
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
C atoms versus Si atoms at the bridgehead positions of phenyl-decorated adamantane-type clusters: influence on the nonlinear optical response
Ferdinand
Ziese
 a,
Stefanie
Dehnen
b and
Simone
Sanna
a
a,
Stefanie
Dehnen
b and
Simone
Sanna
a
aInstitut für Theoretische Physik and Center for Materials Research (LaMa/ZfM), Justus-Liebig-Universität Gießen, 35392 Gießen, Germany. E-mail: ferdinand.ziese@physik.uni-giessen.de; simone.sanna@theo.physik.uni-giessen.de
bKarlsruhe Institute of Technology (KIT), Institute of Nanotechnology, 76131 Karlsruhe, Germany. E-mail: stefanie.dehnen@kit.edu
First published on 2nd December 2025
Abstract
In order to explore the effect of the presence of CPh versus SiPh parts on the optical nonlinearities of phenyl-decorated adamantane-type clusters, the second harmonic generation (SHG) spectra of molecular clusters with the general formula [(CPh)x(SiPh)4−xS6] and x = 0,…, 4, are calculated within (hybrid) density functional theory. The stepwise replacement of C atoms with Si at the bridgehead positions leads to the blue-shift of the first SHG peak and to a redistribution of the spectral weights of the different signatures. The electronic transitions related to the SHG peaks are found to occur mainly at the substituents, which are common to all investigated compounds. However, the peak intensity strongly depends on the substitution induced distortion of the cluster core. Thus, although the cluster core is not directly involved in the genesis of the optical nonlinearities, it heavily impacts their intensity. The presented results suggest that reducing the core symmetry provides a pathway to enhance the SHG response of the clusters and yields a theoretical foundation for the design of nonlinear optical materials with tailored properties.
1 Introduction
Since the pioneer works of Rosemann et al., demonstrating that organotetrel chalcogenide clusters are characterized by outstanding nonlinear optical properties,1,2 a multitude of studies were dedicated to the adamantane-shaped clusters with the general formula [(RT)4E6] (with R = organic group; T = C, Si, Ge, Sn; E = O, S, Se, Te, NH, CH2). Different members of this materials class are efficient broadband and highly directional white-light emitters.3,4 Other adamantane-type clusters with organic substituents are able to upconvert the incident radiation into light of half the wavelength, a phenomenon known as second-harmonic generation (SHG).4,5The clusters can be synthesized in form of glasses,6 which paves the way for potential applications in a multitude of technologies, including devices in every day's life. In particular, the compounds potentially represent the key to a sustainable and ecologically as well as economically friendly technology.7–9
Consequently, the adamantane-type clusters and related compounds came into the focus of the scientific community, and are actually being studied both experimentally10–15 and theoretically.16–18 Existing studies gathered information about the prerequisites for SHG or white-light generation and about the intertwinement of structural features and optical response. Yet, the origin of the nonlinear optical response remains elusive18,19 and the interplay of the many possible degrees of freedom is far from being understood. Chemically and structurally very similar clusters may feature a drastically different linear and nonlinear optical response. Homogeneous vs inhomogeneous core composition and substituents, electron localization, habitus, and external stimuli such as temperature and pressure are known to impact the emission. However, a comprehensive understanding of this cluster class is still missing.
In this work, we focus on a particular aspect of this materials family and investigate whether and to which extent the organic vs inorganic cluster composition affects the nonlinear optical response. Therefore, we calculate the frequency dependence of the SHG spectra of phenyl-decorated adamantane-type clusters with the general formula [(CPh)x(SiPh)4−xS6] and x = 0,…,4, as shown in Fig. 1. Starting with a cluster with x = 4, we substitute stepwise all the C atoms from the bridgehead positions of the cluster core by Si.
 | ||
| Fig. 1 Phenyl-decorated adamantane-type clusters with the general formula [(CPh)x(SiPh)4−xS6] and x = 0,…,4 investigated in this work. | ||
Our calculations reveal that the optical nonlinearities in the technologically relevant energy window between 0 and 3 eV have their main origin in electronic transitions localized at the phenyl substituents. Therefore, all the SHG spectra of the investigated clusters show a dominant peak at about 2.2 eV related to electronic transitions within the substituents. The intensity of this peak correlates with the substitution-induced core distortion. The cluster [(PhC)4S6] features a further low energy SHG peak, which lowers its intensity by the stepwise substitution of C with Si, until it vanishes for the [(PhSi)4S6] cluster. This spectral feature is related to transitions involving the electronic states related to the S orbitals in the cluster core, which is a rather peculiar feature among clusters with strong optical nonlinearities.18 The results provided in the present work demonstrate the possibility to tune the nonlinear optical response of phenyl-decorated adamantane-type clusters, and provide a theoretical foundation for the design of optimized optical sources.
