
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMeasuring the Gibbs free energy of mixing using the concept of an osmotic engine
Dennis Wowern Nielsen *a and
Claus Helix-Nielsenb
*a and
Claus Helix-Nielsenb
aU/Nord, Trollesmindesalle 24, Hillerød 3400, Denmark. E-mail: DWN@Unord.dk
bTechnical University of Denmark, Brovej 118, Kongens Lyngby 2800, Denmark
First published on 7th April 2026
Abstract
This manuscript develops a membrane-thermodynamic framework for quantifying the Gibbs free energy of mixing in real binary solutions. Using an osmotic engine representation under pressure retarded osmosis, the method links static and dynamic membrane-based osmometry to the excess Gibbs free energy of mixing, enabling operational estimation of ΔGE from measurable osmotic and volumetric responses. As an analytical perspective, the van’t Hoff limit produces a simple colligative form that connects the Henry-reference activity coefficient at infinite dilution to the dimensionless osmotic pressure, highlighting the role of reference-state regularisation and the sensitivity to volume assumptions. Beyond this limiting case, the framework provides a natural route to multicomponent mixtures with electrolytes and non-electrolytes, predicts solvent-dependent shifts in reaction equilibrium constants, and supports routine estimation of the Flory–Huggins solvent–polymer interaction parameter.
1 Introduction
In the realm of chemical phenomena taking place in the liquid phase, assessment of Gibbs free mixing energy, ΔGmix, holds a pivotal position. When examining a chemical equilibrium reaction within an inert solvent, one may define a series of steps: firstly, the dissolving of reactants, inducing a change in Gibbs free energy, ΔGreacmix. Subsequently, a reaction must take place, leading to the formation of reaction products, which, in turn, results in a change in Gibbs free energy, ΔGrxn. Finally, the reaction products dissolve within the mixture, leading to yet another term, ΔGprodmix. Thus at equilibrium| −ΔGrxn = ΔGreacmix + ΔGprodmix | (1) |
Hence, the energies of reaction are inseparable from energies of mixing, and like this it is argued that the measurement of the chemical mixing energy is not merely supplementary to, but in many cases as essential as, the knowledge of equilibrium constants for understanding specific chemical reactions.
Consider for example a solution of protein P and ligand L dissolved in a suitable solvent, typically water, entering the chemical addition reaction P + L ⇌ PL. At equilibrium, the chemical affinity constant, Ka, is related to a set of thermodynamic parameters
 | (2) |
Here, n represents the amount of substance of reactants or products, R is the universal gas constant, T is the kelvin temperature, ΔḠmix signifies the change in partial molar Gibbs free mixing energy of the reaction's products or reactants, and μ° represents the standard chemical potential of formation for the reaction's products or reactants. Since the formation potentials remain constant and independent of the inert solvent, mixing energies directly influence Ka, making it a system-dependent constant. This implies that by determining both the mixing energies and formation energies for each reactant and product, the equilibrium constant may be determined.
If the reaction is predominantly shifted towards the product side, eqn (2) can be further simplified as follows
 | (3) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnKa is linearly dependent on ΔḠPLmix. Consequently, if −RTlnKa is determined from ITC measurements in these various solvents and the corresponding ΔḠPLmix values are measured, the typically inaccessible quantity
lnKa is linearly dependent on ΔḠPLmix. Consequently, if −RTlnKa is determined from ITC measurements in these various solvents and the corresponding ΔḠPLmix values are measured, the typically inaccessible quantity  can be estimated.
can be estimated.
In summary, it is proposed that a reliable and precise measurement of ΔGmix across different inert solvents could significantly advance chemical research, and since dG = VdP at constant temperature, this method is recommended for further exploration in the context of osmometry.
