defects such as edge sites and vacancies in carbon materials.113 These defect sites act as additional active centers, creating alternative reaction pathways that enhance catalytic activity and may improve reaction selectivity.113 Li et al. engineered Fe single-atom sites with dual N,O coordination by tuning the coordination environment. XANES and EXAFS results confirm that Fe atoms in Fe–N,O/Graphene are positively charged and bonded to N and O with distinct bond lengths (1.94 and 2.09 Å).114 Density of state (DOS) analysis indicates a upward shift (relative to Fermi level) of the Fe 3d band center in this configuration, suggesting increased unoccupied orbitals that enhance interaction with ORR intermediates and thus boost catalytic activity. Lin et al. developed an asymmetric N,O-coordinated Sn SAC (Sn–N/O–C) with superior ORR activity.115 Zn–air batteries using Sn–N/O–C cathodes demonstrated higher energy density than traditional M–N–C SACs. The theoretical calculation results revealed that the asymmetric Sn–N/O coordination enhances O2 binding and charge transfer compared to symmetric Sn–N4 sites, promoting more efficient ORR kinetics.
Importantly, B doping effectively tailors the electronic structure and activates carbon materials by enhancing oxygen affinity and lowering reaction energy barriers. It also facilitates the formation of catalytic sites like B–C and B–O–C bonds, thereby boosting both the ORR rate and product selectivity.116 Wang et al. introduced a novel boron-doped nickel single-atom catalyst (Ni–B/N–C), where B atoms are strategically positioned next to nickel sites and N atoms, forming a Ni–B1N3 coordination structure.117 This boron incorporation enhances the electronic density at the nickel site, improving the ability of the catalyst to bind with ORR intermediates more effectively than the conventional Ni–N4 configuration. In alkaline conditions, the Ni–B/N–C catalyst shows an impressive half-wave potential of 0.87 V versus the RHE, outperforming commercial Pt/C.
4.3. Axial heteroatom coordination structures
Heteroatoms including N, S, O etc. within conductive carbon substrates can serve as anchoring sites that facilitate axial interactions with M–N4 centers, while isolated heteroatoms may also participate in forming unsaturated axial coordination motifs, often manifesting as terminal or “dangling” atoms.118 These heteroatoms are capable of directly coordinating with the central metal atom in an originally symmetric M–N4 framework, resulting in asymmetrical axial configurations such as M–N5 or M–N4Wx (W = light-element heteroatoms, x = 1, 2). This structural transformation induces a break in local electronic symmetry and significantly modifies the electronic environment of the active site.63 Such alterations enhance intermediate binding affinity and lower the associated activation energy barriers compared to traditional planar M–N4 sites.63 Additionally, the axial heteroatom ligand can itself act as an auxiliary adsorption center, operating synergistically with the in-plane coordination to further facilitate more favorable reaction pathways and improve catalytic performance.63In recent years, metal macrocyclic compounds, particularly metal phthalocyanines (MPc), have attracted considerable attention in electrocatalysis due to their well-defined M–N4 coordination, serving as ideal models for studying ORR mechanisms and tuning SAC activity.119 However, their limited stability remains a challenge.120 To overcome this, researchers have developed catalysts by anchoring metal phthalocyanines with pyridine or imidazole onto carbon nanomaterials, forming M–N5 structures via axial N coordination.121 This strategy enhances ORR performance by enabling rehybridization between metal 3d orbitals and axial ligand orbitals, effectively modifying the electronic and geometric structure of the active site and accelerating the reaction rate.122 Zhang et al. reported that the ORR activity of functionalized multi-walled CNTs-NH2 can be significantly improved by anchoring FePc molecules via axial coordination as shown in Fig. 10a. Specifically, the FePc/CNT-NH2 catalyst features NH2 groups axially coordinated to the Fe center, forming a Fe–N5 configuration (Fig. 10c).122 This structural modification led to a notable enhancement in ORR performance, achieving a high E1/2 of 0.92 V and demonstrating excellent electrocatalytic activity (Fig. 10d). Moreover, Cao et al. achieved the anchoring of FePc onto nitrogen-doped graphene nanonets (NGMs), where the axial coordination between the central Fe–N4 unit in FePc and nitrogen atoms within the NGM substrate led to the formation of an Fe–N5 coordination environment.123 This structural enhancement significantly boosted the electrocatalytic performance toward the ORR, owing to improved electronic interaction and stabilization of the active site. Liu et al. developed Fe–N5/C@graphene featuring axial Fe–N5 coordination sites anchored on monolayer graphene, using FePc as the starting material. During the oxygen reduction reaction, this catalyst exhibited a highly efficient four-electron transfer pathway, highlighting its superior ORR catalytic activity.124 Lin et al. reported the successful fabrication of Fe single-atom catalysts supported on nitrogen-doped carbon (Fe-SAC/N–C) with axial N coordination, forming a Fe–N5 structure that delivered outstanding ORR performance with a E1/2 of 0.89 V and enhanced durability.125 DFT analysis was conducted to investigate the ORR mechanisms on three representative catalyst models, Fe–N4, Fe–N4–OH, and Fe–N4–py (Fig. 10b). The results revealed that axial coordination with pyridine significantly modulates the binding strength between Fe active sites and oxygenated intermediates, thereby promoting higher catalytic efficiency for ORR (Fig. 10e).
 | ||
| Fig. 10 (a) Schematic representation of the self-assembly process of the FePc/CNT-R catalyst, (c) fitting of k3-weighted Fe K-edge EXAFS for FePc/CNT-NH2 and (d) linear scan voltammogram of FePc, FePc/CNT-NH2, and Pt/C in 0.1 M KOH. (Reproduced with permission from ref. 116, copyright 2022, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim). (b) Top-view representations of the three Fe-SAC models: Fe–p4N (left), Fe–p4N–OH (middle), and Fe–p4N–py (right), showcasing their varying coordination environments. Atoms are color-coded: Fe (green), C (grey), N (blue), O (red), H (pink) and (e) free energy diagrams for the ORR on Fe–p4N (black), Fe–p4N–OH (yellow), and Fe–p4N–py (blue) at an overpotential of 0.4 V. The inset shows a volcano plot correlating catalytic activity with the *O binding energy (ΔEO). (Reproduced with permission from ref. 119, copyright 2019, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim). (f) Schematic of the synthetic route for the O–Zr–N–C catalyst, (g) R-space EXAFS fitting analysis for O–Zr–N–C; the inset presents the corresponding local coordination structure of the Zr site, where atomic colors are: C (grey), N (blue), Zr (orange), O (red) and (h) ORR polarization curves measured at 5 mV s−1 and 1600 rpm. (i) Chronoamperometric responses of O–Zr–N–C and Pt/C at 0.70 V vs. RHE; the inset compares their ORR polarization curves before and after accelerated durability tests. (Reproduced with permission from ref. 120, copyright 2022, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim). | ||
Apart from axial N coordination in MPc materials, O-containing ligands also serve as crucial axial modifiers in tailoring SACs configurations.127 These ligands modulate the metal center's electronic environment, thereby fine-tuning the adsorption energies of catalytic intermediates and influencing overall reaction kinetics.127 Wang et al. developed a Zr-based SAC featuring a five-fold coordination structure with an axial oxygen ligand (O–Zr–N4) as illustrated in Fig. 10f and g.126 The incorporation of the axial O ligand lowers the Zr d-band center, which stabilizes the local coordination environment and optimizes intermediate adsorption. Consequently, the O–Zr–N4 catalyst exhibits superior ORR activity compared to commercial Pt/C, achieving an E1/2 of 0.91 V and retaining 92% of its current after 130 hours, demonstrating excellent long-term durability as shown in Fig. 10h and i. Similarly, Zhang et al. synthesized a Co-based SAC (Co–SA@N–CNFs) featuring an axial O-coordinated structure.128 The proposed coordination environment consists of a Co–N4O configuration, where the axial O atom is positioned perpendicular to the Co–N4 plane. This axial design significantly enhanced ORR activity. Notably, the catalyst exhibited a low Tafel slope of 50 mV dec−1, outperforming commercial Pt/C (62 mV dec−1), indicating superior ORR reaction kinetics. Moreover, Fe@Fe/N-graphene 800, a single-atom catalyst featuring axial O–FeN4 sites, was developed by immobilizing FePc molecules onto a graphene-like Fe–N–C material by Xu et al.129 The formation of a characteristic Fe–O–Fe bridge results from the interaction between the iron center of FePc and the oxygen atom in the O–FeN4 site. This bridging structure helps in lowering the ORR overpotential.
Axial S ligands can also modulate the electronic structure of planar M–N4 sites, easing *OH desorption, the key rate-limiting step, and boosting ORR performance.63 Guo et al. developed a chromium-based SAC with a five-coordinated site (S1–Cr1N4–C) by co-pyrolysis with S source and Cr source as shown in Fig. 11a.130 EXAFS analysis revealed four Cr–N bonds (∼1.34 Å) and one axial Cr–S bond (∼2.0 Å), confirming the S-coordinated Cr1N4 configuration (Fig. 11b). This structure delivered enhanced ORR activity, improved methanol tolerance, and excellent stability. Likewise, Li et al. designed an Fe–N–C catalyst with a tailored coordination structure, where the Fe center is bonded to four in-plane nitrogen atoms and one axial sulfur ligand (Fig. 11c and d).131 The DFT calculation revealed that the axial S ligand reduces the electron density on the Fe atom compared to the standard FeN4 site (Fig. 11e), altering its electronic and spin state. This shift increases the number of unpaired electrons, improving the adsorption–desorption dynamics at the active site. Consequently, the catalyst exhibited superior ORR activity, achieving an onset potential of 0.99 V and a E1/2 of 0.88 V, the highest among tested samples (Fig. 11f).