Besides their role as optical sources,20 molecules lacking inversion symmetry and thus featuring a SHG response can be furthermore conveniently detected and recognized by second harmonic spectroscopy.21–25
2 Methods
The cluster geometry as well as the electronic ground state are computed within density functional theory (DFT), as implemented in VASP using projector-augmented wave (PAW) potentials26,27 and the Perdew–Burke–Ernzerhof (PBE) functional.28 All the investigated molecules have been also modeled and structurally optimized within the HSE06 approach, a range-separated hybrid functional including 25% HF exchange in the short-range part of the exchange term.29,30 Geometries calculated within hybrid-DFT closely match the PBE calculated structures and are also included in the SI. For the optical calculations the wave functions are determined using Quantum Espresso employing the freely available SG15 optimized norm-conserving Vanderbilt pseudopotentials,31,32 with exchange and correlation term in the PBE formulation28 as well as within hybrid-DFT, again in the HSE06 formulation. Molecular geometries in the gas phase are optimized within the molecule-in-a-box approach. We employ cubic boxes with a volume of 51179.6 Å3, explicitly designed to decouple periodic images of the clusters. Structural optimization is performed with a Hellmann–Feynman force threshold of 0.005 eV Å−1. The vdW DFT-D3 method with Becke–Johnson damping33,34 is applied to all structural optimizations to account for dispersion forces.SHG spectra are calculated in the time-domain within the independent particle approximation from the evolution the dynamical polarization35 calculated both with the (semi)local PBE XC-functional as well as within hybrid-DFT. This first-principles approach is implemented in the YAMBO code36 and sketched in the SI. Numerical parameters, such as time-step, total propagation time, or spectral broadening, are chosen to ensure convergence of the nonlinear optical spectra. For the spectral broadening, which influences the optical response at resonances, a typical value of 0.2 eV is chosen. Numerical values of all relevant computational parameters as well as the atomic coordinates of the investigated structures are given in the SI.
It is known that the calculation of the nonlinear optical response within semilocal XC-potentials as PBE is affected by some flaws. Although alternative approaches for the calculation of the hyperpolarizability exist, partially based on very accurate computational schemes relying on many-body perturbation theory,37–39 the employed approach has proven to be reliable in establishing chemical trends both for solid state and molecular systems.17,35,40 It furthermore gives access to the frequency dependency of the SHG spectra discussed in this work.
3 Results
The calculated SHG response of all structures from [(PhSi)4S6] to [(PhC)4S6] is plotted in Fig. 2. Solid lines represent DFT-PBE calculations, while the dashed lines represent hybrid-DFT calculations. The transition from [(PhC)4S6] (bottom panel) to [(PhSi)4S6] (top panel) influences massively the optical nonlinearities, modifying both position and intensity of the spectral signatures. | ||
| Fig. 2 SHG response of adamantane-type clusters with the general formula [(CPh)x(SiPh)4−xS6] and x = 0,…,4 calculated by DFT-PBE (solid line) and hybrid-DFT (dashed lines, normalized to the first PBE peak). The main SHG peak is marked by a vertical line of the same color as the spectrum. The spectral signature of the first main peak of [(PhC)4S6] can be found in all clusters other than [(PhSi)4S6], where it vanishes. This signature is marked by the arrow of the same color as the spectrum. The black vertical line indicates the HOMO level. | ||
The first striking difference is the position of the main SHG peak (marked by a vertical line of the same color of the spectrum), which is strongly blueshifted by ca 0.4 eV for all structures containing Si. The change is not gradual but rather abrupt. In the case of [(PhC)4S6] (bottom panel in Fig. 2), the first spectral signature is also the main SHG peak. This is marked by an arrow. This signature is still visible in the spectra of the clusters containing Si (see arrow color-coded as the SHG spectrum). However, its intensity rapidly decreases, finally vanishing for the [(PhSi)4S6] cluster (upper panel in Fig. 2) so that the further spectroscopic signatures (i.e., the second SHG peak) become more intense. A second difference is the SHG intensity of the main spectral signature, which results enhanced for intermediate structures, in particular [(PhSi)3(PhC)S6] (orange plot in Fig. 2.) It can be observed that although the spectral features as calculated within HSE06 are shifted to higher energies due to the gap opening, the form and relative weight of the SHG peaks closely matches that of DFT-PBE in the investigated region. We thus mainly discuss the DFT-PBE results in the remainder of this work. A more detailed analysis of the effect of the employed hybrid functional can be found at the end of this section as well as in the SI.
In order to understand the blueshift of the first SHG peak from compositions comprising exclusively C at the bridgehead positions to compositions that include Si instead, and the corresponding intensity decrease of the [(PhC)4S6] main peak, we explore the electronic structure of the clusters. In particular, the low-energy spectral signatures are generally related to the lowest allowed electronic transitions and may thus involve the HOMO and LUMO levels as well as the neighboring states. The calculated HOMO–LUMO gaps are reported in Fig. 3 in the color coding of Fig. 2. For a direct comparison with the SHG spectra, half of the HOMO–LUMO gap [Egap = (ELUMO − EHOMO)] is plotted. The calculations reveal a “dominant” influence of the C atom in the core in comparison to Si. Starting from the composition [(PhC)4S6] (lhs of the Fig. 3), the stepwise replacement of the C atoms with Si increases Egap. However, the modifications are small unless all the carbon related electronic states disappear from the HOMO–LUMO region and the much larger Egap of [(PhSi)4S6] is obtained.
 | ||
| Fig. 3 DFT calculated HOMO–LUMO energy gaps divided by 2 (for better comparison with the SHG spectra) of the investigated clusters. | ||
The orbital character of the involved HOMO and LUMO states explains this behavior. The LUMO is well localized at the phenyl substituents for all investigated structures. No substantial differences between the LUMO of the different clusters (all of them are shown in the SI) are calculated. The HOMO as calculated by DFT for the different clusters is qualitatively different, instead. Fig. 4 (lhs) shows the HOMO calculated for the [(PhSi)4S6] cluster. It is strongly localized at the S atoms. With the substitution of Si atoms by C, the HOMO is still mainly localized at the S atoms, however, the charge distribution is clearly shifted towards the C atoms and away from the Si atoms. This is illustrated in Fig. 4 (rhs) exemplarily for the [(PhSi)(PhC)3S6] cluster (the DFT calculated HOMO of all clusters are shown in the SI). This can be understood by the larger hybridization of the 3p atomic states of S (−7.11 eV) with the energetically closer 2p states of C (−5.42 eV) than with the 3p atomic states of Si (−4.17 eV). Correspondingly, the HOMO inherits more C character, and rises its energy reducing the electronic gap.