Today's commercial osmometers operate based on three distinct principles: freezing point depression measurement, which relies on osmotically active compounds lowering the freezing point of a solution; vapor pressure measurement, where osmotically active particles reduce the vapor pressure of a solution; and osmotic pressure measurement, which involves isolating a solution from pure solvent using a semipermeable membrane. While techniques such as freezing point depression and vapor pressure osmometry are commonly used to determine osmotic pressure differences, they are insufficient for quantifying the volume displaced at osmotic equilibrium.1 As a result, these methods cannot estimate the work performed on the surroundings, which is represented by the product of the displaced volume and the osmotic pressure of the solution prior to osmosis, arising from the spontaneous osmotic dilution process.
To overcome this limitation, conventional membrane based osmometry is recommended, as it enables the simultaneous measurement of both osmotic pressure and displaced volume. This approach allows for the determination of osmotic work exerted on the surroundings through two distinct methods: the static and dynamic equilibrium methods.2
Pfeffer3 pioneered the development of membrane-based osmometers, enabling the simultaneous measurement of osmotic pressure and displaced volume using the static method, as illustrated in Fig. 1. This approach facilitates the establishment of osmotic equilibrium for evaluating the pressure difference. The resulting pressure, when multiplied by the volume change in the solution compartment, quantifies the work exerted on the surroundings.
 | ||
| Fig. 1 Principal sketch of an arrangement performining static osmometry. | ||
However, highly concentrated, strongly non-ideal solutions can generate osmotic pressures that, at equilibrium, lead to liquid column heights on the order of hundreds of meters, rendering them impractical for standard laboratory settings. Since osmotic equilibrium is a reversible process that may take an indefinite amount of time to establish, accurately measuring osmotic pressure and volume change remains a challenge.
Moreover, this technique is influenced by several factors, including membrane material, surface area, thickness, selectivity, solvent and solute permeability, and the membrane's susceptibility to complete or partial swelling in the solvent. Additionally, ambient pressure and temperature introduce uncertainties. These variables may fluctuate over time, complicating the determination of when osmotic equilibrium is achieved. For instance, the kinetics of osmotic pressure development in aqueous solutions, depending on sucrose solute concentration, vary significantly under two conditions: constant and variable solution volumes.4 The rate of pressure increase is notably higher under constant volume conditions than under variable volume conditions, primarily due to the significantly greater solvent flow in the latter. This effect becomes more pronounced as sucrose concentration increases, exemplifying the behavior of real sucrose solutions.
This challenge underscores the necessity of employing dynamic membrane osmometry, where the extended time required for solvent volume flow is reduced by artificially increasing the pressure difference. Consequently, relying solely on displaced volume and osmotic pressure to evaluate the work in osmotic dilution processes using static membrane osmometry may be insufficient for determining the work done on the surroundings.
In the dynamic equilibrium method, initially pioneered by Berkeley–Hartley,5 a counterpressure is applied to nullify any consequential volume flux between the solvent and solution chambers, see Fig. 2. Although this technique facilitates the expeditious determination of the solution's osmotic pressure, it regrettably fails to furnish the requisite information regarding the volume expansion that an unobstructed osmotic dilution would have incurred. Consequently, the assessment of osmotic displacement work remains unattainable via the dynamic method.
 | ||
| Fig. 2 Principal sketch of an arrangement performing dynamic osmometry. | ||
To address the challenges associated with quantifying both the osmotic pressure and the volume displaced at osmotic equilibrium, this publication introduces the concept of an osmotic engine, as illustrated in Fig. 3. This is a theoretical thermodynamic framework that effectively merges static and dynamic membrane-osmometry techniques.6,7 The approach enables the computation of the Gibbs free energy of mixing of a real binary solution and the corresponding excess Gibbs free energy, and it also allows determination of the solute activity coefficient, among other applications. The concept engine is formulated as a cascade of cylinders that, in each stage, processes an initial volume αVC, where VC is the volume of a single cylinder and 0 < α < 1. To ensure that subsequent physico-chemical conclusions, e.g. expressions for ΔGmix and the solute activity coefficient, are apparatus-independent rather than tied to a particular discrete engine concept design, the continuum limit α → 1 is concidered. In this limit, the per-stage increment becomes vanishingly small and the cascade becomes effectively continuous, corresponding to the idealized limit of infinitely many cylinders.
