Combining classical reactive scattering and the time–energy uncertainty relation
Received
12th November 2025
, Accepted 15th January 2026
First published on 10th February 2026
Abstract
Gaussian binning is a method for analyzing the results of classical dynamical simulations of gas-phase and gas-surface reactions that has been used since the early 2000s in order to improve predictions of state-resolved cross sections and related quantities measured in molecular beam experiments. In this method, classical trajectories are assigned Gaussian statistical weights, with much higher weights given to product energies closer to their quantized values. Here, we extend this idea to the activated complex in two ways: (i) treating it as if it were in a stationary state, just like the products, which requires as narrow a Gaussian as possible, and (ii) considering the activated complex as being in a time-dependent state, broadening the Gaussian so that the statistical properties of the activated complex are consistent with the time–energy uncertainty relation. The latter approach, which builds on a recent semiclassical study of chemical reaction thresholds [L. Bonnet, C. Crespos and M. Monnerville, J. Chem. Phys., 2022, 157, 094114], is coupled with simple calculations of tunneling through adiabatic barriers in the parabolic limit. The resulting methods are then used to calculate the reaction probability for two model processes involving, respectively, long-lived and short-lived activated complexes. The agreement with quantum probabilities appears to be very good. Based on these results, the shapes of quantum probabilities are analysed from the classical dynamics and the time–energy uncertainty relation. The question of zero-point energy violation at the transition state is examined in light of the preceding results.
1 Introduction
Molecular beam experiments provide highly accurate data on the dynamics of chemical reactions and inelastic collisions.1 These data are mainly the state-resolved integral and differential cross sections for gas-phase reactions, and related quantities for gas-surface processes. The most accurate way to reproduce them theoretically is to perform exact quantum scattering calculations.2 More often than not, however, such calculations are unfeasible for gas-phase reactions involving more than three atoms, and for all gas-surface reactions if the surface atoms are allowed to move. Therefore, most polyatomic processes are studied theoretically by means of the quasi-classical trajectory (QCT) method, whose applicability is not limited by the number of atoms.3–8
The QCT method leads to satisfactory predictions for complex-forming reactions if enough quantum states (more than 3 or 4) are available in the separated fragments. QCT calculations are also accurate for barrier reactions if the previous state number constraint is satisfied not only in the separated fragments, but also in the activated complex (AC), defined here as the region around the transition state (TS) in which the vibrational motion orthogonal to the reaction path evolves adiabatically (see Section 2 for more details). Otherwise, the dynamics have a truly quantum nature and QCT predictions may not be realistic. This is typically the case of endothermic barrierless reactions occurring at total energies below the product zero point energy (ZPE). Such processes are quantum mechanically prohibited, but classically allowed.9 This also applies to barrier reactions occurring at total energies below the zero point energy of the activated complex (or TS).10,11 Dissociative chemisorption is another example for which the QCT method may fail. If a molecule in the internal ground state hits a surface with very low kinetic energy and some trajectories corresponding to this event are reflected back into the vacuum, it may happen that most of the reformed reagents have a vibrational energy lower than the ZPE. These unphysical paths artificially enhance the reflection at the expense of the dissociative chemisorption, thus, making the sticking probability too low compared to its quantum value.12
Gaussian binning (GB) is a method of analysing the trajectory outcomes that significantly improves the QCT predictions when the state number constraint is not satisfied in the separated fragments.13 GB is based on the idea, justified by classical S-matrix theory (CSMT),14 that only those trajectories complying with Bohr's quantization condition in the separated fragments should be taken into account in the prediction of the measurements.15,16 Such trajectories start from integer actions of the reagents and end with integer actions of the products (see Chapter 6.2 of ref. 17 for a clear definition of the concept of action; actions are vibrational and rotational for a gas-phase reaction, and vibrational, rotational and diffractional12 for a molecule reflected by a surface). In order to ensure that Bohr's quantization conditions are satisfied, trajectories are launched from the reagents with integer actions (like in the standard QCT method) and should in principle be assigned Dirac delta weights, infinite if the product actions have integer values, zero otherwise.18 Clearly, this approach is intractable as such. Therefore, in practice, trajectories are assigned Gaussian weights normalized to unity such that the closer product actions are to integer values, the higher the statistical weight of the trajectories.12,13,15,19–21 Moreover, the full-width-at-half-maximum of the Gaussians, simply called width from now on, are taken at values much smaller than the unit spacing between two neighboring states, as they are supposed to mimic Dirac deltas. For polyatomic reactions, however, it is more efficient to make Gaussian weights depend on the vibrational energy rather than the actions, as this choice scales much better with dimensionality,22–24 in particular for describing threshold effects.
So far, GB has never been applied to the activated complex. Yet, its states are known to be quantized to some extent, particularly near the reaction threshold.11,25,26 Therefore, one may wonder whether trajectories should be assigned Gaussian weights centered at integer values of the vibrational actions of the activated complex (or at its quantized energies). The only difference with the product states is that the quantized states of the activated complex25 are non-stationary states with an energy width and a lifetime, as shown by the semiclassical developments in Section IV.B.4. of ref. 11. Consequently, Gaussian widths should not be taken as small as possible, like for products whose lifetime is infinite and the quantized energies perfectly defined. Instead, they should be chosen so as to comply with the time–energy uncertainty relation, as suggested by the results in ref. 11. The aim of this study is to show how this can be done in practice within the framework of the Schatz model of H + H2 reaction that mimics the passage from the free rotation of reactants, through bending vibration at the transition state, to the free rotation of products.10 As a preliminary step, however, we consider a recent model reaction, reminiscent of going through a canyon,11 for which the lifetime of the activated complex is long enough for it to be considered as being in a stationary state, and therefore assigned a standard Gaussian weight.
2 Standard Gaussian binning of the activated complex
2.1 Canyon crossing
A detailed study of the quantum, semiclassical and classical dynamics of the present model reaction has recently been published.11 For this reason, we limit ourselves to a brief review of the details that are important for this study.
2.1.1 System and dynamics.
We consider a fixed-plane of the laboratory and a rigid diatom rotating within this plane. Moreover, we force the center-of-mass of the diatom to move on a given R-axis of the plane. The total energy of the rotator in translation is E. The orientation of the diatom with respect to R is determined by the angle ϕ. The classical Hamiltonian of the system is| | 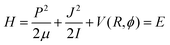 | (1) |
where 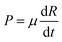 and
and 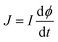 are the momenta conjugate to R and ϕ, respectively, t is the time, μ is the mass associated with motion along the R-axis, I is the moment of inertia of the diatom and V(R,ϕ) is the potential energy. Two values of μ will be considered, 19 amu and 1.9 amu, corresponding to atom–diatom reactions of the heavy plus light-heavy and light plus heavy-light type, respectively. I is equal to mre2 with m = 0.5 amu and re = 0.7414 Å. Our model potential energy surface (PES) is defined by
are the momenta conjugate to R and ϕ, respectively, t is the time, μ is the mass associated with motion along the R-axis, I is the moment of inertia of the diatom and V(R,ϕ) is the potential energy. Two values of μ will be considered, 19 amu and 1.9 amu, corresponding to atom–diatom reactions of the heavy plus light-heavy and light plus heavy-light type, respectively. I is equal to mre2 with m = 0.5 amu and re = 0.7414 Å. Our model potential energy surface (PES) is defined by| | 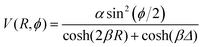 | (2) |
with α = 4.053 eV, Δ = 2 Å and β = 3 Å−1. Its contour level representation is shown in Fig. 1. The right column gives the correspondence between colors and potential energy in eV. The system parameters and the analytical expression of the PES can be found in ref. 11. The PES involves a narrow channel separating the reagent and product planes on the left and right, respectively. An example of reactive trajectory starting from the reagent plane with ϕ = ϕ1 and no rotational excitation is represented by the yellow line. We will indeed assume in the following that the initial diatom is in the rotational ground-state, with a random orientation (−π ≤ ϕ1 ≤ π). The trajectory is started at R1 = −3 Å with 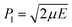 and stopped at R2 = 3 Å (see Section III.A.2 of ref. 11 for more details on the trajectory calculations). Within the channel, there is a periodic orbit (PO) trapped along the ϕ-axis (see the white vertical segment centered at the origin of the (R, ϕ) frame). According to classical mechanics, the transition state (TS) is defined as the phase space surface whose projection on the (R, ϕ) configuration plane coincides with the representation of the PO in the same plane.27 The TS is thus trivially defined in (R, ϕ, P, J) by R = 0. The reactive flux is minimum through the TS which, thus, corresponds to a phase space bottleneck. The reactive trajectory clearly mimics the passage from the free rotation of reactants, through bending vibration at the transition state, to the free rotation of products that occurs in three-dimensional bimolecular reactions. Four white vertical dashed lines define five regions labeled RA, NA and VA above the frame of Fig. 1. The dynamics are rotationally adiabatic in the RA reagent and product half planes (the PES is flat in these domains so that the rotation is free), vibrationally adiabatic in the VA central region encompassing the TS (see further below for a numerical proof of this statement, which is quite obvious from the shape of the trajectory in the VA band), and nonadiabatic in the NA regions due to the presence of strong couplings between the R and ϕ coordinates. We define the activated complex as the system orthogonal to the R-axis in the VA region, whose boundaries are R ≈ −0.75 and 0.75, in Å (see Fig. 1). The activated complex has a lifetime, contrary to the TS which is just crossed by classical trajectories.
