
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular dynamics simulations of pH-dependent ciprofloxacin adsorption to Na-montmorillonite
Rogers E.
Swai
 and
Michael
Holmboe
*
and
Michael
Holmboe
*
Chemistry Department, Umeå University, Umeå, Sweden. E-mail: michael.holmboe@umu.se
First published on 27th March 2026
Abstract
Ciprofloxacin (CIP) is one of the most widely used and environmentally persistent antibiotic compounds, which is increasingly being found in soils, sediments, and natural waters. Because CIP strongly associates with clay minerals, understanding its molecular-scale adsorption energetics, dynamics, and configurations is essential for predicting environmental retention and mobility. For this purpose, a combination of unbiased molecular dynamics simulations, biased umbrella sampling and alchemical free-energy perturbation simulations were used in this study to obtain the free energy of adsorption, orientational and clustering configurations and interlayer density mapping across controlled hydration states. These simulations were applied to the three pH-dependent environmentally relevant CIP species (CIP+, CIP+/−, CIP−) interacting with montmorillonite (MMT) in its Na+ form, at basal surfaces and within variably hydrated interlayers. All CIP species approached similar center-of-mass separations from the MMT surface but adopt distinct orientational preferences: CIP+ binds predominantly flat, CIP+/− partitions between flat and side-on, and CIP− samples multiple poses. The potential of mean force profiles revealed that all species adsorb favourably, but with adsorption strengths steeply increasing in the order CIP− < CIP+/− < CIP+, having standard adsorption free energies of −2.1, −11.8 and −30.0 kJ mol−1, with corresponding sorption Kd values of 6.4, 520, and 1.0 × 106 cm3 g−1, respectively. Unbiased open systems simulations further showed that CIP+ and CIP+/− could be intercalated into 2 W and 3 W hydration states, whereas CIP− failed to intercalate below 4 W. The free energy perturbation simulations revealed the largest hydration free energy for CIP− across bulk, mesopore, and surface environments, while CIP+/− and especially CIP+ experience increasingly favourable hydration near the clay surface due to enhanced water structuring and electrostatic potential. Overall, this study reconciles flat versus tilted pose hypotheses and links pH-driven speciation to MMT retention and mobility. The results explain, on the molecular scale, why clay-rich soils act as strong sinks and how montmorillonite colloids can promote colloid-facilitated transport of antibiotics in real soil-aquifer systems.
1 Introduction
Ciprofloxacin (CIP) is among the most popularly prescribed antibiotics for treating bacterial infections in the world, and for many years it has been included in the World Health Organisation's (WHO) list of essential medicines.1 It is a member of the second-generation quinoline antibiotics containing a fluorine atom, and exhibits a broad spectrum of Gram-negative and Gram-positive inhibitory effects.2–4 Since its discovery in the mid-1960 s, CIP has been increasingly used globally in human and veterinary medicinal applications.5 However, its extensive global use has led to growing environmental concerns, as it does not readily degrade via natural pathways, such as by sunlight (UV-visible)6,7 and biodegradation,8 causing it to accumulate and become pseudo-persistent in the environment.9–12 Similar to many organic contaminants, CIP transport in natural systems is governed not only by its aqueous solubility but also by its strong affinity for mineral and organic colloids. When sorption affinity is high, particularly clay-rich colloids can mobilise antibiotics over aquifer-relevant distances, especially at low ionic strength.13,14 In fact, samples taken from aquatic environments (rivers, lakes, and estuaries) and soils have been shown to contain CIP residues.2,7,9,15–17 Continuous CIP release into the environment poses a severe public health risk, as long-term exposure to the drug via polluted food produce or drinking water can lead to antibiotic resistance.15,18 Thus, to protect public health, most countries impose regulations requiring waste treatment to remove harmful and toxic substances before they are released into the environment. Among the many technologies,2,18–23 adsorption techniques actually involving clay minerals have also been widely applied and sought after, thanks to a good balance between cost and technical implications.4Clays are ubiquitously abundant across the globe,24,25 forming ∼40% of the sedimentary rocks, and ∼16% of the total volume of the Earth's surface.26 Thus, understanding the surface chemistry and reactivity of clay minerals26–29 can provide valuable insight into the fate of organic contaminants in the environment, since clay minerals possess intrinsic properties that enable them to adsorb and retain organic contaminants to a high degree.30–35 Montmorillonite (MMT), which was investigated in this study, is a swelling smectite clay mineral and a common secondary mineral component of topsoil in temperate weathering environments.36,37 Due to its high specific surface area, strong capacity for ion exchange, and the relative ease with which the surface reactivity can be modified,38–41 MMT have been recognised as suitable adsorbent materials for removing organic contaminants from aqueous environments.34,42,43 Several experimental studies have reported good CIP adsorption on MMT across varying pH ranges. At high initial CIP concentrations (1500 and 3000 mg L−1) for Ca and Na-rich MMT, adsorption levels of ∼290–300 mg g−1 at pH 2–8 and ∼200–240 mg g−1 at pH 11 have been reported.44,45 However, at lower initial CIP concentrations (180 and 360 mg L−1), studies reported much lower adsorption capacity for Na–MMT, particularly at higher pH with adsorption levels falling to ∼60–100 mg g−1 at pH 10.18,46
Many factors influence CIP adsorption on MMT, as reflected by the variability of adsorption amounts. A key factor is pH-dependent molecular speciation, which strongly affects how an organic contaminant interacts with mineral surfaces.47 Depending on the solution pH, such compounds may exist in cationic, zwitterionic, or anionic forms, each exhibiting distinct adsorption behaviour. In addition, depending on the origin of the isomorphic substitution (tetrahedra or octahedra), the MMT surface has either diffuse or localised charge sites available for adsorption. Thus, variations in the surface charge of organic contaminants affect their ability to compete with other charged species for adsorption sites. Previous static sorption studies have reported that CIP adsorption capacity on MMT is high when it is cationic (CIP+) at pH < 6. At pH between 6 and 8, for a zwitterion (CIP+/−), adsorption is relatively low. For anionic (CIP−) at pH > 8, adsorption is lower.18,44–46 Additionally, protonation and deprotonation of the functional groups affect the interaction behaviour of the CIP molecule. The CIP+ protonated amine group (NH2) or carboxylic acid (COOH) interacts with the MMT surface mainly via electrostatic interactions. Regarding the transport of organic contaminants, several breakthrough column experiments have shown that MMT colloids are efficient carriers of CIP.14,48 Compared with quartz or kaolinite, Na+-MMT sorbs CIP two orders of magnitude more strongly and mobilises nearly all pre-sorbed CIP within a few pore volumes via co-transport.48,49 Furthermore, while MMT-bound CIP interaction with sand is limited, it remains microbiologically active after transport.13 Using natural soil colloids, the presence of colloids raised CIP breakthrough from ∼4% to 30–40%, with stronger facilitation at lower ionic strength (∼2 mM), conditions that favour colloid stability and transport.14 Mechanistically, desorption hysteresis from MMT (likely due to interlayer fixation/partial intercalation) and the high cation exchange capacity (CEC) of smectites explain why MMT ≫ kaolinite as a carrier; raising NaCl or Ca2+ concentration generally dampens colloidal facilitated transport by aggregating colloids and reducing their mobility.48
Although MMT is known to adsorb a wide range of organic contaminants,18,31,32,50–57 the molecular-scale mechanisms governing CIP adsorption, including the influence of ionic species, hydration, and interlayer diffusion, remain insufficiently understood. Hence, in this study, we employed both unbiased MD simulations, as well as biased simulations using umbrella sampling (US) and free energy perturbation (FEP) simulations, to investigate the adsorption dynamics and energetics, as well as surface configurations of the different CIP species with Na–MMT.