 | ||
| Fig. 11 (a) Synthetic procedure of the Cr single-atom anchored on N, S co-doped porous carbon nanosheet (S1–Cr1N4–C) and (b) Cr K-edge EXAFS fitting analysis of S1–Cr1N4–C in the K space. (Reproduced with permission from ref. 124, copyright 2023, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim). (c) and (d) FT-EXAFS analysis in R space for S-modified Fe–N–C and pristine Fe–N–C (inset: suggested models for S–FeN4 and FeN4), (e) visualized spatial charge density difference for the S–FeN4 and FeN4 structures, depicted as isosurfaces. Regions of electron accumulation and depletion are indicated in yellow and blue, respectively and (f) ORR electrocatalytic performance of S–FeN4, FeN4 and Pt/C exhibited by linear sweep voltammetry (LSV) curves at 1600 rpm. (Reproduced with permission from ref. 125, copyright 2024, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim). (g) The proposed structural model of Fe–N/C-SAC is presented alongside its corresponding EXAFS curve-fitting analysis, with individual Fe–N, Fe–Cl, and Fe–C scattering pathways delineated. The atomic color scheme is Fe (pink), N (blue), Cl (green), and C (gray). (Reproduced with permission from ref. 126, copyright 2022, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim). | ||
In addition to common axial ligands such as N, O, and S, halogens such as chlorine (Cl) can also be coordinated axially in SACs.132 Axial Cl coordination effectively tunes the surface electronic structure, promoting faster 4e− ORR kinetics by optimizing intermediate adsorption.63 Xin et al. developed Fe–N–C–Cl SACs, with EXAFS analysis confirming that each Fe atom resides in a divacancy cavity coordinated to four in-plane N atoms and one axial Cl atom, forming a FeN4–Cl structure embedded in a porous carbon matrix (Fig. 11g).132 The axial Cl ligand significantly boosts ORR activity in alkaline media, achieving a E1/2 of 0.91 V and a kinetic current density of 55 mA cm−2 at 0.85 V, outperforming N/C and Pt/C by factors of 20.8 and 11.5, respectively. Additionally, Ding et al. synthesized Fe–N–C SACs with axial Cl coordination (FeN4Cl SACs) to assess their ORR performance.133 The simulations of different structural models revealed that the FeN4Cl–C configuration, without carbon defects, showed the highest exothermicity and ORR activity. The axial Cl ligand promotes charge transfer from N to Cl at the Fe site, boosting catalytic efficiency. The density of states (DOS) analysis confirmed enhanced orbital interactions near the Fermi level in FeN4Cl–C compared to FeN4–C, consistent with improved ORR activity. Moreover, Hu et al. synthesized SACs by pyrolyzing a 4,5-dichloroimidazole-modified Zn/Fe bimetallic triazole (MET) framework rich in nitrogen.134 This strategy enabled the formation of high-density Fe sites with an FeN4Cl1 structure. The resulting FeN4Cl1/NC catalyst demonstrated strong ORR performance in both acidic and alkaline media. The DFT calculations showed that the axial Cl ligand improves *OH adsorption energy at Fe sites, enhancing ORR kinetics. Notably, this design approach can also activate typically inactive transition metals through axial coordination.
4.4. Dual-metal coordination structures
Incorporating a secondary metal atom into a symmetric SACs framework breaks the original coordination symmetry and forms a bimetallic M1M2–N4 structure, while maintaining atomic-level dispersion.91,135 This modification enables refined control over both the geometric and electronic environments of the active center.91 The cooperative interaction between the host metal (M1) and the introduced dopant (M2) leads to reconfigured electronic states, which can strengthen or weaken adsorption energies of key intermediates as needed.136 Such bimetallic configurations promote synergistic metal–metal effects, offering enhanced catalytic activity and improved reaction kinetics by optimizing intermediate binding and facilitating more efficient charge and mass transfer processes.In general, incorporating a second metal atom near the active site can either create direct metal–metal bonds or induce long-range electronic interactions, modifying the electron density and symmetry around the active center.139 This adjustment enhances the catalyst's overall activity and performance. For instance, Zhang et al. introduced a bimetallic SAC with a covalently linked Ni–Co atomic pair (NiN3–CoN3–NC) embedded in N-doped graphitized carbon (Fig. 12a).137 DFT analysis showed that Co incorporation distorts the Ni 3d orbital distribution, revealing strong interaction between the NiN3 and CoN3 sites (Fig. 12b and c). The Co 3d-band center shifts closer to the Fermi level (1.51 eV), indicating enhanced adsorption of reaction intermediates. This synergistic effect promotes COOH formation, boosting both activity and selectivity for CO2RR by accelerating the reaction kinetics. In addition, Yao et al. studied a Ni–Cu diatomic catalyst (NiCu–NC) supported on N-doped carbon for CO2RR.138 The influence of the adjacent Cu atom on the 3d electronic structure of the Ni site was analyzed. DFT results showed that the introduction of Cu shifted both the peak location (Ep, first peak near Fermi level) the d-band center upward and nearer to the Fermi level (Fig. 12d). Additionally, tuning the distance between Ni–N4 and Cu–N4 to about 5.3 Å (dNiCu-5.3) enhanced electronic interaction, leading to improved CO2RR activity and selectivity (Fig. 12e). Moreover, Pei et al. developed Co–Mn dual-site catalysts with low nitrogen coordination, named L–Co1Mn1–NC, using a post-synthesis co-substitution strategy on MOF-derived N-doped carbon nanorods.140 Charge density difference plots revealed notable charge redistribution at the Co and Mn sites, indicating altered electronic structures that affect catalytic behavior. Further electronic structure analysis showed that the undercoordinated L–Co1Mn1–NC exhibited stronger electron sharing between Co and Mn atoms, as confirmed by electron localization function mapping, enhancing electron transfer and improving CO2RR activity. Similarly, Xie et al. designed a dual-metal catalyst (NiN4–SnN4) with each Ni and Sn atom coordinated by four N atoms.141 The nearby Ni site alters the electron distribution around Sn, lowering the energy barrier for OCHO formation and making the rate-determining step more favorable thermodynamically. Leveraging the synergistic interaction between Ni and Sn sites, the catalyst achieved high formate production selectivity of 86.1% at −0.82 V vs. RHE, outperforming both Sn–SAC (70.4%) and Ni–SAC (<1%). Meanwhile, Hao et al. developed a non-bridged Ni dual-atom catalyst (Ni2N6) featuring a N3–Ni–Ni–N3 coordination for CO2-to-CO conversion.142In situ environmental scanning transmission electron microscopy (STEM) tracked the Ostwald ripening of bulk Ni into atomically dispersed Ni dimers. In situ XAS and DFT analysis revealed that the true active site for efficient CO2RR and suppressed HER is the hydroxyl-modified Ni2N6OH structure. Additionally, an O-bridged Ni2N6 site (O–Ni2N6) has been identified as an effective active site for dynamic CO2 catalysis.143 Other homoatomic dual-atom catalysts, such as non-bridged Ni2N4C2 and Pd2 with PdN2O2 coordination, have also demonstrated exceptional CO selectivity approaching 100%.144,145
 | ||
| Fig. 12 (a) Scheme illustration of the synthetic procedure of NiN3–CoN3–NC and (b) and (c) the PDOS for the Ni 3d and Co 3d orbitals on the NiN3–CoN3–NC. (Reproduced with permission from ref. 137, copyright 2023, Elsevier). (d) The pDOS for the nickel d-orbitals was computed for the sNi, dNiCu-2.6, and dNiCu-5.3 models and (e) stability evaluation of NiCu–NC by chronoamperometric test at −1.07 V versus RHE for continuous 30 h. (Reproduced with permission from ref. 138, copyright 2023, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim). | ||
5. Curvature effects on symmetry breaking of SACs
Curvature quantitatively characterizes the extent to which a surface or structure deviates from flatness.67 It is inversely proportional to the radius of curvature, meaning that as the radius decreases, the degree of curvature increases.67 Thus, a smaller radius corresponds to a more pronounced curvature feature. Materials exhibiting high surface curvature represent a distinct category of functional materials characterized by geometrically curved morphologies and often intricate hierarchical architectures.146 Representative examples include onion-like carbon structures,147 CNTs,148 and curved 2D carbon nanosheets or 2D graphene149 as shown in Fig. 13. These materials are widely employed as electrocatalysts or catalyst supports owing to their unique surface curvature and interfacial properties, which facilitate enhanced electronic interactions, increased active site exposure, and improved mass transport during catalytic processes.150 High-curvature SACs feature isolated metal atoms anchored on curved supports, typically stabilized through coordination with heteroatoms.55,150 The local geometric asymmetry introduced by curvature induces symmetry breaking in the atomic configuration, enhancing the exposure of active sites and promoting efficient interaction with reactants besides strain and defect-triggering symmetry disruption.35,60 These curved architectures facilitate uniform dispersion of metal atoms and generate localized electric fields, which increase the frequency of effective collisions and improve catalytic performance.151 While both curvature-induced electric fields and strain-induced lattice distortion ultimately modify the electronic structure of the single-atom active site, they originate from fundamentally different physical phenomena and thus impact catalytic performance through divergent primary pathways. Strain operates primarily through direct geometric perturbation, compressing or elongating metal–ligand bonds at the angstrom scale, which directly modulates the crystal field splitting, shifts the d-band center of the metal atom, and thereby alters the covalent interaction strength and orbital overlap with adsorbed intermediates, fundamentally tuning the adsorption energetics and activation barriers for specific reaction steps.73,74 In contrast, curvature-induced effects generate intense, localized electric fields at highly curved tip-like or concave regions, which exert a long-range electrostatic influence that can polarize reaction intermediates, concentrate ionic reactants (e.g. OH−, H+, O2-related species) at the interface, and significantly alter the orientation and stabilization of polar transition states, thereby enhancing reaction rates by affecting the pre-exponential factor and local concentration rather than solely the activation energy barrier via the electronic structure.151 Consequently, strain is a strategy for fine-tuning the inherent reactivity of the active site by reshaping its electronic density of states, whereas curvature primarily manipulates the local reaction environment and mass transport, offering a distinct pathway to enhance performance by engineering the electrochemical interface at the nanoscale. Additionally, curvature-induced charge redistribution around the active centers further boosts catalytic activity and selectivity.151 The geometric features of high-curvature SACs, characterized by pronounced bending and confined nanoscale regions, play a pivotal role in modulating catalytic performance.152,153 The curvature of the support introduces structural asymmetry and surface concavity, which enhances the density of exposed active sites and increases contact with reactants.152,153 Simultaneously, narrow channels or confined regions on the support can restrict molecular diffusion, thereby improving selectivity by favoring access to specific reactants while excluding others.152,153 These curvature-induced effects vary across supports of different dimensionalities, enabling tailored optimization of single-atom reactivity and catalytic efficiency. Therefore, curvature engineering of supports of SACs offers a promising strategy for enhancing SAC activity and stability, providing a non-ligand-based approach to control electronic structures at the atomic level. | ||
| Fig. 13 Representative high curvature supports for SACs including curved 2D carbon nanosheets, curved 1D carbon nanotubes and 0D carbon sphere. When supports are curved, the symmetry of SACs is broken, leading to the change of geometric configuration and electrocatalytic performance. | ||
5.1. Zero dimensional supports