 | ||
| Fig. 4 DFT calculated squared wavefunctions associated with the HOMO of the [(PhSi)4S6] cluster (lhs), and with the HOMO of the [(PhSi)(PhC)3S6] cluster (rhs). The isosurface value of 0.001 e Å−3 is shown. The atoms are color-coded by atom type: S atoms are yellow, C atoms are black, Si atoms are gray, and H atoms are light blue. | ||
In any event, the width of the HOMO–LUMO gaps does not explain the position of the first SHG peak, which is evidently not related to HOMO–LUMO transitions. The Egap energy, which we report for convenience as a black line in Fig. 2, marks the onset of the optical nonlinearities, instead. Although the shift of Egap with the Si content qualitatively mirrors the blueshift of the optical spectra, it does not explain the shift in position or intensity of the main SHG peak.
As the HOMO and LUMO states do not seem to be involved in the origin of the optical nonlinearities, we investigate which electronic levels are related to the SHG signatures. A first rough estimate is obtained projecting the partial density of states (PDOS) of different atomic groups onto the SHG response. The projection is performed by dividing the DOS energy scale by a factor of two to match the SHG scale. In practice, this depiction represents an excitation of the electrons from the occupied states into the LUMO.
The result of this procedure is shown in Fig. 5 to estimate the contribution of the core atoms to the electronic transitions related to the SHG signatures. We remark that the goal of this representation is not to compare the SHG intensity of the different compounds (which is done in Fig. 2) but rather to estimate the contribution of certain atomic groups to the SHG response. Therefore, the latter is renormalized with respect to the highest peak and its magnitude is not comparable between the panels. The energy axis is shared by all panels, though. Fig. 5 shows that electronic states localized at the core can only contribute to the onset of the optical nonlinearities as well as to the right side of the main peak.
 | ||
| Fig. 5 Partial density of states of core atoms, color-coded and projected onto the SHG response of the investigated clusters. | ||
The core localization at the onset of the optical nonlinearity corresponds to the previously discussed core localization of the HOMO states. Correspondingly, it shifts to lower energies with the C content, mirroring the mentioned gap reduction. The core localization of states that possibly contribute to the first SHG peak is unusual and not previously predicted for adamantane-type clusters.
To better understand which part of the core contributes to the high partial DOS in correspondence of the first SHG peak, we analyze separately the PDOS of the three different atom types C, Si, and S, present in the core. In Fig. 6, we present the contribution to the PDOS by the S atoms. The distribution is similar to the total PDOS of the core presented in Fig. 5, although a large part of the core atoms is not considered.
 | ||
| Fig. 6 PDOS of the S atoms projected on the SHG response. | ||
This strongly suggests that Si and C atoms at the bridgehead positions of the cluster only yield a minor contribution to the PDOS. Indeed, the plots showing the PDOS related to the Si atoms (in Fig. 7) or C atoms (in Fig. 8) do not show a relevant contribution in the considered energy range, in strong contrast with the S related PDOS. This demonstrates that the entire core contribution to the electronic states in the energy window corresponding to the first SHG peak is due to the S atoms. Again, this is in agreement with the previously discussed orbital character of the HOMO, which is rather localized at the S atoms (see Fig. 4), irrespective of the considered cluster.
 | ||
| Fig. 7 PDOS of the Si atoms at the bridgehead positions projected on the SHG response. | ||
 | ||
| Fig. 8 PDOS of the C atoms at the bridgehead positions projected on the SHG. | ||
Although some contribution from the core atoms (in particular S) is visible at the side flanks of the first SHG signature, the maxima of the SHG spectra do not seem to be related to core-localized electronic states. The projection of the partial DOS related to the substituents is shown in Fig. 9 and confirms this impression. The maximum of the first SHG signature is related to electronic states localized at the substituents. The only exception is [(PhC)4S6], for which the maximum of the second SHG peak is related to a high substituent PDOS. This corroborates our previous assumption that the first SHG peak of all clusters containing Si and the second SHG peak of the cluster containing only C at the bridgehead positions have the same nature, i.e., arise form transitions within corresponding orbital groups.
 | ||
| Fig. 9 Partial DOS of the substituent atoms, color-coded and projected on the SHG response of the investigated clusters. | ||
Summarizing the results presented so far, the analysis of the electronic structure suggests that HOMO–LUMO electronic transitions do not contribute to the SHG response and the analysis of the PDOS suggests that the phenyl substituents play a crucial role in determining the main SHG signature (or the second, in case of [(PhC)4S6]). Yet, a detailed investigation of the electronic transitions is necessary to understand the origin of the optical nonlinearities and the differences between [(PhC)4S6] and the clusters in which C atoms in the cluster core were replaced with Si atoms.
The analysis of the electronic transitions related to the nonlinear optical response is performed by the procedure proposed in ref. 18, which we illustrate while discussing the [(PhC)4S6] cluster. Fig. 10 (left panel) shows the electronic energy levels of the orbital groups in an energy window of about 12 eV embracing the HOMO–LUMO region. The single orbitals are grouped into an orbital group if they are separated by an energy lower than 0.1 eV.
 | ||
| Fig. 10 Orbital group energy values of [(PhSi)(PhC)3S6] with corresponding scaled and shifted SHG response. The energy values of the origin orbital groups are shifted for a direct transition to the respective target orbital group t = 0, t = 1, and t = 2. | ||
We consider the electronic transitions from occupied orbital groups (named origin and labeled by o) to unoccupied orbitals (named target and labeled by t). For the sake of clarity, occupied orbital groups are colored alternately in lighter and darker gray colors, while unoccupied orbital groups are colored individually. The origin orbitals are counted energy downwards from the highest occupied one, while the target orbitals are counted upwards.