2 Methods
2.1 Modelled clay systems
Two main types of clay systems were used in this study. One single-layered and closed periodic system and one three-layered open system with hexagonal clay layers being periodic in the layers’ normal direction, but open in the xy-plane with terminated edges. Both types of systems and components were built with the atom MATLAB library.58 The closed single-layered system was constructed by replicating a single Pyrophyllite unit cell 11 × 6 times along the a and b unit cell vectors. The layer was subjected to isomorphic substitution in 2/3 of all unit cells in the octahedral sheet, resulting in an average unit cell stoichiometry of Na0.66[Si8(Al3.34Mg0.66)O20(OH)4], with Na+ being the counter-cation balancing the negative layer charge.37 Substitutions of Al3+ for Mg2+ sites were performed semi-randomly while ensuring no adjacent substitution sites, obeying Lowenstein's rule.59 A separation of 60 Å was introduced above the layer, creating an orthogonal box with initial dimensions of 56.76 Å × 53.79 Å × 60.00 Å. This closed system was used in both unbiased and biased umbrella sampling simulations, as well as in alchemical FEP simulations.To investigate CIP intercalation into the interlayers of MMT for different basal spacings d001, open-type systems containing three hexagonal layers having the dimensions 120 × 120 × 3d001 Å was used. The layers were obtained by cleaving MMT slabs along edges parallel to the (110), (0–10), and (010) lattice planes, resulting in a diameter of 7 nm.60–63 In order to minimize the number of oxygen edge atomtypes, which are not defined in CLAYFF, isomorphic substitution was performed as previously described albeit avoiding octahedral edge sites. Edge models representing acidic, neutral, or basic conditions in accordance with the systems involving CIP+, CIP+/−, and CIP−, respectively, were obtained by protonating or deprotonating terminating Al(Si)–O sites. Overall, CLAYFF had to be augmented with four additional oxygen atomtypes, whose partial charges were obtained using the charge smearing equation from Lammers et al., 201764 and the existing cationic partial charges in CLAYFF. More in-depth and detailed smectite clay edge models also including SiO(H) and substituted groups have previously been reported by Liu and Tournassat et al., 2023.65 Details on the atom types and example hexagonal MMT layers used in this study can be found in the SI.
2.2 CIP–MMT system setup
 | ||
| Fig. 1 Structures of the CIP molecules considered in the simulations. CIP+ (a), CIP+/− (b), and CIP− (c). | ||
In the biased umbrella sampling simulations in the corresponding closed systems, and to improve sampling, two identical CIP species were placed diagonally from each other in the middle of the simulation box (Fig. S5a and b).
To compare the free energy of hydration for the different CIP species subjected to clay surface adsorption, alchemical FEP simulations of the different CIP species were performed in three different regions of interest, (i) in bulk water (5000 water molecules, without clay), (ii) in the middle of the clay system mesopore, as well (iii) next to the clay surface (Fig. S6). For the system (iv) CIP was placed near the MMT surface at a distance corresponding to the position of the lowest point on the CIP–MMT potential of mean force (PMF) surface obtained from the umbrella sampling simulations.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 18000, 23000, and 28000 water molecules, respectively. In addition to accounting for the CIP charge balance, extra Na+ or Cl− were added to maintain electrical neutrality from edge termination.
000, 18000, 23000, and 28000 water molecules, respectively. In addition to accounting for the CIP charge balance, extra Na+ or Cl− were added to maintain electrical neutrality from edge termination.
2.3 MD simulations
All MMT–water–ion interactions were modelled with ClayFF using the SPC water model.67–69 Partial charges for the new atom types in MMT (resulting from the cleaving) not included in the original ClayFF were calculated following the work of Lammers et al. 2017.70 In addition to the non-bonded interatomic potential parameters of ClayFF, additional angle terms from the Interface force field (IFF) were used in the MMT hexagonal layers, as it improved the overall stability of the modelled unit cells and layer edges in particular.58,71The geometries of the different CIP molecules were optimised by DFT at the B3LYP/6-31G(d) level based on initial CIP structures obtained from the ZINC 15 database.72 They were modelled with forcefield parameters from the General Amber Force Field (GAFF) as obtained from the Bio2byte Acpype server,31,73 with atomic partial charges derived via the RESP-A1 protocol using the PyRED server.74,75
All MD simulations were performed using GROMACS, with a 12 Å cutoff distance for van der Waals (VdW) and Coulombic (Coul) short-range interactions. Long-range electrostatics was handled by the Particle–Mesh Ewald (PME) summation method. The Verlet cutoff scheme was employed to update the short-range neighbour list of 12 Å and a tolerance accuracy of 0.005 kJ mol−1 ps−1, All MD simulations used the velocity-rescaling (V-rescale) thermostat maintained at a temperature of 298 K, using the linear constraint solver (lincs) algorithm to constrain all H-bonded interactions in the simulations. An initial energy minimization step in each simulation system was initiated using the steepest descent algorithm with a convergence criterion of 500 kJ mol−1 nm−1 and a step size of 0.001 Å. Subsequently, a 0.5 ns equilibration run was performed in the canonical NVT ensemble with a 0.1 fs time step. Both the CIP and MMT layers were position-restrained during the energy minimization, while only CIP was restrained during system density equilibration in an isobaric–isothermal (NPT) ensemble for 2.5 ns with a time step of 0.5 fs, using the C-rescale barostat set to 1 bar.
To speed up the umbrella sampling simulations while accurately capturing the most probable adsorption configurations, the umbrella window simulations were extended in four steps (5, 10, 20, 50 ns) based on the distance to the MMT surface (Fig. S5c). Each umbrella window simulation used a 2 fs time step and a biased harmonic potential with a force constant of 1000 kJ mol−1 nm−2.
The free energy profiles from all windows were combined using the Weighted Histogram Analysis Method (WHAM), with an iterative convergence tolerance of 10−6. The first 1 ns of each umbrella window simulation was discarded from the WHAM analysis to ensure equilibration and eliminate transient effects.76,77
For each PMF profile, the standard adsorption free energy for the different CIP species binding to the MMT was computed, using the equilibrium constant for association (KA in units of length) to the unit cell of surface area Auc along the z-direction, as:

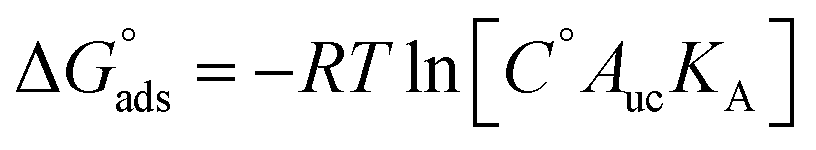 , where the term in brackets is K° and where C° = 1 M = 1/1660 Å−3 and Auc = 0.466 nm2.78,79 To further relate the PMF to the sorption coefficient Kd we also computed the interfacial adsorption coefficient Ki, being in principle a Henry surface–excess length (under dilute conditions) as
, where the term in brackets is K° and where C° = 1 M = 1/1660 Å−3 and Auc = 0.466 nm2.78,79 To further relate the PMF to the sorption coefficient Kd we also computed the interfacial adsorption coefficient Ki, being in principle a Henry surface–excess length (under dilute conditions) as  (units of length).80Ki is related to the Gibbs surface excess as Γ = CKi mol m−2, where C is the molar concentration. From the specific MMT surface area Sm of ∼750 m2 g−1, this Henry surface–excess length then also gives the distribution coefficient by multiplication with the specific surface area (Sm) as Kd = SmKi, (in cm3 g−1). A similar derivation can be found in previous works.62,80
(units of length).80Ki is related to the Gibbs surface excess as Γ = CKi mol m−2, where C is the molar concentration. From the specific MMT surface area Sm of ∼750 m2 g−1, this Henry surface–excess length then also gives the distribution coefficient by multiplication with the specific surface area (Sm) as Kd = SmKi, (in cm3 g−1). A similar derivation can be found in previous works.62,80
3 Results and discussions
3.1 CIP interlayer density profiles and configurations
 | ||
| Fig. 2 Shows the density profiles of CIP species normal to the MMT surface along the z direction. CIP+ exhibits the sharpest and highest adsorption peak, indicative of a well-defined adsorption configuration at the MMT surface. CIP+/− and CIP− both display broader distributions, reflecting similar adsorption behaviour consistent with the overall negative charge character existing on CIP− and about half of CIP+/−, most of it on its carboxylate end. The corresponding density profile for Na+ is shown in Fig. S7. | ||
Surprisingly, despite its net negative charge, CIP− showed significant interactions with the MMT surface. Most of the negative charge of CIP− originates from the O atoms in the carboxylate and ketone groups that interact favourably with surface-associated Na+.