SACs immobilized on small-sized, zero-dimensional (0D) supports can give rise to sharp tip-like features that concentrate surface curvature, thereby generating strong localized electric fields.154,155 These intensified fields enhance the electronic polarization at active sites, significantly promoting catalytic reactivity and accelerating reaction kinetics.155 The inherent curvature also induces structural distortion, manifested as variations in the bond length between the metal atom and the support.67 This bond length modulation alters the local coordination environment and electronic configuration of the active metal center, ultimately tuning its catalytic behavior.67 Furthermore, the pronounced curvature imposes compressive strain on the anchored metal atom, leading to a shift in its d-band center, a phenomenon known as the d-band center effect.156 This electronic modulation directly influences the adsorption energy of reaction intermediates, facilitating both adsorption and desorption processes and thereby improving the overall catalytic efficiency and product selectivity.67 Recently, Wang et al. recently synthesized SACs supported on carbon spheres of varying diameters from 50 to 1000 nm using a high-temperature furnace method.157 The smaller sphere diameters increased surface curvature, which in turn intensified the local electric field (Fig. 14a). Despite identical active sites, catalytic activity could be modulated via curvature effects. Among Fe-based SACs, Fe-SAC-70 (70 nm diameter) showed the highest ORR activity with an E1/2 of 0.91 V, outperforming larger-diameter counterparts (Fig. 14b). Conversely, Co-SAC-350 delivered the best OER performance, indicating that extreme electric field strength is not always beneficial as optimal catalytic performance depends on a balanced field intensity (Fig. 14c). Besides that, onion-like carbon spheres, a class of 0D carbon nanomaterials, feature concentric graphite shells resembling an onion's layers.158,159 Their highly curved, layered structure held by van der Waals forces offers distinct advantages for electrocatalytic applications.158,159 For example, Liang et al. prepared a Co,N-doped carbon catalyst with onion-like carbon (OLC) coatings by pyrolyzing a P123-modified ZIF-67 precursor (Fig. 14e).147 The resulting OLC/Co–N–C material featured curved multilayer nanospheres with clear Co and carbon lattice fringes (Fig. 14d). This structure delivered superior bifunctional ORR/OER performance, with an E1/2 of 0.855 V and an OER overpotential of 344 mV at 10 mA cm−2. The enhanced activity compared to Co–N–C without OLC stems from the hydrophilic curved surface, which promotes OH− adsorption and boosts reactivity near pyridinic nitrogen sites. | ||
Fig. 14 (a) Schematics of modulation of the interfacial electric field by the carbon support sphere diameter, (b) the ORR activity was evaluated using linear sweep voltammetry (LSV) for a series of Fe-SACs with varying sphere diameters, recorded in O2-saturated 0.1 M KOH at a rotation rate of 1600 rpm and (c) LSV curves of Co-SACs with different diameter in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) M KOH for evaluating OER activity. (Reproduced with permission from ref. 151, copyright 2024, Springer Nature). (d) HRTEM image of OLC/Co–N–C and (e) synthesis of OLC/Co–N–C. (Reproduced with permission from ref. 141, copyright 2021, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim). M KOH for evaluating OER activity. (Reproduced with permission from ref. 151, copyright 2024, Springer Nature). (d) HRTEM image of OLC/Co–N–C and (e) synthesis of OLC/Co–N–C. (Reproduced with permission from ref. 141, copyright 2021, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim). | ||
5.2. One dimensional supports
The intrinsic curvature of one-dimensional (1D) supports, such as nanotubes and nanowires, plays a critical role in modulating the structural distortion experienced by single-atom catalytic sites during electrochemical reactions.160,161 This distortion alters the local bonding environment, particularly by varying the bond lengths between the anchored metal atoms and adjacent atoms in the support lattice.160,161 Such changes in local structure directly influence the efficiency of electron transfer from the active sites to adsorbed reactant species, thereby modulating the overall catalytic activity. For example, Han et al. employed single-atom Cu anchored on CNTs with varying diameters to investigate the dynamic evolution of CuN2C2 coordination during in situ ORR, highlighting how substrate-induced strain alters the geometric configuration of active sites and impacts catalytic activity as shown in Fig. 15a.161 Their findings emphasized that both graphene and CNTs serve as ideal model platforms for studying structure–activity relationships in SACs due to their well-defined sp2-hybridized carbon frameworks. In contrast to planar graphene, CNTs contain more sp3-like carbon atoms and inherently strained C–C bonds resulting from their curved geometry.162,163 This curvature-induced strain influences the local coordination environment, leading to distinct dynamic behaviors of the active sites under operating conditions, which in turn regulate the overall electrocatalytic performance.164 Wang et al. explored the nitrate electroreduction (NO3RR) activity of atomically dispersed Cu–N3 sites anchored on highly curved, porous nitrogen-doped CNTs (Cu–N3 SACs/NCNT) (Fig. 15b).165 These unsaturated Cu–N3 single-atom catalysts were synthesized via a sequential approach involving surface polymerization, electrostatic adsorption, and subsequent carbonization. The high-curvature 1D porous CNTs served as structurally favorable support, enhancing the electronic environment around the active sites. This work presents a synergistic design strategy that combines the engineering of low-coordination metal centers with curvature-regulated carbon support, offering a promising route for optimizing NRR electrocatalysts through dual modulation of active site geometry and carrier architecture. Furthermore, Zhou et al. explored how the curvature of M–N–C SACs, anchored on 1D single-walled CNTs with varying diameters, influences their electrocatalytic behavior in ORR and OER processes (Fig. 15c–e).166 They found that the curved geometry weakens the bonding between metal atoms and the N-doped carbon framework, thereby modifying the electronic configuration of the metal centers. This structural distortion causes an upward shift in the metal d-band center, enhancing the adsorption of key reaction intermediates and ultimately tuning the catalytic efficiency for both ORR and OER. | ||
| Fig. 15 (a) The distortion of the CuN2C2 active site is influenced by the carbon substrate, increasing with curvature-induced strain from planar graphene (left) to a small-diameter CNT (right). (Reproduced with permission from ref. 161, copyright 2021, Springer Nature). (b) Schematic illustration of the synthesis of Cu–N3 SACs/NCNT. (Reproduced with permission from ref. 165, copyright 2023, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim). (c) Computational models of transition-metal (TM) single-atoms incorporated into a series of curved nitrogen-doped carbon matrices with varying diameters, and volcano plots of (d) ORR overpotential (ηORR) and (e) OER overpotential (ηOER) versus the descriptor C which refers to the curvature of the carbon matrix. (Reproduced with permission from ref. 166, copyright 2023, The Royal Society of Chemistry). | ||
5.3. Two dimensional supports
The curvature of 2D materials, particularly 2D carbon-based supports, plays a crucial role in tuning the performance of SACs.167 Unlike flat substrates, curved 2D carbon structures such as carbon nanosheets and graphene introduce local strain and modify the hybridization states of surface atoms.167,168 These geometric distortions and symmetry breaking can significantly influence the coordination environment and electronic structure of the anchored metal atoms.167,168 Specifically, increased curvature can induce shifts in the d-band center of metal atoms, alter their electron density, and enhance their interaction with reaction intermediates.168 For example, Cepitis et al. explored how the surface curvature of 2D M–N–C nanosheets with dual-atom sites affects their OER/ORR bifunctional catalytic behavior (Fig. 16a).58 To study this, they designed a series of bimetallic M–N–C models with varying degrees of curvature inside the micropores, represented by radii of 4, 6, 8, and ∞ Å (where ∞ Å means the surface is flat). DFT results revealed that the highest overall catalytic activity was achieved when the radius reached 8.5 Å as shown in Fig. 16b and c. By developing models that allowed curvature modulation at the dual-metal active sites during the reaction, they provided a mechanistic understanding of how nanoscale surface curvature of 2D materials impacts catalytic efficiency for ORR (Fig. 16d). Furthermore, Tang et al. developed more realistic curved graphene models and constructed a series of transition metal (Co, Ni)-based SACs anchored on these curved structures.169 Their study revealed that compressing the lattice constant of graphene to 95% introduces a suitable curvature that improves both the structural stability and catalytic properties of the materials. Compared to flat graphene, the curved models exhibited enhanced catalytic activity. Specifically, curvature notably boosted HER performance across all systems. Similarly, Ni-based curved SACs also showed substantial improvement in ORR and OER performance, highlighting the critical role of curvature in optimizing electrocatalytic efficiency. Similarly, Wang et al. designed a series of tetracoordinated transition metal–N-graphene SACs by compressing the graphene lattice to 90%, covering configurations such as M–C4, M–C3N1, M–C2N2 (three types), M–C1N3, and M–N4 as shown in Fig. 16e.170 They assessed their NRR electrocatalytic performance by evaluating the stability of key intermediates. Out of 406 combinations (29 metals, 7 coordination types, and two N2 adsorption modes), five catalysts were identified with limiting potentials below 0.55 V. Further analysis comparing NRR selectivity confirmed Fe–C2N2-3 (side-on), Mn–C1N3 (end-on-2), Os–C1N3 (end-on-2), V–C2N2-1 (side-on), and V–N4 (side-on) as the most promising NRR candidates. | ||
| Fig. 16 (a) A model of in-pore dual-atom site M–N–C, exhibiting the effect of surface curvature effect on ORR, (d) schematics of ORR through associative and dissociative mechanisms, and ORR intermediate adsorption energies in (b) the CoCo model and (c) the CoNi model. The green line indicates the ideal adsorption energy. (Reproduced with permission from ref. 52, copyright 2023, American Chemical Society). (e) Computational models of seven N-doped graphene-supported SACs with varying coordination environments. Transition metal, carbon, and nitrogen atoms are represented by purple, yellow, and white spheres, respectively. (Reproduced with permission from ref. 164, copyright 2024, Elsevier). | ||
5.4. The principal and influence of curvature radius on SACs
As previously discussed, support materials with high surface curvature can significantly enhance catalytic performance by modulating the electronic environment and structure–activity relationship of metal single-atom sites.64 This enhancement is closely associated with alterations in the electronic structure, which are governed by factors such as the Fermi level of the metal atom and the electronic interactions with the underlying support.67 These effects collectively influence charge redistribution and catalytic reactivity at the atomic scale.67 The curvature of carbon-based supports plays a critical role in shaping the catalytic behavior of SACs.150 Even minor variations in curvature can induce substantial changes in the local electronic environment of metal centers, which in turn influence their binding affinity and activation ability toward reactant molecules.171 Studies on materials such as graphene, CNTs, and N-doped CNTs have demonstrated that modulating surface curvature effectively tunes their electronic configurations and catalytic performance.161,165,170 Interestingly, the catalytic response to curvature is not universal, some systems favor highly curved substrates, while others achieve optimal activity at lower curvature levels.171 This discrepancy highlights the need for tailored curvature control based on the catalyst system.172 By engineering the curvature of the support, one can alter coordination geometries, including bond lengths and angles between metal atoms and neighboring heteroatoms, thereby shifting the d-band center and breaking local symmetry.171,173,174 Thus, effective catalyst design must consider multiple interconnected factors: (1) metal coordination environment; (2) atomic arrangement; and (3) local bond characteristics. Together, they define catalytic efficiency and provide essential insight for the rational development of advanced SACs.246. Conclusion
