Gaunt and Breit two-electron contributions to mean-field transformations and fine structure splitting
Received
2nd April 2026
, Accepted 20th May 2026
First published on 3rd June 2026
Abstract
Materials utilized by novel energy systems are often studied using weakly correlated mean-field theories. However, if these systems incorporate heavy elements, relativistic effects must be included. Therefore, a Kramers unrestricted coupled cluster with singles and doubles excitation formalism within a molecular mean-field exact two-component framework (X2Cmmf) using a four-component Dirac–Hartree–Fock (DHF) reference state is presented. The exact X2Cmmf transformed normal-order Hamiltonian incorporates all one-electron and two-electron (2e) contributions from the Coulomb, Gaunt, and Breit operators and is used with the equation of motion method to calculate the excitation energies of the alkali group of elements. Using this framework, the effects of 2e Gaunt and Breit integrals are studied. Results demonstrate growing contributions from these integrals to the generated X2Cmmf mean-fields and electronic fine structure calculations with increasing atomic number. Overall, this paper outlines the method, its effect within the X2Cmmf approach, and lays the foundation for future theoretical development of relativistic calculations within this framework.
Introduction
Materials relied on by novel energy systems, e.g., generation IV nuclear reactors,1 are often studied using weakly correlated mean-field theories. In weakly correlated electron systems, the direct Coulomb interaction can be modeled by an effective single-particle potential, e.g., density functional theory. When strong correlations dominate, these mean-field theories can be used as a reference state for various strongly correlated approaches.2–5 Relativistic effects are typically added perturbatively,6–8 but this framework is not adequate for systems composed of heavy elements9–15 and the vast unexplored space of topological materials.16,17 These deficiencies require us to adjust existing theories to account for a four-component (4c) Dirac reference state18 utilizing increasingly accurate relativistic Hamiltonians, e.g., the Dirac–Coulomb (DC), Dirac–Coulomb–Gaunt (DCG), and Dirac–Coulomb–Breit (DCB) Hamiltonians. In the no-pair approximation, exact two-component (X2C) transformation theory can be used to decouple the positive energy (pe) and negative energy (ne) spectrum of the Dirac Hamiltonian for wave function-based correlation methods.19–23 A variety of X2C flavors have been developed most of which fall under the one-electron (1e) X2C framework.24,25 In the case of the 1eX2C Hamiltonian scheme, the two-electron (2e) interaction terms are omitted from the defining 4c Dirac Hamiltonian resulting in a two-component (2c) Hamiltonian that is “exact” only in the 1e terms. Because the 2e interaction terms are often left untransformed in the 1eX2C basis set, 2e picture-change effects (2ePCEs) will appear.25 In contrast to the 1eX2C transformation, performed before the self-consistent field (SCF) cycle, molecular mean-field X2C (X2Cmmf) transformations utilize a unitary decoupling of the 4c molecular mean-field Fock matrix after having converged the 4c SCF DHF equations.23 Formally, X2Cmmf transformation are exact. However, due to the computational cost associated with recomputing and transforming 2e integrals associated with Coulomb (GLLLL, GLSLS, GSLSL, GSSSS) and Gaunt/Breit integrals (GSSLL, GSLLS, GLSSL, GLLSS), previous work using X2Cmmf transformations have largely only considered the large component Coulomb integrals in the 2e terms of the X2Cmmf transformed normal-order Hamiltonian and often in untransformed form (G+ ++ + ≈ GLLLL). Neglecting or using untransformed 2e interaction terms results in only an approximate X2Cmmf transformation.23,25–27
This work demonstrates that the Gaunt and Breit 2e integral contribution to the generated mean-field grows as elements become heavier. Furthermore, variations of the X2Cmmf transformations including the exact effective 1e DC, DCG, and DCB Fock matrix in-conjunction with the approximate and exact 2e transformed Coulomb, Coulomb–Gaunt, and Coulomb–Breit interaction23 are computed. Using these X2Cmmf transformed normal-order Hamiltonians, we apply Kramers unrestricted coupled cluster (CC) with singles and doubles (SD) excitations.28–31 The X2Cmmf-CCSD ground state wave function is then used as a reference for the equation of motion (EOM) method to obtain the excited wave function of the alkali elements.26,32,33 Results offer insights into the importance of the Gaunt and Breit 2e integral contributions to the 2e terms of the exact X2Cmmf transformed normal-order Hamiltonian used in post 4c SCF correlation-excitation steps as elements become heavier.