 | ||
| Fig. 3 Spatial distributions of key functional groups along the z-direction during unbiased MD simulations for (a) CIP−, (b) CIP+/−, and (c) CIP+ moieties in the interlayer normal to MMT surface. MMT, 4-quinolone, COO−, COOH (carboxylate and carboxyl, respectively), NH, and NH2+ (piperazinyl secondary and primary amines, respectively). The corresponding densities are shown on the right side of each figure. An expanded view is shown in Fig. S8. | ||
Away from the surface, the negative carboxylate group of CIP− interacted with the Na+ counter-ions closer to the surface, however, upon adsorption, the surface's electrostatic repulsion caused the negative carboxylate group to rotate, pointing to the bulk (the spatial density in Fig. 3a and b). Overall, the spatial distributions normal to the MMT surface showed that the secondary amine, 4-quinolone, and carboxylate groups were reasonably separated (Fig. 3a). Thus, at the surface, the piperazinyl group stayed relatively near the MMT surface, with the 4-quinolone in the middle, whereas the negatively charged ketone and carboxylate ends were repelled and stayed further away. Given that CIP− piperazinyl group is deprotonated, attractive electrostatic interactions can not account for the close proximity of NH. Instead, short-range VdW forces and likely hydrogen bonds come into play.
The spatial distribution analysis also showed that in some instances, CIP− completely desorbs from the MMT surface, as observed at 100 ns in Fig. 3a. On the contrary, CIP+/− and CIP+ interact strongly with the MMT surface since both the 4-quinolone and the piperazinyl with a primary amine (NH2+) are attracted to the surface (Fig. 3b and c). For CIP+/− the spatial distribution of the end groups (Fig. 3b) showed that they are all very close to the surface compared to the CIP−. CIP+/− did, however, partially detach from the MMT surface on a few occasions, as observed from 60 ns to 140 ns. Despite the change in configuration, CIP+/− was not completely desorbed due to the significant attraction of the 4-quinolone and piperazinyl moieties to the surface. For CIP+ the primary amine, 4-quinolone, and carboxyl (COOH) strongly adsorb to the MMT surface. All monitored functional groups lie flat at nearly the same horizontal plane above the surface due to attractive interactions between the MMT surface and the CIP+ functional groups. Furthermore, because CIP+ has more positively charged atoms, it immediately migrated to the surface from the middle of the mesopore upon starting the simulations, where the strong attraction of CIP+ to the negative MMT surface makes spontaneous desorption highly unlikely.
 | ||
| Fig. 4 2D Concentration maps of 6 CIP molecules at varying water content. The top to bottom rows show the corresponding 1 W, 2 W, 3 W, and 4 W states, and the left to right columns the corresponding CIP+, CIP+/−, CIP− species, respectively. | ||
Moreover, because the surface adsorption energy was lower than that of CIP+, CIP+/− exhibited greater configurational flexibility and mobility upon adsorption. Experimental studies have in fact reported that maximum adsorption occurs at pKa1 (∼6.1–6.4), where the solution contains nearly equal proportions of CIP+ and CIP+/− species.18,44–46,82,83 This indicates that CIP can adsorb to MMT to a slightly higher amount than the corresponding CEC, suggesting that CIP+ and CIP+/− can synergistically be co-adsorbed.83 In contrast, the negative CIP− molecule showed a greater tendency to adsorb to one or two adjacent MMT layers edges via its ketone and carboxylate groups, interacting with the protonated edge sites. This dominant mode of interaction was greatly facilitated by the strong charge asymmetry on CIP−, evident from the electrostatic surface potential (ESP) of the carboxylate–ketone end of CIP−, which is ∼7 times more negative than at the piperazinyl end (Fig. S29c).
As a consequence of the strong edge interactions, CIP− exhibited limited ability to intercalate the MMT, even at high water content compared to CIP+ and CIP+/− (Fig. 4f, i and l). At 1 W edge adsorption contributed significantly to the total adsorption for CIP−. On the other hand, for CIP+ and CIP+/− at 2 W–4 W, most of the adsorption occurs on the basal planes, edge contribution therefore becomes insignificant as the edges only account for ∼2–5% of the total external surface area.84
000 configurations sampled from unbiased NVT closed system simulations. The RMSD correlation analysis showed that CIP− fluctuated the most, followed by CIP+/−, while CIP+ showed the highest structural stability upon adsorption. Fig. 5 presents the pairwise RMSD results projected onto a 2D plane, where each point represents an individual simulation configuration. High-density regions of the scattered population identify the dominant, recurrently visited adsorption configurations, while sparse regions correspond to transient or rarely visited geometries.
 | ||
| Fig. 5 Correlation density maps of equilibrated CIP structures core (4-quinolone) RMSD to the reference cross, side, and flat configurations. CIP− (a)–(c), CIP+/− (d)–(f), and CIP+ (g)–(i). The square marks at the axes represent the fit between the reference structures themselves. Selected clusters discussed in the text are indicated by numbered arrows prefixed with a C. | ||
From the configurational correlation density analysis, CIP− displayed the highest orientational heterogeneity with five identifiable clustered densities (Fig. 5c), compared to the CIP+/− and CIP+, which displayed two and one density clusters, respectively (Fig. 5f and i). Overall, representative CIP configurations extracted from frames in the density cluster analysis showed that all CIP molecules preferred to stay near the MMT surface (<5 Å) in a mainly side or flat (or in between side and flat) configuration (Fig. S10). In order to further investigate the interaction energies of the different configurations from the correlation density maps, pairwise potential energies between the different CIP molecules and MMT, water, and Na+, were decomposed into short-range electrostatic (Coulombic) and Lennard-Jones (LJ) contributions (Table 1). For CIP− in particular, a variation of both favourable and unfavourable interactions with the MMT surface was found. Density cluster C0 and C4 exhibited the more favourable Lennard-Jones interactions but unfavourable intramolecular Coulombic repulsive energies, compared to density clusters C1, C2, and C3, which demonstrated attractive Coulombic interactions (Fig. 5c). Despite the repulsive interaction between the deprotonated carboxylate group of CIP− and the negatively charged MMT surface, a net favourable interaction between MMT–CIP− was found, driven by solvation and Na+-mediated charge screening. However, aside from the possible screening effect, no apparent clear trend of the independent roles of water and Na+ in MMT–CIP− adsorption, involving the Coulombic and Lennard-Jones interactions was found.