Symmetry breaking has been identified as an effective approach in optimizing the atomic configurations and physicochemical characteristics of SACs to facilitate exceptional electrocatalytic performance.39 Nevertheless, attaining comprehensive understanding of the intricate structural configurations and exerting fine-tuned regulation over these engineered alterations towards symmetry breaking of SACs continues to present significant scientific and technical challenges.91 This work provides a systematic overview of recent progress in symmetry breaking architectures of SACs, detailing modulation methodologies and their implementation in energy-related electrocatalytic processes to understand the multifaceted structure–property correlations. Symmetry-breaking strategies in SACs, including strain-engineered, defect-mediated, and curvature-induced symmetry destabilization, lead to atomic-scale geometric distortions, generating asymmetric lattice deformations, undercoordinated sites, planar/axial heteroatom coordination motifs, bimetallic coordination networks, and high-curvature surface topologies.55,62,161,162,166,174 For the deeper understanding on mechanism of symmetry breaking of SACs toward electrocatalysis, DFT calculation is a very critical tool to comprehensively investigate nature and optimization of SACs based on structural symmetry disruption.175 While static DFT calculations have been invaluable in establishing structure–activity relationships and identifying the initial electronic effects of symmetry breaking, they indeed face significant limitations in representing the true dynamic nature of electrocatalysis, primarily because they model catalysts in a vacuum-like, ground-state equilibrium without accounting for the dynamic interface under operational conditions.175 Specifically, static DFT cannot capture the potential-dependent evolution of the coordination environment, such as the transient formation of metal-intermediate complexes that further distort local symmetry during the reaction, the solvent and electric field effects that can stabilize certain asymmetric configurations, or the dynamic interconversion between different coordination states under applied potential as suggested by in situ studies.176 For instance, while static DFT might identify a strained M–N4 site as optimal, it cannot predict the potential-induced ligand dissociation that could transform it into a more active but less stable M–N3 site during operation, nor can it accurately describe the entropic contributions and the dynamic charge transfer at the electrochemical interface that fundamentally alter the free energy landscape. These limitations highlight the critical need to integrate more advanced computational approaches, such as AIMD with explicit solvent models and constant-potential DFT methods, to bridge the gap between static models and the dynamically evolving, potential- and environment-dependent nature of symmetry-broken active sites in real electrocatalytic systems.175,176 In recent years, the machine learning is emerging as a powerful new tool for electrocatalyst development, offering a data-driven alternative to the intuition and trial-and-error that have traditionally guided the field.177 While DFT has been indispensable for elucidating the fundamental mechanisms behind symmetry–activity relationships, machine learning models can dramatically accelerate and enhance this research paradigm by addressing several key limitations of traditional computational approaches. Specifically, machine learning can leverage the extensive DFT datasets of structural descriptors, such as metal–ligand bond lengths, coordination numbers, d-band centers, and charge transfer values, to establish high-dimensional correlations that may be overlooked in conventional analysis, enabling the rapid prediction of optimal symmetry-broken configurations for target reactions without performing exhaustive DFT calculations for each candidate. For instance, by training graph neural networks on atomic structures of various M–NxXy sites, one could instantly screen thousands of potential symmetry-broken configurations to identify those with ideal adsorption energies for specific intermediates, effectively bypassing the computational cost of DFT while capturing the complex, non-linear relationships between geometric distortion and electronic structure. Moreover, machine learning models can integrate experimental synthesis parameters (e.g., pyrolysis temperature, precursor types) with theoretical descriptors to predict not only catalytic activity but also synthetic feasibility and stability, thus providing a holistic design framework that bridges the gap between atomic-scale symmetry breaking and macroscale catalyst performance.177 This synergistic DFT-machine learning approach would allow researchers to navigate the vast design space of symmetry-broken SACs more efficiently, moving from retrospective explanation to truly predictive design of next-generation electrocatalysts with tailored asymmetric active sites.177 To systematically compare the three major symmetry-breaking strategies in SACs, we summarize their structural characteristics, electronic effects, and catalytic influences in Table 1. This comparison highlights how each approach uniquely tunes the local coordination environment, electronic structure, and catalytic performance, offering clear guidance for the rational design of high-efficiency SACs in various electrocatalytic applications.| Strategy | Structural features | Electronic effects | Catalytic influence | Typical reactions with ref. | Key challenges |
|---|---|---|---|---|---|
| Strain-induced | 1. Bond length/angle alteration 2. Planar M–N4 distortion | 1. Shift in d-band center 2. Modified orbital hybridization | 1. Optimized intermediate adsorption 2. Enhanced activity/selectivity | ORR,80–82,84,87 CO2RR,77,82,83 HER,75 OER78,81 | 1. Precise strain control 2. Stability under operation |
| Defect-induced | 1. Low coordination (M–Nx, x < 4) 2. Heteroatom doping | 1. Local charge polarization 2. Asymmetric electron distribution | 1. Improved intermediate stabilization 2. Broken scaling relations | ORR,107,108,111,112 CO2RR,38,99,101,137,138 NRR102 | 1. Synthesis reproducibility 2. Dynamic structural changes |
| Curvature-induced | 1. Non-planar support (0D–2D) 2. Curved atomic sites | 1. Local electric field 2. Local strain effect; 3. Charge redistribution | 1. Enhanced mass/charge transfer 2. Exposed active sites | ORR,58,147,161,166 OER,58,147,166 NO3RR,165 HER,164 CO2RR,157 NRR170 | 1. Curvature uniformity 2. Scalability of curved supports |
These engineered asymmetries create electronic asymmetry, strengthen preferred intermediate binding, reduce activation energies, and amplify charge transfer kinetics, culminating in augmented electrocatalytic performance.39 This review elucidates how symmetry breaking fundamentally reconfigures electrocatalytic systems, enhancing electrocatalytic efficiency of oxidative and/or reductive reactions through optimized charge transfer dynamics and thermodynamic stabilization. While symmetry breaking strategies in SACs have demonstrated substantial potential, knowledge on such strategies needs to be deepened through systematic explorations in the following fundamental directions shown in Fig. 17.39
 | ||
| Fig. 17 Schematic illustration of the perspectives for symmetry breaking of SACs in electrocatalysis. | ||
1. Architectural precision challenges in SAC symmetry engineering: implementing symmetry breaking in SACs demands atomically resolved synthetic control, presenting intricate challenges requiring advanced nanoscale fabrication and operando characterizations. While ex situ characterization can only provide a static snapshot, the correlation of transient symmetry distortion with reaction kinetics is achievable in practice through a concerted methodology that combines operando spectroscopy, electrochemical analysis, and computational modeling. Specifically, operando XAS conducted simultaneously with electrochemical measurements, such as cyclic voltammetry or chronoamperometry, allows for the direct, real-time monitoring of changes in the metal center's coordination number, bond lengths, and oxidation state under varying applied potentials or current densities, thereby capturing the dynamic evolution of symmetry breaking as it occurs. By quantitatively analyzing these operando XANES/EXAFS data, for instance, tracking the intensity of pre-edge features indicative of symmetry-lowering or the contraction/elongation of metal–ligand bonds, and correlating these structural descriptors directly with the measured activity (e.g., turnover frequency) or the kinetic current density at each potential, one can establish a functional relationship between the degree of asymmetry and the catalytic rate. Furthermore, this experimental correlation is powerfully reinforced by DFT calculations, which can model the potential-induced structural distortion observed in the operando spectra and calculate the corresponding changes in the activation energy barriers for the rate-determining step, thereby providing a mechanistic bridge that directly connects the transient symmetry distortion, as a structural descriptor, to the observed reaction kinetics and overall catalytic performance. This necessitates paradigm-shifting methodologies combining atomically precise deposition, computational screening platforms, and directed growth protocols.61 Apart from engineering of coordination, strain, and curvature, more strategic interventions such as crystallographic facet engineering, vacancy/dopant implantation, and charge polarization engineering enable atomic-scale tailoring of non-equilibrium coordination states, establishing robust frameworks for rationally designed SAC architectures with optimized catalytic landscapes.
2. Stability implications of symmetry-engineered SAC systems: symmetric SAC architectures exhibit inherent thermodynamic destabilization effects, particularly under electrochemical operational stressors where prolonged exposure induces metastable structural transitions and catalytic attenuation.68 The remission strategies involve stabilization protocols through protective surface passivation layers or durable heteroatom integration to reinforce coordination thermodynamics. Advanced atomic layer deposition protocols enable resistance to surface reconstruction, while topologically engineered SAC configurations, featuring inherent symmetry-broken electronic configurations, demonstrate exceptional thermal/mechanical resilience under extreme operational conditions, presenting transformative potential for durable electrocatalytic systems. In addition to the static structural features of symmetry-broken SACs, their dynamic evolution under operational conditions plays a decisive role in long-term catalytic performance. During electrocatalytic reactions, the local coordination environment and strain state of symmetry-broken sites can undergo significant reconstruction due to interactions with reaction intermediates, applied potentials, and local chemical environments. In past years, in situ and operando characterization have revealed that metal centers in asymmetric coordination motifs such as M–N3 and M–N2 may experience reversible ligand rearrangement or axial coordination with electrolyte species, which can either stabilize active sites or lead to gradual metal leaching. Similarly, strain states, whether tensile or compressive, can relax or intensify under cyclic electrochemical loading, influencing both electronic structure and mechanical integrity. These dynamic processes may initially enhance activity by optimizing intermediate adsorption, but they can also accelerate degradation pathways such as site aggregation or support corrosion if not properly managed. Therefore, understanding and controlling the dynamic behavior of symmetry-broken sites, through operando spectroscopy, computational modeling, and rational material design, is essential for developing SACs that combine high activity with long-term operational stability.