Theory and methodology
In this work, PySCF's34,35 implementation of 4c DHF36,37 using the DC, DCG, and DCB Hamiltonian was used (see Fock matrix in eqn (1)). The 4c DHF calculation enforced the restricted kinetic balance condition36,37 and used the finite-size nuclear model with a convergence tolerance of 10−10 and a max direct inversion of the iterative subspace38 (DIIS) of 8.| |
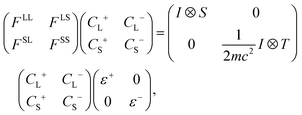 | (1) |
We implemented Kramers unrestricted CCSD (using NumPy)39,40 employing DIIS following standard CCSD methods for post-SCF wavefunction correlation calculations. The family of relativistic ANO basis sets41,42 used for this work are obtained from basis set exchange.43 We note that in 4c DHF, the 2e integral ĝ operator takes the following form:
| |
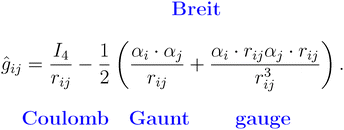 | (2) |
The first term in this expression is identified as the typical Coulomb interaction, with the second as Gaunt, and the third as a gauge. The second and third terms are often collectively called the Breit term and are associated with spin–orbit interactions and retardation effects. As dictated by an X2Cmmf transformation, after converging the 4c DHF equation, the Hamiltonian for the valence electrons can be written in normal-ordered form,
| |
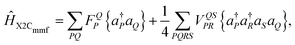 | (3) |
for further use in wave function-based correlation methods.
21,23 The brackets indicate normal ordering with respect to occupied (hole) and virtual (particle) orbitals. The effective 1e terms of the normal-ordered Hamiltonian are elements of the Fock matrix,
FQP. The anti-symmetrized electron–electron integrals,
VQSPR =
GQSPR −
GSQPR, represent the 2e contributions to the normal-ordered Hamiltonian. The summation is formally restricted to pe orbitals but in practice, it is further reduced due to truncations of the occupied and virtual space commonly employed in correlated calculations. In this form, it is observed that the matrix elements of the Fock matrix
FQP can be obtained from exact decoupling of the corresponding converged 4c molecular Fock matrix. If the basis chosen for the correlation calculation is taken to be the canonical HF orbitals, the list of nonzero matrix elements reduces to the orbital energies
FQP =
εP+δPQ.
23 Similarly, we restricted ourselves to the pe 2e interaction terms,
G+ ++ +. These can be obtained for the Coulomb integrals as
G+ ++ + =
C†,+LC†,+LGLLLLCL+CL+ +
C†,+LC†,+SGLSLSCL+CS+ +
C†,+SC†,+LGSLSLCS+CL+ +
C†,+SC†,+SGSSSSCS+CS+ and for Gaunt/Breit as
G+ ++ + =
C†,+LC†,+LGSSLLCS+CS+ +
C†,+LC†,+SGSLLSCS+CL+ +
C†,+SC†,+LGLSSLCL+CS+ +
C†,+SC†,+SGLLSSCL+CL+.
23 All X2C
mmf transformed normal-order Hamiltonians used in this study are exact in the 1e Fock term (labeled-1e). X2C
mmf transformed normal-order Hamiltonians that are also exact in the 2e interaction terms are labeled-1e2e and account for all Coulomb integrals (
GLLLL,
GLSLS,
GSLSL,
GSSSS) and Gaunt/Breit integrals (if used at 4c SCF level,
GSSLL,
GSLLS,
GLSSL,
GLLSS). Nonexact 2e interaction terms in the X2C
mmf transformed normal-ordered Hamiltonians account only for the transformed Coulomb integrals (
GLLLL,
GLSLS,
GSLSL,
GSSSS). These variations of X2C
mmf transformed normal-order Hamiltonians are defined in
Table 1.
Table 1 X2Cmmf transformation used in study as well as their contributions to 1-electron and 2-electron terms
| X2Cmmf type |
1-Electron |
2-Electron contribution |
| DC-1e2e |
Coulomb |
Coulomb |
| DCG-1e |
Coulomb–Gaunt |
Coulomb |
| DCB-1e |
Coulomb–Breit |
Coulomb |
| DCG-1e2e |
Coulomb–Gaunt |
Coulomb–Gaunt |
| DCB-1e2e |
Coulomb–Breit |
Coulomb–Breit |
After X2Cmmf, Kramers unrestricted CC theory is used to approximate the true wave function by systematically mixing in excited configurations. The wave function in CC theory is defined as |Ψ〉 = e![[T with combining circumflex]](https://www.rsc.org/images/entities/i_char_0054_0302.gif) |ΦX2Cmmf,0〉 where |ΦX2Cmmf,0〉 is the single determinant wave function.21,44 For this work, the cluster operator was truncated to the SD hole-particle excitations = 1 + 2 where
|ΦX2Cmmf,0〉 where |ΦX2Cmmf,0〉 is the single determinant wave function.21,44 For this work, the cluster operator was truncated to the SD hole-particle excitations = 1 + 2 where 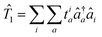 and
and 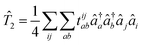 . Using the similarity transformed Hamiltonian,
. Using the similarity transformed Hamiltonian, ![[H with combining tilde]](https://www.rsc.org/images/entities/i_char_0048_0303.gif) = e−ĤX2Cmmfe, one can solve for the SD excitation amplitudes, tia and tijab through various subspace projections listed in eqn (4)–(6),21,39,40,45–48
= e−ĤX2Cmmfe, one can solve for the SD excitation amplitudes, tia and tijab through various subspace projections listed in eqn (4)–(6),21,39,40,45–48
| | |
〈ΦX2Cmmf,0||ΦX2Cmmf,0〉 = E,
| (4) |
| | |
〈ΦX2Cmmf,0|â†iâa|ΦX2Cmmf,0〉 = 0,
| (5) |
| | |
〈ΦX2Cmmf,0|â†iâ†jâbâa|ΦX2Cmmf,0〉 = 0.