| Pair | Density cluster | MMT (kJ mol−1) | Water (kJ mol−1) | Na+ (kJ mol−1) | |||
|---|---|---|---|---|---|---|---|
| Coul-SR | LJ-SR | Coul-SR | LJ-SR | Coul-SR | LJ-SR | ||
| CIP− | C0 | 26.91 | −64.92 | −191.91 | −47.41 | −369.76 | 290.57 |
| C1 | −21.33 | −40.81 | −221.94 | −55.76 | −331.91 | 235.88 | |
| C2 | −23.45 | −36.77 | −245.95 | −58.76 | −311.05 | 224.28 | |
| C3 | −21.40 | −38.71 | −226.73 | −59.03 | −332.52 | 238.87 | |
| C4 | 9.76 | −53.02 | −229.51 | −49.78 | −332.20 | 249.35 | |
| CIP+/− | C0 | −16.84 | −67.97 | −257.30 | −42.28 | −303.36 | 241.81 |
| C1 | −12.01 | −69.42 | −222.83 | −44.25 | −341.34 | 275.57 | |
| CIP+ | C0 | −63.67 | −75.18 | −216.26 | −51.71 | −20.91 | 25.64 |
CIP+/− had similar energetics across its two density clusters (Fig. 5f), suggesting energetically equivalent but geometrically distinct adsorption modes. This can be attributed to its distinct charge polarity, having both negatively and positively charged ends. However, both density clusters displayed weaker Coulombic energy contributions but stronger Lennard-Jones contributions than the favourable energies for the CIP− (C1, C2, and C3). This observation shows that CIP− could favourably interact with the MMT surface in specific configurational and adsorption modes. CIP+ exhibited the strongest Coulombic and Lennard-Jones interactions to MMT, but the least Na+ interaction. CIP+ also had the most restricted configurational landscape, with a single density cluster (Fig. 5i) exhibiting the highest electrostatic interaction energy. These results show that CIP adsorption on the MMT surface was governed by electrostatic interactions.
The corresponding relative free energy differences between the different density clusters for each CIP molecule are shown in Fig. 6. The free energy differences were obtained from the inverted Boltzmann distributions relative to the un-clustered population configurations for each CIP molecule. Overall, the single density cluster of CIP+ had the highest negative free energy, representing 89.1% of the total configurations. The two configurational density clusters of CIP+/− comprised in total 58.1%, and had both higher energy than the un-clustered reference population. The nearly equal distributions suggest an equilibrium among the structured configurations of C0 and C1 and the transient configurations. For CIP− the corresponding energy landscape spanned 2.16 kJ mol−1 (from −0.28 to 1.88 kJ mol−1) with 20.5% of the configurations in un-clustered transient states. This analysis demonstrate the heterogeneous nature of the adsorbed configurations of both CIP+/− and CIP−, compared with that of adsorbed CIP+.
 | ||
| Fig. 6 CIP free energy differences (ΔΔG) for cationic (CIP+, left), zwitterionic (CIP+/−, middle), and anionic (CIP−, right) from the correlation density clusters from Fig. 5, right column. | ||
 | ||
| Fig. 7 Structural and coordination characteristics of CIP adsorption at the MMT surface. (a) CIP–water oxygen RDF. (b) Representative Na+-mediated adsorption geometry between CIP and the MMT surface, showing Na+ (green) bridging the carboxylate and the surface oxygens (red) (c) functional group density distributions along the surface normal and, (d) mean CIP–Na+ coordination numbers for: carboxyl (I), quinolone (II), and piperazinyl (III), for the determined cluster populations identified in Fig. 5, right column. | ||
No direct perpendicular bridging was observed when the ∠CIP−(carboxylate oxygen)–Na+–MMT arrangement approached collinearity (∼180°), as in this geometry the Na+ lie along the shortest MMT–CIP− axis without lateral displacement. A moderate bridging was observed at intermediate angles (120°–140°), where the three-point geometry begins to adopt a bent configuration. The most frequent Na+–CIP− interactions occurred at 90°–120° angles (Fig. 7b), indicating existence of only suboptimal Na+-mediated association between CIP− and the MMT surface. The CIP− piperazinyl group showed less frequent Na+ coordination than the quinolone and carboxylate groups (Fig. 7d(III)). This coordination environment explained why the proximity of CIP− to the MMT surface is the closest in cluster C0, followed by cluster C4, as shown in spatial distributions (Fig. 7c). CIP− configurational implications to its coordination behaviour and distribution remained persistent regardless of the choice of reference structure RMSD fitting projections (Fig. S17–S19).
CIP+/− followed a similar water and Na+ interaction trend, with cluster C0 populations showing higher water oxygen RDF peaks but less Na+ coordination (Fig. 7). Cluster C1, exhibiting higher Na+ interaction energies, suggests a geometry where the negative end of CIP+/− is near the MMT surface. CIP+ was dominated by a single stable density cluster, stronger Coulombic and Lennard-Jones interactions with the MMT. Overall, CIP+ had the lowest and demonstrated unfavourable interaction with Na+. This is reflected in the lowest mean coordination of Na+ across all functional groups (Fig. 7d), indicating that CIP+ species will compete with Na+ for adsorption sites on the MMT surface. Moreover, CIP+ showed the lowest hydration peak intensity (Fig. 7a), along with the presence of an NH2 group, resulting in much more stable surface adsorption via a combination of inner-sphere complexation and H-bonding. The rest of the reference structures RMSD fitted projection results are provided in Fig. S17–S27.
3.2 Free energy calculation
 | ||
| Fig. 8 Potential of mean force (PMF) profiles for CIP− (red), CIP+/− (black) and CIP+ (green) adsorption to the MMT along the surface normal. Error bars represent a 95% confidence interval from 10 independent simulations. Labelled positions are discussed in the text. | ||
Between positions 1 and 2 (Fig. 8), the PMF profiles for all CIP molecules exhibited a potential barrier characterised by a pronounced peak. For CIP+ and CIP+/−, this barrier feature was ∼0.88 nm from the surface, attributed to the increased repulsion between the increasing density of Na+ counter-ions and approaching positive CIP atoms. The barrier was slightly shifted towards the surface (∼0.78 nm at position 2′) for the CIP− as the proximity to the octahedral negative charge was further increased. Moreover, the sharp decrease following this barrier indicates a rapid reorientation of CIP−, where the positively charged piperazinyl group aligned toward the surface while the negatively charged carboxylate group points away. This orientation minimised electrostatic repulsion and enhanced attraction, aided by the shielding effect of nearby Na+ counter-ions. CIP+/− PMF demonstrated a characteristic double minimum with distinctive energy minima at ∼0.34 nm (−18.88 kJ mol−1) and 0.51 nm (−18.29 kJ mol−1) from the surface (positions 5 and 3 in Fig. 8, respectively). The computed CIP+/− ESP distribution showed a distinctive surface charge separation, with the negative and positive surfaces covering nearly equal surface areas, with ratios 50.41% and 49.59% (Table S2 and Fig. S29b). Thus, the barrier in PMF profiles observed at position 4 (Fig. 8) originated from the repulsive interactions between the MMT surface and the CIP+/− negative carboxylate end. For CIP+ the global energy minimum was −44.43 kJ mol−1 at ∼0.34 nm (Fig. 8, position 5) from the surface. This higher adsorption energy of CIP+ was primarily attributed to stronger Coulombic attraction to the negatively charged MMT surface. Additional open systems simulations, performed by starting with a CIP in the interlayer region and spanning a total simulation time of over 250 ns, showed that all CIPs remained in the interlayer, even for the CIP−, which demonstrated, indeed, CIP− favourable interaction with the surface.