3. Scale-up manufacturing challenges in symmetry-engineered SAC architectures: while symmetry breaking in SACs is readily achievable under controlled laboratory settings, attempts at industrial-scale replication may be met with challenges in defect topology conservation and catalytic fidelity retention.68 The atomically precise fabrication protocols incur prohibitive energy expenditures, critically impacting technoeconomic viability. This necessitates concurrent technoeconomic analysis during developmental phases to identify scalable material-process synergies. While the fundamental benefits of symmetry breaking at the atomic level, such as optimized adsorption energies and enhanced intrinsic activity, are clearly established in idealized laboratory settings, their ultimate validation lies in translating these gains to practical device performance. For instance, in fuel cells and water electrolyzers, the improved turnover frequency of symmetry-broken SACs must be effectively leveraged within the complex architecture of a catalyst layer, where factors like mass transport of reactants and products, proton/electron conductivity, and flooding management become critically important. Strategies such as engineering asymmetric coordination or strain can be tailored not only to lower overpotentials but also to improve stability under harsh operating conditions, thereby directly enhancing the energy efficiency and durability of these devices. Furthermore, the design of high-curvature supports or defect-rich matrices for symmetry breaking can simultaneously improve the dispersion of active sites and create hierarchical pore structures, which facilitate better mass transport and accessibility, addressing key limitations in conventional catalyst layers. Therefore, the future trajectory of symmetry-breaking research must increasingly bridge the gap between atomic-scale electronic modulation and the macro-scale engineering of electrode interfaces to realize the full potential of SACs in next-generation energy conversion technologies. Furthermore, although the atomic-scale precision required for symmetry control presents undeniable scalability challenges, several of the discussed approaches show promising pathways toward industrial implementation, albeit with varying degrees of feasibility. Defect-induced symmetry breaking, particularly through heteroatom doping or the creation of low-coordination sites, is arguably the most scalable, as it can be integrated into established bulk pyrolysis methods and precursor-controlled syntheses, similar to current industrial production of M–N–C catalysts, where the statistical distribution of asymmetric sites can be reproducibly achieved even if atomic-level uniformity remains imperfect. Strain engineering, while more challenging, can be scaled through substrate-driven approaches, such as using pre-strained carbon supports or leveraging the intrinsic thermal contraction of supports during pyrolysis, a process that can be standardized in continuous reactor systems. The primary bottlenecks for true industrial translation are the reproducible creation of specific asymmetric configurations like M–N3 or axial M–N5 sites at a large scale and the maintenance of structural uniformity across batches, which currently relies heavily on precise control of precursor ratios, pyrolysis temperatures, and gas atmospheres, those parameters that are manageable in lab settings but require sophisticated process engineering for cubic-meter-scale production. Nevertheless, the demonstrated performance enhancements in key reactions like ORR and CO2RR, coupled with the emerging use of template-assisted and flow synthesis methods, provide a strong impetus for further development, suggesting that with advanced manufacturing protocols and statistical quality control, the most robust symmetry-breaking strategies, particularly those based on defect and strain engineering, can indeed transition toward industrial electrocatalytic applications, provided that the cost-benefit analysis of the enhanced performance justifies the more complex synthesis. Accelerating progress demands focused innovation in advancing research toward energy-efficient synthetic routes for symmetry-breaking-oriented SACs while establishing unified manufacturing frameworks integrating modular symmetry-engineering modules, ensuring atomic-scale reproducibility across production volumes.63
4. Versatile SACs from symmetry breaking in sustainable energy systems: the emergence of polyfunctional catalytic architectures represents a paradigm shift in clean energy materials science. Asymmetry-driven engineering enables polyfunctional active site integration within unitary SAC frameworks, providing interconnected reaction pathways in modern energy systems. Atomic-scale asymmetry modulates adsorption energetics, transition state stabilization, and intermediate flux management, enhancing turnover frequencies while geometric/electronic confinement effects mitigate parasitic pathways, yielding highly selective product streams. The fundamental principles of symmetry breaking established for SACs, where controlled disruption of local symmetry enables precise tuning of electronic properties through degeneracy lifting, orbital polarization, and charge redistribution, provide a powerful conceptual framework that can be directly translated to the quantum design of other functional materials. For photocatalysts, strategic symmetry breaking could be engineered to create asymmetric charge localization centers that enhance the separation of photogenerated electron–hole pairs, while simultaneously tailoring band edge positions via d-band modulation to optimize visible-light absorption and redox potentials, much like how asymmetric coordination in SACs optimizes intermediate adsorption. In spintronic devices, the same principles that enable spin-state control in symmetry-broken SACs through crystal field splitting could be harnessed to design materials with tailored magnetic anisotropy or asymmetric spin–orbit coupling interfaces, potentially enabling more efficient spin injection and manipulation. Furthermore, the curvature- and strain-induced symmetry breaking strategies discussed could be adapted to create built-in electric fields in heterostructures or to break inversion symmetry in non-centrosymmetric materials, thereby enhancing Rashba effects or piezoelectric responses. Thus, the symmetry-breaking paradigm developed for SACs serves as a versatile quantum design blueprint that transcends electrocatalysis, offering systematic pathways to engineer wavefunction localization, spin configuration, and charge dynamics across a wide spectrum of advanced materials by strategically controlling atomic-scale asymmetry. These capabilities position symmetry-engineered SACs as transformative platforms for deployment across CO2 capture, photocatalysis, rechargeable batteries, spintronic devices, environmental remediation, and hybrid energy application platforms.39 Meanwhile, while the fundamental principles of symmetry breaking, such as using strain, coordination engineering, or curvature effects to modify electronic structure, can be conceptually extended to multi-site or cluster catalysts, maintaining true atomic precision becomes significantly more challenging due to the inherent complexity of these systems. In SACs, symmetry breaking targets a single, well-defined metal center, allowing for precise control through tailored coordination environments; however, in multi-site systems, the introduction of metal–metal bonds and ensemble effects creates a more complex energy landscape where symmetry breaking may affect multiple sites simultaneously and often non-uniformly, making it difficult to achieve the same level of design control. For instance, while the strain and ligand effects could theoretically be applied to a cluster, the resulting structural distortion would likely be distributed across several metal atoms and the support interface, leading to a spectrum of slightly different local environments rather than a single, precisely defined asymmetric site. In other words, certain subclasses like dual-atom catalysts represent a promising intermediate case, where the symmetry-breaking principles can be applied with relatively high precision to create tailored asymmetric dimers, but as the nuclearity increases further to clusters or nanoparticles, the atomic-level precision inevitably gives way to statistical distributions of surface sites, meaning that while symmetry breaking remains a powerful strategy to tune catalytic properties, its application transitions from atomic-level precision to a more averaged, ensemble-level design approach.
In summary, methodological exploration of symmetry breaking in SACs establishes new paradigms in electrocatalytic science. Through strategic atomic engineering of non-centrosymmetric architectures, multidimensional active site integration, and operational sustainability optimization, symmetry-modified SACs demonstrate enhanced electrocatalytic metrics (e.g., activity, selectivity, and durability), advancing transformative electrocatalytic platforms for next-generation energy conversion systems.
Conflicts of interest
The authors declare no conflict of interest.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Acknowledgements
This work was supported by the National Research Foundation Singapore, and A*STAR (Agency for Science, Technology and Research) under its LCER Phase 2 Programme 1st Emerging Technology (Award No. U2411D4006), Key Laboratory of Functional Inorganic Material Chemistry (Heilongjiang University), Ministry of Education, P. R. China and Key Laboratory of Applied Surface and Colloid Chemistry (Shaanxi Normal University), Ministry of Education, P.R. China.Notes and references
- S.-L. Li and Q. Xu, Energy Environ. Sci., 2013, 6, 1656–1683 RSC.
- Z. Yan, J. L. Hitt, J. A. Turner and T. E. Mallouk, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12558–12563 CrossRef CAS PubMed.
- H. Mistry, A. S. Varela, S. Kühl, P. Strasser and B. R. Cuenya, Nat. Rev. Mater., 2016, 1, 16009 CrossRef CAS.
- Y. Li, A. Feng, L. Dai, B. Xi, X. An, S. Xiong and C. An, Adv. Funct. Mater., 2024, 34, 2316296 CrossRef CAS.
- S. Mitchell, E. Vorobyeva and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2018, 57, 15316–15329 CrossRef CAS PubMed.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- M. Miceli, P. Frontera, A. Macario and A. Malara, Catalysts, 2021, 11, 591 CrossRef CAS.
- J. G. de Vries and S. D. Jackson, Catal. Sci. Technol., 2012, 2, 2009 RSC.
- A. Jamma, C. S. Vennapoosa, H. V. Annadata, B. Ghosh, R. Govu, H. Aggarwal, M. Ahmadipour, B. M. Abraham, X. Wang and U. Pal, ACS Appl. Mater. Interfaces, 2024, 16, 64681–64690 CrossRef CAS PubMed.
- N. Chanda, S. Gonuguntla, S. K. Verma, A. K. Basak, Y. Soujanya, M. Ahmadipour, S. Bojja and U. Pal, Int. J. Hydrogen Energy, 2023, 48, 37715–37724 CrossRef CAS.
- N. Chanda, B. Jaksani, S. Saha, A. Singh, H. Joshi, N. Sharma, S. Pakhira, U. Pal and M. Ahmadipour, Int. J. Hydrogen Energy, 2025, 140, 36–44 CrossRef CAS.
- R. Garg, S. Gonuguntla, S. Sk, M. S. Iqbal, A. O. Dada, U. Pal and M. Ahmadipour, Adv. Colloid Interface Sci., 2024, 330, 103203 CrossRef CAS PubMed.
- M. Ahmadipour, M. S. Iqbal, M. A. Saeed, A. A. Hamzah, A. Ramzan, A. Bhattacharya, U. Pal, M. Ahmadipour, A. L. Pang and M. Satgunam, Results Eng., 2025, 28, 107168 CrossRef CAS.
- L. L. Chng, N. Erathodiyil and J. Y. Ying, Acc. Chem. Res., 2013, 46, 1825–1837 CrossRef CAS PubMed.
- Š. Kment, A. Bakandritsos, I. Tantis, H. Kmentová, Y. Zuo, O. Henrotte, A. Naldoni, M. Otyepka, R. S. Varma and R. Zbořil, Chem. Rev., 2024, 124, 11767–11847 CrossRef PubMed.
- S. Mehmood, S. Sk, B. M. Abraham, M. Ahmadipour, U. Pal and J. Dutta, Adv. Funct. Mater., 2025, 35, 2418602 CrossRef CAS.
- Z. Li, D. Wang, Y. Wu and Y. Li, Natl. Sci. Rev., 2018, 5, 673–689 CrossRef CAS.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- T. Zhang, Nano Lett., 2021, 21, 9835–9837 CrossRef CAS.
- G. Luo, M. Song, Q. Zhang, L. An, T. Shen, S. Wang, H. Hu, X. Huang and D. Wang, Nano-Micro Lett., 2024, 16, 241 CrossRef PubMed.
- J. Xu, S. Zhang, H. Liu, S. Liu, Y. Yuan, Y. Meng, M. Wang, C. Shen, Q. Peng, J. Chen, X. Wang, L. Song, K. Li and W. Chen, Angew. Chem., Int. Ed., 2023, 62, e202308044 CrossRef CAS PubMed.
- K.-M. Zhao, S. Liu, Y.-Y. Li, X. Wei, G. Ye, W. Zhu, Y. Su, J. Wang, H. Liu, Z. He, Z.-Y. Zhou and S.-G. Sun, Adv. Energy Mater., 2022, 12, 2103588 CrossRef CAS.
- J. Li, M. Lin, W. Huang, X. Liao, Y. Ma, L. Zhou, L. Mai and J. Lu, Small Methods, 2023, 7, 2201664 CrossRef CAS PubMed.
- R. Li and D. Wang, Nano Res., 2022, 15, 6888–6923 CrossRef CAS.
- X. Q. Mu, S. L. Liu, M. Y. Zhang, Z. C. Zhuang, D. Chen, Y. R. Liao, H. Y. Zhao, S. C. Mu, D. S. Wang and Z. H. Dai, Angew. Chem., Int. Ed., 2024, 63, e202319618 CrossRef CAS PubMed.
- L. Wang, H. Chen, Y. Wang, X. Liu, C. Li, J. He and T. Yao, Nano Res., 2023, 16, 8596–8613 CrossRef CAS.
- X. Zhao, R. M. Young, C. Tang, G. Wu, K. R. Peinkofer, Y. Han, S. Yang, Y.-K. Xing, H. Han, H. Wu, X. Li, Y. Feng, R. Zhang, C. L. Stern, M. R. Wasielewski and J. F. Stoddart, Chem, 2025, 11, 102248 CAS.
- Q. N. Nguyen, C. Wang, Y. Shang, A. Janssen and Y. Xia, Chem. Rev., 2023, 123, 3693–3760 CrossRef CAS PubMed.
- H. Li, J. Zhao, L. Luo, J. Du and J. Zeng, Acc. Chem. Res., 2021, 54, 1454–1464 CrossRef CAS PubMed.
- F. Gao, M. Fang, S. Zhang, M. Ni, Y. Cai, Y. Zhang, X. Tan, M. Kong, W. Xu and X. Wang, Appl. Catal., B, 2022, 316, 121664 CrossRef CAS.
- J. Lin, W. Tian, H. Zhang, H. Sun and S. Wang, Acc. Chem. Res., 2024, 57, 2303–2315 CrossRef CAS PubMed.
- X. Zhu, Y. Jia, Y. Liu, J. Xu, H. He, S. Wang, Y. Shao, Y. Zhai and Y. Zhu, Angew. Chem., Int. Ed., 2024, 63, e202405962 CrossRef CAS PubMed.