| (6) |
It is important to note that the asymmetric CCSD equations do not satisfy the variational principle due to the truncation of the cluster operators. This means that the calculated energy will not necessarily be an upper bound for the exact energy.49 The Kramers unrestricted CCSD implementation was benchmarked using PySCF implementation of CCSD. Both the developed CCSD code and PySCF's implementation used the same X2Cmmf transformed normal-ordered Hamiltonian as the single determinant reference state. The results of these calculations can be seen in Tables S7–S10 in the supplementary information, which used a convergence tolerance of 10−11 and max DIIS of 8). From these tables it is observed that all converged values match to convergence tolerance. Using the CCSD ground-state wave function and noting that is not Hermitian, one can define the excited wave function using
| |
 | (7) |
| |
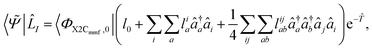 | (8) |
where
I represents the
Ith excited state.
27,32,33 Similar to CCSD, this procedure truncates the operators and assumes that the CCSD ground state captures the dominant dynamical correlations such that the excited states can be represented as excitations on top of the correlated vacuum. These bra and ket states, although not orthonormal among themselves, satisfy biorthogonality 〈
ΦX2Cmmf,0|
![[L with combining circumflex]](https://www.rsc.org/images/entities/i_char_004c_0302.gif) I
Ie
−e
![[R with combining circumflex]](https://www.rsc.org/images/entities/i_char_0052_0302.gif) J
J|
ΦX2Cmmf,0〉 =
CδIJ and by choosing
C to be unity to one can enforce normalization. Defining the normal-ordered similarity transformed Hamiltonian as
![[H with combining macron]](https://www.rsc.org/images/entities/i_char_0048_0304.gif)
=
−
E one can solve for the expansion coefficients
r0,
rai,
rabij,
l0,
lia, and
lijab using
eqn (9) and (10),
| | |
I|ΦX2Cmmf,0〉 = ωII|ΦX2Cmmf,0〉,
| (9) |
| | |
〈ΦX2Cmmf,0|I = 〈ΦX2Cmmf,0|IωI,
| (10) |
where
ωI is the difference in energy between the
Ith excited state and
E. In this study, PySCF
34,35 implementation of EOM was used and we refer the reader to their open source code for details of their implementation. Using EOM-CCSD-X2C
mmf-DHF, we calculated the
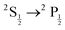
excitations for the alkali elements. Results of these calculations are consistent with experimental values, see in Table S1 and
Table 2, and a similar study
32 using the Kramers unrestricted EOM-CCSD-X2C
mmf-DHF, a point-nucleus charge, and the X2C
mmf untransformed DC-1e, and DCB-1e Hamiltonian for Na, K, and Rb in the ANO basis sets.
Table 2 Electronic fine structure splitting  for varying atom types, basis types, and X2Cmmf transformation types, calculated using EOM-CCSD-X2Cmmf-DHF and compared to experiment (units are eV)
for varying atom types, basis types, and X2Cmmf transformation types, calculated using EOM-CCSD-X2Cmmf-DHF and compared to experiment (units are eV)
| Atom |
Basis |
Experiment |
DC-1e2e |
DCG-1e |
DCB-1e |
DCG-1e2e |
DCB-1e2e |
| Na |
ANO-RCC-MB |
0.0021 |
0.0030 |
0.0028 |
0.0028 |
0.0028 |
0.0028 |
| Na |
ANO-RCC-VDZ |
0.0021 |
0.0023 |
0.0022 |
0.0022 |
0.0022 |
0.0022 |
| Na |
ANO-RCC-VDZP |
0.0021 |
0.0024 |
0.0022 |
0.0022 |
0.0022 |
0.0022 |
| Na |
ANO-RCC-VTZP |
0.0021 |
0.0023 |
0.0022 |
0.0022 |
0.0022 |
0.0022 |
| Na |
ANO-RCC-VQZP |
0.0021 |
0.0023 |
0.0022 |
0.0022 |
0.0022 |
0.0022 |
| K |
ANO-RCC-MB |
0.0072 |
0.0074 |
0.0072 |
0.0072 |
0.0072 |
0.0072 |
| K |
ANO-RCC-VDZ |
0.0072 |
0.0066 |
0.0064 |
0.0064 |
0.0064 |
0.0064 |
| K |
ANO-RCC-VDZP |
0.0072 |
0.0077 |
0.0075 |
0.0075 |
0.0075 |
0.0075 |
| K |
ANO-RCC-VTZP |
0.0072 |
0.0076 |
0.0074 |
0.0074 |
0.0074 |
0.0074 |
| Rb |
ANO-RCC-MB |
0.0295 |
0.0220 |
0.0220 |
0.0219 |
0.0220 |
0.0219 |
| Rb |
ANO-RCC-VDZ |
0.0295 |
0.0235 |
0.0232 |
0.0232 |
0.0232 |
0.0232 |
| Rb |
ANO-RCC-VDZP |
0.0295 |
0.0240 |
0.0237 |
0.0237 |
0.0237 |
0.0237 |
| Cs |
ANO-RCC-MB |
0.0687 |
0.0508 |
0.0513 |
0.0512 |
0.0513 |
0.0512 |
| Cs |
ANO-RCC-VDZ |
0.0687 |
0.0529 |
0.0529 |
0.0529 |
0.0529 |
0.0529 |