To obtain KA and the free energy of adsorption per-site and 1 M standard state  , each PMF profile was integrated from the middle of the mesopore to the distance of closest approach, arbitrarily defined as F < 0 since beyond this point the PMF do not contribute much to the adsorption free energy. To compare with sorption coefficients Kd used in environmental contexts, we also evaluated the interfacial adsorption coefficient as a Henry surface–excess length (Ki) from the same PMFs and reported Kd. Together, the obtained
, each PMF profile was integrated from the middle of the mesopore to the distance of closest approach, arbitrarily defined as F < 0 since beyond this point the PMF do not contribute much to the adsorption free energy. To compare with sorption coefficients Kd used in environmental contexts, we also evaluated the interfacial adsorption coefficient as a Henry surface–excess length (Ki) from the same PMFs and reported Kd. Together, the obtained  and Kd. derived here quantify (i) the intrinsic per-site binding strength and (ii) the macroscopic partitioning expected for MMT, assuming access to its full surface area of ∼750 m2 g−1. Table 2 shows that the species tendency to adsorb to the MMT surface increases in the order CIP+ > CIP+/− > CIP−. Values in Table 2 show that CIP+ has several orders of magnitude higher affinity for the MMT surface than CIP+/− and CIP−.
and Kd. derived here quantify (i) the intrinsic per-site binding strength and (ii) the macroscopic partitioning expected for MMT, assuming access to its full surface area of ∼750 m2 g−1. Table 2 shows that the species tendency to adsorb to the MMT surface increases in the order CIP+ > CIP+/− > CIP−. Values in Table 2 show that CIP+ has several orders of magnitude higher affinity for the MMT surface than CIP+/− and CIP−.
| Molecule | Ads. free energy ΔG° (kJ mol−1) | Sorption coefficient Kd (cm3 g−1) |
|---|---|---|
| CIP− | −2.1 ± 1.0 | 6.43 ± 3.80 |
| CIP+/− | −11.8 ± 1.1 | 518 ± 212 |
| CIP+ | −30.0 ± 0.8 | 1.04 × 106 ± 0.35 × 106 |
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) in the carboxylate and ketone groups and its high negative partial charge, increasing its water affinity (Fig. 8). CIP+/− exhibited very similar hydration behaviour to CIP−, with slightly lower hydration energy due to having a less negative partial charge on the CO. On the other hand, CIP+ had the smallest hydration energy, which can be attributed to the protonation of the carboxylate group, thereby decreasing its affinity for water. The hydration energies of CIP− showed no clear deterministic trend. However, the CIP+ and CIP+/− hydration energies steadily increased from the homogeneous water box to the Na–MMT mesopore and at the surface, due to increased water density and organisation near the surface (Fig. S7). Moreover, the increase in negative electrostatic potential near the surface increases the polarisation of water molecules, which favours an increase in hydration of the zwitterionic and positively charged species.85
O) in the carboxylate and ketone groups and its high negative partial charge, increasing its water affinity (Fig. 8). CIP+/− exhibited very similar hydration behaviour to CIP−, with slightly lower hydration energy due to having a less negative partial charge on the CO. On the other hand, CIP+ had the smallest hydration energy, which can be attributed to the protonation of the carboxylate group, thereby decreasing its affinity for water. The hydration energies of CIP− showed no clear deterministic trend. However, the CIP+ and CIP+/− hydration energies steadily increased from the homogeneous water box to the Na–MMT mesopore and at the surface, due to increased water density and organisation near the surface (Fig. S7). Moreover, the increase in negative electrostatic potential near the surface increases the polarisation of water molecules, which favours an increase in hydration of the zwitterionic and positively charged species.85
 | ||
| Fig. 9 Hydration free energy for the CIP molecules obtained via alchemical free-energy perturbation in three environments: bulk water, mesopore (bulk-like), and near the MMT surface (adsorbed region). Error bars represent a 95% confidence interval obtained from 3 repeated simulations in both forward and backward directions. | ||
4. Conclusion
This work provides a molecular-level description of the adsorption energetics and configurations of CIP species to Na+-exchanged MMT clay surfaces, across environmentally relevant pH conditions. The preferred configuration analyses show that, regardless of the CIP ionic charge, the dominant adsorption configuration is primarily entirely or nearly flat. This contradicts postulates in previous literature that tilt and cross orientations are the main configurations.44,82,84 Additionally, the configurations from 200 ns simulation revealed that the significance of the bridging effect of the interlayer cation on CIP− adsorption to MMT has been slightly exaggerated in the literature. Our results show that the impact of negative surface charge screening by counter-ions was evident only during the initial CIP surface migration. On the surface, Na+–carboxylate contacts are present in multiple configurations, but the Na+ does not adopt a collinear MMT–Na+–CIP− perpendicular bridging geometry; instead, the bridging angles are consistently below 140°, reflecting a suboptimal screening effect. Once adsorbed to the surface, CIP− interacts mainly from its 4-quinolone core and the H atoms in the cyclopropyl and the piperazinyl groups. However, the net Coulombic contribution is configuration-dependent: some adsorption configurations exhibit favourable pair-wise Coulombic interactions, indicating that direct electrostatic stabilisation is not universally unfavourable for CIP−. The density profiles of the different CIP+, CIP+/− and CIP− molecules corroborated these results, as all species display similar preferred distances from the surface. Under basic conditions, when speciation is dominated by CIP−, adsorption occurs by interacting with the protonated edge groups on MMT and, to a not insignificant extent, the negative MMT basal surface. However, CIP− interaction with the basal surface depends on the hydration state (interlayer separation) and concentration, as seen in the concentration maps generated from the open system simulations. Moreover, given the layered structure of MMT, the contribution of edge adsorption alone cannot account for the high adsorption capacities reported in experiments,44 suggesting that the reported amounts of adsorption may have originated from basal surface adsorption.Cluster-derived dominant configurations analysis from the RMSD-derived fitting correlations to the reference structures confirms that all three CIP species preferentially adsorb in flat or near-flat configurations at the MMT surface. However, adsorption stability is strongly dependent on speciation. CIP+ is locked into a single dominant electrostatically stabilised configuration state, whereas CIP+/− samples two energetically accessible geometries, and CIP− remains the most heterogeneous with adsorption driven by a combination of Na+-mediated Coulombic interactions and VdW dispersion.
The adsorption free energy profiles show that CIP+ exhibits strong electrostatic attraction to the surface, supporting cation exchange as the primary driver of CIP+ adsorption.82 Notably, CIP− also exhibited favourable adsorption energies comparable to those reported for MMT–Na+ interactions (Fig. S28).86 Thus, while it may not be due to cation-exchange effects, we cannot completely disregard the possibility that the CIP− can intercalate the MMT interlayer at hydration states higher than 1 W. Further exploration of factors, such as the layer charge and charge origin characteristics (octahedral/tetrahedral) of the MMT TOT layers, specific edge sites pKa65,87,88 as well as the type of counter-ion, may prove crucial in shedding light on the cation exchange role in CIP species adsorption.
Overall, the standard adsorption free energies follow the sequence CIP− (ΔG° ≈ −2.1 kJ mol−1) < CIP+/− (−11.8 kJ mol−1) < CIP+ (−30.0 kJ mol−1). Because the adsorption free energy enters exponentially in the calculation of the sorption coefficients, modest differences in ΔG° result in orders-of-magnitude variation in Kd. Indeed, the corresponding sorption coefficients span six orders of magnitude (Kd values of 6.4, 520, and 1.0 × 106 cm3 g−1), demonstrating how sensitive macroscopic retention is to small differences in the PMF profiles.
The FEP simulations revealed that CIP− possesses higher hydration energy compared to CIP+ and CIP+/−. In addition to the Na+ screening effects, this strong hydration potentially mitigated electrostatic repulsion during approach, explaining why CIP− despite its net negative charge could still access the basal surface under certain hydration conditions.
These results further suggest that counter-ion identity and MMT layer charge origin (octahedral versus tetrahedral isomorphic substitution) may significantly influence CIP–surface interactions, which should be explored in future work.
Overall, this study unifies molecular configurations, hydration energetics, and free-energy landscapes into a coherent mechanistic picture of CIP sorption to smectite clay particles, and provide a quantitative foundation for understanding sorption, intercalation, and colloid-facilitated transport of CIP in clay-rich soils and aquifer systems.