- L. Li, H. Ying, P. Qiao, W. Liu, S. Shang, W. Shao, H. Wang, X. Zhang and Y. Xie, Nano Lett., 2024, 24, 14412–14419 CrossRef CAS PubMed.
- D. Liu, Q. He, S. Ding and L. Song, Adv. Energy Mater., 2020, 10, 2001482 CrossRef CAS.
- L. Du, T. Hasan, A. Castellanos-Gomez, G.-B. Liu, Y. Yao, C. N. Lau and Z. Sun, Nat. Rev. Phys., 2021, 3, 193–206 CrossRef CAS.
- Y. Li, H. Sun, L. Ren, K. Sun, L. Gao, X. Jin, Q. Xu, W. Liu and X. Sun, Angew. Chem., Int. Ed., 2024, 63, e202405334 CrossRef CAS PubMed.
- X. Mu, X. Gu, S. Dai, J. Chen, Y. Cui, Q. Chen, M. Yu, C. Chen, S. Liu and S. Mu, Energy Environ. Sci., 2022, 15, 4048–4057 RSC.
- J. Dong, Y. Liu, J. Pei, H. Li, S. Ji, L. Shi, Y. Zhang, C. Li, C. Tang, J. Liao, S. Xu, H. Zhang, Q. Li and S. Zhao, Nat. Commun., 2023, 14, 6849 CrossRef CAS PubMed.
- P. Cao, X. Mu, F. Chen, S. Wang, Y. Liao, H. Liu, Y. Du, Y. Li, Y. Peng, M. Gao, S. Liu, D. Wang and Z. Dai, Chem. Soc. Rev., 2025, 54, 3848–3905 RSC.
- S. Zhang, J. Chen, B. Wei, H. Zhou, K. Hua, X. Liu, H. Wang and Y. Sun, J. Am. Chem. Soc., 2024, 146, 6037–6044 CrossRef CAS PubMed.
- J. Wang, H. Zhang, Y. Nian, Y. Chen, H. Cheng, C. Yang, Y. Han, X. Tan, J. Ye and T. Yu, Adv. Funct. Mater., 2024, 34, 2406549 CrossRef CAS.
- Z. Qi, Y. Zhou, R. Guan, Y. Fu and J.-B. Baek, Adv. Mater., 2023, 35, 2210575 CrossRef CAS PubMed.
- Y. Liu, Z. Zhuang, Y. Liu, N. Liu, Y. Li, Y. Cheng, J. Yu, R. Yu, D. Wang and H. Li, Angew. Chem., Int. Ed., 2024, 63, e202411396 CAS.
- J. Hao, Z. Zhuang, J. Hao, K. Cao, Y. Hu, W. Wu, S. Lu, C. Wang, N. Zhang, D. Wang, M. Du and H. Zhu, ACS Nano, 2022, 16, 3251–3263 CrossRef CAS PubMed.
- Z. Zhuang, L. Xia, J. Huang, P. Zhu, Y. Li, C. Ye, M. Xia, R. Yu, Z. Lang, J. Zhu, L. Zheng, Y. Wang, T. Zhai, Y. Zhao, S. Wei, J. Li, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2023, 62, e202212335 CrossRef CAS PubMed.
- L. Lv, H. Tan, Y. Kong, B. Tang, Q. Ji, Y. Liu, C. Wang, Z. Zhuang, H. Wang, M. Ge, M. Fan, D. Wang and W. Yan, Angew. Chem., Int. Ed., 2024, 63, e202401943 CrossRef CAS PubMed.
- Z. Fang, J. Xiao, S. Tan, C. Deng, G. Wang and S. X. Mao, Sci. Adv., 2022, 8, eabn3785 CrossRef CAS PubMed.
- Z. Zhuang, Y. Li, Y. Li, J. Huang, B. Wei, R. Sun, Y. Ren, J. Ding, J. Zhu, Z. Lang, L. V. Moskaleva, C. He, Y. Wang, Z. Wang, D. Wang and Y. Li, Energy Environ. Sci., 2021, 14, 1016–1028 RSC.
- Y. Zhang, J. Yang, R. Ge, J. Zhang, J. M. Cairney, Y. Li, M. Zhu, S. Li and W. Li, Coord. Chem. Rev., 2022, 461, 214493 CrossRef CAS.
- C. Wang, Z. Wang, S. Mao, Z. Chen and Y. Wang, Chin. J. Catal., 2022, 43, 928–955 Search PubMed.
- Y. Shen, Y. Pan, H. Xiao, H. Zhang, C. Zhu, Q. Fang, Y. Li, L. Lu, L. Ye and S. Song, J. Mater. Chem. A, 2024, 12, 9075–9087 RSC.
- G. Li, J. Zhang, Z. Wu, H. Li, Y. Song and S. Zhang, Mater. Today, 2025, 85, 141–170 Search PubMed.
- X. Hou, J. Ding, W. Liu, S. Zhang, J. Luo and X. Liu, Nanomaterials, 2023, 13, 309 Search PubMed.
- S. Zhang, J. Huang, L. Ma, D. Zhai, B. Wei, H. Yang and C. He, Energy Rev., 2024, 3, 100075 CrossRef.
- J.-K. Li, H. Zhao, Y. Zhang, J.-J. Ma, F.-F. Wang, S.-N. Zhao, J. Li and S.-Q. Zang, Adv. Sci., 2025, 12, 2412387 Search PubMed.
- T. Wang, J. Wang, C. Lu, K. Jiang, S. Yang, Z. Ren, J. Zhang, X. Liu, L. Chen, X. Zhuang and J. Fu, Adv. Mater., 2023, 35, 2205553 CrossRef CAS PubMed.
- X. Li, X. Wu, Y. Zhao, Y. Lin, J. Zhao, C. Wu, H. Liu, L. Shan, L. Yang, L. Song and J. Jiang, Adv. Mater., 2023, 35, 2302467 CrossRef CAS PubMed.
- R. Cepitis, N. Kongi, J. Rossmeisl and V. Ivaništšev, ACS Energy Lett., 2023, 8, 1330–1335 CrossRef CAS PubMed.
- Y. Zhu, Y. Jiang, H. Li, D. Zhang, L. Tao, X.-Z. Fu, M. Liu and S. Wang, Angew. Chem., Int. Ed., 2024, 63, e202319370 CrossRef CAS PubMed.
- J. Li, X. Li and H. Zhu, Mater. Today, 2024, 75, 187–209 Search PubMed.
- G. Yun and W. Dingsheng, CCS Chem., 2023, 6, 833–855 Search PubMed.
- X.-F. Cheng, J.-H. He, H.-Q. Ji, H.-Y. Zhang, Q. Cao, W.-J. Sun, C.-L. Yan and J.-M. Lu, Adv. Mater., 2022, 34, 2205767 CrossRef CAS PubMed.
- L. Zhang, N. Jin, Y. Yang, X.-Y. Miao, H. Wang, J. Luo and L. Han, Nano-Micro Lett., 2023, 15, 228 CrossRef CAS PubMed.
- J. Li, W. Xia, Y. Guo, R. Qi, X. Xu, D. Jiang, T. Wang, Y. Sugahara, J. He and Y. Yamauchi, Chem. Eng. J., 2023, 477, 146841 CrossRef CAS.
- J. Yang, Z. Wang, C.-X. Huang, Y. Zhang, Q. Zhang, C. Chen, J. Du, X. Zhou, Y. Zhang, H. Zhou, L. Wang, X. Zheng, L. Gu, L.-M. Yang and Y. Wu, Angew. Chem., Int. Ed., 2021, 60, 22722–22728 CrossRef CAS PubMed.
- Y. Wang, Q. Wang, J. Wu, X. Zhao, Y. Xiong, F. Luo and Y. Lei, Nano Energy, 2022, 103, 107815 CrossRef CAS.
- C. Chen, Z. Zhang, L. Dai, B. Li and Z. Li, Appl. Catal., A, 2025, 694, 120160 CrossRef CAS.
- Y. Liu, Y. Luo, M. Zhang, A. Zhang and L. Wang, Small Methods, 2025, 9, 2401067 CrossRef CAS PubMed.
- Z. Zhang, H. Zhao, S. Xi, X. Zhao, X. Chi, H. Bin Yang, Z. Chen, X. Yu, Y.-G. Wang, B. Liu and P. Chen, Nat. Commun., 2025, 16, 1301 CrossRef CAS PubMed.
- Z. Zhang, Z. Zhang, C. Chen, R. Wang, M. Xie, S. Wan, R. Zhang, L. Cong, H. Lu, Y. Han, W. Xing, Z. Shi and S. Feng, Nat. Commun., 2024, 15, 2556 CrossRef CAS PubMed.
- F. Zhang, Z. Tang, T. Zhang, H. Xiao, H. Zhuang, P. Han, L. Zheng, L. Jiang and Q. Gao, Angew. Chem., Int. Ed., 2025, 64, e202418749 CrossRef CAS PubMed.
- R. Xu, J. Ren, X. Shen, Y. Zhu, Y. Shan and C.-G. Shi, ACS Omega, 2022, 7, 18826–18833 CrossRef CAS PubMed.
- L. Qi and J. Guan, Sci. Bull., 2025, 70, 1856–1871 CrossRef CAS PubMed.
- Y. Ni, Y. Lu, W. Xie and J. Chen, ChemElectroChem, 2025, 12, e202400535 CrossRef CAS.
- X. Gao, Y. Zhou, Y. Tan, B. Yang, Z. Cheng, Z. Shen and J. Jia, Appl. Surf. Sci., 2019, 473, 770–776 CrossRef CAS.
- K. Jiang, M. Luo, Z. Liu, M. Peng, D. Chen, Y.-R. Lu, T.-S. Chan, F. M. F. de Groot and Y. Tan, Nat. Commun., 2021, 12, 1687 CrossRef CAS PubMed.
- Y. Li, N. M. Adli, W. Shan, M. Wang, M. J. Zachman, S. Hwang, H. Tabassum, S. Karakalos, Z. Feng, G. Wang, Y. C. Li and G. Wu, Energy Environ. Sci., 2022, 15, 2108–2119 RSC.
- Y. Wang, S. Meng and M. Liu, J. Phys. Chem. Lett., 2023, 14, 9918–9925 CrossRef CAS PubMed.
- A. Kumar, S. Ibraheem, T. Anh Nguyen, R. K. Gupta, T. Maiyalagan and G. Yasin, Coord. Chem. Rev., 2021, 446, 214122 CrossRef CAS.
- Z. Miao, X. Wang, Z. Zhao, W. Zuo, S. Chen, Z. Li, Y. He, J. Liang, F. Ma, H.-L. Wang, G. Lu, Y. Huang, G. Wu and Q. Li, Adv. Mater., 2021, 33, 2006613 CrossRef CAS.
- C. Z. Loyola, G. Abarca, S. Ureta-Zañartu, C. Aliaga, J. H. Zagal, M. T. Sougrati, F. Jaouen, W. Orellana and F. Tasca, J. Mater. Chem. A, 2021, 9, 23802–23816 RSC.
- P. Zhao, Q. Zhang, Y. Liu, Z. Yin, Y. Wang, X. Zheng, H. Wang, Y. Deng and X. Fan, ACS Appl. Mater. Interfaces, 2024, 16, 49286–49292 CrossRef CAS PubMed.