The convolution o ∗ t of all origin orbital groups with a single target orbital group is calculated to model electronic transitions. o ∗ t is color-coded as the considered unoccupied orbital group, and represented in histogram form in the right panel. The transparency roughly mirrors the density of energy values.
To represent transitions into higher traget states, the origin orbital groups as well as the convolution are shifted downwards on the energy axis by the amount corresponding to the distance of the considered target orbital group to the LUMO. The calculated SHG spectrum is reported as a blue solid line in the right panel as well, to correlate the spectral features to the electronic transitions.†
Peaks of the convolution o ∗ t in correspondence of peaks of the SHG spectrum allow to identify electronic transitions contributing to the SHG signatures. The spatial overlap of the electronic orbitals involved in these transitions are also plotted in real space to gain more insight. Obviously, a large overlap favors the corresponding o → t transition. Moreover, it shows in which part of the cluster the transitions leading to the SHG features have their origin.
Due to the large amount of possible electronic excitations, we report in the following only on the relevant transitions, i.e., those with an energy corresponding to the calculated SHG signatures. Our analysis reveals that all Si containing clusters are characterized by a similar behavior. We therefore discuss the [(PhSi)(PhC)3S6] cluster as a prototypical example.
The lower energy transitions such as the HOMO–LUMO transitions have an energy much lower than the energy of the main SHG peak, whose origin must be sought for in more energetic transitions. Fig. 10 shows the the electronic states with the highest convolution in correspondence of the first SHG (double) peak. This spectral feature correlates best with transitions from o = 3 to t = 0, t = 1, and t = 2. In particular, the transition from o = 3 to t = 0 has exactly the energy of the central peak, the transition o = 3 to t = 2 coincides with the side peak, and o = 3, t = 0 is in-between. The overlaps o = 3 with t = 0, 1, 2 are shown in Fig. 11. A strong localization on the substituent structures is observed, suggesting that the first SHG peak has its origin at the phenyl substituents. We remark, that these transitions have by far the largest overlap in comparison to others transitions which could possibly contribute due to their energy (for example in comparison to the overlaps depicted in Fig. 12).
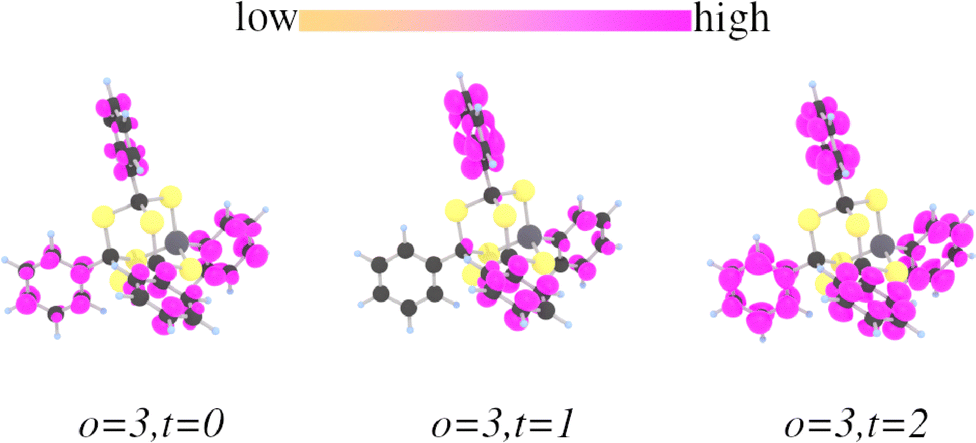 | ||
| Fig. 11 Overlaps ρ of o = 3 with t = 0, 1, 2 for [(PhSi)(PhC)3S6]. The atoms are color-coded by atom type: S atoms are yellow, C atoms are black, the Si atom is grey, and H atoms are light blue. | ||
 | ||
| Fig. 12 Overlaps ρ of o = 0, 1 with t = 5 for [(PhSi)(PhC)3S6]. The electronic levels (not shown here) and their color coding used for the convolution are those of Fig. 10. The atoms are color-coded by atom type: S atoms are yellow, C atoms are black, the Si atom is grey, and H atoms are light blue. | ||
In Fig. 12, we report the overlap of transitions from o = 0, 1 to higher target states, which also are characterized by energies correlating with the maximum of the first SHG peak. These transitions have a much lower convolution and a much lower overlap than the previously discussed transitions from o = 3 to t = 0, 1, 2 and are expected to contribute to a lesser extent to the SHG of the cluster. These transition with a smaller contribution are localized at the S atoms and play a more central role in the case of the [(PhC)4S6] cluster.
It must be pointed out that we do not show the same isosurface value to represent the overlap between o and t groups. This would result in a very large volume, e.g., for o = 3, t = 0 and a vanishing overlap for, e.g., o = 0, t = 5. Instead, we arbitrarily chose a isosurface value so that the overlap region is well visible, and color code the isosurface to represent its magnitude.
The SHG spectrum of [(PhC)4S6] differs from that of clusters containing Si atoms due to the presence of a first main peak not present in the other spectra (see Fig. 2). In order to explore the origin of this peak, we again correlate the electronic transitions with the SHG response. Again, we we will not present the full set of data, rather only result relevant to the discussion. The first electronic transition with energy corresponding to the first SHG peak is due to transitions to the target orbital t = 3, shown in Fig. 13.