and stopped at R2 = 3 Å (see Section III.A.2 of ref. 11 for more details on the trajectory calculations). Within the channel, there is a periodic orbit (PO) trapped along the ϕ-axis (see the white vertical segment centered at the origin of the (R, ϕ) frame). According to classical mechanics, the transition state (TS) is defined as the phase space surface whose projection on the (R, ϕ) configuration plane coincides with the representation of the PO in the same plane.27 The TS is thus trivially defined in (R, ϕ, P, J) by R = 0. The reactive flux is minimum through the TS which, thus, corresponds to a phase space bottleneck. The reactive trajectory clearly mimics the passage from the free rotation of reactants, through bending vibration at the transition state, to the free rotation of products that occurs in three-dimensional bimolecular reactions. Four white vertical dashed lines define five regions labeled RA, NA and VA above the frame of Fig. 1. The dynamics are rotationally adiabatic in the RA reagent and product half planes (the PES is flat in these domains so that the rotation is free), vibrationally adiabatic in the VA central region encompassing the TS (see further below for a numerical proof of this statement, which is quite obvious from the shape of the trajectory in the VA band), and nonadiabatic in the NA regions due to the presence of strong couplings between the R and ϕ coordinates. We define the activated complex as the system orthogonal to the R-axis in the VA region, whose boundaries are R ≈ −0.75 and 0.75, in Å (see Fig. 1). The activated complex has a lifetime, contrary to the TS which is just crossed by classical trajectories.
 |
| | Fig. 1 Potential energy surface of the canyon-crossing model and a typical classical reactive path. See the text for details. | |
Although the TS is a classical concept, it can nevertheless be treated “artificially” in a quantum framework. For our process, the quantum Hamiltonian of the TS is  (
( where h is Planck's constant) and its spectrum can be obtained by diagonalization methods (see ref. 28 and references therein). Alternately, it can be semiclassically estimated as follows. Calling
where h is Planck's constant) and its spectrum can be obtained by diagonalization methods (see ref. 28 and references therein). Alternately, it can be semiclassically estimated as follows. Calling  the energy of the TS, its vibrational action over one cycle is given by
the energy of the TS, its vibrational action over one cycle is given by
| |  | (3) |
with
| |  | (4) |
(see Chapter 6.2 of ref. 17). In the harmonic limit, valid for our system in the energy range that we will be considering, V(0,ϕ) is equal to  and eqn (3) and (4) lead to
and eqn (3) and (4) lead to
| |  | (5) |
This illustrates clearly that N‡ is the classical analog of the vibrational quantum number. The quantized energies E‡n of the TS, where n is a positive integer or 0, are given by eqn (5) with N‡ replaced by n (the latter cannot be too large, because the harmonic approximation of V(0,ϕ) is valid for moderate values of |ϕ|). E‡0 is equal to 705 cm−1 (2E‡0 typically corresponds to a bending vibration quantum) and E‡n = (2n + 1)E‡0.
The vibrational action can also be calculated along the trajectories inside the channel. It is still given by eqn (3) and (4), except that in the latter, E‡ and V(0,ϕ) are replaced by [E − P2/(2μ)] and V(R,ϕ), respectively. This action, now called N, is represented in terms of R in Fig. 2 for four randomly selected reactive trajectories at the energy H = E = 1.5E‡0 (at this energy, the system is bounded along the ϕ-axis for |R| < 1.5, so N is given by eqn (3) and (4); for larger values of |R|, the rotational motion is hindered and the expression of the action along the ϕ-axis is different). As a matter of fact, N does not vary significantly between R ≈ −0.75 and R ≈ 0.75 compared to the unit spacing between two vibrational states. In other words, the nature of the vibrational motion does not change within the previous boundaries, i.e., the dynamics are vibrationally adiabatic within the VA band (see Fig. 1). In contrast, Fig. 2 shows that N fluctuates significantly within the NA bands, due to strong couplings between the R and ϕ coordinates. We will call N‡(ϕ1) the value of N at the TS for the reactive trajectory starting from ϕ1.
 |
| | Fig. 2 Variation, for the canyon crossing model, of the vibrational action N along the reaction coordinate R, given in Å, for some reactive trajectories at E = 1.5E‡0. | |
2.1.2 Reaction probabilities.
The quantum mechanical (QM) and QCT reaction probabilities are represented in Fig. 3 for the heavy and light masses (details of the QM and QCT calculations can be found, respectively, in Sections III.A.1 and III.A.2 of ref. 11; see also ref. 29). Within the energy range considered, the quantum probabilities (solid lines) involve two thresholds which coincide with the quantized TS energies E‡0 and E‡2, respectively. It is known from the research by Truhlar and co-workers25 that these energies are associated with quantized states of the activated complex that ensure the passage of reactive flux as soon as they are accessible. These states are discussed further below within the framework of semiclassical reactive scattering theory. We note that there is no threshold at E‡1, for the following reason: the initial rotational state, given by the constant value  , is even. Moreover, the ϕ-dependence of the potential energy is also even. Therefore, the E-resolved scattering state is necessarily symmetric with respect to the R-axis. Consequently, the coefficients of its development on the vibrational states Ψn(ϕ) of the TS are necessarily 0 for odd values of n (for these states are antisymmetric). The QCT probabilities (dashed lines in Fig. 3) are in poor agreement with QM ones. This is especially true around the thresholds, which classical mechanics obviously ignores. Nevertheless, we see that the disagreement decreases when the total energy increases, as it should be. In particular, QM probabilities perform damped “oscillations” around QCT probabilities in such a way that both are nearly equal at the middle of the jumps for the threshold associated with the TS excited states n ≥ 2 (only the one corresponding to n = 2 can be seen in Fig. 3). We will take advantage of this observation later.
, is even. Moreover, the ϕ-dependence of the potential energy is also even. Therefore, the E-resolved scattering state is necessarily symmetric with respect to the R-axis. Consequently, the coefficients of its development on the vibrational states Ψn(ϕ) of the TS are necessarily 0 for odd values of n (for these states are antisymmetric). The QCT probabilities (dashed lines in Fig. 3) are in poor agreement with QM ones. This is especially true around the thresholds, which classical mechanics obviously ignores. Nevertheless, we see that the disagreement decreases when the total energy increases, as it should be. In particular, QM probabilities perform damped “oscillations” around QCT probabilities in such a way that both are nearly equal at the middle of the jumps for the threshold associated with the TS excited states n ≥ 2 (only the one corresponding to n = 2 can be seen in Fig. 3). We will take advantage of this observation later.
 |
| | Fig. 3 QM and QCT reaction probabilities for the heavy and light masses in terms of the collision energy E; QM: solid lines, QCT: dashed lines. | |
We now consider the GB procedure applied to the activated complex. We found that at E = 1.5E‡0, the ratio of the average lifetime of the activated complex (time to cross the VA band) and its vibrational period is roughly equal to 12 and 4 for the heavy and light masses, respectively. Therefore, the activated complex survives for a pretty long time, making it reasonable to consider it in a stationary state. Consequently, classical trajectories may be assigned Gaussian weights centered at integer values of the bending vibrational action at the TS. The GB-QCT reaction probability (also called the GB probability in the following) is then formally given by16
| |  | (6) |
with
| |  | (7) |
[note that
G(
x) = 2
δ(
x) in the limit where
ε tends toward 0]. Reactive trajectories start from (
R1,
ϕ1) with
ϕ1 within the interval [0,
ϕ>] (we limit ourselves to positive angles since trajectories are initially run parallel to the
R-axis (
J = 0) and the potential is symmetric with respect to the latter).