- T. Liu, T. Xu, T. Li and Y. Jing, J. Am. Chem. Soc., 2024, 146, 24133–24140 CrossRef CAS PubMed.
- J. Li, H. Zhang, W. Samarakoon, W. Shan, D. A. Cullen, S. Karakalos, M. Chen, D. Gu, K. L. More, G. Wang, Z. Feng, Z. Wang and G. Wu, Angew. Chem., Int. Ed., 2019, 58, 18971–18980 CrossRef CAS PubMed.
- Y. Yuan, Q. Zhang, L. Yang, L. Wang, W. Shi, P. Liu, R. Gao, L. Zheng, Z. Chen and Z. Bai, Adv. Funct. Mater., 2022, 32, 2206081 CrossRef CAS.
- X. Li, Q. Zhou, S. Wang, Y. Li, Y. Liu, Q. Gao and Q. Wu, J. Phys. Chem. C, 2021, 125, 11963–11974 CrossRef CAS.
- L. Li, J. Zhu, F. Kong, Y. Wang, C. Kang, M. Xu, C. Du and G. Yin, Matter, 2024, 7, 1517–1532 CrossRef CAS.
- Y. Duan, Y. Xia, Y. Ling, S. Zhou, X. Liu, Y. Lan, X. Yin, Y. Yang, X. Yan, M. Liang, S. Hong, L. Zhang and L. Wang, ACS Nano, 2024, 18, 21326–21335 CrossRef CAS PubMed.
- W. Zhang, A. Mehmood, G. Ali, H. Liu, L. Chai, J. Wu and M. Liu, Angew. Chem., Int. Ed., 2025, 64, e202424552 CrossRef CAS PubMed.
- Q. Song, Z. Gong, J. Liu, K. Huang, G. Ye, S. Niu and H. Fei, Adv. Sci., 2025, 12, 2415665 CrossRef CAS PubMed.
- J. Bai, Z. Sun, H. Zhang, Y. Lian, Y. Deng, M. Xiang and Y. Su, Adv. Funct. Mater., 2025, 35, 2417013 CrossRef CAS.
- J. W. Sun, X. Wu, P. F. Liu, J. Chen, Y. Liu, Z. X. Lou, J. Y. Zhao, H. Y. Yuan, A. Chen, X. L. Wang, M. Zhu, S. Dai and H. G. Yang, Nat. Commun., 2023, 14, 1599 CrossRef CAS PubMed.
- Z. Wang, Y. Yan, Y. Zhang, Y. Chen, X. Peng, X. Wang, W. Zhao, C. Qin, Q. Liu, X. Liu and Z. Chen, Carbon Energy, 2023, 5, e306 CrossRef CAS.
- W. Zhou, H. Su, Y. Li, M. Liu, H. Zhang, X. Zhang, X. Sun, Y. Xu, Q. Liu and S. Wei, ACS Energy Lett., 2021, 6, 3359–3366 CrossRef CAS.
- R. Cheng, Y. Min, H. Li and C. Fu, Nano Energy, 2023, 115, 108718 CrossRef CAS.
- H. Wang, T. Yang, J. Wang, Z. Zhou, Z. Pei and S. Zhao, Chem, 2024, 10, 48–85 CAS.
- Y.-Y. Duan, Y. Zhang, Y.-Z. Cheng, X. Bao, K.-F. Gao, B.-R. Huang, S. Li, H. Bai, Y. Gong, D.-H. Yang, X. Ding, F. Li, Q. Zheng, X. Liu and B.-H. Han, Appl. Catal., B, 2025, 368, 125117 CrossRef CAS.
- Y. Duan, D. Chen, R. Zhang, Y. Du and L. Wang, Coord. Chem. Rev., 2025, 522, 216243 CrossRef CAS.
- L. Yuan, S. Zeng, G. Li, Y. Wang, K. Peng, J. Feng, X. Zhang and S. Zhang, Adv. Funct. Mater., 2023, 33, 2306994 CrossRef CAS.
- C. Xie, L. Lin, L. Huang, Z. Wang, Z. Jiang, Z. Zhang and B. Han, Nat. Commun., 2021, 12, 4823 CrossRef CAS PubMed.
- S. Li, S. Zhao, X. Lu, M. Ceccato, X.-M. Hu, A. Roldan, J. Catalano, M. Liu, T. Skrydstrup and K. Daasbjerg, Angew. Chem., Int. Ed., 2021, 60, 22826–22832 CrossRef CAS PubMed.
- Z. Gao, H. Huang, S. Xu, L. Li, G. Yan, M. Zhao, W. Yang and X. Zhao, Mol. Catal., 2020, 493, 111091 CAS.
- Y.-N. Gong, L. Jiao, Y. Qian, C.-Y. Pan, L. Zheng, X. Cai, B. Liu, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2020, 59, 2705–2709 CrossRef CAS.
- H. Shang, W. Sun, R. Sui, J. Pei, L. Zheng, J. Dong, Z. Jiang, D. Zhou, Z. Zhuang, W. Chen, J. Zhang, D. Wang and Y. Li, Nano Lett., 2020, 20, 5443–5450 CrossRef CAS PubMed.
- J. Liu, J. Zhu, H. Xu and D. Cheng, ACS Catal., 2024, 14, 6952–6964 CrossRef CAS.
- X. Xie, H. Peng, G. Ma, Z. Lei and Y. Xu, Mater. Chem. Front., 2023, 7, 2595–2619 RSC.
- X. Bai, Y. Wang, J. Han, X. Niu and J. Guan, Appl. Catal., B, 2023, 337, 122966 CrossRef CAS.
- H. Shang, X. Zhou, J. Dong, A. Li, X. Zhao, Q. Liu, Y. Lin, J. Pei, Z. Li, Z. Jiang, D. Zhou, L. Zheng, Y. Wang, J. Zhou, Z. Yang, R. Cao, R. Sarangi, T. Sun, X. Yang, X. Zheng, W. Yan, Z. Zhuang, J. Li, W. Chen, D. Wang, J. Zhang and Y. Li, Nat. Commun., 2020, 11, 3049 CrossRef CAS PubMed.
- K. Yuan, D. Lützenkirchen-Hecht, L. Li, L. Shuai, Y. Li, R. Cao, M. Qiu, X. Zhuang, M. K. H. Leung, Y. Chen and U. Scherf, J. Am. Chem. Soc., 2020, 142, 2404–2412 CrossRef CAS.
- Y. Zhou, R. Lu, X. Tao, Z. Qiu, G. Chen, J. Yang, Y. Zhao, X. Feng and K. Müllen, J. Am. Chem. Soc., 2023, 145, 3647–3655 CrossRef CAS PubMed.
- J. Roh, A. Cho, S. Kim, K.-S. Lee, J. Shin, J. S. Choi, J. Bak, S. Lee, D. Song, E.-J. Kim, C. Lee, Y. R. Uhm, Y.-H. Cho, J. W. Han and E. Cho, ACS Catal., 2023, 13, 9427–9441 CrossRef CAS.
- W. Xue, Q. Zhou, X. Cui, J. Zhang, S. Zuo, F. Mo, J. Jiang, X. Zhu and Z. Lin, Angew. Chem., Int. Ed., 2023, 62, e202307504 CrossRef CAS PubMed.
- Y. Jia, K. Jiang, H. Wang and X. Yao, Chem, 2019, 5, 1371–1397 CAS.
- Y. Li, Y. Ding, B. Zhang, Y. Huang, H. Qi, P. Das, L. Zhang, X. Wang, Z.-S. Wu and X. Bao, Energy Environ. Sci., 2023, 16, 2629–2636 RSC.
- X. Lin, X. Zhang, D. Liu, L. Shi, L. Zhao, Y. Long and L. Dai, Adv. Energy Mater., 2024, 14, 2303740 CrossRef CAS.
- J. Wang, J. Guoa, Y.-Y. Liua, P. Lia, Q. Fanga, X.-C. Li and W. Song, ChemPhysChem, 2024, 25, e202400414 CrossRef CAS PubMed.
- F. Wang, R. Zhang, Y. Zhang, Y. Li, J. Zhang, W. Yuan, H. Liu, F. Wang and H. L. Xin, Adv. Funct. Mater., 2023, 33, 2213863 CrossRef CAS.
- Y. Zhao, W.-J. Jiang, J. Zhang, E. C. Lovell, R. Amal, Z. Han and X. Lu, Adv. Mater., 2021, 33, 2102801 CrossRef CAS PubMed.
- J. Barrio, A. Pedersen, S. Ch Sarma, A. Bagger, M. Gong, S. Favero, C.-X. Zhao, R. Garcia-Serres, A. Y. Li, Q. Zhang, F. Jaouen, F. Maillard, A. Kucernak, I. E. L. Stephens and M.-M. Titirici, Adv. Mater., 2023, 35, 2211022 CrossRef CAS PubMed.
- M. P. Oyarzún, N. Silva, D. Cortés-Arriagada, J. F. Silva, I. O. Ponce, M. Flores, K. Tammeveski, D. Bélanger, A. Zitolo, F. Jaouen and J. H. Zagal, Electrochim. Acta, 2021, 398, 139263 CrossRef.
- R. Venegas, F. J. Recio, J. Riquelme, K. Neira, J. F. Marco, I. Ponce, J. H. Zagal and F. Tasca, J. Mater. Chem. A, 2017, 5, 12054–12059 RSC.
- W. Zhang, E. J. Meeus, L. Wang, L.-H. Zhang, S. Yang, B. de Bruin, J. N. H. Reek and F. Yu, ChemSusChem, 2022, 15, e202102379 CrossRef CAS PubMed.
- R. Cao, R. Thapa, H. Kim, X. Xu, M. Gyu Kim, Q. Li, N. Park, M. Liu and J. Cho, Nat. Commun., 2013, 4, 2076 CrossRef PubMed.
- F. Liu, N. Yan, G. Zhu, Z. Liu, S. Ma, G. Xiang, S. Wang, X. Liu and W. Wang, New J. Chem., 2021, 45, 13004–13014 RSC.
- Y. Lin, P. Liu, E. Velasco, G. Yao, Z. Tian, L. Zhang and L. Chen, Adv. Mater., 2019, 31, 1808193 CrossRef PubMed.
- X. Wang, Y. An, L. Liu, L. Fang, Y. Liu, J. Zhang, H. Qi, T. Heine, T. Li, A. Kuc, M. Yu and X. Feng, Angew. Chem., Int. Ed., 2022, 61, e202209746 CrossRef CAS PubMed.
- Q. Wang, G. Long, X. Gao, J. Chen, C. You, X. Tian, X. Wang, D. Kong and W. Fan, Appl. Catal., B, 2023, 337, 123009 CrossRef CAS.
- S. Zhang, Q. Zhou, L. Fang, R. Wang, T. Lu, Q. Zhao, X. Gu, S. Tian, L. Xu, H. Pang, J. Yang, Y. Tang and S. Sun, Appl. Catal., B, 2023, 328, 122489 CrossRef CAS.
- C. Xu, Y. Si, B. Hu, X. Xu, B. Hu, Y. Jiang, H. Chen, C. Guo, H. Li and C. Chen, Inorg. Chem. Front., 2022, 9, 3306–3318 RSC.
