Mechanisms and regio- and stereoselectivities in NHC-catalyzed [3+3] annulations for the synthesis of axially and centrally chiral dihydropyridinones
Received
16th September 2025
, Accepted 24th November 2025
First published on 25th November 2025
Abstract
Organocatalytic annulation strategies serve as an ideal approach for constructing molecules integrating both axial and central chirality, yet theoretical studies on the origin of stereoselectivity remain insufficient. In this study, the NHC-catalyzed [3+3] annulation between cinnamaldehyde and 2-aminomaleate was systematically investigated through density functional theory (DFT) calculations. The catalytic cycle involves eight key steps: nucleophilic addition, 1,2-proton transfer, oxidation, Michael addition, protonation, deprotonation, ring closure, and catalyst dissociation. Deprotonation is identified as the rate-determining step, with an overall energy barrier of 19.7 kcal mol−1. The Michael addition step is the stereoselectivity-controlling step that preferentially generates the R-configuration product, consistent with the experimental results. The calculated enantiomeric excess value of 89.3% aligns well with experimental observations (90% ee). Noncovalent interaction (NCI) analysis revealed that the stability of the key stereoselective transition state can be traced to LP⋯π, C–H⋯O and C–H⋯N interactions. The mechanistic insights should be helpful for understanding the origins of stereoselectivity in NHC-catalyzed atroposelective annulations.
1. Introduction
Chirality is a fundamental property of nature1 and includes various forms such as central chirality, axial chirality, and face chirality. One of the most cutting-edge fields in organic synthesis is the enantioselective synthesis of chiral compounds by asymmetric catalysis.2 Especially in the last decade, a class of axially chiral molecules have been synthesized using asymmetric catalytic strategies that can serve as important structural units for drugs, chiral materials and bioactive molecules.3–8 Among them, atropisomers combining both axial and central chirality can serve as fundamental scaffolds for organocatalysts and chiral ligands in asymmetric synthetic reactions, thus attracting increasing attention.9–14 Previous studies showed that organocatalysts or ligands integrating central and axial chirality can significantly enhance chiral transfer in diverse enantioselective transformations.15–18 Therefore, the synthesis of atropisomers bearing central and axial chiral elements is of great importance.
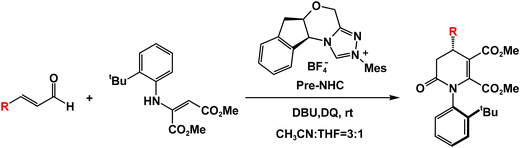
As a highly efficient and environmentally benign organocatalyst, N-heterocyclic carbene (NHC) exhibits unique activation modes and provides diverse asymmetric synthetic pathways for constructing chiral molecules.19–23NHC-catalyzed annulation/addition reactions, kinetic resolution (KR) and desymmetrization are practical methods for constructing atropisomers.24–29 It is worth noting that the lower stability of the C–N axis renders the construction of atropisomers around this bond particularly challenging.30–34 Most reported NHC-catalyzed strategies predominantly construct axial chirality alone, with only a few examples achieving construction of C–N axial chirality with central chirality simultaneously. In this context, theoretical investigations to elucidate detailed reaction mechanisms become critically important for developing novel, efficient, and sustainable methods to synthesize atropisomers concurrently bearing both axial and point chirality. Recently, Qi's group35 reported the NHC-catalyzed [3+3] annulation reaction between cinnamaldehyde and 2-aminomaleate, which afforded dihydropyridinones bearing both axial and central chirality with high yield (74%) and stereoselectivity (90%). Scheme 1 shows the details of the experiment.
 |
| | Scheme 1 Reaction model of cinnamaldehyde and 2-aminomaleate. | |
In recent years, numerous computational studies have been reported to investigate NHC-catalyzed asymmetric synthesis reactions for understanding their detailed mechanism.36–42 However, the reaction developed by Qi's group has not yet been theoretically investigated, and several critical questions remain unresolved: (1) what is the catalytic mechanism of this reaction? (2) What are the key factors controlling stereoselectivity? (3) Four potential mechanisms for oxidation were proposed: hybrid transfer to the oxygen/carbon (HTO/HTC),43–46 single electron transfer followed by hydrogen atom transfer (SET-HAT)47–50 and hydrogen atom transfer followed by single electron transfer (HAT-SET).51–54 Which one is more preferred? (4) What factors are responsible for maintaining axial chirality? In this work, we used density functional theory (DFT) computations to examine the catalytic reaction in order to answer these fundamental problems.
2. Computational details
All calculations in this study are based on density functional theory (DFT) and have been performed using the Gaussian16 program (Revision A03).55 Density functional theory has shown excellent performance in reactions such as transition metal catalysis56–62 and organocatalysis.63–69 Geometry optimization of reactants, transition states, intermediates and products was carried out using the M06-2X functional70 with the 6-31G(d,p) basis set. The M06-2X functional has been employed in many previous theoretical studies of NHC-catalyzed reactions71–74 owing to its good performance in describing thermodynamics, kinetics and noncovalent interactions. The mixed solvents (CH3CN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :THF = 3:1) were simulated using the IEF-PCM75,76 implicit solvation model. In this model, the mixed solvent is treated as a continuous medium, characterized by the dielectric constant (ε = 28.622 and ε∞ = 1.847), which embeds a molecular-shape cavity containing the solute. Frequency analyses were subsequently conducted at the same level to confirm the nature of stationary points: minima (reactants, intermediates, and products) showed no imaginary frequencies, while transition states exhibited one imaginary frequency. Afterward, intrinsic reaction coordinate (IRC)77,78 calculations were performed to verify the connectivity between each transition state and its associated reactants/products. For more accurate energy evaluations, single point energy calculations were carried out using the M06-2X-D3 functional with the 6-311++G(2df,2dp) basis set and the IEF-PCM implicit solvation model. To validate the computational level, alternative DFT methods (ωB97X-D79 and B3LYP-D380), basis sets (6-311++G(d,p) and 6-31G(d,p)), and solvation models (IEF-PCM and SMD81) were tested for the stereoselectivity-controlling step, with detailed discussions provided in Tables S1 and S2, SI. Analysis of the noncovalent interaction (NCI)82 was conducted using Multiwfn software83,84 to elucidate the origins of stereoselectivity. Optimized geometries were visualized using CYLView.85 SambVca86 was employed to calculate the buried volume and generate topographic steric maps of the products.