ϕ> is the maximum angle leading to reaction at
E (between
ϕ> and π, trajectories are non-reactive).
kmax is the maximum value of
k consistent with
E, and
ε should be taken at a value much lower than 1. Here,
ε is taken at 0.06, its usual value,
13 except in the vicinity of the thresholds where it needs to be significantly reduced for better accuracy—right at a threshold, however, the Gaussian, in addition to be very narrow, must also be centered at a value slightly smaller than the integer action corresponding to the threshold, so it can fully overlap a range of available vibrational actions; if the Gaussian is centered exactly at an integer action, it covers only half of the available action range, and the GB probability therefore amounts to only half of its actual value. A few hundred (thousands) trajectories are far (near) enough from thresholds. Note the presence of a factor of 2 in the numerator of the Gaussian weight (
eqn (7)) in order to compensate for the fact that only even states of the activated complex need be taken into account. A similar doubling is used in the QCT approach of homonuclear diatom scattering with a third body; the parity of the rotational state of the diatom is indeed conserved throughout the collision. An illuminating semiclassical analysis of this question can be found in ref.
30. QM and GB-QCT reaction probabilities are compared in
Fig. 4. The agreement between both approaches is quantitatively satisfactory, but qualitatively excellent. As a matter of fact, Gaussian binning the vibrational action of the activated complex considerably improves the shape of the reaction probability (compare the dashed curves in
Fig. 3 and 4). We note in the QM curve for the light mass a slight smoothing of the second step, due to tunneling through the adiabatic barrier
11 of state
n = 2. Such smoothing is obviously absent from the GB curve which assumes that the activated complex is in a perfectly stationary state.
 |
| | Fig. 4 QM and GB-QCT reaction probabilities for the heavy and light masses in terms of the collision energy E; QM: solid lines, GB-QCT: dashed lines. | |
2.1.3 Shape of the quantum probability.
We now use eqn (6) and (7) to explain the shape of the quantum probability. In the limit where ε tends toward 0, these equations become| |  | (8) |
Let us consider the large value of μ and two energies Ea and Eb slightly larger and slightly lower than E‡0 and E‡2, respectively. For both Ea and Eb, the TS vibrational energy is necessarily lower than E‡2. Now, the latter corresponds to N‡(ϕ1) = 2 (see eqn (5) for the harmonic case). As a consequence, N‡(ϕ1) is strictly lower than 2 for both Ea and Eb, and the sum in eqn (8) reduces to δ(N‡(ϕ1)). N‡(ϕ1) is shown in Fig. 5 for Ea = 1.1E‡0 and Eb = E‡2 − 0.1E‡0. For ϕ1 = 0, the trajectories follow the R-axis and N‡(ϕ1) takes the minimum value of −1/2 [see eqn (3)]. Above ϕ1 = 0, the larger ϕ1, the larger the amplitude of the vibrational motion within the channel (see Fig. 1), and the larger N‡(ϕ1). The maximum angle ϕ> leading to reaction at Ea is labelled ϕa> (see the right end of the blue solid curve; note for the following that if one decreases the energy from Ea to E‡0, the right end of blue solid curve ends up touching the dashed green line). For each energy, only one value of ϕ1 leads to N‡(ϕ1) = 0. These values, called ϕa1 and ϕb1, respectively (see Fig. 5), lead to the blue and red trajectories represented in Fig. 6. Using the fact that for Ea,
| |  | (9) |
 |
| | Fig. 5
ϕ
1-dependence of the vibrational action at the transition state for Ea = 1.1E‡0 (blue solid curve) and Eb = E‡2 − 0.1E‡0 (red solid curve). The highest action available at Ea, given by the height of the dashed blue line, is reached at the angle ϕa>. A dotted black straight line is represented within the action range [−0.3, 0.3], showing that the red line is nearly linear within this range. A Gaussian weight centered on the dashed green line, i.e. at 0, is represented in magenta. The action is 0 at ϕa1 and ϕb1 for Ea and Eb, respectively. | |
 |
| | Fig. 6 Trajectories leading to N‡(ϕ1) = 0 at Ea (blue path), Eb (red path) and E = +∞ (green path). | |
(see eqn (1.158) in ref. 31), the value Pa of P at Ea, deduced from eqn (8), is
| |  | (10) |
The value Pb of P at Eb is given by the same expression with ϕb1 substituted for ϕa1. Fig. 6 shows that the potential at the entrance of the interaction region acts as a converging lens that focuses the reactive trajectories. We note, however, that the trajectory is more deviated for the lowest energy (blue curve) than for the highest (red curve). This is logical since for an infinite energy, the trajectory leading to N‡(ϕ1) = 0 is the straight line defined by ϕ∞1 (green curve), not deviated at all. Therefore, ϕa1 is larger than ϕb1, so the slope of N‡(ϕ1) is stronger for Eb than for Ea (see Fig. 5). As a consequence, the reaction probability, proportional to the inverse of the slope (see eqn (10)), decreases with E. However, it approaches a plateau at high energies for which all the trajectories converge towards the green trajectory. This justifies the shapes of each TS vibrational state contribution to the reaction probability in Fig. 4.
A comment on the value of ε in eqn (7) is in order. It can be shown that the integral of G(f(ϕ1)) does not depend on ε if f(ϕ1) is linear (the proof is straightforward). Now, at Eb, N‡(ϕ1) is relatively close to linearity within the range [−0.3, 0.3] (see the solid red curve and dotted black straight line in Fig. 5). The width of G(N‡(ϕ1)) is equal to  . The value of ε corresponding to the width of the previous range is ∼0.36. Any value of ε lower than 0.36 is thus expected to make P, as obtained from eqn (6) and (7), very close to Pb. Moreover, numerical calculations show that even for ε ∼ 0.6, corresponding to the unit width (i.e., the classical limit32), P remains relatively close to Pb. On the other hand, the situation is different at Ea. The maximum vibrational action N‡(ϕa>) is slightly above 0 (see the right end of the blue curve in Fig. 5). Therefore, G(N‡(ϕ1)) must be very narrow for eqn (6) and (7) to agree with eqn (10). Otherwise, G(N‡(ϕ1)) will be non 0 for all the actions greater than N‡(ϕa>), which are not available, so eqn (6) and (7) will significantly minimize the reaction probability. This would for instance be the case with the magenta Gaussian in Fig. 5 which partly overlaps the region of unavailable actions (above the horizontal dashed blue line). We will come back to this point in Section 3.3.
. The value of ε corresponding to the width of the previous range is ∼0.36. Any value of ε lower than 0.36 is thus expected to make P, as obtained from eqn (6) and (7), very close to Pb. Moreover, numerical calculations show that even for ε ∼ 0.6, corresponding to the unit width (i.e., the classical limit32), P remains relatively close to Pb. On the other hand, the situation is different at Ea. The maximum vibrational action N‡(ϕa>) is slightly above 0 (see the right end of the blue curve in Fig. 5). Therefore, G(N‡(ϕ1)) must be very narrow for eqn (6) and (7) to agree with eqn (10). Otherwise, G(N‡(ϕ1)) will be non 0 for all the actions greater than N‡(ϕa>), which are not available, so eqn (6) and (7) will significantly minimize the reaction probability. This would for instance be the case with the magenta Gaussian in Fig. 5 which partly overlaps the region of unavailable actions (above the horizontal dashed blue line). We will come back to this point in Section 3.3.
2.2 Schatz model of the H + H2 reaction
The Hamiltonian is the same as previously (see eqn (1)), with μ = 1224.7 a.u. and I = 3600 a.u. The analytical expression of Schatz PES is| |  | (11) |
with B = 1.63 eV and D = a0/2.10 The system parameters are roughly appropriate to the H + H2 reaction. Its contour level representation is shown in Fig. 7. Compared to the canyon-crossing model (Fig. 1), the interaction region is narrow. The spatial extension of the activated complex is thus necessarily shorter than previously, as well as its average lifetime. Fig. 8 shows the variation of the vibrational action N along the R-axis for four sets of a dozen trajectories having the following properties: the blue, red, green and orange trajectories are run at E = 3E‡0/2, E‡2 + E‡0/2, E‡4 + E‡0/2 and E‡6 + E‡0/2, respectively. Moreover, they cross the TS with N‡ close to 0, 2, 4 and 6, respectively, thus implying that semiclassically, they strongly contribute to the reactivity (see Section IV.B.3 of ref. 11 and Section 3.1 in the present work). The rectangles delimit the VA zones, where N is nearly constant. The length of these zones, indicated above each rectangle, is obtained from the trajectories that cross the transition state with N equal to an integer. This length, hereafter denoted L, appears to decrease with N. This is because the couplings between the R and ϕ coordinates near the ϕ-axis become stronger as ϕ increases, and the larger the value of N, the greater the range of ϕ that can be accessed. Interestingly, the zones are not necessarily centered exactly at R = 0.
 |
| | Fig. 7 Potential energy surface of the model H + H2 reaction. ϕ is limited to the range [−π/2, π/2]. | |
 |
| | Fig. 8 Variation, for the model H + H2 reaction, of the vibrational action N along the reaction coordinate R, given in Å, for some reactive trajectories slightly above the threshold energies E‡0, E‡2, E‡4 and E‡6. Actions are represented by dots every 0.1 fs. Black rectangles locate the activated complex, or the VA region, where actions do not vary. The lengths of these regions are indicated above the rectangles. | |
The calculations show that the ratio of the lifetime of the activated complex and its vibrational period is less to ∼1/3. Consequently, unlike the canyon case, the activated complex survives for a negligible amount of time. As a result, we cannot consider it in a stationary state and use eqn (6) and (7) with ε ∼ 0. If we do so, we obtain the dashed blue curve in Fig. 9 (GB results), which shows poor agreement with the solid green curve (QM results), in contrast with the canyon case (see Fig. 4). Therefore, we need to go beyond standard GB.
 |
| | Fig. 9 Energy dependence of the reaction probability for the Schatz model of the H + H2 reaction obtained by means of three approaches: QM calculations (solid green), the standard QCT method (solid red), and the GB-QCT method (dashed blue). | |
3 Uncertainty-based Gaussian binning of the activated complex
We base our new method on the semiclassical analysis of chemical reaction thresholds published in ref. 11. We first review some key results of this study and then present the method.