| Fr |
ANO-RCC-VDZ |
0.2091 |
0.1543 |
0.1566 |
0.1563 |
0.1566 |
0.1563 |
Analysis of results
The influence of the Gaunt and Breit 2e integrals on their respective DHF–DCG and DHF–DCB mean-fields is quantified using the mean squared displacement of the pe eigenvalue spectrum (ε+) of the 4c Fock (labeled εDHF+) and decoupled 2c Fock matrix. The εDCG-1e+, εDCB-1e+ represent decoupled 2c pe Fock spectrum which are exact in all one-body terms but neglect two-body Gaunt and Breit integrals respectively in decoupling while εDC-1e2e+, εDCG-1e2e+, εDCB-1e2e+ represent the exactly decoupled 2c pe Fock spectrum. Results of these calculations are seen in Fig. 1 (Fig. S1 in the supplementary information demonstrates results are independent of basis choice). Here, we find ε+,2cDCG-1e vs. εDHF–DCG+ and εDCB-1e+ vs. εDHF–DCB+ to exhibit a growing discrepancy in the pe eigenvalue spectrum while the exactly decoupled 2c have consistent negligible error. This confirms that Gaunt and Breit integrals increasingly contribute to their respective 4c DHF mean-field as elements become heavier.
 |
| | Fig. 1 Mean squared displacement of positive energy spectrum after X2Cmmf using DC-1e2e, DCG-1e, and DGB-1e, DCG-1e2e, and DCB-1e2e transformation versus the respective positive energy spectrum from 4c DHF using DC, DCG, and DCB Hamiltonian in ANO-RCC-VDZ basis set (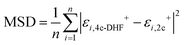 , units are Hartree). , units are Hartree). | |
A similar procedure can be performed to quantify discrepancies in 4c DHF mean fields using higher order relativistic corrections (DC, DCG, and DCB) with increasing atomic number. This is accomplished using the mean squared displacement of the pe eigenvalue spectrum (ε+) of the 4c DHF–DCB Fock (labeled εDHF–DCB+) and various decoupled 2c Fock introduced above (εDCG-1e+, εDCB-1e+, εDC-1e2e+, εDCG-1e2e+, εDCB-1e2e+). Fig. 2 shows the results of mean squared displacement analysis while Fig. S2 (see supplementary information) shows results are independent of basis choice. Observing a growing displacement in pe eigenvalue spectrum neglecting Gaunt or gauge (εDHF–DCB+ vs. εDCG-1e+, εDCB-1e+, εDC-1e2e+, εDCG-1e2e+), a lower displacement in spectrum exact in Gaunt (εDHF–DCB+ vs. εDCG-1e2e+), and a negligible displacement in the exactly decoupled spectrum including both Gaunt and gauge (εDHF–DCB+ vs. εDCB-1e2e+), confirms an increasing discrepancy in pe mean field obtained using 4c DHF–DC, DHF–DCG, or DHF–DCB with increasing atomic number. This result emphasizes the need for increasingly accurate Hamiltonian as atomic number increases.
 |
| | Fig. 2 Mean squared displacement of positive energy spectrum after X2Cmmf using DC-1e2e, DCG-1e, and DGB-1e, DCG-1e2e, and DCB-1e2e transformation versus the absolute value of positive energy spectrum from 4c DHF using the DCB Hamiltonian in ANO-RCC-VDZ basis set (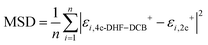 , units are Hartree). , units are Hartree). | |
Moving on to the EOM-CCSD-X2Cmmf-DHF calculations, it is of interest to understand the impact of the Gaunt and Breit integrals on the 1e and 2e terms of X2Cmmf transformed normal-ordered Hamiltonians used in post 4c SCF correlation-excitations steps. Table 2 shows electronic fine structure (EFS, 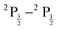 ) for varying atom types, basis types, and X2Cmmf transformation types, calculated using EOM-CCSD-X2Cmmf-DHF and compared to experiment. Table 2 demonstrates that error of the predicted and experimental EFS decreases with increasingly accurate basis sets (ANO-RCC-VDZ → ANO-RCC-VDZP → ANO-RCC-VTZP → ANO-RCC-VQZP). Table 2 further demonstrates that as elements increase in atomic number, predictions deviate from experiment. These deviations can likely be attributed the neglect of important higher-order excitations necessary for modeling heavier elements.50 However, for small elements (Na,K), where a CCSD framework is sufficient to provide a reasonable reconstruction of correlation, it is noted that more accurate X2Cmmf transformed normal-ordered Hamiltonians provide a better prediction of the EFS.