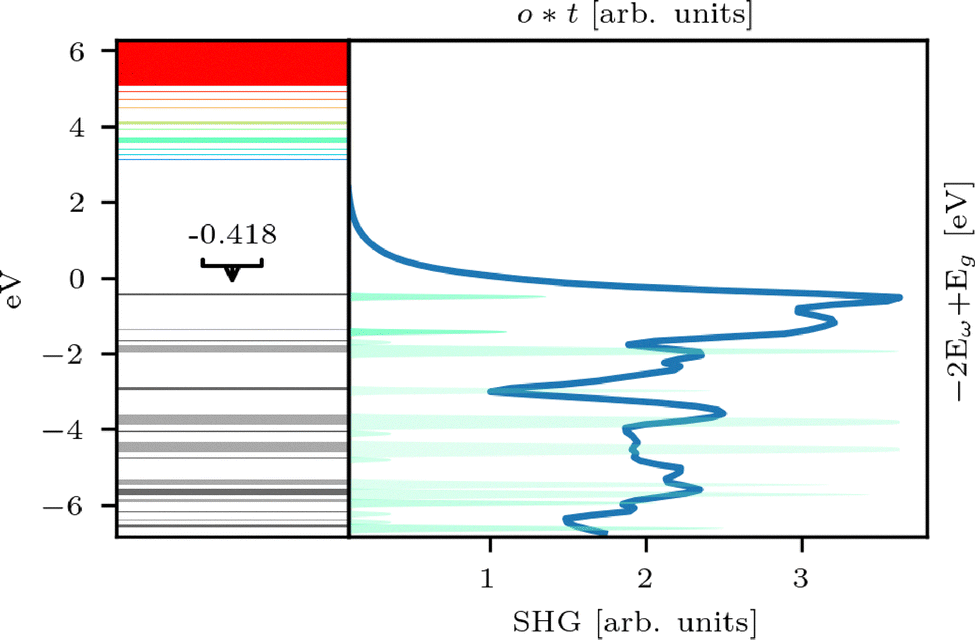 | ||
| Fig. 13 Orbital group energy values of [(PhC)4S6] with corresponding scaled and shifted SHG response. The energy values of the origin (occupied) orbital groups are shifted for a direct transition to the respective target (unoccupied) orbital group 3. | ||
The target orbital groups t = 0, 1, 2 have energies lower and the target groups t = 4, 5 have energies higher than the first SHG peak. The overlap of all the target states with o = 0 is depicted in Fig. 14. All overlaps have a large localization at the S atoms, however, in the case of o = 0, t = 3 a certain localization at the substituents is observed. This corroborates the hypothesis that the substituents are the dominant factor in defining the SHG characteristics and magnitude.
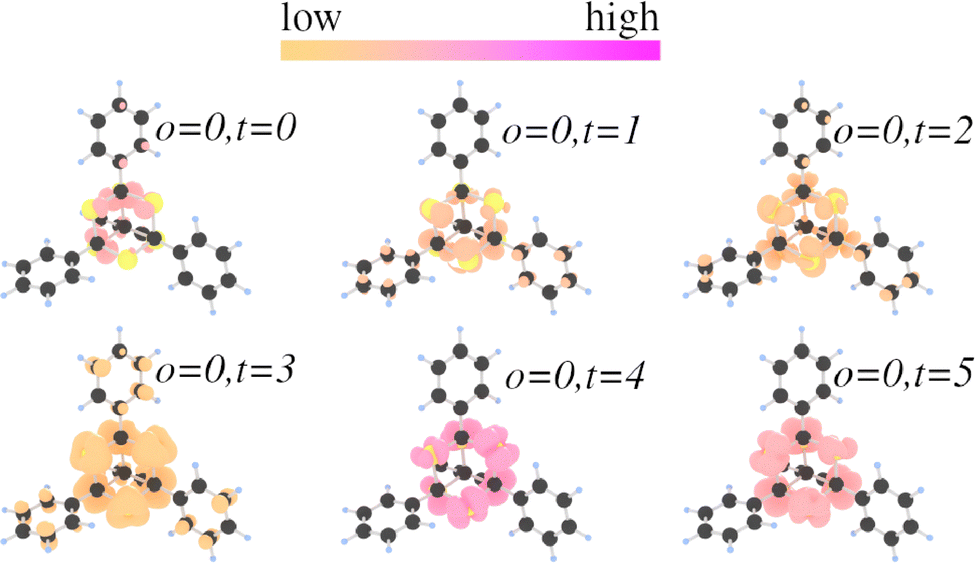 | ||
| Fig. 14 Overlaps ρ of o = 0 with t = 0, 1, 2, 3, 4, 5 within [(PhC)4S6]. The atoms are color-coded by atom type: S atoms are yellow, C atoms are black, and H atoms are light blue. | ||
The second peak in the SHG spectrum of [(PhC)4S6] originates from electronic transitions of lower origin orbitals to the target groups t = 0, 1, 2, 3. The corresponding overlaps are shown in the Fig. 15–18. The color coding of the convolution is chosen according to the target orbital groups shown in Fig. 13. The isosurface choice is arbitrary, however, the scale for the isosurface value are shared. The analysis of the transitions leading to the second SHG peak shows cleary that the highest convolution in correspondence of the SHG peak ist given by transitions characterized by a large overlap at the phenyl substituents, exactly as in the case of the clusters comprising Si atoms at the bridgehead positions. Transitions with some overlap at the S atoms are present, however, their contribution to the SHG peak is less pronounced.