3.1 Quantization of the activated complex from the point of view of classical S-matrix theory
In ref. 11, we applied CSMT to the canyon-crossing model in order to calculate the probability P00 that reactants in the rotational ground state yield products in the same state. P00 is equal to |S00|2 where the S-matrix element S00 is approximated by the CSMT expression| |  | (12) |
The terms of the sum are the probability amplitudes assigned to the infinite number of real-time trajectories starting from the reagent plane with J = 0 and reaching the product plane with J = 0 (see the blue line in Fig. 3 of ref. 11, where the final value of J is referred to as J2 and the initial angle ϕk1 of the kth trajectory leading to J2 = 0 is referred to as ![[small phi, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d6.gif) k1). In eqn (12), the modulus of each amplitude is the square root of the classical statistical weight pclk of the corresponding trajectory,33 while its phase Ωk/ħ is the action integral in the momentum space along the trajectory, expressed in units of ħ (this phase includes the so-called Maslov index, which plays a crucial role in explaining the origin of quantum reaction thresholds11,18). Quantum mechanics comes into play in eqn (12)via the sum of complex numbers, which is the mathematical expression of the superposition principle, and via ħ, in the denominator of the phases. In particular, the sum creates interferences between probability amplitudes when taking the squared modulus of S00 (we will simply say that trajectories “interfere”).
k1). In eqn (12), the modulus of each amplitude is the square root of the classical statistical weight pclk of the corresponding trajectory,33 while its phase Ωk/ħ is the action integral in the momentum space along the trajectory, expressed in units of ħ (this phase includes the so-called Maslov index, which plays a crucial role in explaining the origin of quantum reaction thresholds11,18). Quantum mechanics comes into play in eqn (12)via the sum of complex numbers, which is the mathematical expression of the superposition principle, and via ħ, in the denominator of the phases. In particular, the sum creates interferences between probability amplitudes when taking the squared modulus of S00 (we will simply say that trajectories “interfere”).
We found that for E = 1.2E‡0, for example, the phase Ωk/ħ is stationary for a given value k0 of k, and the Ωk's are close to Ωk0—with respect to ħ—for a range of values [k0 − K, k0 + K] where K is of the order of ∼10. For these values, the probability amplitudes are nearly aligned in the complex plane, so they interfere constructively. Outside this range, however, Ωk/ħ increases rapidly with |k − k0| in such a way that the corresponding trajectories interfere destructively. Consequently, only ∼2 K trajectories actually contribute to P00 (a few tens at E = 1.2E‡0, more if E is closer to E‡0; details are given in ref. 11, in particular in Section IV.B.3). For each of these trajectories—hereafter referred to as dominant paths (the blue trajectories in Section IV.B of ref. 11)—we calculated the vibrational energy at the TS, E‡v = J2/(2I) + V(0,ϕ), and the lifetime T‡ of the activated complex, given by
| |  | (13) |
The denominator of this expression is the velocity along the R-axis within the VA band. The dependence of L on N was extrapolated from the 4 lengths indicated in Fig. 8.
We then calculated the standard deviations ΔE‡v and ΔT‡ of E‡v and T‡ for the dominant paths (see eqn (61)–(64) in Section IV.B.4 of ref. 11). A first remarkable result of these calculations is that E‡v is distributed around E‡0 in such a way that its mean value Ē‡v is very close to E‡0 (property I). This also corresponds to N‡ distributed about 0, with mean value ![[N with combining macron]](https://www.rsc.org/images/entities/i_char_004e_0304.gif) ‡ ∼ 0. A second remarkable result is that the product ΔE‡vΔT‡ is distributed around ħ/2 so that it roughly satisfies the time-energy uncertainty relation (property II).34 In other words, the activated complex is in a time-dependent quantum state whose average energy is E‡0. This corroborates the findings of Truhlar and co-workers.25 The previous observations are valid whatever E within the range [E‡0, E‡2]. This is also the case for E within the range [E‡2, E‡4], except that a few tens of trajectories cross the TS with E‡v ∼ E‡0, and a few other tens with E‡v ∼ E‡2. A similar scenario is valid for the following energy intervals, with more states of the activated complex involved in the reaction. The trajectories represented in Fig. 8 in the (R, N) plane are dominant paths.
‡ ∼ 0. A second remarkable result is that the product ΔE‡vΔT‡ is distributed around ħ/2 so that it roughly satisfies the time-energy uncertainty relation (property II).34 In other words, the activated complex is in a time-dependent quantum state whose average energy is E‡0. This corroborates the findings of Truhlar and co-workers.25 The previous observations are valid whatever E within the range [E‡0, E‡2]. This is also the case for E within the range [E‡2, E‡4], except that a few tens of trajectories cross the TS with E‡v ∼ E‡0, and a few other tens with E‡v ∼ E‡2. A similar scenario is valid for the following energy intervals, with more states of the activated complex involved in the reaction. The trajectories represented in Fig. 8 in the (R, N) plane are dominant paths.
Below E‡0, the situation is similar to that within the range [E‡0, E‡2], except that the trajectories contributing to P00 must be integrated in imaginary time (see Section IV.B.3 in ref. 11). This allows them to tunnel through the adiabatic ground state barrier with a negative kinetic energy P2/(2μ) and a vibrational energy E‡v greater than E, also distributed around E‡0 and equal on average to E‡0. Furthermore, the product ΔE‡vΔT‡, where T‡ is now the modulus of the imaginary time, is again close to ħ/2. Similar findings are found for tunneling through adiabatic excited state barriers.
Since the semiclassical reaction probability is mainly determined by the dominant paths, one may wonder whether the QCT probability computed using only these paths could be more accurate than the standard QCT probability. However, the rigorous identification of the dominant paths (DP) is difficult for full-dimensional reactions. Consequently, in the following section we propose an approximate identification based on GB in order to estimate the DP-based QCT probability.
3.2 Method and results
For simplicity's sake, we only consider trajectories in real time, as in general, running trajectories in imaginary time poses real problems. We first assume that the total energy is slightly above the first threshold, E‡0, so the only available state of the activated complex is the ground one. Ntraj trajectories are run parallel to the R-axis (J = 0) from R1 = −3 Å and regularly spaced values of ϕ1. Nreac trajectories are reactive. For the kth reactive trajectory, the vibrational action and energy at the TS as well as the activated complex lifetime are denoted by N‡k, E‡vk and T‡k, respectively [the kth trajectory here has no connection with the kth trajectory in eqn (12)].
We have seen previously that the dominant paths cross the TS with N‡ distributed around 0 and ‡ ∼ 0. We now assume that, if reactive trajectories are assigned the weight G(N‡) given by eqn (7) and if ε is varied to satisfy both properties I and II, the resulting probability should be in reasonably good agreement with the DP-based QCT probability, and hopefully with the quantum one. To check the validity of this assumption, we proceed as follows.