) for varying atom types, basis types, and X2Cmmf transformation types, calculated using EOM-CCSD-X2Cmmf-DHF and compared to experiment. Table 2 demonstrates that error of the predicted and experimental EFS decreases with increasingly accurate basis sets (ANO-RCC-VDZ → ANO-RCC-VDZP → ANO-RCC-VTZP → ANO-RCC-VQZP). Table 2 further demonstrates that as elements increase in atomic number, predictions deviate from experiment. These deviations can likely be attributed the neglect of important higher-order excitations necessary for modeling heavier elements.50 However, for small elements (Na,K), where a CCSD framework is sufficient to provide a reasonable reconstruction of correlation, it is noted that more accurate X2Cmmf transformed normal-ordered Hamiltonians provide a better prediction of the EFS.
Fig. 3 presents results of the absolute difference between the EFS 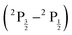 predicted by DC-1e2e, DCG-1e, DCB-1e, and DCG-1e2e versus the EFS predicted by DCB-1e2e transformation (supplementary Fig. S3 demonstrates results are independent of basis choice). Fig. 3 and Table 2 both indicate that even for elements with small atomic number, including only the Coulomb integrals is insufficient, the Gaunt integrals in both the 1e and 2e terms of the X2Cmmf transformed normal ordered Hamiltonian contribute in 3rd–4th decimal of EFS prediction within the EOM-CCSD-X2Cmmf-DHF framework. Additionally, as elements increase in atomic number, the contributions of the gauge integrals in the 1e and 2e term of the X2Cmmf transformed normal ordered Hamiltonian contribute in 4th–5th decimal of EFS prediction.
predicted by DC-1e2e, DCG-1e, DCB-1e, and DCG-1e2e versus the EFS predicted by DCB-1e2e transformation (supplementary Fig. S3 demonstrates results are independent of basis choice). Fig. 3 and Table 2 both indicate that even for elements with small atomic number, including only the Coulomb integrals is insufficient, the Gaunt integrals in both the 1e and 2e terms of the X2Cmmf transformed normal ordered Hamiltonian contribute in 3rd–4th decimal of EFS prediction within the EOM-CCSD-X2Cmmf-DHF framework. Additionally, as elements increase in atomic number, the contributions of the gauge integrals in the 1e and 2e term of the X2Cmmf transformed normal ordered Hamiltonian contribute in 4th–5th decimal of EFS prediction.
 |
| | Fig. 3 Absolute difference of fine structure splitting predicted using the DC-1e2e, DCG-1e, DCB-1e, and DCG-1e2e transformation versus the DCB-1e2e transformation for various basis sets calculated using EOM-CCSD-X2Cmmf-DHF in ANO-RCC-VDZ basis set (units are eV). | |
Closing remarks
As higher fidelity electronic structure is needed, the introduction of relativistic theories becomes increasingly important to characterize certain systems. In order to address this issue, we developed a CCSD code using the 4c DHF ground state and X2Cmmf transformed normal-order Hamiltonian incorporating all 1e and 2e contributions from the Coulomb, Gaunt, and Breit operators. Results confirm that Gaunt and Breit integrals increasingly contribute to the DHF solutions causing deviations with increasing atomic number in the mean fields generated with varying orders of relativistic corrections (DC, DCG, and DCB). It is also shown that for elements with small atomic number, the Gaunt integrals in both the 1e and 2e terms of the X2Cmmf transformed normal ordered Hamiltonian contribute significantly to EFS predictions within the EOM-CCSD-X2Cmmf-DHF framework while those elements with increasing atomic number have non-negligible contributions of the gauge integrals. Overall, this work outlines limitations of various X2Cmmf transformations, and lays the ground work for more studies utilizing the DCB Hamiltonian within an exact X2C mean-field approach.
Conflicts of interest
There are no conflicts to declare.
Data availability
The data supporting the conclusions of this article are contained within the manuscript and its supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d6cp01222a. This contains additional calculation results and extended analysis.
Acknowledgements
This work was carried out under the auspices of the U.S. Department of Energy (DOE) National Nuclear Security Administration (NNSA) under Contract No. 89233218CNA000001. It was supported by the G.T Seaborg Institute for Transactium Science at Los Alamos National Laboratory (L.M.), the LANL LDRD program (C. L. and R. M. T), and in part by the Center for Integrated Nanotechnologies, a DOE BES user facility, in partnership with the LANL Institutional Computing Program for computational resources.
References
- L. Murg, S. C. Lee and V. F. Grizzi, et al., Enhanced exploration of LiF–NaF thermal conductivity through transferable equivariant graph neural networks, J. Appl. Phys., 2025, 137, 064701 Search PubMed.
- B. Himmetoglu, A. Floris, S. De Gironcoli and M. Cococcioni, Hubbard-corrected DFT energy functionals: The LDA+U description of correlated systems, Int. J. Quantum Chem., 2014, 114, 14–49 Search PubMed.
- A. Georges, G. Kotliar, W. Krauth and M. J. Rozenberg, Dynamical mean-field theory of strongly correlated fermion systems and the limit of infinite dimensions, Rev. Mod. Phys., 1996, 68, 13 CrossRef CAS.