 | ||
| Fig. 15 Overlaps ρ of o = 1, 2, 3 with t = 0, related to the second peak of the SHG spectrum for [(PhC)4S6]. The atoms are color-coded by atom type: S atoms are yellow, C atoms are black, and H atoms are light blue. | ||
 | ||
| Fig. 16 Overlaps ρ of o = 1, 2 with t = 1, related to the second peak of the SHG spectrum for [(PhC)4S6]. The atoms are color-coded by atom type: S atoms are yellow, C atoms are black, and H atoms are light blue. | ||
 | ||
| Fig. 17 Overlaps ρ of o = 1, 2 with t = 2, related to the second peak of the SHG spectrum for [(PhC)4S6]. The atoms are color-coded by atom type: S atoms are yellow, C atoms are black, and H atoms are light blue. | ||
 | ||
| Fig. 18 Overlaps ρ of o = 2 with t = 3, related to the second peak of the SHG spectrum for [(PhC)4S6]. The atoms are color-coded by atom type: S atoms are yellow, C atoms are black, and H atoms are light blue. | ||
Summarizing, we assign the first peak of all Si containing clusters as well as to the second peak of [(PhC)4S6] to electronic transitions localized at the substituents. The first SHG peak of [(PhC)4S6] is due to different transitions, also involving the S atoms in the cluster core. By the stepwise replacement of the C atoms in the core, the probability of these transitions decreases, correspondingly the intensity of the related SHG peak drops and the peak becomes a more or less pronounced shoulder in [(PhSi)(PhC)3S6], [(PhSi)2(PhC)2S6] and [(PhSi)3(PhC)S6], before it completely vanishes in [(PhSi)4S6].
As previously discussed, electronic transitions from the HOMO and neighboring electronic levels do not contribute to the SHG response in the technologically relevant region from 0 to ca. 3 eV. The analysis of the electronic transitions reveals that this spectral region is determined by transitions from a peculiar orbital group consisting of 11 energy levels. This group, as calculated within DFT-PBE is highlighted in Fig. 19 for all the investigated clusters. In the picture, the origin of the energy scala (the 0.0 eV level) is the HOMO level of each structure. The energy levels in this orbital group drift further apart with the C content. Correspondingly, the SHG intensity in this region is spread over a broader region, and the first SHG peak is less high toward [(PhSi)4S6], with the latter being the only exception. Corresponding calculations performed within hybrid-DFT (see SI) also confirm this interpretation. The electronic states within the mentioned orbital group are spread over a slightly larger energy window (by ca. 0.15 eV, depending on the cluster), otherwise they closely match the PBE calculations.
 | ||
| Fig. 19 Electronic energy values of the investigated structures. The orbital group responsible for the SHG response in the region below 3 eV is highlighted. It contains 11 individual electronic levels and widens from left to right. The 0.0 eV level is the HOMO level. Only occupied orbitals are plotted. | ||
Another way to interpret the enhanced SHG intensity of the intermediate structures is based on their geometry. Indeed, previous studies41 have revealed that the SHG intensity of molecular clusters decreases with the core symmetry. To verify this assumption for the investigated materials family, we examine some geometrical features in the following. In Table 1 the average T–S bond lengths (T = C, Si) are listed. The average bond length grows as expected with the number of Si atoms, closely mirroring the sum of the covalent radii. Correspondingly, the core size increases.
| Structure | min. [Å] | max. [Å] | avg. [Å] |
|---|---|---|---|
| [(SiPh)4S6] | 2.140 | 2.153 | 2.15 |
| [(SiPh)3(CPh)S6] | 1.840 | 2.153 | 2.07 |
| [(SiPh)2(CPh)2S6] | 1.839 | 2.148 | 2.00 |
| [(SiPh)(CPh)3S6] | 1.825 | 2.133 | 1.92 |
| [(CPh)4S6] | 1.827 | 1.837 | 1.83 |
In order to estimate the cluster core volume, a convex hull's volume of the adamantane-like core is considered (excluding H atoms). The resulting volumes are shown in Table 2. Along with the volume expansion, a distortion of the core is observed. The distortion, quantified by the difference between minimal and maximal bond length enhances the deviation of the cluster symmetry from the ideal tetragonal symmetry. A visual representation of the relative distortion of the investigated molecular clusters can be found in the SI. The largest deviation δ from the average bond length is predicted for the [(SiPh)3(CPh)S6] (see Table 2), which indeed is the cluster featuring the highest SHG peak. This might be interpreted either by a symmetry related modification of the selection rules for the optically active transitions or by a geometry related redistribution of the electronic levels in a narrower energy region.
| Structure | Core volume [Å3] | % | δ [Å] |
|---|---|---|---|
| [(SiPh)4S6] | 25.8 | 100 | 0.007 |
| [(SiPh)3(CPh)S6] | 23.1 | 89 | 0.228 |
| [(SiPh)2(CPh)2S6] | 20.6 | 79 | 0.156 |
| [(SiPh)(CPh)3S6] | 18.3 | 70 | 0.218 |
| [(CPh)4S6] | 16.1 | 62 | 0.006 |
As a matter of fact, the DOS of the unoccupied sates of [(PhSi)4S6] and [(PhC)4S6] in a region between the LUMO and LUMO+0.3 eV is much more similar than for the intermediate structures. This similarity can be quantified by the distance
 | (1) |
Independently from the physical origin of the mechanism leading to this effect, also in the investigated cluster family the reduced core symmetry results in an enhanced SHG intensity.