:THF = 3:1) were simulated using the IEF-PCM75,76 implicit solvation model. In this model, the mixed solvent is treated as a continuous medium, characterized by the dielectric constant (ε = 28.622 and ε∞ = 1.847), which embeds a molecular-shape cavity containing the solute. Frequency analyses were subsequently conducted at the same level to confirm the nature of stationary points: minima (reactants, intermediates, and products) showed no imaginary frequencies, while transition states exhibited one imaginary frequency. Afterward, intrinsic reaction coordinate (IRC)77,78 calculations were performed to verify the connectivity between each transition state and its associated reactants/products. For more accurate energy evaluations, single point energy calculations were carried out using the M06-2X-D3 functional with the 6-311++G(2df,2dp) basis set and the IEF-PCM implicit solvation model. To validate the computational level, alternative DFT methods (ωB97X-D79 and B3LYP-D380), basis sets (6-311++G(d,p) and 6-31G(d,p)), and solvation models (IEF-PCM and SMD81) were tested for the stereoselectivity-controlling step, with detailed discussions provided in Tables S1 and S2, SI. Analysis of the noncovalent interaction (NCI)82 was conducted using Multiwfn software83,84 to elucidate the origins of stereoselectivity. Optimized geometries were visualized using CYLView.85 SambVca86 was employed to calculate the buried volume and generate topographic steric maps of the products.
3. Results and discussion
3.1. Reaction mechanism
Based on the experimental results of Qi's group35 and our DFT computations, we proposed a mechanism for the NHC-catalyzed oxidative [3+3] annulation reaction involving cinnamaldehyde and 2-aminomaleate. The key steps of the catalytic cycle are described in Scheme 2. The precatalyst Pre-NHC undergoes deprotonation using base 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) to generate the actual catalyst NHC and DBU·H+ (see Fig. S1, SI). Additionally, Pre-R2 undergoes deprotonation to yield R2 (see Fig. S2, SI). The catalytic cycle consists of the following eight steps: (1) nucleophilic attack of NHC to R1 to generate intermediate M1, (2) the 1,2-proton transfer process to form the Breslow intermediate M2, (3) the oxidation process to produce intermediate M3, (4) addition of M3 to R2 to produce intermediate M4, (5) protonation of M4 to produce intermediate M5, (6) deprotonation to produce intermediate M6, (7) ring-closure to form intermediate M7, and (8) dissociation of NHC from the final product P. In the subsequent section, we will present the mechanism in terms of eight steps.
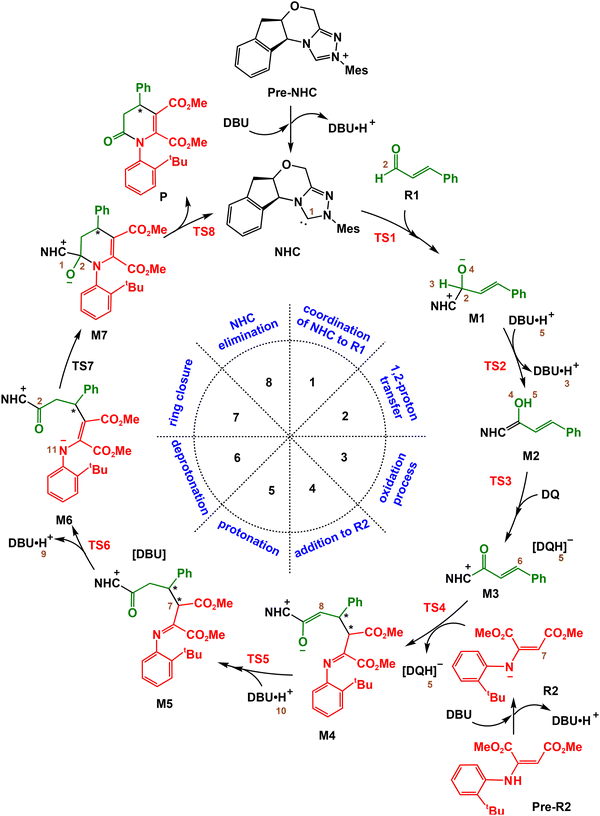 |
| | Scheme 2 The possible catalytic cycle for [3+3] cyclization. | |
3.1.1. First step: nucleophilic addition.
In this step, the attack of the C1 atom of NHC on the re/si-face of the C2 atom of R1 results in the formation of the intermediate re/si-M1 (Scheme 2). The calculated energy barrier of 12.3/12.8 kcal mol−1 for re/si-TS1 indicates that the nucleophilic addition is feasible under the experimental conditions (Fig. 1). By comparison, we found that the energy barrier difference between re-TS1 and si-TS1 is within the error margin of the computational method used, and re-M1 (6.2 kcal mol−1) and si-M1 (6.3 kcal mol−1) exhibit similar stability. Therefore, we considered both transformation pathways. The C1–C2 bond is reduced from 2.03/2.04 Å in re/si-TS1 to 1.54/1.54 Å in re/si-M1, as shown in Fig. 2 and Fig. S3, indicating the complete complexation between the catalyst and the reactant.
 |
| | Fig. 1 Free energy profiles (kcal mol−1) of the catalytic cycle. | |
 |
| | Fig. 2 Optimized geometries of intermediates and transition states (distance in Å). | |
3.1.2. Second step: 1,2-proton transfer.
The 1,2-proton transfer of M1 leads to the formation of the Breslow intermediate M2, wherein proton H3 migrates from the C2 atom to the O4 atom. As illustrated in Scheme 3, three possible mechanisms were examined to identify the most plausible pathway. Computational results revealed that the direct proton transfer process exhibits a high energy barrier of 40.0/42.2 kcal mol−1 (re/si-TS2D, Fig. S4, SI), rendering this pathway unlikely. This result aligns with previous studies that direct proton transfer involves a high barrier.36,41,42 Further investigation focused on the proton transfer assisted by DBU. The presence of a DBU molecule significantly reduced the energy barrier to 17.7/17.7 kcal mol−1 (re/si-TS2B, Fig. S4, SI). Additionally, the energy barriers were further reduced with the introduction of DBU·H+ as a mediator, reaching 13.0 and 18.4 kcal mol−1viare-TS2 and si-TS2, respectively (Fig. 1).
 |
| | Scheme 3 Possible mechanism for M1 → M2 conversion (energies in kcal mol−1). | |
In summary, under the assistance of DBU·H+, the pathway of generating intermediate re-M2 through 1, 2-proton transfer has a significantly lower energy barrier and is the most feasible reaction pathway. This result is consistent with previous studies that protic mediators play crucial roles in proton transfer.38–42 Alternative [1,2]-proton transfer mechanisms can thus be excluded.