 | ||
| Fig. 3 Principal sketch of a concept arrangement which combines static and dynamic osmometry. | ||
2 Theory
In the following, the binary Test solution is treated as a practically incompressible liquid, so that volume changes during osmotic dilution are ascribed to changes in composition rather than to elastic compression. This keeps the modelling of ΔGTestmix focused on mixing non-ideality rather than on the ideal or non-ideal elasticity of the liquid.A binary solution consisting of solvent A and solute B at constant temperature T and at the standard pressure P° is considered. This concentrated Test solution containing n(A) and n(B) is osmotically diluted to equilibrium by adding an additional amount nDil(A) of A, yielding a more dilute solution containing n(A) + nDil(A) and n(B), hereafter denoted TestDil.
The Test solution is regarded as a high-concentration state, whereas the final state TestDil represents a low-concentration state. The controlled dilution process connecting these two states is viewed as a chemical analogue of a Carnot process: the difference in mixing free energy between the concentrated and the dilute state is converted into mechanical work by an osmotic engine, while the Gibbs free energy of mixing of the solution changes.
The purpose of this section is to establish a simple operational relation between ΔGmix(n(A), n(B)) = ΔGmix(Test) = ΔGTestmix, the Gibbs free energy of mixing in the initial Test solution, ΔGmix(n(A) + nDil(A), n(B)) = ΔGmix(TestDil) = ΔGfinalmix, the Gibbs free energy of mixing in the final osmotically diluted solution, and the reversible work associated with the osmotic dilution.
2.1 Energy balance in the osmotic engine
The two-state balance is written as (arguments are omitted for brevity):| ΔGTestmix − ΔGFinalmix = −ΔGDilsys | (4) |
For convenience, the quantity
| ΔGDil ≡ −ΔGDilsys | (5) |
Throughout this work, a distinction is made between the system free-energy change upon dilution and the work extracted by the osmotic engine. The system free-energy change is defined as ΔGDilsys ≡ GFinal − GTest, and the maximum reversible work delivered is defined as ΔGDil ≡ −ΔGDilsys. With this convention, ΔGDil > 0 is obtained for spontaneous dilution, and
| ΔGTestmix(n(A), n(B)) = ΔGFinalmix(n(A) + nDil(A), n(B)) + ΔGDil | (6) |
This relation constitutes a chemical analogue of the Carnot balance between two reservoirs and a machine: the decrease in the system Gibbs free energy upon dilution is converted into reversible mechanical work delivered by the osmotic engine.
The dilution from (n(A), n(B)) to (n(A) + nDil(A), n(B)) is assumed to be implemented in an osmotic engine. On the B-side of an ideal semipermeable membrane, permeable only to A, the solution is present, whereas pure A is present on the opposite side. On the B-side, the pressure is increased by an externally imposed counterpressure ΔP, such that the total pressure is given by P° + ΔP. The volume of the solution in the B-chamber is denoted V, with V(Test) corresponding to the Test state and V(Final) corresponding to the final diluted state. During an infinitesimal dilution step, the volume on the B-side is increased by dVExp due to osmotic influx of A.10,11
According to the Kedem–Katchalsky formulation of volumetric flux through a semipermeable membrane, combined with Onsager's linear irreversible thermodynamics12–14 and Gibbs' maximum work theorem at constant T and P°, the reversible mechanical work delivered by the osmotic engine in such a step is given by
| δWrev = ΔP·dVExp | (7) |
| δWrev = −dGsys | (8) |
Along a quasi-reversible Kedem–Katchalsky path from V(Test) to V(Final), for which JV → 0 is satisfied at each intermediate state and the actual osmotic pressure, Δπ = [Δπ], approaches the applied counterpressure ΔP, integration yields
 | (9) |
 | (10) |
By insertion into the two-state balance for the mixing free energy, the central operational expression is obtained as
 | (11) |