We first set ε at a very small value, say, 0.001. G(N‡) is thus an extremely narrow Gaussian centered at 0 and it is obvious that property I is satisfied, i.e., Ē‡v = E‡0. On the other hand, there is no dispersion in the vibrational actions and energies and, consequently, no dispersion in the lifetimes [see eqn (13)]. Therefore, both ΔE‡v and ΔT‡ tend toward 0, and property II is not satisfied. We then increase ε by a small amount, so that the Gaussian becomes slightly wider; the dispersion of both the vibrational energies and the lifetimes increases slightly, and ΔE‡vΔT‡ increases accordingly. We repeat this iterative calculation until ΔE‡vΔT‡ exceeds ħ/2—we checked that property I is still satisfied—and use the value of ε corresponding to the last iteration to calculate the reaction probability according to
| |  | (14) |
with
G‡k =
G(
N‡k). The subscript on the left-hand-side of
eqn (14) will remind us that its right-hand-side is the expression used above
E‡0. For an energy larger than
E‡2 but lower than
E‡4, the same approach is applied twice, once for the ground state of the activated complex, as before, and once for its second excited state, with GB weights
G‡k =
G(
N‡k − 2). For the next interval, three states of the activated complex are considered, and so on. We will call UGB this uncertainty-based GB procedure. We wish to emphasize that during the iterative calculation,
ε must not exceed the classical limit of 0.6.
32
Now, what about the reaction probability P< below the minimum threshold E‡0? As previously stated, we wish to avoid running trajectories in imaginary time, as this leads to tricky calculations. However, we have recently demonstrated by means of the saddle point method that, in the case of a simplified canyon-crossing model with analytically tractable dynamics, the reaction probability obtained from eqn (12) is proportional to the probability of tunneling through the adiabatic ground state barrier (see eqn (60) in ref. 11). We now approximate this probability as follows. The second order development in ϕ of the right-hand-side of eqn (11) can be written as Iω(R)2ϕ2/2 with  . The adiabatic ground state barrier E0(R) is given by the sum of the ZPE at R and the potential energy along the reaction path. The latter being 0, we have E0(R) = ħω(R)/2 = E‡0/cosh(R/D) with
. The adiabatic ground state barrier E0(R) is given by the sum of the ZPE at R and the potential energy along the reaction path. The latter being 0, we have E0(R) = ħω(R)/2 = E‡0/cosh(R/D) with  . If we approximate E0(R) with a parabolic barrier, we obtain a very satisfying approximation of the tunneling probability immediately below E‡0 from35
. If we approximate E0(R) with a parabolic barrier, we obtain a very satisfying approximation of the tunneling probability immediately below E‡0 from35
| |  | (15) |
with
| | 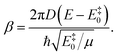 | (16) |
Calling P>(E‡0) the value of P> at E‡0, and taking into account the fact that (i) P< is proportional to Ptun and (ii) P> and P< must have the same value at E‡0, we find
Eqn (14) with ΔE‡vΔT‡ = ħ/2 and eqn (17) lead to the dashed curve in Fig. 10. It appears that the slope is discontinuous at E‡0, which constitutes a non-physical result. However, we found that this issue disappears when ΔE‡vΔT‡ is reduced to ∼ħ/8. Should we be concerned about choosing ħ/8 instead of ħ/2? We do not believe so, for the following reason: while the reaction probabilities obtained from both CSMT and UGB are based on roughly the same set of trajectories, the dominant paths that satisfy properties I and II, the calculations leading to these probabilities differ fundamentally in nature. Indeed, the former accounts for constructive interference between trajectories as well as quantization of the final rotational motion, whereas the latter involves a straightforward summation of UGB weights and no quantization of the final rotational motion. Thus, only a qualitative agreement between the two probabilities can be expected from the standard uncertainty relation ΔE‡vΔT‡ = ħ/2. It was thus tempting to tune the degree of uncertainty by varying α in therelation ΔE‡vΔT‡ = αħ/2 so as to ensure the continuity of the slope of P< and P> at E‡0 and make the reaction probability more realistic in this respect. This is what we did and this led to the solid curve in Fig. 10, corresponding to α = 1/4. We then repeated the same calculations for all the excited states of the activated complex available at E. The sum of the resulting probabilities, along with that of the ground state (solid curve in Fig. 10), is plotted in magenta in Fig. 11. As in the case of the long-lived activated complex (Fig. 4), the qualitative agreement with the quantum probability is pleasing. In particular, both the period of the oscillations and the decay of their amplitudes with energy are well reproduced (Fig. 11). Moreover, the quantitative agreement is also satisfactory.
 |
| | Fig. 10 Energy dependence of the reaction probability around E‡0 for the Schatz model of the H + H2 reaction obtained from a treatment combining, first, the UGB-QCT method [green curves corresponding to P>, given by eqn (14)] and, second, a simple calculation of tunneling through the adiabatic ground state barrier in the parabolic limit [blue curves corresponding to P<, given by eqn (17)]. Dashed and solid curves correspond to ΔE‡vΔT‡ taken at ħ/2 and ħ/8, respectively. Only the second value makes the slope of the reaction probability continuous at E‡0. | |
 |
| | Fig. 11 Energy dependence of the reaction probability for the Schatz model of the H + H2 reaction obtained by means of QM calculations (green curve), and the UGB-QCT + tunneling method (magenta curve). | |
To explain the structure of the semiclassical probability, and thereby gain insight into that of the quantum probability, we consider in Fig. 12 the TS ground state contribution to the UGB, tunneling, and GB probabilities, as well as the value of the Gaussian width (W) after the last iteration of the UGB procedure. W increases from 0.27, at E‡0, to 1 (the classical limit) at 0.21 eV (vertical dashed line), beyond which it plateaus. A width of 0.27 is small enough to consider that the activated complex is quantized. Nevertheless, the UGB probability at E‡0 is much smaller than the GB one which, yet, results from “exact” quantization (in the sense of Bohr). Therefore, such a difference may not seem to be logical. However, in UGB, only half of the Gaussian overlaps the region of available vibrational actions (right at E‡0), unlike in our application of GB which necessarily leads to a higher probability than UGB [see, a few lines after eqn (7), the discussion on the width and position of the Gaussian in the GB curves shown in this work]. When E increases from E‡0 to 0.21 eV, the Gaussian widens (W increases from 0.27 to 1). Concomitantly, N‡(ϕ1), which is below the solid blue line in Fig. 5 at E‡0, moves upwards (at 0.21 eV, N‡(ϕ1) is an intermediate curve between the blue and red lines in Fig. 5; note that the latter concerns the canyon crossing but the scenario is similar for the model H + H2 reaction). These two effects combine in a subtle way that creates the hill of the UGB probability (Fig. 12) resembling that of the quantum probability (Fig. 11). Once on the plateau (E ≥ 0.21 eV), the classical limit is reached, since W = 1. Nevertheless, the GB probability remains close to the UGB probability for the reasons discussed in the penultimate paragraph of Section 2.1 (quasi-linearity of N‡(ϕ1) over the range of actions overlapped by the Gaussian). We therefore conclude that quantization of the activated complex is significant only around the threshold E‡0. In fact, all the trajectories contributing to the UGB curve in Fig. 12 cross the TS with vibrational energies close to E‡0. As a result, the translational energy remains small only when the total energy E is sufficiently close to E‡0. In this regime, the activated complex tends to have a relatively long average lifetime, and the standard deviation of its lifetime distribution is significant.36 This, in turn, implies a small standard deviation in its vibrational energy (according to the uncertainty relation), meaning that the vibrational energy is effectively quantized. However, once E reaches the plateau, the translational energy becomes too large for the lifetime distribution to retain a significant spread. Consequently, the standard deviation of the vibrational energy is large and the activated complex is no longer quantized.
 |
| | Fig. 12 Energy dependence of the contribution of the TS ground state to the reaction probability for the Schatz model of the H + H2 reaction obtained by means of the UGB-QCT method (green curve), the tunneling model (red curve) and the GB-QCT method (dashed blue curve). The value of the Gaussian width after the last iteration of the UGB procedure is also shown (orange curve). | |
The green, blue and red curves in Fig. 13 show the value of W after the final iteration of the UGB procedure for the TS states n = 2, 4 and 6 (the one for n = 0 is repeated here for comparison). For n = 2 and 4, there is still some quantization of the activated complex at the threshold, though the Gaussians are much broader than for the ground state. On the other hand, quantization no longer occurs for n = 6. The reason why threshold quantization is less and less effective as n increases can be found in Fig. 8: the size of the activated complex along the R-axis decreases with n, which reduces the standard deviation of the lifetimes36 and consequently, increases the spreading of the vibrational energies; the activated complex is thus less and less quantized.