- Y. Guo, H. Yin, F. Cheng, M. Li, S. Zhang, D. Wu, K. Wang, Y. Wu, B. Yang and J.-N. Zhang, Small, 2023, 19, 2206861 CrossRef CAS PubMed.
- S. Li, L. Xia, J. Li, Z. Chen, W. Zhang, J. Zhu, R. Yu, F. Liu, S. Lee, Y. Zhao, L. Zhou and L. Mai, Energy Environ. Mater., 2024, 7, e12560 CrossRef CAS.
- C. Xin, W. Shang, J. Hu, C. Zhu, J. Guo, J. Zhang, H. Dong, W. Liu and Y. Shi, Adv. Funct. Mater., 2022, 32, 2108345 CrossRef CAS.
- S. Ding, J. A. Barr, Q. Shi, Y. Zeng, P. Tieu, Z. Lyu, L. Fang, T. Li, X. Pan, S. P. Beckman, D. Du, H. Lin, J.-C. Li, G. Wu and Y. Lin, ACS Nano, 2022, 16, 15165–15174 CrossRef CAS PubMed.
- L. Hu, C. Dai, L. Chen, Y. Zhu, Y. Hao, Q. Zhang, L. Gu, X. Feng, S. Yuan, L. Wang and B. Wang, Angew. Chem., Int. Ed., 2021, 60, 27324–27329 CrossRef CAS PubMed.
- Z. Han, S. Chai, Y. Zhu, X. Yao, L. Yang, C. Du, X. Ma, C. Cao and M. Zou, Chem. Eng. J., 2024, 502, 157943 CrossRef CAS.
- Y. Tian, R. Chen, X. Liu, Z. Mao, H. Tan, D. Yang, F. Hou, X. Liu, L. Yin, X. Yan and J. Liang, Carbon Energy, 2025, 7, e667 CrossRef CAS.
- Q. Zhang, D. Liu, Y. Zhang, Z. Guo, M. Chen, Y. Chen, B. Jin, Y. Song and H. Pan, J. Energy Chem., 2023, 87, 509–517 CrossRef CAS.
- D. Yao, C. Tang, X. Zhi, B. Johannessen, A. Slattery, S. Chern and S.-Z. Qiao, Adv. Mater., 2023, 35, 2209386 CrossRef CAS PubMed.
- Y. Chen, J. Lin, Q. Pan, X. Liu, T. Ma and X. Wang, Angew. Chem., Int. Ed., 2023, 62, e202306469 CrossRef CAS PubMed.
- J. Pei, G. Zhang, J. Liao, S. Ji, H. Huang, P. Wang, P. An, S. Chu and J. Dong, J. Mater. Chem. A, 2024, 12, 13694–13702 RSC.
- W. Xie, H. Li, G. Cui, J. Li, Y. Song, S. Li, X. Zhang, J. Y. Lee, M. Shao and M. Wei, Angew. Chem., Int. Ed., 2021, 60, 7382–7388 CrossRef CAS PubMed.
- Q. Hao, H. Zhong, J. Wang, K. Liu, J. Yan, Z. Ren, N. Zhou, X. Zhao, H. Zhang, D. Liu, X. Liu, L. Chen, J. Luo and X. Zhang, Nat. Synth., 2022, 1, 719–728 CrossRef CAS.
- T. Ding, X. Liu, Z. Tao, T. Liu, T. Chen, W. Zhang, X. Shen, D. Liu, S. Wang, B. Pang, D. Wu, L. Cao, L. Wang, T. Liu, Y. Li, H. Sheng, M. Zhu and T. Yao, J. Am. Chem. Soc., 2021, 143, 11317–11324 CrossRef CAS PubMed.
- N. Zhang, X. Zhang, Y. Kang, C. Ye, R. Jin, H. Yan, R. Lin, J. Yang, Q. Xu, Y. Wang, Q. Zhang, L. Gu, L. Liu, W. Song, J. Liu, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 13388–13393 CrossRef CAS PubMed.
- X. Cao, L. Zhao, B. Wulan, D. Tan, Q. Chen, J. Ma and J. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202113918 CrossRef CAS PubMed.
- C. Li and Y. Yamauchi, ACS Cent. Sci., 2024, 10, 16–18 CrossRef CAS PubMed.
- Z. Liang, N. Kong, C. Yang, W. Zhang, H. Zheng, H. Lin and R. Cao, Angew. Chem., Int. Ed., 2021, 60, 12759–12764 CrossRef CAS PubMed.
- Y. Wang, T. Yang, X. Fan, Z. Bao, A. Tayal, H. Tan, M. Shi, Z. Liang, W. Zhang, H. Lin, R. Cao, Z. Huang and H. Zheng, Angew. Chem., Int. Ed., 2024, 63, e202313034 CrossRef CAS PubMed.
- J. Liu, J. Xiao, Z. Wang, H. Yuan, Z. Lu, B. Luo, E. Tian and G. I. N. Waterhouse, ACS Catal., 2021, 11, 5386–5395 CrossRef CAS.
- Q. Wang, L. Lyu, X. Hu, W. Fan, C. Shang, Q. Huang, Z. Li, Z. Zhou and Y.-M. Kang, Angew. Chem., Int. Ed., 2025, 64, e202422920 CrossRef CAS PubMed.
- F. Gong, Y. Liu, Y. Zhao, W. Liu, G. Zeng, G. Wang, Y. Zhang, L. Gong and J. Liu, Angew. Chem., Int. Ed., 2023, 62, e202308091 CrossRef CAS PubMed.
- Z. Lu, D. Gao, D. Yi, Y. Yang, X. Wang and J. Yao, ACS Cent. Sci., 2020, 6, 1451–1459 CrossRef CAS PubMed.
- Y. Feng, C.-Q. Cheng, C.-Q. Zou, X.-L. Zheng, J. Mao, H. Liu, Z. Li, C.-K. Dong and X.-W. Du, Angew. Chem., Int. Ed., 2020, 59, 19297–19303 CrossRef CAS PubMed.
- Y. Zhang, Y. He, Y. Jiao, G. Yang, Y. Pu, Z. Wan, S. Li, Y. Wu, W. Liao and J. Guo, Chem. Sci., 2025, 16, 3479–3489 RSC.
- B. Wang, S. Chen, Z. Zhang and D. Wang, SmartMat, 2022, 3, 84–110 CrossRef CAS.
- W. Yan, W.-Y. He, Z.-D. Chu, M. Liu, L. Meng, R.-F. Dou, Y. Zhang, Z. Liu, J.-C. Nie and L. He, Nat. Commun., 2013, 4, 2159 CrossRef PubMed.
- B. Wang, M. Wang, Z. Fan, C. Ma, S. Xi, L. Chang, M. Zhang, N. Ling, Z. Mi, S. Chen, W. R. Leow, J. Zhang, D. Wang and Y. Lum, Nat. Commun., 2024, 15, 1719 CrossRef CAS PubMed.
- K. Guo, Z. He, S. Lu, P. Zhang, N. Li, L. Bao, Z. Yu, L. Song and X. Lu, Adv. Funct. Mater., 2023, 33, 2302100 CrossRef CAS.
- M. Zikhali, T. Matthews, C. T. Selepe, S. P. Mbokazi and N. W. Maxakato, Chem. Mater., 2024, 36, 6637–6650 CrossRef CAS.
- W. Liu, K. Guo, Y. Xie, S. Liu, L. Chen and J. Xu, Sci. Rep., 2023, 13, 9926 CrossRef CAS PubMed.
- G. Han, X. Zhang, W. Liu, Q. Zhang, Z. Wang, J. Cheng, T. Yao, L. Gu, C. Du, Y. Gao and G. Yin, Nat. Commun., 2021, 12, 6335 CrossRef CAS PubMed.
- J. Moreno, S. Aspera, M. David and H. Kasai, Carbon, 2015, 94, 936–941 CrossRef CAS.
- V. Eckert, E. Haubold, S. Oswald, S. Michel, C. Bellmann, P. Potapov, D. Wolf, S. Hampel, B. Büchner, M. Mertig and A. Leonhardt, Carbon, 2019, 141, 99–106 CrossRef CAS.
- L. Zhao, S. Guo, H. Liu, H. Zhu, S. Yuan and W. Guo, ACS Appl. Nano Mater., 2018, 1, 6258–6268 CrossRef CAS.
- Y. Wang, W. Zhang, W. Wen, X. Yu, Y. Du, K. Ni, Y. Zhu and M. Zhu, Adv. Funct. Mater., 2023, 33, 2302651 CrossRef CAS.
- X. Zhou, Z. Jin, J. Zhang, K. Hu, S. Liu, H.-J. Qiu and X. Lin, Nanoscale, 2023, 15, 2276–2284 RSC.
- N. Wei, Y. Ding, J. Zhang, L. Li, M. Zeng and L. Fu, Natl. Sci. Rev., 2023, 10, nwad145 CrossRef CAS PubMed.
- F. Li, Y. Anjarsari, J. Wang, R. Azzahiidah, J. Jiang, J. Zou, K. Xiang, H. Ma and Arramel, Carbon Lett., 2023, 33, 1321–1331 CrossRef CAS.
- S. Tang, L. Xu, K. Dong, Q. Wang, J. Zeng, X. Huang, H. Li, L. Xia and L. Wang, Appl. Surf. Sci., 2023, 615, 156357 CrossRef CAS.
- J. Wang, N. Li, Y. Jiang, H. Sheng and M. Sun, Appl. Surf. Sci., 2024, 658, 159858 CrossRef CAS.
- Y. Wang, Z. Bao, M. Shi, Z. Liang, R. Cao and H. Zheng, Chem. – Eur. J., 2022, 28, e202102915 CrossRef CAS PubMed.
- J. Yang, S. H. Kim, S. K. Kwak and H.-K. Song, ACS Appl. Mater. Interfaces, 2017, 9, 23302–23308 CrossRef CAS PubMed.
- N. Ma, Y. Zhang, Y. Wang, J. Zhao, B. Liang, Y. Xiong, S. Luo, C. Huang and J. Fan, J. Colloid Interface Sci., 2024, 654, 1458–1468 CrossRef CAS PubMed.
- Y. Zhao, J. Wan, C. Ling, Y. Wang, H. He, N. Yang, R. Wen, Q. Zhang, L. Gu, B. Yang, Z. Xiang, C. Chen, J. Wang, X. Wang, Y. Wang, H. Tao, X. Li, B. Liu, S. Zhang and D. Wang, Nature, 2025, 644, 668–675 CrossRef CAS PubMed.
- Z. W. Chen, L. X. Chen, Z. Wen and Q. Jiang, Phys. Chem. Chem. Phys., 2019, 21, 23782–23802 RSC.
- X. Liao, R. Lu, L. Xia, Q. Liu, H. Wang, K. Zhao, Z. Wang and Y. Zhao, Energy Environ. Mater., 2022, 5, 157–185 CrossRef CAS.
- R. Ding, J. Chen, Y. Chen, J. Liu, Y. Bando and X. Wang, Chem. Soc. Rev., 2024, 53, 11390–11461 RSC.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2026 |