4 Conclusions
The nonlinear optical response of phenyl-decorated clusters with general formula [(CPh)x(SiPh)4−xS6] and x = 0,…,4 has been calculated from first principles within DFT-PBE. Corresponding hybrid-DFT calculations confirm the chemical trends identified in this investigation and the interpretation of the results. All clusters feature SHG spectra characterized by corresponding optical signatures, with the exception of an intense SHG peak for [(CPh)4S6], which loses intensity by the stepwise replacement of C with Si, and vanishes in the [(SiPh)4S6] cluster. The spectra are dominated by a main peak at energies of about 2.2 eV, substantially higher than half of the HOMO–LUMO gap. The SHG intensity related to this peak grows with the substitution induced distortion of the cluster core. The analysis of the electronic transitions associated with the main SHG peak reveals that the involved orbitals are localized at the substituents. Thus, the origin of the optical nonlinearities in the technologically relevant energy interval from 0 to 3 eV is spatially located at the phenyl rings. On the one hand, this explains why many phenyl-decorated adamantane-type clusters show similar spectra.5 On the other hand, this confirms the assumption that the presence of delocalized π-orbitals at the substituents is a prerequisite for the high nonlinear optical activity of such molecular clusters.4 However, in comparison with previously investigated clusters' extreme nonlinear optical properties,18 where the origin of the optical response was attributed exclusively to the substituents, we predict in this work a contribution of the S atoms of the core, in particular for [(CPh)4S6] cluster. A further difference with previously investigated clusters is that transitions between HOMO and LUMO states do not contribute to the optical nonlinearities.To put the nonlinear optical response of the molecular clusters presented in this work into the broader context of other adamantane-shaped molecular clusters with strong nonlinear optical properties, we compare the end compounds of our investigation, [(PhC)4S6] and [(PhSi)4S6] with their S-free counterparts. The clusters [(PhC)4(CH2)6] and [(PhSi)4(CH2)6], in which the S atoms in the core are replaced by CH2, had been previously investigated.18 The corresponding SHG spectra are shown in Fig. 20.
 | ||
| Fig. 20 Calculated SHG response of adamantane-type clusters [(PhC)4S6] and [(PhSi)4S6] with their S-free counterparts [(PhC)4(CH2)6] and [(PhSi)4(CH2)6]. | ||
The intensity of the nonlinear response is of the same order of magnitude for all the investigated molecular clusters, with the clusters containing S atoms showing in general higher intensities. In the 0–3 eV energy range, all the spectra are dominated by the peak originating from electronic transitions within the substituents. Yet, for the molecular clusters containing S atoms, the peak occurs at lower energies, closer to the usually employed excitation laser frequencies. Moreover, the presence of S atoms strongly enhances the SHG response of the prototypical AdPh4 molecular cluster (here labeled as [(PhC)4(CH2)6]).
Summarizing, the nonlinear optical response of the molecular clusters investigated in the present work is generally higher (yet comparable) in magnitude to that of corresponding molecular clusters without S atoms. Moreover, due to the redshift of the spectral features and (in the case of AdPh4) to an enhanced SHG intensity, the molecular clusters containing S atoms are better emitters if the excitation is achieved by lasers in the visible range.
The present investigation represents a crucial step towards the understanding of the nonlinear optical response of the materials class with more general formula [(RT)4E6] with R = organic group; T = C, Si, Ge, Sn; E = O, S, Se, Te, NH, CH2 and provides a theoretical foundation for the design of materials with tailored nonlinear optical properties.
Author contributions
F. Z. and S. S. conceived the project. F. Z. performed the calculations and analysis with support from S. S. and S. D. The manuscript was written by F. Z. and S. S. and contributed to by all authors. All authors reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
Atom coordinates are provided in the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5cp03750f.An overview of how computational parameters were chosen for non linear optical calculations is provided in the supplementary information. Further computational details will be provided upon reasonable request. To do so please contact Ferdinand Ziese.
Acknowledgements
This work is supported by the German Research Foundation (DFG) through the research group FOR2824 (grant no. 398143140, project SA1948/2-2). Calculations for this research were conducted on the Lichtenberg high performance computer of the TU Darmstadt and at the Höchstleistungrechenzentrum Stuttgart (HLRS). The authors furthermore acknowledge the computational resources provided by the HPC Core Facility and the HRZ of the Justus-Liebig-Universität Gießen.References
- N. W. Rosemann, J. P. Eußner, E. Dornsiepen, S. Chatterjee and S. Dehnen, J. Am. Chem. Soc., 2016, 138, 16224–16227 CAS.
- N. W. Rosemann, J. P. Eußner, A. Beyer, S. W. Koch, K. Volz, S. Dehnen and S. Chatterjee, Science, 2016, 352, 1301–1304 CrossRef CAS PubMed.
- D. Li, W. Hu, J. Wang, Q. Zhang, X.-M. Cao, X. Ma and H. Tian, Chem. Sci., 2018, 9, 5709–5715 RSC.
- S. Dehnen, P. R. Schreiner, S. Chatterjee, K. Volz, N. W. Rosemann, W.-C. Pilgrim, D. Mollenhauer and S. Sanna, ChemPhotoChem, 2021, 5, 1033–1041 CrossRef CAS.
- N. Rinn, I. Rojas-León, B. Peerless, S. Gowrisankar, F. Ziese, N. W. Rosemann, W.-C. Pilgrim, S. Sanna, P. R. Schreiner and S. Dehnen, Chem. Sci., 2024, 15, 9438–9509 RSC.
- I. Rojas-León, J. Christmann, S. Schwan, F. Ziese, S. Sanna, D. Mollenhauer, N. W. Rosemann and S. Dehnen, Adv. Mater., 2022, 34, 2203351 CrossRef.
- F. Schütt, M. Zapf, S. Signetti, J. Strobel, H. Krüger, R. Röder, J. Carstensen, N. Wolff, J. Marx, T. Carey, M. Schweichel, M.-I. Terasa, L. Siebert, H.-K. Hong, S. Kaps, B. Fiedler, Y. K. Mishra, Z. Lee, N. M. Pugno, L. Kienle, A. C. Ferrari, F. Torrisi, C. Ronning and R. Adelung, Nat. Commun., 2020, 11, 1437 CrossRef.
- J. Wu, G. Zheng, X. Liu and J. Qiu, Chem. Soc. Rev., 2020, 49, 3461–3483 RSC.