 |
| | Fig. 13 Energy dependence of the Gaussian widths at the end of the iterative procedure of the UGB-QCT method for the Schatz model of the H + H2 reaction. The orange, green, blue and red curves correspond respectively to the TS vibrational states n = 0, 2, 4 and 6. The red plateau at 1 covers the blue which covers the green which covers the orange. | |
3.3 Semiempirical correction of the reaction probability
We return to the observation that the QCT and QM probabilities converge to the same values for large values of E, as it should be. For the canyon crossing, this necessarily makes the two probabilities nearly equal at the middle of the jumps corresponding to the thresholds of the TS excited states (see Fig. 3). We now propose a simple modification of the GB probability that enables it to satisfy the previous requirement. We consider the GB-QCT and QCT probabilities at E‡2, E‡4 and E‡6, divide the latter by the former, and perform a linear fitting of the obtained ratios (we found them to be nearly aligned; we thus considered the straight line through the first and third energies). The GB probabilities multiplied by this fitting, called GB′, are compared in Fig. 14 with the QM probabilities. This time, the quantitative agreement is very satisfactory. Note that for the present process, SCIVR calculations turned out to be more tricky, more time-consuming and less accurate than the present calculations.
 |
| | Fig. 14 QM and GB′-QCT reaction probabilities for the heavy and light masses in terms of the collision energy E; QM: solid lines, and GB′-QCT: dashed lines. | |
For the H + H2 model reaction, the analog of the previous method consists in modifying the UGB probability in such a way that the new UGB′ probability oscillates around the QCT probability in the limit of large energies. To this aim, we have found the straight line around which the UGB + tunneling curve oscillates for energies larger than E > E‡2 (the dashed blue line in Fig. 15). Moreover, we have fitted the QCT probability by a straight line in the same energy range (the dashed red line in Fig. 15). The mathematical expressions of these lines are given in the caption of Fig. 15. Dividing the second by the first and multiplying by the UGB + tunneling probability leads to the UGB′ + tunneling probability. The latter is compared with the quantum probability in Fig. 16. The agreement is now very satisfactory.
 |
| | Fig. 15 Energy dependence of the reaction probability for the Schatz model of the H + H2 reaction obtained by means of the UGB-QCT + tunneling method (solid blue curve) and the standard QCT method (solid red curve). The dashed blue line is a straight line chosen in such a way that the blue probability performs damped oscillations around it for E > E‡2. The dashed red line is a linear fit of the red probability within the same energy range. These lines are given by 0.29 + 0.365E and 0.2355 + 0.3575E, respectively. Dividing the red dashed line by the blue dashed line and multiplying by the UGB + tunneling probability leads to the UGB′ + tunneling probability (magenta curve). | |
 |
| | Fig. 16 Energy dependence of the reaction probability for the Schatz model of the H + H2 reaction obtained by means of QM calculations (green curve), and the UGB′-QCT + tunneling method (magenta curve). | |
4 Conclusion
In this paper, we have introduced the idea that, in classical simulations of chemical reaction dynamics, trajectories can be assigned Gaussian weights centered at integer values of the vibrational actions of the activated complex, defined as the region around the transition state in which the vibrational motion orthogonal to the reaction path evolves adiabatically (alternatively, an energy-based Gaussian weighting scheme may be employed). The Gaussian width is then adjusted so as to satisfy the time-energy uncertainty relation ΔE‡vΔT‡ ∼ ħ/2 (in a loose sense), where ΔE‡v is the standard deviation of the vibrational energy of the activated complex and ΔT‡ is the standard deviation of its lifetime. In this approach, called UGB, the weight of each trajectory depends on the weight of all the others. Therefore, trajectories “feel” each other, a bit like in semiclassical approaches where trajectories are assigned phases that allow them to interfere. Consequently, UGB has a more quantum spirit than GB, which considers each trajectory independently on the others (like standard binning). Its application to the calculation of the reaction probability for a model H + H2 reaction yields results in very good agreement with quantum scattering calculations.
A current limitation of UGB is that it cannot be applied below adiabatic thresholds if one uses real-time trajectories. To remedy this flaw, the approach must be coupled with a treatment of tunneling through adiabatic barriers. It would also be of interest to run imaginary-time trajectories and assess whether such calculations are not only feasible but also yield accurate UGB probabilities well below adiabatic barrier tops. Even so, a tunneling treatment would have to be applied right at the top of the adiabatic ground state barrier in order to set the value of α in the UGB uncertainty relation ΔE‡vΔT‡ = αħ/2. Note that at this energy, even a tunneling treatment within the parabolic limit is expected to be accurate. Of course, we intend to apply these ideas to realistic three-dimensional processes in the near future.37
In the past, several so-called active methods have been proposed to prevent ZPE leakage along a trajectory.38–46 The present study tends to show that there is no reason why one should prevent ZPE leakage if the vibrational motion occurs below the ZPE a period of time shorter than the vibrational period (see Fig. 12 and the related discussion). Therefore, we believe that the only case where ZPE leakage is an issue is when a trajectory slowly crosses a TS below its ZPE, or reaches the products (or the reformed reagents) below their ZPE. Standard GB solves the second issue and a treatment of tunneling can help solve the first—or possibly, UGB with imaginary-time trajectories, as suggested above.
Conflicts of interest
The authors have no conflicts to disclose.
Data availability
The data that support the findings of this study are available from the authors upon reasonable request.
Appendices
The standard deviations ΔE‡v and ΔT‡ of E‡v and T‡ are given by| |  | (A.1) |
with| |  | (A.2) |
and| | 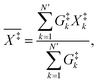 | (A.3) |
X = Ev, T. Note that these expressions are different from eqn (61)–(64) in Section IV.B.4 of ref. 11.
References
- N. Kotsina, P. D. Lane, J. G. Leng, K. E. Moncrieff, M. J. Roman and L. Saalbach, Viewpoints on the 2019 International Symposium on Molecular Beams, J. Phys. Chem. A, 2019, 123, 9247 CrossRef CAS PubMed.
- B. Fu, X. Shan, D. H. Zhang and D. C. Clary, Recent advances in quantum scattering calculations on polyatomic bimolecular reactions, Chem. Soc. Rev., 2017, 46, 7625 RSC.
- J. Li, B. Zhao, D. Xie and H. Guo, Advances and New Challenges to Bimolecular Reaction Dynamics Theory, J. Phys. Chem. Lett., 2020, 11, 8844 CrossRef CAS PubMed.
- L. Bonnet, J. C. Corchado and J. Espinosa-Garcia, Pair-correlated speed distributions for the OH + CH4/CD4 reactions: Further remarks on their classical trajectory calculations in a quantum spirit, C. R. Chim., 2016, 19, 571 CrossRef CAS.
- L. Zhang, J. Chen and B. Jiang, Vibrational State-to-State Scattering of Water from Cu(111): Comparison of Quantum and Quasiclassical Methods with Normal Mode and Adiabatic Switching Sampling, J. Phys. Chem. C, 2021, 125, 4995 CrossRef CAS.
- J. Espinosa-Garcia and C. Rangel, The CN(X2Σ+) + C2H6 reaction: Dynamics study based on an analytical full-dimensional potential energy surface, J. Chem. Phys., 2023, 159, 124307 CrossRef CAS PubMed.
- G. Czakó, B. Gruber, D. Papp, V. Tajti, D. Tasi and C. Yin, First-principles mode-specific reaction dynamics, Phys. Chem. Chem. Phys., 2024, 26, 15818 RSC.
- P. L. Houston, C. Qu, B. Fu and J. M. Bowman, Calculations of Dissociation Dynamics of CH3OH on a Global Potential Energy Surface Reveal the Mechanism for the Formation of HCOH; Roaming Plays a Role, J. Phys. Chem. Lett., 2024, 15, 9994 CrossRef CAS PubMed.
- P. G. Jambrina, F. J. Aoiz, N. Bulut, S. C. Smith, G. G. Balint-Kurti and M. Hankel, The dynamics of the H+ + D2 reaction: a comparison of quantum mechanical wavepacket, quasi-classical and statistical-quasi-classical results, Phys. Chem. Chem. Phys., 2010, 12, 1102 Search PubMed.
- G. C. Schatz, Tunnelling in bimolecular collisions, Chem. Rev., 1987, 81, 87 Search PubMed.
- L. Bonnet, C. Crespos and M. Monnerville, Chemical reaction thresholds according to classical-limit quantum dynamics, J. Chem. Phys., 2022, 157, 094114 CrossRef CAS PubMed.
- A. Rodriguez-Fernandez, L. Bonnet, C. Crespos, P. Larregaray and R. D. Muino, When classical trajectories get to quantum accuracy: The scattering of H2 on Pd(111), J. Phys. Chem. Lett., 2019, 10, 7629 CrossRef CAS PubMed.
- L. Bonnet, Classical dynamics of chemical reactions in a quantum spirit, Int. Rev. Phys. Chem., 2013, 32, 171 Search PubMed.
- W. H. Miller, Classical S Matrix: Numerical Application to Inelastic Collisions, J. Chem. Phys., 1970, 53, 3578 CrossRef CAS.
- L. Bonnet and J. C. Rayez, Quasiclassical trajectory method for molecular scattering processes: necessity of a weighted binning approach, Chem. Phys. Lett., 1997, 277, 183 Search PubMed.