In this representation, the analogy to a Carnot process is rendered transparent: ΔGTestmix and ΔGFinalmix play the role of two chemical levels, whereas the integral  represents the work term, analogous to the area under a pressure–volume curve in a mechanical cycle.20–24 For some systems, ΔP(V), which coincides with the osmotic pressure Δπ(V) at equilibrium, can be represented analytically. Examples include van't Hoff ideality in the dilute limit,
represents the work term, analogous to the area under a pressure–volume curve in a mechanical cycle.20–24 For some systems, ΔP(V), which coincides with the osmotic pressure Δπ(V) at equilibrium, can be represented analytically. Examples include van't Hoff ideality in the dilute limit,
 | (12) |
 | (13) |
In the van't Hoff ideal limit, the mixing free energy of the Test solution can thus be expressed in terms of the osmotic work as
 | (14) |
At osmotic equilibrium, ΔP(V) = Δπ(V) is obtained, and the dimensionless dilution factor
 | (15) |
| V(Final) = V(Test) + ΔV(Test) = (1 + ψ)·V(Test) | (16) |
Accordingly, the work integral is written as
 | (17) |
| ΔGvHmix(n(A), n(B)) = ΔGFinalmix(n(A) + nDil(A), n(B)) + n(B)RT·ln(1 + ψ) | (18) |
The final, strongly osmotically diluted solution is used as a residual osmotic reference state, i.e. a state for which negligible remaining osmotic working capacity is present. To avoid confusion with the absolute mixing free energy, the extractable dilution work relative to the final state is therefore introduced as
 | (19) |
| Wavail(Test → Final) = ΔGTestmix − ΔGFinalmix | (20) |
It is emphasized that Wavail is extracted work, i.e. path-dependent, whereas ΔGmix is a state function.
For residual osmotic dilutions, a strongly diluted final state is obtained and the excess contribution is reduced. In this limit, ΔGfinalE ≈ 0 is assumed, such that ΔGFinalmix ≈ ΔGFinal,idealmix is obtained. The ideal mixing free energy of this residual osmotic reference composition is defined as
| ΔGrefmix ≡ ΔGFinal,idealmix | (21) |
Here the superscript ref refers to the composition of the residual osmotic reference state and the ideal-mixture approximation, and no new thermodynamic potential is introduced. Accordingly, an absolute estimate of ΔGTestmix is obtained as
| ΔGTestmix ≈ Wavail(Test → Final) + ΔGrefmix | (22) |
|
ΔGrefmix = (nfinalA + n(B))RT·(xfinalAlnxfinalA + xfinalBlnxfinalB)
| (23) |
In the residual-dilution limit considered here, ΔGFinalmix is therefore replaced by ΔGrefmix in the van't Hoff expressions, i.e. in the regime where activity coefficients approach unity. By use of the van't Hoff relation for the Test volume, eqn (18) yields
| ΔGvHmix(n(A), n(B)) = ΔGrefmix + n(B)RT·ln(1 + ψ) | (24) |
| ΔGvHmix(n(A), n(B)) = ΔGrefmix + ΔπvH(Test)·V(Test)·ln(1 + ψ) | (25) |
For sufficiently dilute Test solutions, a small osmotic pressure is obtained, the osmotic influx is limited, and hence ψ = ΔV(Test)/V(Test) ≪ 1 is obtained, such that ln(1 + ψ) ≈ ψ is obtained. In this limit,
| ΔGvHmix(Test) ≈ ΔGrefmix + ΔπvH(Test)·V(Test)·ψ = ΔGrefmix + ΔπvH(Test)·ΔV(Test) | (26) |
2.2 Riemann approximation and experimental implementation
In most practical systems, Δπ(V) is not available in a simple closed analytical form. The dilution in the osmotic engine is therefore implemented as a scanning, nearly reversible Kedem–Katchalsky process; please refer to eqn (46). A sequence of counterpressures ΔPk, with k = 1, 2, 3 …, is chosen. For each ΔPk, the system is allowed to evolve until the volumetric flux approaches zero, JV → 0, thereby defining a quasi-equilibrium at this pressure. The solution volume in the B-chamber changes from Vk−1 to Vk, and the corresponding volume increment is| ΔVExp,k = Vk − Vk−1 | (27) |
In practice, the increments ΔVExp,k can be fixed mechanically by a well-defined displacement of a piston or crank mechanism, so that the volume steps are determined by the geometry of the apparatus. The braking torque, or applied load, is then adjusted until the crank is at rest and the measured pressure difference satisfies JV ≈ 0 at the new volume. When the crank is at rest, the counterpressure ΔPk is read, and ΔVExp,k is obtained from simple geometrical relations. In this way, a set of paired data (ΔPk, ΔVExp,k) is generated along a Kedem–Katchalsky, nearly reversible path from V(Test) to V(Final).