- A. Wodynski and M. Kaupp, Local hybrid functional applicable to weakly and strongly correlated systems, J. Chem. Theory Comput., 2022, 18, 6111–6123 CrossRef CAS PubMed.
- P. Rivero, I. D. P. Moreira, G. E. Scuseria and F. Illas, Description of magnetic interactions in strongly correlated solids via range-separated hybrid functionals, Phys. Rev. B:Condens. Matter Mater. Phys., 2009, 79, 245129 Search PubMed.
- B. A. Heß, C. M. Marian, U. Wahlgren and O. Gropen, A mean-field spin-orbit method applicable to correlated wavefunctions, Chem. Phys. Lett., 1996, 251, 365–371 CrossRef.
- P. Schwerdtfeger, Relativistic Electronic Structure Theory, Part 1: Fundamentals, Elsevier Science, 2002, pp. 1–926 Search PubMed.
- K. Klein and J. Gauss, Perturbative calculation of spin-orbit splittings using the equation-of-motion ionization-potential coupled-cluster ansatz, J. Chem. Phys., 2008, 129, 194106 Search PubMed.
- R. M. Tutchton, W.-T. Chiu, R. C. Albers, G. Kotliar and J.-X. Zhu, Electronic correlation induced expansion of Fermi pockets in δ-plutonium, Phys. Rev. B, 2020, 101, 245156 Search PubMed.
- P. Pyykko, Relativistic effects in structural chemistry, Chem. Rev., 1988, 88, 563–594 Search PubMed.
- T. Saue, Relativistic Hamiltonians for chemistry: A primer, ChemPhysChem, 2011, 12, 3077–3094 Search PubMed.
- J. Autschbach, Perspective: relativistic effects, J. Chem. Phys., 2012, 136, 150902 CrossRef PubMed.
- P. Pyykkö, Relativistic effects in chemistry: more common than you thought, Annu. Rev. Phys. Chem., 2012, 63, 45–64 Search PubMed.
- W. Liu, Advances in relativistic molecular quantum mechanics, Phys. Rep., 2014, 537, 59–89 CrossRef.
- D. Jayatilaka and T. J. Lee, The form of spin orbitals for open-shell restricted Hartree–Fock reference functions, Chem. Phys. Lett., 1992, 199, 211–219 CrossRef CAS.
- T. Fukui and Y. Hatsugai, Topological aspects of the quantum spin-Hall effect in graphene: Z 2 topological order and spin Chern number, Phys. Rev. B:Condens. Matter Mater. Phys., 2007, 75, 121403 CrossRef.
- J. G. Rau, E. K. H. Lee and H. Y. Kee, Spin-orbit physics giving rise to novel phases in correlated systems: Iridates and related materials, Annu. Rev. Condens. Matter Phys., 2016, 7, 195–221 CrossRef CAS.
- B. Swirles, The relativistic self-consistent field, Proc. R. Soc. London, Ser. A, 1935, 152, 625–649 Search PubMed.
- J. Liu, Y. Shen, A. Asthana and L. Cheng, Two-component relativistic coupled-cluster methods using mean-field spin-orbit integrals, J. Chem. Phys., 2018, 148, 034106 Search PubMed.
- A. Asthana, J. Liu and L. Cheng, Exact two-component equation-of-motion coupled-cluster singles and doubles method using atomic mean-field spin-orbit integrals, J. Chem. Phys., 2019, 150, 074102 CrossRef PubMed.
- J. V. Pototschnig, A. Papadopoulos, D. I. Lyakh, M. Repisky, L. Halbert, A. Severo Pereira Gomes, H. J. A. Jensen and L. Visscher, Implementation of relativistic coupled cluster theory for massively parallel GPU-accelerated computing architectures, J. Chem. Theory Comput., 2021, 17, 5509–5529 CrossRef CAS PubMed.
- P. Tecmer, A. Severo Pereira Gomes, S. Knecht and L. Visscher, Communication: Relativistic Fock-space coupled cluster study of small building blocks of larger uranium complexes, J. Chem. Phys., 2014, 141, 041107 Search PubMed.
- J. Sikkema, L. Visscher, T. Saue and M. Iliaš, The molecular mean-field approach for correlated relativistic calculations, J. Chem. Phys., 2009, 131, 124116 CrossRef PubMed.
- D. Peng and M. Reiher, Exact decoupling of the relativistic Fock operator, Theor. Chem. Acc., 2012, 131, 1081 Search PubMed.
- S. Knecht, M. Repisky, H. J. A. Jensen and T. Saue, Exact two-component Hamiltonians for relativistic quantum chemistry: Two-electron picture-change corrections made simple, J. Chem. Phys., 2022, 157, 114106 CrossRef CAS PubMed.
- T. Saue, R. Bast, A. S. P. Gomes, H. J. A. Jensen, L. Visscher, I. A. Aucar, R. Di Remigio, K. G. Dyall, E. Eliav and E. Fasshauer, et al., The DIRAC code for relativistic molecular calculations, J. Chem. Phys., 2020, 152, 204104 Search PubMed.