3.1.3. Third step: oxidation.
In the third step of the catalytic cycle, the Breslow intermediate re-M2 undergoes a hydride transfer to oxygen (HTO) process. As illustrated in Fig. 1, the transfer of hydride H5 from M2 to 3,3′,5,5′-tetra-tert-butyldiphenoquinone (DQ) generates intermediate M3via the transition state TS3. This step requires overcoming a remarkably low energy barrier of 1.7 kcal mol−1via the transition state TS3, yielding intermediate M3 with a relative energy of −20.1 kcal mol−1. Fig. 2 demonstrates the formation of the O1′–H5 bond, as evidenced by the shortening of its bond length from 1.25 Å in the transition state TS3 to 0.96 Å in intermediate M3. Additionally, three other possible mechanisms including the hydride transfer to carbon pathway (HTC), the single electron transfer-hydrogen atom transfer pathway (SET-HAT), and the hydrogen atom transfer-single electron transfer pathway (HAT-SET) were evaluated. As shown in Schemes S1 and S2, SI, all these pathways required overcoming relatively higher energy barriers compared to the HTO pathway and were thus excluded from further consideration. These results are consistent with previous studies showing that the HTO pathway is the most feasible pathway for oxidation.87,88
3.1.4. Fourth step: addition of M3 to R2.
In the Michael addition step, the re/si face of reactant R2 can react with the re/si face of intermediate M3, leading to the formation of four stereoisomeric transition states (Fig. 3). As shown in Fig. 1, after passing through the transition states TS4(RR) (5.4 kcal mol−1), TS4(RS) (11.6 kcal mol−1), TS4(SR) (5.1 kcal mol−1), and TS4(SS) (3.7 kcal mol−1), the intermediates M4(RR), M4(RS), M4(SR), and M4(SS) were formed with relative energies of −29.1, −26.2, −30.4, and −30.9 kcal mol−1, respectively. Notably, conformational analysis of TS4 (Fig. S5 and Table S3, SI) confirms that the selected conformers (TS4(RR/RS/SR/SS)) in Fig. 1 exhibit the lowest energy. By comparison, the reaction pathway viaTS4(SS) is energetically more favorable compared to the other three pathways. Therefore, only the SS-configuration pathway will be discussed in the main text, and the pathways of the RR/RS/SR-configuration can be found in Fig. S6, SI. As shown in Fig. 2, the C6–C7 bond decreases from 2.39 Å in TS4(SS) to 1.60 Å in M4(SS). This change confirms the formation of the C6–C7 bond.
 |
| | Fig. 3 Possible stereochemistry of Michael addition. | |
3.1.5. Fifth step: protonation.
In the fifth step, the proton H10 of DBU·H+ is transferred to the C8 atom of intermediate M4(SS)via transition state TS5(SS), yielding intermediate M5(SS) (Scheme 4). As illustrated in Fig. 1, the proton transfer requires overcoming an energy barrier of 12.5 kcal mol−1. As shown in Fig. 2, from TS5(SS) to M5(SS), the length of the N15–H10 bond is prolonged from 1.40 to 2.06 Å, whereas the C8–H10 bond is reduced from 1.33 to 1.11 Å. These changes in the bond length confirm the dissociation of the N15–H10 bond and the formation of the C8–H10 bond.
 |
| | Scheme 4 Possible mechanism for M4 → M6 conversion (energies in kcal mol−1). | |
3.1.6. Sixth step: deprotonation.
In the sixth step, the proton H9 is transferred from the C7 atom of intermediate M5(SS) to DBUvia transition state TS6(SS), generating intermediate M6(R). This process requires overcoming an energy barrier of 10.9 kcal mol−1 (Fig. 1). The deprotonation (M4(SS) → TS6(SS)) is also found to be the rate-determining step, with an overall energy barrier of 19.7 kcal mol−1. It is noteworthy that after deprotonation, the C7 atom becomes achiral, and the C6 atom experiences chirality inversion. For the sake of clarity, the R/S in the name of the stationary points (M6 → P) indicates the chirality of the C6 atom. As demonstrated in Fig. 2, the C7–H9 bond length increases from 1.09 Å in intermediate M5(SS) to 1.41 Å in transition state TS6(SS), indicating the cleavage of the C7–H9 bond.
For the transformation of M4(SS) to M6(R), the direct and DBU-assisted pathways were also examined (Scheme 4). However, these pathways can be excluded due to the related high energy barriers. For details, see Fig. S7, SI.
3.1.7. Seventh step: ring-closure process.
In the seventh step, the N11 atom in intermediate M6(R) attacks the C2 atom viaTS7(R), forming the six-membered ring intermediate M7(R). This process requires overcoming an energy barrier of 10.8 kcal mol−1 (Fig. 1). As shown in Fig. 2, the C2–N11 bond decreases from 1.99 Å in TS7(R) to 1.58 Å in M8(R), indicating the formation of the C2–N11 bond.
3.1.8. Eighth step: elimination of NHC.
In the last step, intermediate M7(R) undergoes C1–C2 bond cleavage through transition state TS8(R), yielding product P(R) and regenerating the NHC catalyst. Energy calculations (Fig. 1) indicate that this step proceeds via a low energy barrier of 1.3 kcal mol−1. As shown in Fig. 2, the length of the C1–C2 bond increases from 1.60 Å in M7(R) to 1.93 Å in TS8(R), indicating bond dissociation.
3.2. Other regioselective reaction pathways
In addition to the Michael addition of R2 and M3 discussed above, computational investigations were extended to the C2–C7 and C2–N11 bond-forming pathways. In the C2–C7 bond formation pathway, the re/si face of reactant R2 reacts with the re/si face of intermediate M3 generating intermediates NM4(RR), NM4(RS), NM4(SR), and NM4(SS) (Fig. S8). The related energy barriers are 12.0, 11.8, 8.7 and 9.0 kcal mol−1 for NTS4(RR/RS/SR/SS), which are much higher than the 3.7 kcal mol−1 for TS4SS (Michael addition), ruling out these pathways.