- L. Bonnet and J. C. Rayez, Gaussian weighting in the quasiclassical trajectory method, Chem. Phys. Lett., 2004, 397, 106–109 Search PubMed.
-
M. Tabor, Chaos and Integrability in Non linear Dynamics, John Wiley and Sons, New York, 1989 Search PubMed.
- L. Bonnet and C. Crespos, Phase-index problem in the semiclassical description of molecular collisions, Phys. Rev. A:At., Mol., Opt. Phys., 2008, 78, 062713 CrossRef.
- L. Banares, F. J. Aoiz, P. Honvault, B. Bussery-Honvault and J. M. Launay, Quantum mechanical and quasi-classical trajectory study of the C(1D) + H2 reaction dynamics, J. Chem. Phys., 2003, 118, 565 CrossRef CAS.
- T. Xie, J. Bowman, J. W. Duff, M. Braunstein and B. Ramachandran, Quantum and quasiclassical studies of the O(3P) + HCl → OH + Cl(2P) reaction using benchmark potential surfaces, J. Chem. Phys., 2005, 122, 014301 CrossRef PubMed.
- M. Braunstein and L. Bonnet, A quasi-classical study in a quantum spirit of
mode specificity of the H + HOD abstraction reaction, Phys. Chem. Chem. Phys., 2024, 26, 26084 RSC.
- G. Czakó and J. M. Bowman, Quasiclassical trajectory calculations of correlated product distributions for the F + CHD3 (v1 = 0, 1) reactions using an ab initio potential energy surface, J. Chem. Phys., 2009, 131, 244302 Search PubMed.
- L. Bonnet and J. Espinosa-Garcia, The method of Gaussian weighted trajectories. V. On the 1GB procedure for polyatomic processes, J. Chem. Phys., 2010, 133, 164108 CrossRef CAS PubMed.
- L. Bonnet and J. Espinosa-Garcia, Simulation of the experimental imaging results for the OH + CHD3 reaction with a simple and accurate theoretical approach, Phys. Chem. Chem. Phys., 2017, 19, 20267 Search PubMed.
- D. C. Chatfield, R. S. Friedman, D. G. Truhlar, B. C. Garrett and D. W. Schwenke, Global control of suprathreshold reactivity by quantized transition states, J. Am. Chem. Soc., 1991, 113, 486 CrossRef CAS.
- M. Gustafsson and R. T. Skodje, The state-to-state-to-state model for direct chemical reactions: Application to D + H2 → HD + H, J. Chem. Phys., 2006, 124, 144311 CrossRef PubMed.
- P. Pechukas and E. Pollak, Transition states, trapped trajectories, and classical bound states embedded in the continuum, J. Chem. Phys., 1978, 69, 1218 CrossRef.
- J. M. Bowman, S. Carter and X. Huang, MULTIMODE: A code to calculate rovibrational energies of polyatomic molecules, Int. Rev. Phys. Chem., 2003, 22, 533 Search PubMed.
- In ref. 11, the QM curves shown in Fig. 6 do not correspond to the masses 19 (upper panel) and 1.9 (lower panel), but to one tenth of these masses. The SCIVR results had to be arbitrarily divided by 2 to be in good agreement with the (wrong) QM results and we attributed that to a failure of the SCIVR approach. This attribution was in fact erroneous since doubling the height of the green dots in Fig. 6 of ref. 11 makes them in satisfactory agreement with the (correct) QM results of the present work. Importantly, none of the analyses or conclusions of ref. 11 are affected by this mistake.
- W. H. Miller, Classical-limit quantum mechanics and the theory of molecular collisions, Adv. Chem. Phys., 1974, 25, 69 CrossRef.
-
H. J. Weber and G. B. Arfken, Essential Mathematical Methods for Physicists, Academic Press, 2003 Search PubMed.
- In the usual GB procedure applied to a triatomic exchange reaction, each reactive trajectory is assigned the comb of narrow Gaussians
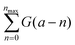 where a is the product vibrational action (assuming that the rotational motion is classically treated). As previously stated, ε is often taken at 0.06, which corresponds to a width of 10%, i.e., one tenth of the spacing between two neighbouring quantum states. Therefore, the classical limit is reached for ε taken at 0.6, for which the comb slightly oscillates around 1 (a little less on the edges). Therefore, all the trajectories have nearly the same unit weight, just like with the standard binning procedure.
where a is the product vibrational action (assuming that the rotational motion is classically treated). As previously stated, ε is often taken at 0.06, which corresponds to a width of 10%, i.e., one tenth of the spacing between two neighbouring quantum states. Therefore, the classical limit is reached for ε taken at 0.6, for which the comb slightly oscillates around 1 (a little less on the edges). Therefore, all the trajectories have nearly the same unit weight, just like with the standard binning procedure.
- More exactly, pkcl is the contribution of path k to the classical density of probability that the system start from the reagent plane with J = 0 and reach the product plane with J = 0.
- M. Bauer and P. A. Mello, The Time-Energy Uncertainty Relation, Ann. Phys., 1978, 111, 38 Search PubMed.
- W. H. Miller, Tunneling Corrections to Unimolecular Rate Constants, with Application to Formaldehyde, J. Am. Chem. Soc., 1979, 101, 6810 CrossRef CAS.
- Let us call NAR and VAR the nonadiabatic entrance region and vibrationally adiabatic region, respectively. Moreover, consider two reactive trajectories of radial velocities having the same value at the entrance of the NAR. The crossing of the latter will make these velocities different at the entrance of the VAR. Now, let us neglect the variation of these velocities throughout the VAR. Then, the difference ΔT of activated complex lifetimes for the two trajectories satisfies ΔT = L(v2 − v1)/(v1v2) while the average lifetime satisfies Tav = L(v2 + v1)/(2v1v2). Therefore, ΔT is proportional to Tav. This argument supports the idea that the spreading of the lifetimes and, thus, their standard deviation, increase with the average lifetime.
- In three-dimensional simulations of the dynamics, the tunneling treatment must take into account the curvature of the reaction path at small energies. This can be done, for instance, using the toolkit developed by Truhlar and coworkers to treat tunneling via corner cutting within variational transition-state theory (see J. L. Bao and D. G. Truhlar, Variational transition state theory: theoretical framework and recent developments, Chem. Soc. Rev., 2017, 46, 7548 RSC ).
- J. M. Bowman, B. Gazdy and Q. Sun, Method to Constrain Vibrational Energy in Quasiclassical Trajectory Calculations, J. Chem. Phys., 1989, 91, 2859 CrossRef CAS.
- W. H. Miller, W. L. Hase and C. L. Darling, A Simple Model for Correcting the Zero Point Energy Problem in Classical Trajectory Simulations of Polyatomic Molecules, J. Chem. Phys., 1989, 91, 2863 CrossRef CAS.
- G. Peslherbe and W. L. Hase, Analysis and Extension of a Model for Constraining Zero-point Energy Flow in Classical Trajectory Simulations, J. Chem. Phys., 1994, 100, 1179 Search PubMed.
- A. J. C. Varandas and J. M. C. Marques, Method for Quasiclassical Trajectory
Calculations on Potential Energy Surfaces Defined from Gradients and Hessians, and Model to Constrain the Energy in Vibrational Modes, J. Chem. Phys., 1994, 100, 1908 CrossRef CAS.
- Z. Xie and J. M. Bowman, Zero-Point Energy Constraint in Quasi-Classical Trajectory Calculations, J. Phys. Chem. A, 2006, 110, 5446 CrossRef CAS PubMed.
- D. Bonhommeau and D. G. Truhlar, Mixed Quantum/Classical Investigation of the Photodissociation of NH3(A) and a Practical Method for Maintaining Zero-Point Energy in Classical Trajectories, J. Chem. Phys., 2008, 129, 014302 CrossRef PubMed.
- G. Czakó, A. L. Kaledin and J. M. Bowman, A Practical Method to Avoid Zero-Point Leak in Molecular Dynamics Calculations: Application to the Water Dimer, J. Chem. Phys., 2010, 132, 164103 CrossRef PubMed.
- K. L. K. Lee, M. S. Quinn, S. J. Kolmann, S. H. Kable and M. J. T. Jordan, Zero-Point Energy Conservation in Classical Trajectory Simulations: Application to H2CO, J. Chem. Phys., 2018, 148, 194113 CrossRef PubMed.