The continuous integral for the dilution work,
 | (28) |
 | (29) |
Substitution into the operational expression for the mixing free energy in the Test state yields the experimentally accessible approximation
 | (30) |
For residual osmotic dilutions, the final, osmotically diluted solution is used as a residual osmotic reference state for which the remaining osmotic working capacity is negligible and activity coefficients approach unity. In this limit one may approximate ΔGFinalmix ≈ ΔGrefmix, refer to eqn (21), and therefore
 | (31) |
The quality of the Riemann approximation depends on the magnitudes of the steps ΔVExp,k and the resulting changes in ΔPk. If the steps become too large, the path becomes both numerically coarse and thermodynamically irreversible, with non-negligible entropy production in each step. In practice, small and mechanically well-controlled volume steps and a sensitive mechanical gearing are therefore advantageous for maintaining JV ≈ 0 and a nearly reversible path. As a consistency check, V(Final) can be obtained either from direct volume readout or as  .
.
An electric brake engine or generator can be employed to provide the required braking torque on the shaft of the osmotic engine. For each step k, the braking current is adjusted until the volumetric flux vanishes and the crank is at rest, JV → 0. The corresponding counterpressure ΔPk is then determined, the volume increment ΔVExp,k is obtained from the geometry of the apparatus, and the final volume V(Final) of the diluted solution is measured or inferred from the step sum. In this manner, all quantities required to evaluate the Riemann approximation and thus ΔGTestmix(n(A), n(B)) for a given practically incompressible binary solution are obtained experimentally.
Moreover, if the paired values of the braking current Ibrake and the corresponding electromotive force Ubrake are recorded for each step k, the electric brake engine can simultaneously be operated as a generator. In that case, the PRO dilution step and the corresponding RO concentration step may be viewed as two branches of a closed electro-osmotic cycle, formally analogous to cyclic voltammetry. This opens the possibility of representing the osmotic engine as a cyclic voltammogram in the (Ubrake, Ibrake)-plane. In the quasi-reversible limit and neglecting electrical losses, or after calibration, the electrical work delivered over a closed electro-osmotic cycle equals the osmotic work, so that the area enclosed by the loop in the (Ubrake, Ibrake)-plane is proportional to Wavail and hence to −ΔGDilsys. Such a representation may be particularly appealing when the solute B undergoes conformation changes or other structural rearrangements during dilution; in such systems, modifications of the loop shape or hysteresis could provide an experimental handle on slow, conformation-dependent contributions to ΔGTestmix(n(A), n(B)).
2.3 The conceptual osmotic engine
An idealized, conceptual osmotic engine is considered, consisting of two reservoirs separated by a semipermeable membrane. The engine comprises at least three cylinders, each with a fixed volume VC, all connected to a common crankshaft. At least three cylinders are used to ensure continuous torque delivery on the crankshaft while one cylinder is in the working stage.The engine operates through an osmotic cycle with three distinct stages. In the filling stage, F, a cylinder is partially charged with a volume αVC of the Test solution with density ρ(Test). During the working stage, W, this volume is osmotically diluted while mechanical work, W, is delivered to the crankshaft and thereby to the surroundings. In the exhaust stage, E, the osmotically diluted Test solution with density ![[small rho, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e4.gif) (Test) is discharged, and the cycle is repeated. The cylinder sequence can thus be summarized as (F, W, E), with only one cylinder in the working stage at any given time.