In the C2–N11 bond formation pathway, the reactant R2 attacks the re/si face of the C2 atom in intermediate M3. Fig. S9 shows that after passing through transition states re-TS4 (17.0 kcal mol−1) and si-TS4 (14.2 kcal mol−1), the intermediates re-M4 and si-M4 are formed with relative energies of −11.2 and −8.1 kcal mol−1, respectively. Compared with the Michael addition pathway (ΔG‡ = 3.7 kcal mol−1, TS4(SS)), these two pathways require higher energy barriers, and the stabilities of re/si-M4 are significantly lower than that of intermediate M4(SS). Therefore, these two pathways are excluded.
3.3. Origin of stereoselectivity
Two chiral centers (C6 and C7) were generated in the Michael addition of M3 to R2, indicating that this step is the stereoselectivity-controlling step. Based on the preceding discussion, the si-face of R2 reacts with the re-face of the acyl intermediate M3viaTS4SS yielding the R-configuration product, which is energetically most favorable. As shown in Fig. 1, the energy barrier difference between transition states TS4(RR) and TS4(SS) is 1.7 kcal mol−1, corresponding to an enantiomeric excess (ee) of 89.3%, in excellent agreement with the experimentally observed value of 90% ee.35
To elucidate the origin of stereoselectivity, noncovalent interaction (NCI) analysis was performed to investigate the nature of interactions between R2 and M3. Fig. 4 shows that transition states TS4(RR), TS4(SR) and TS4(SS) exhibit three C–H⋯O, one C–H⋯N, three C–H⋯π and one π⋯π interactions. Although the lone pair LP⋯π interaction exists in these three transition states, the LP⋯π interaction in TS4(SS) is stronger than that in TS4(RR)/TS4(SR) (3.065 and 3.312 versus 3.022/3.090). Additionally, the interactions (two C–H⋯O, two C–H⋯π, and two LP⋯π interactions) in TS4(RS) are weaker than those in TS4(SS). Overall, the noncovalent interactions in TS4(SS) are stronger than those in TS4(RR/RS/SR), which leads to the pathway associated with TS4(SS) having a lower energy barrier.
 |
| | Fig. 4 NCI plots for TS4(RR/RS/SR/SS) (isosurface = 0.008). Distances are given in Å. The blue, green, and red regions represent strong, weak, and repulsive interactions, respectively. | |
3.4. Substituent effects
Experimental studies35 showed that the substituents on cinnamaldehyde could affect the stereoselectivity in the annulation reaction of 2-aminomaleate with cinnamaldehyde. To understand this result, we calculated the energy barrier differences of key transition states in the stereoselectivity-controlling step, as summarized in Table 1. The energy barrier differences of entries 1–5 are in good agreement with the enantiomeric excess values reported in experiments, demonstrating that our results are reliable.
Table 1 The computed energy barrier differences for the stereoselectivity-controlling step
|
|
| Entry |
R |
ΔΔG‡ (kcal mol−1) |
ee (%) in the experiment |
| 1 |
3-MeC6H4 |
2.1 |
93 |
| 2 |
2-OMeC6H4 |
2.5 |
96 |
| 3 |
β-Heteroaryl |
1.3 |
85 |
| 4 |
Me |
1.9 |
90 |
| 5 |
β-Alkynyl |
3.1 |
97 |
3.5. Stabilization of axial chirality
To elucidate the factors responsible for maintaining the high axial chirality, the racemization process of products was investigated. As shown in Fig. 5, the energy barrier for the racemization process through the transition state TS9 is 31.1 kcal mol−1, making the racemization reaction unlikely to occur under the experimental conditions. In other words, the product exhibits stable axial chirality. In addition, we also calculated the energy barriers and the buried volumes of different substituents (Fig. S10, SI). The results indicate that the larger buried volumes of substituents lead to higher energy barriers of racemizations.
 |
| | Fig. 5 Energy barrier and optimized structures of the racemization transition state TS9. | |
3.5. The role of the catalyst
To understand the role of the catalyst, we conducted global reactivity index (GRI) analysis.89–93 As shown in Table 2, the values of the nucleophilicity index of R1, re-M1 and re-M2 are 2.453, 3.373 and 4.824 eV, respectively, indicating that the nucleophilicity of R1 is strengthened after coordination with the NHC. Subsequently, the nucleophilic re-M2 will convert to electrophilic M3 through an oxidation process. The nucleophilicity and electrophilicity indexes of M3 are 2.118 and 2.477 eV, respectively. At this point, M3 serves as an electrophile to react with R2.
Table 2 Electronic potential (µ, a.u.), chemical hardness (η, a.u.), global electrophilicity (ω, eV), and global nucleophilicity (N, eV) of R1, re-M1, re-M2, M3 and R2
| Species |
η
|
µ
|
ω
|
N
|
|
R1
|
0.242 |
−0.166 |
1.542 |
2.453 |
|
re-M1
|
0.253 |
−0.126 |
0.860 |
3.373 |
|
re-M2
|
0.194 |
−0.102 |
0.737 |
4.824 |
|
M3
|
0.208 |
−0.195 |
2.477 |
2.118 |
|
R2
|
0.173 |
−0.117 |
1.079 |
4.707 |
4. Conclusions
We conducted a comprehensive DFT study on NHC-catalyzed [3+3] cycloaddition of cinnamaldehyde with 2-aminomaleate. The most preferred mechanism involves eight steps: (1) nucleophilic addition of NHC to cinnamaldehyde, (2) DBU·H+-assisted 1,2-proton transfer forming the Breslow intermediate, (3) Breslow intermediate oxidation, (4) Michael addition of acyl intermediate to 2-aminomaleate, (5) protonation mediated by DBU·H+, (6) deprotonation mediated by DBU·H+, (7) ring-closure process, and (8) NHC dissociation. The Michael addition step determines the stereoselectivity of the entire catalytic cycle, affording the R-configured product. The calculated enantiomeric excess value of 89.3% aligns well with the experimental observations (90% ee). Further NCI analysis revealed that weak interactions involved in the stereo-controlling transition states should be responsible for the observed stereoselectivity. Further calculations of the racemization energy barriers for the axially chiral products with different substituents revealed that the buried volume is crucial for maintaining high axial chirality.