- S. Mukherjee and M. Barbatti, A Hessian-Free Method to Prevent Zero-Point Energy Leakage in Classical Trajectories, J. Chem. Theory Comput., 2022, 18, 4109 CrossRef CAS PubMed.
|
| This journal is © the Owner Societies 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Maurice
Monnerville
*b
*a and
Maurice
Monnerville
*b

 and
and  are the momenta conjugate to R and ϕ, respectively, t is the time, μ is the mass associated with motion along the R-axis, I is the moment of inertia of the diatom and V(R,ϕ) is the potential energy. Two values of μ will be considered, 19 amu and 1.9 amu, corresponding to atom–diatom reactions of the heavy plus light-heavy and light plus heavy-light type, respectively. I is equal to mre2 with m = 0.5 amu and re = 0.7414 Å. Our model potential energy surface (PES) is defined by
are the momenta conjugate to R and ϕ, respectively, t is the time, μ is the mass associated with motion along the R-axis, I is the moment of inertia of the diatom and V(R,ϕ) is the potential energy. Two values of μ will be considered, 19 amu and 1.9 amu, corresponding to atom–diatom reactions of the heavy plus light-heavy and light plus heavy-light type, respectively. I is equal to mre2 with m = 0.5 amu and re = 0.7414 Å. Our model potential energy surface (PES) is defined by
 and stopped at R2 = 3 Å (see Section III.A.2 of ref. 11 for more details on the trajectory calculations). Within the channel, there is a periodic orbit (PO) trapped along the ϕ-axis (see the white vertical segment centered at the origin of the (R, ϕ) frame). According to classical mechanics, the transition state (TS) is defined as the phase space surface whose projection on the (R, ϕ) configuration plane coincides with the representation of the PO in the same plane.27 The TS is thus trivially defined in (R, ϕ, P, J) by R = 0. The reactive flux is minimum through the TS which, thus, corresponds to a phase space bottleneck. The reactive trajectory clearly mimics the passage from the free rotation of reactants, through bending vibration at the transition state, to the free rotation of products that occurs in three-dimensional bimolecular reactions. Four white vertical dashed lines define five regions labeled RA, NA and VA above the frame of Fig. 1. The dynamics are rotationally adiabatic in the RA reagent and product half planes (the PES is flat in these domains so that the rotation is free), vibrationally adiabatic in the VA central region encompassing the TS (see further below for a numerical proof of this statement, which is quite obvious from the shape of the trajectory in the VA band), and nonadiabatic in the NA regions due to the presence of strong couplings between the R and ϕ coordinates. We define the activated complex as the system orthogonal to the R-axis in the VA region, whose boundaries are R ≈ −0.75 and 0.75, in Å (see Fig. 1). The activated complex has a lifetime, contrary to the TS which is just crossed by classical trajectories.
and stopped at R2 = 3 Å (see Section III.A.2 of ref. 11 for more details on the trajectory calculations). Within the channel, there is a periodic orbit (PO) trapped along the ϕ-axis (see the white vertical segment centered at the origin of the (R, ϕ) frame). According to classical mechanics, the transition state (TS) is defined as the phase space surface whose projection on the (R, ϕ) configuration plane coincides with the representation of the PO in the same plane.27 The TS is thus trivially defined in (R, ϕ, P, J) by R = 0. The reactive flux is minimum through the TS which, thus, corresponds to a phase space bottleneck. The reactive trajectory clearly mimics the passage from the free rotation of reactants, through bending vibration at the transition state, to the free rotation of products that occurs in three-dimensional bimolecular reactions. Four white vertical dashed lines define five regions labeled RA, NA and VA above the frame of Fig. 1. The dynamics are rotationally adiabatic in the RA reagent and product half planes (the PES is flat in these domains so that the rotation is free), vibrationally adiabatic in the VA central region encompassing the TS (see further below for a numerical proof of this statement, which is quite obvious from the shape of the trajectory in the VA band), and nonadiabatic in the NA regions due to the presence of strong couplings between the R and ϕ coordinates. We define the activated complex as the system orthogonal to the R-axis in the VA region, whose boundaries are R ≈ −0.75 and 0.75, in Å (see Fig. 1). The activated complex has a lifetime, contrary to the TS which is just crossed by classical trajectories.

 (
( where h is Planck's constant) and its spectrum can be obtained by diagonalization methods (see ref. 28 and references therein). Alternately, it can be semiclassically estimated as follows. Calling
where h is Planck's constant) and its spectrum can be obtained by diagonalization methods (see ref. 28 and references therein). Alternately, it can be semiclassically estimated as follows. Calling  the energy of the TS, its vibrational action over one cycle is given by
the energy of the TS, its vibrational action over one cycle is given by

 and eqn (3) and (4) lead to
and eqn (3) and (4) lead to

 , is even. Moreover, the ϕ-dependence of the potential energy is also even. Therefore, the E-resolved scattering state is necessarily symmetric with respect to the R-axis. Consequently, the coefficients of its development on the vibrational states Ψn(ϕ) of the TS are necessarily 0 for odd values of n (for these states are antisymmetric). The QCT probabilities (dashed lines in Fig. 3) are in poor agreement with QM ones. This is especially true around the thresholds, which classical mechanics obviously ignores. Nevertheless, we see that the disagreement decreases when the total energy increases, as it should be. In particular, QM probabilities perform damped “oscillations” around QCT probabilities in such a way that both are nearly equal at the middle of the jumps for the threshold associated with the TS excited states n ≥ 2 (only the one corresponding to n = 2 can be seen in Fig. 3). We will take advantage of this observation later.
, is even. Moreover, the ϕ-dependence of the potential energy is also even. Therefore, the E-resolved scattering state is necessarily symmetric with respect to the R-axis. Consequently, the coefficients of its development on the vibrational states Ψn(ϕ) of the TS are necessarily 0 for odd values of n (for these states are antisymmetric). The QCT probabilities (dashed lines in Fig. 3) are in poor agreement with QM ones. This is especially true around the thresholds, which classical mechanics obviously ignores. Nevertheless, we see that the disagreement decreases when the total energy increases, as it should be. In particular, QM probabilities perform damped “oscillations” around QCT probabilities in such a way that both are nearly equal at the middle of the jumps for the threshold associated with the TS excited states n ≥ 2 (only the one corresponding to n = 2 can be seen in Fig. 3). We will take advantage of this observation later.









 . The value of ε corresponding to the width of the previous range is ∼0.36. Any value of ε lower than 0.36 is thus expected to make P, as obtained from eqn (6) and (7), very close to Pb. Moreover, numerical calculations show that even for ε ∼ 0.6, corresponding to the unit width (i.e., the classical limit32), P remains relatively close to Pb. On the other hand, the situation is different at Ea. The maximum vibrational action N‡(ϕa>) is slightly above 0 (see the right end of the blue curve in Fig. 5). Therefore, G(N‡(ϕ1)) must be very narrow for eqn (6) and (7) to agree with eqn (10). Otherwise, G(N‡(ϕ1)) will be non 0 for all the actions greater than N‡(ϕa>), which are not available, so eqn (6) and (7) will significantly minimize the reaction probability. This would for instance be the case with the magenta Gaussian in Fig. 5 which partly overlaps the region of unavailable actions (above the horizontal dashed blue line). We will come back to this point in Section 3.3.
. The value of ε corresponding to the width of the previous range is ∼0.36. Any value of ε lower than 0.36 is thus expected to make P, as obtained from eqn (6) and (7), very close to Pb. Moreover, numerical calculations show that even for ε ∼ 0.6, corresponding to the unit width (i.e., the classical limit32), P remains relatively close to Pb. On the other hand, the situation is different at Ea. The maximum vibrational action N‡(ϕa>) is slightly above 0 (see the right end of the blue curve in Fig. 5). Therefore, G(N‡(ϕ1)) must be very narrow for eqn (6) and (7) to agree with eqn (10). Otherwise, G(N‡(ϕ1)) will be non 0 for all the actions greater than N‡(ϕa>), which are not available, so eqn (6) and (7) will significantly minimize the reaction probability. This would for instance be the case with the magenta Gaussian in Fig. 5 which partly overlaps the region of unavailable actions (above the horizontal dashed blue line). We will come back to this point in Section 3.3.






 . The adiabatic ground state barrier E0(R) is given by the sum of the ZPE at R and the potential energy along the reaction path. The latter being 0, we have E0(R) = ħω(R)/2 = E‡0/cosh(R/D) with
. The adiabatic ground state barrier E0(R) is given by the sum of the ZPE at R and the potential energy along the reaction path. The latter being 0, we have E0(R) = ħω(R)/2 = E‡0/cosh(R/D) with  . If we approximate E0(R) with a parabolic barrier, we obtain a very satisfying approximation of the tunneling probability immediately below E‡0 from35
. If we approximate E0(R) with a parabolic barrier, we obtain a very satisfying approximation of the tunneling probability immediately below E‡0 from35