Author contributions
R. S.: simulations and data analysis, original drafting and revision. M. H.: conceptualization, funding acquisition, editing and revision.Conflicts of interest
All authors reviewed the manuscript, and no potential conflict of interest was reported.Data availability
The supplementary information (SI) contains CIP atomtypes and their partial charges, details about simulation system setup and descriptions, and references to software used for data analysis. The hexagonal MMT structural files are provided separately as a zip file. Any additional MD source data for this article are available upon request from the authors. The supplementary information is available at DOI: https://doi.org/10.1039/d5cp04597e.Acknowledgements
The authors acknowledge the constructive comments on the MD simulations from Sébastien Le Crom. The authors also acknowledge the High Performance Computing Centre North (HPC2N) at Umeå University for providing computational resources. This work was supported by the Swedish Research Council (grant 2019-04733).References
- World Health Organization. WHO model list of essential medicines – 22nd list, 2021. Tech Doc, 2021, 2021. Available from: https://www.who.int/publications/i/item/WHO-MHP-HPS-EML-2021.02.
- A. K. Al-Buriahi, M. M. Al-shaibani, R. M. S. R. Mohamed, A. A. Al-Gheethi, A. Sharma and N. Ismail, Ciprofloxacin removal from non-clinical environment: a critical review of current methods and future trend prospects, J. Water Process Eng., 2022, 47, 102725 CrossRef.
- L. M. Fisher, J. M. Lawrence, I. C. Josty, R. Hopewell, E. E. C. Margerrison and M. E. Cullen, Ciprofloxacin and the fluoroquinolones, Am. J. Med., 1989, 87(5), S2–S8 CrossRef PubMed.
- M. Bizi and F. E. El Bachra, Evaluation of the ciprofloxacin adsorption capacity of common industrial minerals and application to tap water treatment, Powder Technol., 2020, 362, 323–333 CrossRef CAS.
- U. W. E. Petersen, K. Grohe and K. G. M. Hans-Joachim Zeiler, United States Patent (19), 4547503, USA, 1985. Available from: https://patentimages.storage.googleapis.com/58/51/f6/04bba0cfdfbd27/US4547503.pdf Search PubMed.
- A. P. Toolaram, T. Haddad, C. Leder and K. Kümmerer, Initial hazard screening for genotoxicity of photo-transformation products of ciprofloxacin by applying a combination of experimental and in-silico testing, Environ Pollut., 2016, 211, 148–156 CrossRef CAS PubMed.
- R. Andreozzi, R. Marotta and N. Paxéus, Pharmaceuticals in STP effluents and their solar photodegradation in aquatic environment, Chemosphere, 2003, 50(10), 1319–1330 CrossRef CAS PubMed.
- C. Girardi, J. Greve, M. Lamshöft, I. Fetzer, A. Miltner and A. Schäffer, et al., Biodegradation of ciprofloxacin in water and soil and its effects on the microbial communities, J. Hazard Mater., 2011, 198, 22–30 CrossRef CAS PubMed.
- M. Patel, R. Kumar, K. Kishor, T. Mlsna, C. U. Pittman and D. Mohan, Pharmaceuticals of emerging concern in aquatic systems: chemistry, occurrence, effects, and removal methods, Chem. Rev., 2019, 119(6), 3510–3673 CrossRef CAS PubMed.
- S. I. Polianciuc, A. E. Gurzău, B. Kiss, M. Georgia Stefan and F. Loghin, Antibiotics in the environment: causes and consequences, Med. Pharm. Rep., 2020, 93(3), 231–240 CrossRef PubMed.
- V. Rilstone, Y. Filion and P. Champagne, Study on the persistence of ciprofloxacin and sulfamethoxazole in simulated drinking water systems, Environ. Syst. Res. (Heidelb)., 2025, 14, 7 CrossRef PubMed.
- R. M. P. Leal and A. K. Sarmah, Modelling sorption and dissipation kinetics of ciprofloxacin and enrofloxacin antibiotics in New Zealand pastoral soils, Soil Environ. Health, 2025, 3(1), 100125 CrossRef CAS.
- H. Chen, B. Gao, L. Y. Yang and L. Q. Ma, Montmorillonite enhanced ciprofloxacin transport in saturated porous media with sorbed ciprofloxacin showing antibiotic activity, J. Contam. Hydrol., 2015, 173, 1–7 CrossRef CAS PubMed.
- Y. Xing, X. Chen, X. Chen and J. Zhuang, Colloid-Mediated Transport of Pharmaceutical and Personal Care Products through Porous Media, Sci. Rep., 2016, 6, 1–10 CrossRef PubMed.
- A. L. Córdova-Kreylos and K. M. Scow, Effects of ciprofloxacin on salt marsh sediment microbial communities, ISME J., 2007, 1(7), 585–595 CrossRef PubMed.
- H. Nakata, K. Kannan, P. D. Jones and J. P. Giesy, Determination of fluoroquinolone antibiotics in wastewater effluents by liquid chromatography-mass spectrometry and fluorescence detection, Chemosphere, 2005, 58(6), 759–766 CrossRef CAS PubMed.
- E. M. Golet, A. C. Alder, A. Hartmann, T. A. Temes and W. Giger, Trace determination of fluoroquinolone antibacterial agents in urban wastewater by solid-phase extraction and liquid chromatography with fluorescence detection, Anal. Chem., 2001, 73(15), 3632–3638 CrossRef CAS PubMed.
- H. Zhang, F. Zhao, M. Xia and F. Wang, Microscopic adsorption mechanism of montmorillonite for common ciprofloxacin emerging contaminant: molecular dynamics simulation and Multiwfn wave function analysis, Colloids Surf., A, 2021, 614, 126186 CrossRef CAS.
- Z. B. Gönder, Y. Kaya, I. Vergili and H. Barlas, Optimization of filtration conditions for CIP wastewater treatment by nanofiltration process using Taguchi approach, Sep. Purif. Technol., 2010, 70(3), 265–273 CrossRef.
- Y. Wang, Q. Nie, B. Huang, H. Cheng, L. Wang and Q. He, Removal of ciprofloxacin as an emerging pollutant: a novel application for bauxite residue reuse, J. Cleaner Prod., 2020, 253, 120049 CrossRef CAS.
- X. Ma and Z. Wang, Removal of Ciprofloxacin from Wastewater by Ultrasound/Electric Field/Sodium Persulfate (US/E/PS), Processes, 2022, 10(1), 24 CrossRef.
- J. H. Al-Rifai, C. L. Gabelish and A. I. Schäfer, Occurrence of pharmaceutically active and non-steroidal estrogenic compounds in three different wastewater recycling schemes in Australia, Chemosphere, 2007, 69(5), 803–815 CrossRef CAS PubMed.
- A. H. Zyoud, S. H. Zyoud and A. Amireh, An innovative and sustainable approach for water purification: thermal decomposition of tetracycline contaminant adsorbed on thermally stable montmorillonite, Case Stud. Chem. Environ. Eng., 2024, 9, 100631 Search PubMed.
- A. Ito and R. Wagai, Data Descriptor: global distribution of clay-size minerals on land surface for biogeochemical and climatological studies, Sci. Data, 2017, 4, 1–11 Search PubMed.
- Q. Hu, C. Li, B. Yang, X. Fang, H. Lü and X. Shi, et al., Clay mineral distribution characteristics of surface sediments in the South Mid-Atlantic Ridge, J. Oceanol Limnol., 2023, 41(3), 897–908 Search PubMed.
- T. Al-Ani and O. Sarapaa, in Clay and clay mineralogy, ed. T. Al-Ani and O. Sarapaa, Helsinki, 2008 Search PubMed.
- S. Guggenheim and R. T. Martin, Definition of clay and clay mineral: joint report of the AIPEA and CMS Nomenclature Committees, Clay Miner., 1995, 30(3), 257–259 Search PubMed.