Conflicts of interest
The authors declare no conflicts of interest.
Data availability
The data underlying this study are available in the published article and its supporting information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5cp03567h.
Acknowledgements
DFT computations were carried out on the cluster at USTL.
References
- M. H. Liu, L. Zhang and T. Y. Wang, Supramolecular chirality in self-assembled systems, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS PubMed.
- H. C. Shen, Asymmetric synthesis of chiral chromans, Tetrahedron, 2009, 65, 3931–3952 CrossRef CAS.
- S. R. LaPlante, L. D. Fader, K. R. Fandrick, D. R. Fandrick, O. Hucke, R. Kemper, S. P. Miller and P. J. Edwards, Assessing atropisomer axial chirality in drug discovery and development, J. Med. Chem., 2011, 54, 7005–7022 CrossRef CAS.
- E. Kumarasamy, R. Raghunathan, M. P. Sibi and J. Sivaguru, Nonbiaryl and heterobiaryl atropisomers: molecular templates with promise for atropselective chemical transformations, Chem. Rev., 2015, 115, 11239–11300 CrossRef CAS.
- J. E. Smyth, N. M. Butler and P. A. Keller, A twist of nature-the significance of atropisomers in biological systems, Nat. Prod. Rep., 2015, 32, 1562–1583 RSC.
- Z. Y. Wang, L. Y. Meng, X. X. Liu, L. Z. Zhang, Z. J. Yu and G. Z. Wu, Recent progress toward developing axial chirality bioactive compounds, Eur. J. Med. Chem., 2022, 243, 114700 CrossRef CAS.
- S. S. Ranganathappa, B. S. Dehury, G. K. Singh, S. Shee and A. T. Biju, Atroposelective Synthesis of N–N Axially Chiral Indoles and Pyrroles via NHC-Catalyzed Diastereoselective (3 + 3) Annulation Strategy, ACS Catal., 2024, 14, 6965–6972 CrossRef CAS.
- Z. Q. Liu, C. X. Li, S. M. Zhang and D. Du, Divergent Synthesis of Axially Chiral 2-Pyranones and Fused 2-Pyridones via N-Heterocyclic Carbene-Catalyzed Atroposelective [3 + 3] Annulation, J. Org. Chem., 2025, 90, 1934–1939 CrossRef CAS PubMed.
- Y. K. Kang and D. Y. Kim, Organocatalytic highly enantio-and diastereoselective Mannich reaction of β-Ketoesters with N-Boc-aldimines, J. Org. Chem., 2009, 74, 5734–5737 CrossRef CAS.
- X. F. Bai, J. F. Zou, M. Y. Chen, Z. Xu, L. Li, Y. M. Cui, Z. J. Zheng and L. W. Xu, Lewis-Base-Mediated Diastereoselective Silylations of Alcohols: Synthesis of Silicon-Stereogenic Dialkoxysilanes Controlled by Chiral Aryl BINMOLs, Chem. – Asian J., 2017, 12, 1730–1735 CrossRef CAS.
- S. C. Lu, J. Y. Ong, H. Yang, S. B. Poh, X. Liew, C. S. D. Seow, M. W. Wong and Y. Zhao, Diastereo-and atropselecive synthesis of bridged biaryls bearing an eight-membered lactone through an organocatalytic cascade, J. Am. Chem. Soc., 2019, 141, 17062–17067 CrossRef CAS PubMed.
- P. Yang, L. Zhang, K. Y. Fu, Y. X. Sun, X. H. Wang, J. Y. Yue, Y. Ma and B. Tang, Nickel-catalyzed asymmetric transfer hydrogenation and α-selective deuteration of N-sulfonyl imines with alcohols: access to α-deuterated chiral amines, Org. Lett., 2020, 22, 8278–8284 CrossRef CAS.
- H. Q. Wang, S. F. Wu, J. R. Yang, Y. C. Zhang and F. Shi, Design and organocatalytic asymmetric synthesis of indolyl-pyrroloindoles bearing both axial and central chirality, J. Org. Chem., 2022, 88, 7684–7702 CrossRef.
- M. W. Liu, Y. H. Zhang, X. Y. Ke, S. F. Ni and P. F. Li, Asymmetric Organocatalytic 1, 6-Conjugate Addition of Alkynyl 8-Methylenenaphthalen-2(8H)-one Formed In Situ: Synergistic Construction of Axial and Central Chirality, Org. Lett., 2025, 27, 1271–1275 CrossRef CAS.
- G. Gao, F. L. Gu, J. X. Jiang, K. Z. Jiang, C. Q. Sheng, G. Q. Lai and L. W. Xu, Neighboring Lithium-Assisted [1,2]- Wittig Rearrangement: Practical Access to Diarylmethanol-Based 1,4-Diols and Optically Active BINOL Derivatives with Axial and sp3-Central Chirality, Chem. – Eur. J., 2011, 17, 2698–2703 CrossRef CAS PubMed.
- X. F. Bai, T. Song, Z. Xu, C. G. Xia, W. S. Huang and L. W. Xu, Aromatic Amide-Derived Non-Biaryl Atropisomers as Highly Efficient Ligands in Silver-Catalyzed Asymmetric Cycloaddition Reactions, Angew. Chem., 2015, 127, 5344–5348 CrossRef.
- W. C. Yang, X. B. Chen, K. L. Song, B. Wu, W. E. Gan, Z. J. Zheng, J. Cao and L. W. Xu, Pd-catalyzed enantioselective tandem C-C bond activation/cacchi reaction between cyclobutanones and o-ethynylanilines, Org. Lett., 2021, 23, 1309–1314 CrossRef CAS PubMed.
- H. H. Zhang, T. Z. Li, S. J. Liu and F. Shi, Catalytic asymmetric synthesis of atropisomers bearing multiple chiral elements: an emerging field, Angew. Chem., Int. Ed., 2024, 63, e202311053 CrossRef CAS.
- X. Y. Chen, Z. H. Gao and S. Ye, Bifunctional N-heterocyclic carbenes derived from l-pyroglutamic acid and their applications in enantioselective organocatalysis, Acc. Chem. Res., 2020, 53, 690–702 CrossRef CAS.