- T. Zhang, S. Banerjee, L. N. Koulias, E. F. Valeev, A. E. DePrince III and X. Li, Dirac-Coulomb-Breit Molecular Mean-Field Exact-Two-Component Relativistic Equation-of-Motion Coupled-Cluster Theory, J. Phys. Chem. A, 2024, 128, 3408–3418 CrossRef CAS PubMed.
- J. Čížek, On the correlation problem in atomic and molecular systems. Calculation of wavefunction components in Ursell-type expansion using quantum-field theoretical methods, J. Chem. Phys., 1966, 45, 4256–4266 CrossRef.
- J. Čížek, On the use of the cluster expansion and the technique of diagrams in calculations of correlation effects in atoms and molecules, Adv. Chem. Phys., 1969, 14, 35–89 CrossRef.
- J. Čížek and J. Paldus, Correlation problems in atomic and molecular systems III. Rederivation of the coupled-pair many-electron theory using the traditional quantum chemical methodst, Int. J. Quantum Chem., 1971, 5, 359–379 CrossRef.
- R. J. Bartlett and M. Musiał, Coupled-cluster theory in quantum chemistry, Rev. Mod. Phys., 2007, 79, 291–352 CrossRef CAS.
- S. H. Yuwono, R. R. Li, T. Zhang, X. Li and A. E. DePrince, Two-component relativistic equation-of-motion coupled cluster for electron ionization, J. Chem. Phys., 2025, 162, 084110 Search PubMed.
- J. F. Stanton and R. J. Bartlett, The equation of motion coupled-cluster method. A systematic biorthogonal approach to molecular excitation energies, transition probabilities, and excited state properties, J. Chem. Phys., 1993, 98, 7029–7039 CrossRef CAS.
- Q. Sun, T. C. Berkelbach, N. S. Blunt, G. H. Booth, S. Guo, Z. Li, J. Liu, J. D. McClain, E. R. Sayfutyarova and S. Sharma, et al., PySCF: the Python-based simulations of chemistry framework, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1340 Search PubMed.
- Q. Sun, X. Zhang, S. Banerjee, P. Bao, M. Barbry, N. S. Blunt, N. A. Bogdanov, G. H. Booth, J. Chen and Z.-H. Cui, et al., Recent developments in the PySCF program package, J. Chem. Phys., 2020, 153, 024109 CrossRef CAS PubMed.
- S. Sun, T. F. Stetina, T. Zhang, H. Hu, E. F. Valeev, Q. Sun and X. Li, Efficient Four-Component Dirac-Coulomb-Gaunt Hartree-Fock in the Pauli Spinor Representation, J. Chem. Theory Comput., 2021, 17, 3388–3402 CrossRef CAS PubMed.
- S. Sun, J. Ehrman, Q. Sun and X. Li, Efficient evaluation of the Breit operator in the Pauli spinor basis, J. Chem. Phys., 2022, 157, 064112 CrossRef CAS PubMed.
- P. Császár and P. Pulay, Geometry optimization by direct inversion in the iterative subspace, J. Mol. Struct., 1984, 114, 31–34 CrossRef.
- L. Visscher, K. G. Dyall and T. J. Lee, Kramers-restricted closed-shell CCSD theory, Int. J. Quantum Chem., 1995, 56, 411–419 CrossRef.
- L. Visscher, T. J. Lee and K. G. Dyall, Formulation and implementation of a relativistic unrestricted coupled-cluster method including noniterative connected triples, J. Chem. Phys., 1996, 105, 8769–8776 CrossRef CAS.
- B. O. Roos, R. Lindh, P. Å. Malmqvist, V. Veryazov and P. O. Widmark, Main group atoms and dimers studied with a new relativistic ANO basis set, J. Phys. Chem. A, 2004, 108, 2851–2858 CrossRef CAS.
- B. O. Roos, R. Lindh, P. Å. Malmqvist, V. Veryazov and P. O. Widmark, New relativistic ANO basis sets for transition metal atoms, J. Phys. Chem. A, 2005, 109, 6575–6579 CrossRef CAS PubMed.
- B. P. Pritchard, D. Altarawy, B. Didier, T. D. Gibson and T. L. Windus, New basis set exchange: An open, up-to-date resource for the molecular sciences community, J. Chem. Inf. Model., 2019, 59, 4814–4820 CrossRef CAS PubMed.
- I. Shavitt and R. J. Bartlett, Many-body methods in chemistry and physics: MBPT and coupled-cluster theory, Cambridge University Press, 2009 Search PubMed.
- R. J. Bartlett, Coupled-cluster theory and its equation-of-motion extensions, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 126–138 Search PubMed.
- J. Cizek, On the Correlation Problems in Atomic and Molecular Systems. Calculation of Wavefunction Components in Ursell-Type Expansion Using Quantum-Field Theoretical Methods, J. Chem. Phys., 1966, 45(11), 4256–4266 CrossRef CAS.
- G. D. Purvis and R. J. Bartlett, A full coupled-cluster singles and doubles model: The inclusion of disconnected triples, J. Chem. Phys., 1982, 76, 1910–1918 CrossRef CAS.