- S. Guggenheim, J. M. Adams, D. C. Bain, F. Bergaya, M. F. Brigatti and V. A. Drits, et al., Summary of recommendations of nomenclature committees relevant to clay mineralogy: report of the Association Internacionale pour l’Etude des Argiles (AIPEA) nomenclature committee for 2006, Clays Clay Miner., 2006, 54(6), 761–772 CrossRef CAS.
- F. Uddin, Clays, nanoclays, and montmorillonite minerals, Metall. Mater. Trans. A, 2008, 39(12), 2804–2814 CrossRef.
- M. Szczerba, Y. Deng and M. Kowalik-Hyla, Molecular Modeling to Predict the Optimal Mineralogy of Smectites as Binders of Aflatoxin, Clays Clay Miner., 2022, 70(6), 824–836 Search PubMed.
- G. Corbin, E. Vulliet, B. Lanson, A. Rimola and P. Mignon, Adsorption of pharmaceuticals onto smectite clay minerals: a combined experimental and theoretical study, Minerals, 2021, 11(1), 1–12 Search PubMed.
- P. H. Chang, Z. Li, W. T. Jiang and B. Sarkar, Clay minerals for pharmaceutical wastewater treatment. Modified Clay and Zeolite Nanocomposite Materials: Environmental and Pharmaceutical Applications, Elsevier Inc., 2018, pp. 167–196 Search PubMed.
- A. Pasket, H. Zhang, Y. Wang, M. Krzmarzick, J. E. Gustafson and S. Deng, Clay Content Played a Key Role Governing Sorption of Ciprofloxacin in Soil. Front, Soil Sci., 2022, 2, 1–11 Search PubMed.
- T. De Oliveira, M. Boussafir, L. Fougère, E. Destandau, Y. Sugahara and R. Guégan, Use of a clay mineral and its nonionic and cationic organoclay derivatives for the removal of pharmaceuticals from rural wastewater effluents, Chemosphere, 2020, 259, 127480 Search PubMed.
- G. J. Churchman, W. P. Gates, B. K. G. Theng and G. Yuan, Chapter 11.1 Clays and Clay Minerals for Pollution Control, Dev. Clay Sci., 2006, 1(C), 625–675 CAS.
- C. S. Ross and S. B. Hendricks, Minerals of the Montmorillonite Group Their Origin and Relation to Soils and Clays, Geol. Surv., 1945,(Professional Paper 205-B), 71 Search PubMed.
- G. Sposito, The chemistry of soils. Second. Science, Oxford University Press, New York, 2008 Search PubMed.
- A. M. Awad, S. M. R. Shaikh, R. Jalab, M. H. Gulied, M. S. Nasser and A. Benamor, et al., Adsorption of organic pollutants by natural and modified clays: a comprehensive review, Sep. Purif Technol., 2019, 228, 115719 CrossRef CAS.
- D. Ewis, M. M. Ba-Abbad, A. Benamor and M. H. El-Naas, Adsorption of organic water pollutants by clays and clay minerals composites: a comprehensive review, Appl. Clay Sci., 2022, 229, 106686 CrossRef CAS.
- S. Barakan and V. Aghazadeh, The advantages of clay mineral modification methods for enhancing adsorption efficiency in wastewater treatment: a review, Environ. Sci. Pollut. Res., 2021, 28(3), 2572–2599 CrossRef CAS PubMed.
- Advances in the Development and Applications of Clay-Based Composites, in Clay Composites Advances in Material Research and Technology, ed. M. Vithanage, G. Lazzara, A. Upamali R, 2023, pp. 75–88 Search PubMed.
- J. Akhtar, N. A. S. Amin and K. Shahzad, A review on removal of pharmaceuticals from water by adsorption, Desalin. Water Treat., 2016, 57(27), 12842–12860 CrossRef CAS.
- G. Sheng, C. T. Johnston, B. J. Teppen and S. A. Boyd, Adsorption of dinitrophenol herbicides from water by montmorillonites, Clays Clay Miner., 2002, 50(1), 25–34 CrossRef CAS.
- C. J. Wang, Z. Li, W. T. Jiang, J. S. Jean and C. C. Liu, Cation exchange interaction between antibiotic ciprofloxacin and montmorillonite, J. Hazard Mater., 2010, 183(1–3), 309–314 CrossRef CAS PubMed.
- Q. Wu, Z. Li, H. Hong, K. Yin and L. Tie, Adsorption and intercalation of ciprofloxacin on montmorillonite, Appl. Clay Sci., 2010, 50(2), 204–211 CrossRef CAS.
- M. E. Roca Jalil, M. Baschini and K. Sapag, Influence of pH and antibiotic solubility on the removal of ciprofloxacin from aqueous media using montmorillonite, Appl. Clay Sci., 2015, 114, 69–76 CrossRef CAS.
- G. W. Bailey and J. L. White, Factors influencing the adsorption, desorption, and movement of pesticides in soi, Residue Rev., 1970, 32(3656), 29–92 Search PubMed.
- H. Zhang, T. Lu, R. Zhang, M. Wang, S. Krishnan and S. Liu, et al., Effects of clay colloids on ciprofloxacin transport in saturated quartz sand porous media under different solution chemistry conditions, Ecotoxicol. Environ. Saf., 2020, 199, 110754 CrossRef CAS PubMed.
- H. Chen, B. Gao, L. Yang and L. Q. Ma, Montmorillonite enhanced ciprofloxacin transport in saturated porous media with sorbed ciprofloxacin showing antibiotic activity, J. Contam. Hydrol., 2015, 173, 1–7 CrossRef CAS PubMed.
- R. A. Al-Bayati and A. S. Ahmed, Adsorption – Desorption of Trimethoprim Antibiotic Drug from Aqueous Solution by Two Different Natural Occurring Adsorbents, Int. J. Chem., 2011, 3(3), 21–30 CAS.
- L. K. Ong, F. E. Soetaredjo, A. Kurniawan, A. Ayucitra, J. C. Liu and S. Ismadji, Investigation on the montmorillonite adsorption of biocidal compounds incorporating thermodynamical-based multicomponent adsorption isotherm, Chem. Eng. J., 2014, 241, 9–18 CrossRef CAS.
- A. Septian, S. Oh and W. S. Shin, Sorption of antibiotics onto montmorillonite and kaolinite: competition modelling, Environ. Technol., 2019, 40(22), 2940–2953 CrossRef CAS PubMed.
- Q. Xie, J. Xie, Z. Wang, D. Wu, Z. Zhang and H. Kong, Adsorption of organic pollutants by surfactant modified zeolite as controlled by surfactant chain length, Microporous Mesoporous Mater., 2013, 179, 144–150 Search PubMed.
- R. A. Figueroa, A. Leonard and A. A. Mackay, Modeling Tetracycline Antibiotic Sorption to Clays, Environ. Sci. Technol., 2004, 38(2), 476–483 CrossRef CAS PubMed.
- V. O. Shikuku, R. Zanella, C. O. Kowenje, F. F. Donato, N. M. G. Bandeira and O. D. Prestes, Single and binary adsorption of sulfonamide antibiotics onto iron-modified clay: linear and nonlinear isotherms, kinetics, thermodynamics, and mechanistic studies, Appl. Water Sci., 2018, 8, 175 Search PubMed.
- T. Thiebault, M. Boussafir, L. Le Forestier, C. Le Milbeau, L. Monnin and R. Guégan, Competitive adsorption of a pool of pharmaceuticals onto a raw clay mineral, RSC Adv., 2016, 6(69), 65257–65265 Search PubMed.
- L. Aristilde, C. Marichal, J. Miéhé-Brendlé, B. Lanson and L. Charlet, Interactions of oxytetracycline with a smectite clay: a spectroscopic study with molecular simulations, Environ. Sci. Technol., 2010, 44(20), 7839–7845 Search PubMed.