- P. Bellotti, M. Koy, M. N. Hopkinson and F. Glorius, Recent advances in the chemistry and applications of N-heterocyclic carbenes, Nat. Rev. Chem., 2021, 5, 711–725 CrossRef CAS.
- B. H. Liu, J. Qi, Y. T. Wu, J. H. Li, Y. T. Li and X. Y. Duan, The N-heterocyclic carbene-catalyzed [3 + 2] annulation of isoindigos with enals: the enantioselective construction of three contiguous stereogenic centers, Org. Chem. Front., 2022, 9, 3552–3557 RSC.
- M. Zhang, X. Q. Yang, X. L. Peng, X. Y. Li and Z. C. Jin, Asymmetric construction of axial and planar chirality with N-heterocyclic carbene(NHC) organocatalysis, Sci. China: Chem., 2024, 68, 1–11 CAS.
- Z. Q. Zhu, T. Z. Li, S. J. Liu and F. Shi, Advances in organocatalytic asymmetric [3 + 3] cycloadditions: synthesis of chiral six-membered (hetero) cyclic compounds, Org. Chem. Front., 2024, 11, 5573–5604 RSC.
- L. Candish, A. Levens and D. W. Lupton, N-Heterocyclic carbene catalysed redox isomerisation of esters to functionalised benzaldehydes, Chem. Sci., 2015, 6, 2366–2370 RSC.
- A. Borissov, T. Davies, S. Ellis, T. Fleming, M. Richardson and D. Dixon, Organocatalytic enantioselective desymmetrisation, Chem. Soc. Rev., 2016, 45, 5474–5540 RSC.
- D. H. Guo, Q. P. Peng, B. Zhang and J. Wang, Atroposelective dynamic kinetic resolution via in situ hemiaminals catalyzed by N-heterocyclic carbene, Org. Lett., 2021, 23, 7765–7770 CrossRef CAS PubMed.
- S. M. Zhang, X. X. Wang, L. L. Han, J. B. Li, Z. Liang, D. H. Wei and D. Du, Atroposelective Synthesis of Triaryl α-Pyranones with 1,2-Diaxes by N-Heterocyclic Carbene Organocatalysis, Angew. Chem., Int. Ed., 2022, 61, e202212005 CrossRef CAS.
- S. C. Zhang, S. P. Liu, X. Wang, S. J. Wang, H. Yang, L. Li, B. M. Yang, M. W. Wong, Y. Zhao and S. C. Lu, Enantioselective access to triaryl-2-pyrones with monoaxial or contiguous C-C diaxes via oxidative NHC catalysis, ACS Catal., 2023, 13, 2565–2575 CrossRef CAS.
- X. X. Hou and D. H. Wei, Mechanism and origin of stereoselectivity for the NHC-catalyzed desymmetrization reaction for the synthesis of axially chiral biaryl aldehydes, J. Org. Chem., 2024, 89, 3133–3142 CrossRef CAS PubMed.
- I. Takahashi, Y. Suzuki and O. Kitagawa, Asymmetric synthesis of atropisomeric compounds with an N-C chiral axis, Org. Prep. Proced. Int., 2014, 46, 1–23 CrossRef CAS.
- Z. S. Liu, P. P. Xie, Y. Hua, C. G. Wu, Y. Y. Ma, J. W. Chen, H. G. Cheng, X. Hong and Q. H. Zhou, An axial-to-axial chirality transfer strategy for atroposelective construction of C-N axial chirality, Chem, 2021, 7, 1917–1932 CAS.
- P. Rodríguez-Salamanca, R. Fernández, V. Hornillos and J. M. Lassaletta, Asymmetric synthesis of axially chiral C-N atropisomers, Chem. – Eur. J., 2022, 28, e202104442 CrossRef.
- J. S. Sweet and P. C. Knipe, Catalytic enantioselective synthesis of C-N atropisomeric heterobiaryls, Synthesis, 2022, 2119–2132 CAS.
- O. Kitagawa, Structural Chemistry of C–N Axially Chiral Compounds, J. Org. Chem., 2024, 89, 11089–11099 CrossRef CAS.
- Y. T. Li, X. Y. Duan, C. X. Yang, Y. M. Wei, J. H. Li, X. J. Ren and J. Qi, Atroposelective Access to Dihydropyridinones with C-N Axial and Point Chirality via NHC-Catalyzed [3 + 3] Annulation, J. Org. Chem., 2023, 88, 11299–11309 CrossRef CAS PubMed.
- T. Liu, S. M. Han, Y. P. Li and S. W. Bi, Theoretical insight into the mechanisms and regioselectivity of [4 + 3] and [4 + 1] annulations of enals with azoalkenes catalyzed by n-heterocyclic carbenes, J. Org. Chem., 2016, 81, 9775–9784 CrossRef CAS.
- Y. Qiao, Y. Xiao, M. Zhao, X. Li and J. B. Chang, Mechanisms and origin of regioselectivity on N-heterocyclic carbene-catalyzed [3 + 2]/[4 + 2] annulations of C60 with α, β-unsaturated aldehydes, Mol. Catal., 2020, 493, 111045 CAS.
- Y. Wang, Y. Qiao, Y. Lan and D. H. Wei, Predicting the origin of selectivity in NHC-catalyzed ring opening of formylcyclopropane: a theoretical investigation, Catal. Sci. Technol., 2021, 11, 332–337 RSC.
- Y. L. Kang, Y. Li and Z. Q. Zhang, Mechanism, regioselectivity and stereoselectivity of NHC-catalyzed [12 + 2] annulation of 5H-benzo[a]-pyrrolizine-3-carbaldehydes and cyclic sulfonic imines: a DFT study, New J. Chem., 2025, 49, 15623–15630 RSC.
- K. H. Chen, J. M. Zhang, Q. Q. Shi, L. L. Han, D. M. Fu, D. H. Wei and Y. Y. Zhu, NHC-catalyzed enantioselective radical reactions of enal and pyridinium salt: mechanism and origin of regio-and stereoselectivities, Catal. Sci. Technol., 2023, 13, 5259–5266 RSC.
- Y. Li, M. C. Zhang and Z. Q. Zhang, Mechanisms and origins of stereoselectivity in the NHC-catalyzed oxidative reaction of enals and pyrroles: a density functional theory study, Phys. Chem. Chem. Phys., 2024, 26, 28112–28123 RSC.