- J. Gotta, T. B. Shalom, S. Aslanoglou, A. Cifuentes-Rius, N. H. Voelcker, R. Elnathan, O. Shoseyov and S. Richter, Adv. Funct. Mater., 2018, 28, 1706967 CrossRef.
- J. R. Stellhorn, S. Hayakawa, B. D. Klee, B. Paulus, J. Link Vasco, N. Rinn, I. Rojas León, C. A. Hosier, S. Dehnen and W.-C. Pilgrim, Adv. Opt. Mater., 2023, 11, 2201932 CrossRef CAS.
- B. Peters, N. Lichtenberger, E. Dornsiepen and S. Dehnen, Chem. Sci., 2020, 11, 16–26 CAS.
- J. M. T. Thompson and J. M. Cole, Philos. Trans. R. Soc., A, 2003, 361, 2751–2770 CrossRef PubMed.
- E. Dornsiepen, F. Dobener, N. Mengel, O. Lenchuk, C. Dues, S. Sanna, D. Mollenhauer, S. Chatterjee and S. Dehnen, Adv. Opt. Mater., 2019, 7, 1801793 Search PubMed.
- M. J. Müller, F. Ziese, J. Belz, F. Hüppe, S. Gowrisankar, B. Bernhardt, S. Schwan, D. Mollenhauer, P. R. Schreiner, K. Volz, S. Sanna and S. Chatterjee, Opt. Mater. Express, 2022, 12, 3517–3529 Search PubMed.
- J. Belz, J. Haust, M. J. Müller, K. Eberheim, S. Schwan, S. Gowrisankar, F. Hüppe, A. Beyer, P. R. Schreiner, D. Mollenhauer, S. Sanna, S. Chatterjee and K. Volz, J. Phys. Chem. C, 2022, 126, 9843–9854 CAS.
- S. Schwan, A. J. Achazi, F. Ziese, P. R. Schreiner, K. Volz, S. Dehnen, S. Sanna and D. Mollenhauer, J. Comput. Chem., 2023, 44, 843–856 CAS.
- K. Eberheim, C. Dues, C. Attaccalite, M. J. Müller, S. Schwan, D. Mollenhauer, S. Chatterjee and S. Sanna, J. Phys. Chem. C, 2022, 126, 3713–3726 CAS.
- F. Ziese, J. Wang, I. Rojas León, S. Dehnen and S. Sanna, J. Phys. Chem. A, 2024, 128, 8360–8372 CAS.
- K. Hanau, S. Schwan, M. R. Schäfer, M. J. Müller, C. Dues, N. Rinn, S. Sanna, S. Chatterjee, D. Mollenhauer and S. Dehnen, Angew. Chem., Int. Ed., 2021, 60, 1176–1186 CAS.
- M. A. Bonnin, L. Bayarjargal, S. Wolf, V. Milman, B. Winkler and C. Feldmann, Inorg. Chem., 2021, 60, 15653–15658 CAS.
- J. Salafsky, Chem. Phys. Lett., 2001, 342, 485–491 CrossRef CAS.
- E. Donohue, S. Khorsand, G. Mercado, K. M. Varney, P. T. Wilder, W. Yu, A. D. MacKerell, P. Alexander, Q. N. Van, B. Moree, A. G. Stephen, D. J. Weber, J. Salafsky and F. McCormick, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 17290–17297 CrossRef CAS PubMed.
- L. Persechini and J. F. McGilp, Phys. Status Solidi B, 2012, 249, 1155–1159 CrossRef CAS.
- B. Moree, G. Yin, D. F. Lázaro, F. Munari, T. Strohäker, K. Giller, S. Becker, T. F. Outeiro, M. Zweckstetter and J. Salafsky, J. Biol. Chem., 2015, 290, 27582–27593 CrossRef CAS.
- S. Chen, Y. Hou, B. Xu, S. Chen, Q. Yuan and W. Gan, Colloids Surf., A, 2024, 683, 133004 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 224106 CrossRef PubMed.
- D. R. Hamann, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 085117 CrossRef.
- M. Schlipf and F. Gygi, Comput. Phys. Commun., 2015, 196, 36–44 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- C. Attaccalite and M. Grüning, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 235113 CrossRef.
- D. Sangalli, A. Ferretti, H. Miranda, C. Attaccalite, I. Marri, E. Cannuccia, P. Melo, M. Marsili, F. Paleari, A. Marrazzo, G. Prandini, P. BonfÃ, M. O. Atambo, F. Affinito, M. Palummo, A. Molina-Sánchez, C. Hogan, M. Grüning, D. Varsano and A. Marini, J. Phys.: Condens. Matter, 2019, 31, 325902 CrossRef CAS PubMed.
- L. E. Johnson, L. R. Dalton and B. H. Robinson, Acc. Chem. Res., 2014, 47, 3258–3265 CrossRef CAS PubMed.
- D. Jacquemin, E. A. Perpète, M. Medved’, G. Scalmani, M. J. Frisch, R. Kobayashi and C. Adamo, J. Chem. Phys., 2007, 126, 191108 CrossRef PubMed.
- F. Castet and B. Champagne, J. Chem. Theory Comput., 2012, 8, 2044–2052 CrossRef CAS PubMed.
- F. Ziese and S. Sanna, ACS Omega, 2024, 9, 49816–49824 CrossRef CAS PubMed.
- J. Wang, I. Rojas-León, N. Rinn, L. Guggolz, F. Ziese, S. Sanna, N. W. Rosemann and S. Dehnen, Angew. Chem., Int. Ed., 2024, e202411752 CAS.
Footnote |
| † The spectrum is scaled by a factor of 2 to share the energy axis of the electronic energy values |
| This journal is © the Owner Societies 2026 |