- M. Holmboe, atom: A Matlab Package for Manipulation of Molecular Systems, Clays Clay Miner., 2019, 67(5), 419–426 Search PubMed.
- W. Loewenstein, The Distribution of Aluminum in the Tetra-Hedra of Silicates and Aluminates, Minerol Soc. Am., 1954, 39, 92–96 CAS.
- G. N. White and L. W. Zelazny, Analysis and Implications of the Edge Structure of Dioctahedral Phyllosilicates, Clays Clay Miner., 1988, 36(2), 141–146 CrossRef CAS.
- L. P. Lavikainen, J. T. Hirvi, S. Kasa, T. Schatz and T. A. Pakkanen, Stability of dioctahedral 2:1 phyllosilicate edge structures based on pyrophyllite models, Theor. Chem. Acc., 2015, 134(9), 1–8 Search PubMed.
- J. A. R. Willemsen, S. C. B. Myneni and I. C. Bourg, Molecular Dynamics Simulations of the Adsorption of Phthalate Esters on Smectite Clay Surfaces, J. Phys. Chem. C, 2019, 13624–13636 Search PubMed.
- X. Shen and I. C. Bourg, Interaction between Hydrated Smectite Clay Particles as a Function of Salinity (0–1 M) and Counterion Type (Na, K, Ca), J. Phys. Chem. C, 2022, 20990–20997 CrossRef CAS PubMed.
- L. N. Lammers, I. C. Bourg, M. Okumura, K. Kolluri, G. Sposito and M. Machida, Molecular dynamics simulations of cesium adsorption on illite nanoparticles, J. Colloid Interface Sci., 2017, 490, 608–620 CrossRef CAS PubMed.
- P. Gao, X. Liu, Z. Guo and C. Tournassat, Acid–Base Properties of Cis-Vacant Montmorillonite Edge Surfaces: A Combined First-Principles Molecular Dynamics and Surface Complexation Modeling Approach, Environ. Sci. Technol., 2022, 1342–1352 Search PubMed.
- M. Holmboe, S. Wold and M. Jonsson, Porosity investigation of compacted bentonite using XRD profile modeling, J. Contam Hydrol., 2012, 128(1–4), 19–32 Search PubMed.
- R. T. Cygan, J. J. Liang and A. G. Kalinichev, Molecular Models of Hydroxide, Oxyhydroxide, and Clay Phases and the Development of a General Force Field, J. Phys. Chem. B, 2004, 108(4), 1255–1266 Search PubMed.
- H. J. C. Berendsen, J. R. Grigera and T. P. Straatsma, The missing term in effective pair potentials, J. Phys. Chem., 1987, 91(24), 6269–6271 CrossRef CAS.
- T. I. Mizan, P. E. Savage and R. M. Ziff, Comparison of rigid and flexible simple point charge water models at supercritical conditions, J. Comput. Chem., 1996, 17(15), 1757–1770 CrossRef CAS.
- L. N. Lammers, I. C. Bourg, M. Okumura, K. Kolluri, G. Sposito and M. Machida, Molecular dynamics simulations of cesium adsorption on illite nanoparticles, J. Colloid Interface Sci., 2017, 490, 608–620 Search PubMed.
- H. Heinz, T. J. Lin, R. Kishore Mishra and F. S. Emami, Thermodynamically consistent force fields for the assembly of inorganic, organic, and biological nanostructures: the INTERFACE force field, Langmuir, 2013, 29(6), 1754–1765 Search PubMed.
- T. Sterling and J. J. Irwin, ZINC 15 – Ligand Discovery for Everyone, J. Chem. Inf. Model., 2015, 55(11), 2324–2337 Search PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, Development and testing of a general Amber force field, J. Comput. Chem., 2004, 25(9), 1157–1174 Search PubMed.
- F.-Y. Dupradeau, A. Pigache, C. S. Thomas Zaffran, R. Lelong, N. Grivel, D. Lelong, W. Rosanski and P. Cieplak, The R.E.D. Tools: advances in RESP and ESP charge derivation, Phys. Chem. Chem. Phys., 2010, 77, 114–122 Search PubMed.
- R. J. Woods and R. Chappelle, Restrained electrostatic potential atomic partial charges for condensed-phase simulations of carbohydrates, Theochem., 2000, 527(1–3), 149–156 Search PubMed.
- S. Kumar, D. Bouzida, R. H. Swendsen, P. A. Kollman and J. M. Rosenberg, The weighted histogram analysis method for free-energy calculations on biomolecules, J. Comput. Chem., 1992, 13(8), 1011–1021 CrossRef CAS.
- M. R. Shirts and D. L. Mobley, An introduction to best practices in free energy calculations, Methods Mol. Biol., 2013, 924, 271–311 Search PubMed.
- E. G. Brandt and A. P. Lyubartsev, Molecular Dynamics Simulations of Adsorption of Amino Acid Side Chain Analogues and a Titanium Binding Peptide on the TiO2(100) Surface, J. Phys. Chem. C, 2015, 119(32), 18126–18139 CrossRef CAS.
- J. Wang, Y. Deng and B. Roux, Absolute binding free energy calculations using molecular dynamics simulations with restraining potentials, Biophys. J., 2006, 91(8), 2798–2814 Search PubMed.
- A. C. Lemay, E. J. Sontarp, D. Martinez, P. Maruri, R. Mohammed and R. Neapole, et al., Molecular Dynamics Simulation Prediction of the Partitioning Constants (KH, Kiw, Kia) of 82 Legacy and Emerging Organic Contaminants at the Water–Air Interface, Environ. Sci. Technol., 2023, 6296–6308 CrossRef CAS PubMed.
- C. H. Bennett, Efficient estimation of free energy differences from Monte Carlo data, J. Comput. Phys., 1976, 22(2), 245–268 CrossRef.
- C. J. Wang, Z. Li and W. T. Jiang, Adsorption of ciprofloxacin on 2:1 dioctahedral clay minerals, Appl. Clay Sci., 2011, 53(4), 723–728 CrossRef CAS.
- A. K. Helmy, S. G. De Bussetti and E. A. Ferreiro, Adsorption of quinoline from aqueous solutions by some clays and oxides, Clays Clay Miner., 1983, 31(1), 29–36 CrossRef CAS.
- A. J. Carrasquillo, G. L. Bruland, A. A. Mackay and D. Vasudevan, Sorption of ciprofloxacin and oxytetracycline zwitterions to soils and soil minerals: influence of compound structure, Environ. Sci. Technol., 2008, 42(20), 7634–7642 CrossRef CAS PubMed.
- H. Y. Wen and M. Hedström, Molecular Dynamics Simulations of NaCl Permeation in Bihydrated Montmorillonite Interlayer Nanopores, J. Phys. Chem. C, 2015, 119, 17352–17361 CrossRef.
- T. Underwood, V. Erastova and H. Chris Greenwell, Ion adsorption at Clay-Mineral surfaces: the Hofmeister series for hydrated smectite minerals, Clays Clay Miner., 2016, 64(4), 472–487 CrossRef CAS.
- X. Liu, X. Lu, M. Sprik, J. Cheng, E. J. Meijer and R. Wang, Acidity of edge surface sites of montmorillonite and kaolinite, Geochim. Cosmochim. Acta, 2013, 117, 180–190 CrossRef CAS.
- E. Tertre, S. Castet, G. Berger, M. Loubet and E. Giffaut, Surface chemistry of kaolinite and Na-montmorillonite in aqueous electrolyte solutions at 25 and 60 °C: experimental and modeling study, Geochim. Cosmochim. Acta, 2006, 70(18), 4579–4599 CrossRef CAS.
| This journal is © the Owner Societies 2026 |