- M. C. Zhang, Z. Q. Zhang and Y. Li, Mechanisms and Origins of Regio-and Stereoselectivities in NHC-Catalyzed [3 + 3] Annulation of α-Bromoenals and 5-Aminoisoxazoles: A DFT Study, J. Org. Chem., 2024, 89, 10748–10759 CrossRef CAS.
- B. Chan and L. Radom, Uncatalyzed Transfer Hydrogenation of Quinones and Related Systems: A Theoretical Mechanistic Study, J. Phys. Chem. A, 2007, 111, 6456–6467 CrossRef CAS PubMed.
- V. S. Batista, R. H. Crabtree, S. J. Konezny, O. R. Luca and J. M. Praetorius, Oxidative Functionalization of Benzylic C-H Bonds by DDQ, New J. Chem., 2012, 36, 1141–1144 RSC.
- S. Yamabe, S. Yamazaki and S. Sakaki, A DFT Study of Hydride Transfers to the Carbonyl Oxygen of DDQ, Int. J. Quantum Chem., 2015, 115, 1533–1542 CrossRef CAS.
- X. Guo, H. Zipse and H. Mayr, Mechanisms of Hydride Abstractions by Quinones, J. Am. Chem. Soc., 2014, 136, 13863–13873 CrossRef CAS.
- J. Mo, L. Shen and Y. R. Chi, Direct β-Activation of Saturated Aldehydes to Formal Michael Acceptors through Oxidative NHC Catalysis, Angew. Chem., Int. Ed., 2013, 52, 8588–8591 CrossRef CAS PubMed.
- O. R. Shehab and A. M. Mansour, Charge Transfer Complexes of 2-Arylaminomethyl-1H-Benzimidazole with 2,3-Dichloro-5,6-Dicyano-1,4- Benzo-quinone: Experimental and DFT Studies, J. Mol. Struct., 2013, 1047, 121–135 CrossRef CAS.
- A. K. Turek, D. J. Hardee, A. M. Ullman, D. G. Nocera and E. N. Jacobsen, Activation of Electron-Deficient Quinones through Hydrogen-Bond-Donor-Coupled Electron Transfer, Angew. Chem., Int. Ed., 2016, 55, 539–544 CrossRef CAS.
- F. Liu, Z. Yang, Y. Yu, Y. Mei and K. N. Houk, Bimodal Evans−Polanyi Relationships in Dioxirane Oxidations of sp3 C−H: Non-Perfect Synchronization in Generation of Delocalized Radical Intermediates, J. Am. Chem. Soc., 2017, 139, 16650–16656 CrossRef CAS PubMed.
- C. Hofler and C. Ruchardt, Bimolecular Formation of Radicals by Hydrogen Transfer, 10. On the Mechanism of Quinone Dehydrogenations, Liebigs Ann., 1996, 1996, 183–188 CrossRef.
- C. Ruchardt, M. Gerst and J. Ebenhoch, Uncatalyzed Transfer Hydrogenation and Transfer Hydrogenolysis: Two Novel Types of Hydrogen-Transfer Reactions, Angew. Chem., Int. Ed. Engl., 1997, 36, 1406–1430 CrossRef CAS.
- D. A. Pratt, J. S. Wright and K. U. Ingold, Theoretical Study of Carbon−Halogen Bond Dissociation Enthalpies of Substituted Benzyl Halides. How Important Are Polar Effects?, J. Am. Chem. Soc., 1999, 121, 4877–4882 CrossRef CAS.
- H. H. Jung and P. E. Floreancig, Mechanistic Analysis of Oxidative C−H Cleavages Using Inter- and Intramolecular Kinetic Isotope Effects, Tetrahedron, 2009, 65, 10830–10836 CrossRef CAS PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc, Wallingford, CT, 2016 Search PubMed.
- J. M. Fraile, J. I. García, V. Martínez-Merino, J. A. Mayoral and L. Salvatella, Theoretical (DFT) insights into the mechanism of copper-catalyzed cyclopropanation reactions. Implications for enantioselective catalysis, J. Am. Chem. Soc., 2001, 123, 7616–7625 CrossRef CAS PubMed.
- X. Y. Chen, Y. X. Zhao and S. G. Wang, Relativistic DFT study on the reaction mechanism of second-row transition metal Ru with CO2, J. Phys. Chem. A, 2006, 110, 3552–3558 CrossRef CAS PubMed.
- I. D. Gridnev, M. Watanabe, H. Wang and T. Ikariya, Mechanism of Enantioselective C−C Bond Formation with Bifunctional Chiral Ru Catalysts: NMR and DFT Study, J. Am. Chem. Soc., 2010, 132, 16637–16650 CrossRef CAS.
- C. W. Liu, Q. Y. Li, J. Zhang, Y. G. Jin, D. R. MacFarlane and C. H. Sun, Conversion of dinitrogen to ammonia on Ru atoms supported on boron sheets: a DFT study, J. Mater. Chem. A, 2019, 7, 4771–4776 RSC.
- J. Y. Sun, Z. Y. Yu and T. Liu, Theoretical Investigation on the Rhodium-Catalyzed Annulation of 2-Phenyl-1 H-indole with Ethyl 2-Diazo-3-oxo-3-phenylpropanoate, Russ. J. Phys. Chem. A, 2021, 95, 2573–2577 CrossRef CAS.
- J. L. Cai, C. Peng and Y. Wang, DFT Insights into the mechanism of Ru (II) Catalyzed C7-selective amidation of N-pivaloylindole, J. Organomet. Chem., 2022, 982, 122534 CrossRef CAS.
- L. N. Geng, M. C. Zhang, Z. Q. Zhang and Y. Li, Production of carbon monoxide and hydrogen from methanol using a ruthenium pincer complex: a DFT study, Dalton Trans., 2023, 52, 13653–13661 RSC.
- L. R. Domingo, R. J. Zaragozá, J. A. Saéz and M. Arnó, Understanding the mechanism of the intramolecular Stetter reaction. A DFT study, Molecules, 2012, 17, 1335–1353 CrossRef CAS.
- G. T. Huang, M. H. Hsieh and J. S. K. Yu, Formation of Breslow Intermediates under Aprotic Conditions: A Computational Study, J. Org. Chem., 2022, 87, 2501–2507 CrossRef CAS.