- R. J. Bartlett, Coupled-cluster approach to molecular structure and spectra: a step toward predictive quantum chemistry, J. Phys. Chem., 1989, 93, 1697–1708 CrossRef CAS.
- T. D. Crawford and H. F. Schaefer III, An introduction to coupled cluster theory for computational chemists, Rev. Comput. Chem., 2007, 14, 33–136 Search PubMed.
- I. Magoulas, J. Shen and P. Piecuch, Addressing strong correlation by approximate coupled-pair methods with active-space and full treatments of three-body clusters, Mol. Phys., 2022, 120, e2057365 CrossRef.
|
| This journal is © the Owner Societies 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Christopher Lane
*ab,
Christopher Lane


 and
and  . Using the similarity transformed Hamiltonian,
. Using the similarity transformed Hamiltonian, 

 excitations for the alkali elements. Results of these calculations are consistent with experimental values, see in Table S1 and Table 2, and a similar study32 using the Kramers unrestricted EOM-CCSD-X2Cmmf-DHF, a point-nucleus charge, and the X2Cmmf untransformed DC-1e, and DCB-1e Hamiltonian for Na, K, and Rb in the ANO basis sets.
excitations for the alkali elements. Results of these calculations are consistent with experimental values, see in Table S1 and Table 2, and a similar study32 using the Kramers unrestricted EOM-CCSD-X2Cmmf-DHF, a point-nucleus charge, and the X2Cmmf untransformed DC-1e, and DCB-1e Hamiltonian for Na, K, and Rb in the ANO basis sets.
 for varying atom types, basis types, and X2Cmmf transformation types, calculated using EOM-CCSD-X2Cmmf-DHF and compared to experiment (units are eV)
for varying atom types, basis types, and X2Cmmf transformation types, calculated using EOM-CCSD-X2Cmmf-DHF and compared to experiment (units are eV)

 , units are Hartree).
, units are Hartree).
 , units are Hartree).
, units are Hartree). ) for varying atom types, basis types, and X2Cmmf transformation types, calculated using EOM-CCSD-X2Cmmf-DHF and compared to experiment. Table 2 demonstrates that error of the predicted and experimental EFS decreases with increasingly accurate basis sets (ANO-RCC-VDZ → ANO-RCC-VDZP → ANO-RCC-VTZP → ANO-RCC-VQZP). Table 2 further demonstrates that as elements increase in atomic number, predictions deviate from experiment. These deviations can likely be attributed the neglect of important higher-order excitations necessary for modeling heavier elements.50 However, for small elements (Na,K), where a CCSD framework is sufficient to provide a reasonable reconstruction of correlation, it is noted that more accurate X2Cmmf transformed normal-ordered Hamiltonians provide a better prediction of the EFS.
) for varying atom types, basis types, and X2Cmmf transformation types, calculated using EOM-CCSD-X2Cmmf-DHF and compared to experiment. Table 2 demonstrates that error of the predicted and experimental EFS decreases with increasingly accurate basis sets (ANO-RCC-VDZ → ANO-RCC-VDZP → ANO-RCC-VTZP → ANO-RCC-VQZP). Table 2 further demonstrates that as elements increase in atomic number, predictions deviate from experiment. These deviations can likely be attributed the neglect of important higher-order excitations necessary for modeling heavier elements.50 However, for small elements (Na,K), where a CCSD framework is sufficient to provide a reasonable reconstruction of correlation, it is noted that more accurate X2Cmmf transformed normal-ordered Hamiltonians provide a better prediction of the EFS. predicted by DC-1e2e, DCG-1e, DCB-1e, and DCG-1e2e versus the EFS predicted by DCB-1e2e transformation (supplementary Fig. S3 demonstrates results are independent of basis choice). Fig. 3 and Table 2 both indicate that even for elements with small atomic number, including only the Coulomb integrals is insufficient, the Gaunt integrals in both the 1e and 2e terms of the X2Cmmf transformed normal ordered Hamiltonian contribute in 3rd–4th decimal of EFS prediction within the EOM-CCSD-X2Cmmf-DHF framework. Additionally, as elements increase in atomic number, the contributions of the gauge integrals in the 1e and 2e term of the X2Cmmf transformed normal ordered Hamiltonian contribute in 4th–5th decimal of EFS prediction.
predicted by DC-1e2e, DCG-1e, DCB-1e, and DCG-1e2e versus the EFS predicted by DCB-1e2e transformation (supplementary Fig. S3 demonstrates results are independent of basis choice). Fig. 3 and Table 2 both indicate that even for elements with small atomic number, including only the Coulomb integrals is insufficient, the Gaunt integrals in both the 1e and 2e terms of the X2Cmmf transformed normal ordered Hamiltonian contribute in 3rd–4th decimal of EFS prediction within the EOM-CCSD-X2Cmmf-DHF framework. Additionally, as elements increase in atomic number, the contributions of the gauge integrals in the 1e and 2e term of the X2Cmmf transformed normal ordered Hamiltonian contribute in 4th–5th decimal of EFS prediction.