- R. Rowshanpour, M. Gravel and T. Dudding, N-heterocyclic carbene organocatalyzed redox-active/ring expansion reactions: Mechanistic insights unveiling base cooperativity, J. Org. Chem., 2022, 87, 16785–16793 CrossRef CAS PubMed.
- Q. Y. Zhang, X. Li, J. Luo, X. Li, J. H. Song and D. H. Wei, Cofactor-free dioxygenases-catalyzed reaction pathway via proton-coupled electron transfer, J. Phys. Chem. B, 2022, 127, 95–103 CrossRef PubMed.
- S. Gallardo Fuentes, L. Lodeiro, R. Matute and I. Fernández, Mechanistic Insights into the DABCO-Catalyzed Cloke-Wilson Rearrangement: A DFT Perspective, J. Org. Chem., 2023, 88, 15902–15912 CrossRef CAS.
- P. Jin, P. J. Liu, Y. T. Chong, S. Pruksawan, L. Li, Y. Q. Wen, H. J. Wei and F. K. Wang, A DFT study on possible mechanisms for Aza-Baeyer-Villiger rearrangement reaction of cyclobutanone with aminodiphenylphosphonate, Comput. Theor. Chem., 2024, 1237, 114635 CrossRef CAS.
- B. Chen, T. T. Feng, D. G. Zhou and L. J. Yang, Mechanisms of C (sp3)-H Functionalization of Acetonitrile or Acetone with Alkynes: A DFT Investigation, J. Chem. Inf. Model., 2025, 65, 1953–1966 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- M. Pareek and R. B. Sunoj, Cooperative Asymmetric Catalysis by N-Heterocyclic Carbenes and Brønsted Acid in γ-Lactam Formation: Insights into Mechanism and Stereoselectivity, ACS Catal., 2016, 6, 3118–3126 Search PubMed.
- P. L. Han, Y. J. Hou, S. X. Ge, Z. F. Yang, Y. L. Yan, H. D. Guo and C. H. Liu, DFT study on the mechanism and origin of stereoselectivity of an NHC-catalyzed activation/transformation of an α-bromo enal, New J. Chem., 2025, 49, 6374–6379 RSC.
- S. S. Yu, W. X. Zhou, Y. M. Jiang, H. Y. Wang, X. Y. Zhou and S. W. Yang, DFT Insights into NHC-Catalyzed Switchable [3 + 4] and [3 + 2] Annulations of Isatin-Derived Enals and N-Sulfonyl Ketimines: Mechanism, Regio- and Stereoselectivity, Molecules, 2025, 30, 4218 CrossRef CAS PubMed.
- K. H. Chen, J. M. Zhang, Q. Q. Shi, L. L. Han, D. M. Fu, D. H. Wei and Y. Y. Zhu, NHC-catalyzed enantioselective radical reactions of enal and pyridinium salt: mechanism and origin of regio- and stereoselectivities, Catal. Sci. Technol., 2023, 13, 5259–5266 RSC.
- B. Mennucci and J. Tomasi, Continuum solvation models: A new approach to the problem of solute's charge distribution and cavity boundaries, J. Chem. Phys., 1997, 106, 5151–5158 CrossRef CAS.
- V. Barone and M. Cossi, Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- K. Fukui, Formulation of the reaction coordinate, J. Phys. Chem., 1970, 74, 4161–4163 CrossRef CAS.
- K. Fukui, The path of chemical reactions - the IRC approach, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- J. D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the Damping Function in Dispersion Corrected Density Functional Theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, Revealing noncovalent interactions, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS.
- T. Lu and F. W. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
- T. Lu and Q. X. Chen, Independent gradient model based on Hirshfeld partition: A new method for visual study of interactions in chemical systems, J. Comput. Chem., 2022, 43, 539–555 CrossRef CAS PubMed.
-
C. Y. Legault, CYLview20. http://www.cylview.org (accessed February 2023).
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Towards the online computer-aided design of catalytic pockets, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed.
- J. M. Zhang, Q. Y. Qiao, Z. J. Wu, Z. Pang, Q. Q. Shi, Y. Y. Wang, Y. Qiao and D. H. Wei, The mechanism and origin of selectivities for NHC-catalyzed synthesis of axially chiral benzothiophene/benzofuran-fused biaryls, Org. Biomol. Chem., 2022, 20, 1662 RSC.
- X. Li, S. J. Li, Y. Y. Wang, Y. Wang, L. B. Qu, Z. J. Li and D. H. Wei, Insights into NHC-catalyzed oxidative α-C(sp3)−H activation of aliphatic aldehydes and cascade [2 + 3] cycloaddition with azomethine imines, Catal. Sci. Technol., 2019, 9, 2514–2522 RSC.
- D. Yepes, J. S. Murray, P. Pérez, L. R. Domingo, P. Politzer and P. Jaque, Complementarity of reaction force and electron localization function analyses of asynchronicity in bond formation in Diels-Alder reactions, Phys. Chem. Chem. Phys., 2014, 16, 6726–6734 RSC.
- L. R. Domingo and P. Pérez, Global and local reactivity indices for electrophilic/nucleophilic free radicals, Org. Biomol. Chem., 2013, 11, 4350–4358 RSC.
- E. Chamorro, P. Pérez and L. R. Domingo, On the nature of Parr functions to predict the most reactive sites along organic polar reactions, Chem. Phys. Lett., 2013, 582, 141–143 CrossRef CAS.
- L. R. Domingo, M. T. Picher and J. A. Sáez, Toward an Understanding of the Unexpected Regioselective Hetero-Diels-Alder Reactions of Asymmetric Tetrazines with Electron-Rich Ethylenes: A DFT Study, J. Org. Chem., 2009, 74, 2726–2735 CrossRef CAS PubMed.
- L. R. Domingo, J. A. Saéz, R. J. Zaragozá and M. Arnó, Understanding the Participation of Quadricyclane as Nucleophile in Polar [2σ + 2σ + 2π] Cycloadditions toward Electrophilic π Molecules, J. Org. Chem., 2008, 73, 8791–8799 CrossRef CAS PubMed.
|
| This journal is © the Owner Societies 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 * and
Zhiqiang
Zhang
* and
Zhiqiang
Zhang