(Test) is discharged, and the cycle is repeated. The cylinder sequence can thus be summarized as (F, W, E), with only one cylinder in the working stage at any given time.
The pressure contribution is associated with the actual osmotic pressure of the Test solution, and the change in cylinder volume, ΔVC, is confined to
| ΔVC = (1 − α)·VC | (32) |
The first reservoir is filled with the pure solvent A, whereas the second reservoir contains solute B dissolved in A to form a non-reactive binary Test solution. Initially, the Test solution occupies the volume αVC within the working cylinder. The system is characterized by an observed osmotic pressure difference Δπobs(Test) relative to the pure solvent. Operationally, in the quasi-reversible limit, Δπobs(V) denotes the counterpressure ΔP required to reach a quasi-equilibrium condition JV → 0 at the corresponding cylinder volume V, so that Δπobs(V) = ΔP(V) along the quasi-reversible path.
A hypothetical free-expansion equilibrium volume VeqC is defined as the volume that would be attained if the piston were allowed to expand freely against the surroundings, i.e. with vanishing applied counterpressure. In addition, a critical stall osmotic pressure Δπobscri(Test) is defined as the counterpressure at which the piston cannot advance further within the admissible expansion range between αVC and VC under quasi-equilibrium conditions JV → 0, i.e. the limiting mechanical equilibrium where the net driving pressure across the membrane is balanced by the applied counterpressure and the piston velocity vanishes.
Moreover, it is observed, see SI: the concept of the osmotic engine, that Δπobscri(Test) → Δπobs(TestDil), the residual osmotic pressure, as α → 1. For a more detailed description of the osmotic engine, reference is made to Fig. 4 and 5, as well as to the SI: the concept of the osmotic engine.
 | ||
| Fig. 4 (a) In this embodiment, it is preferable that the membrane area, AM, exceeds the piston cross-section, AC. (b) By adding a pressure-levelling nozzle, the condition (ΔPk)JV=0 can be measured. | ||
 | ||
| Fig. 5 (a) Star engine configuration: when α = 1/3 the engine has three cylinders. (b) Inline engine viewed from below: when α = 1/2 it has four cylinders. The pistons move downward as in Fig. 4(b) when the system acts as a membrane osmometer. For clarity, pressure nozzles, manometer and crankshaft are omitted. | ||
2.4 Non-ideal solutions
At osmotic equilibrium, the corresponding change in cylinder volume of the Test solution, ΔVeqC(Test), is given by| ΔVeqC(Test) = VeqC(Test) − αVC | (33) |
A convenient determination of VeqC(Test) can be achieved by recirculating the exhaust volume back into the Test solution, so that the solution is gradually diluted. At a certain dilution step, the critical osmotic pressure, Δπobscri(Test), becomes equal to the hydrostatic pressure associated with a column height between the initial cylinder volume αVC and the final cylinder volume VC, at which point the engine stalls.
When the system is brought to osmotic equilibrium, the resulting membrane flux is reduced to JV → 0, and the actual osmotic pressure is obtained from the measured counterpressure, ΔPk, in the cylinder compartment. This measured pressure is paired with the corresponding volume increment ΔVExp,k, determined from straightforward analytical geometry. Accordingly, using eqn (30), the measured Gibbs free energy of mixing can be expressed in the residual-dilution approximation as
 | (34) |
Here the cumulative experimentally imposed volume change is identified with the equilibrium expansion,
 | (35) |
| VFinal = V(Test) + ΔVeqC(Test) = (1 + ψ)·V(Test) | (36) |
If the residual-dilution approximation is not invoked, the corresponding expression is instead written as
 | (37) |
From the above, it follows that when VeqC(Test) < VC, the operational cylinder is brought to rest, the osmotic cycle is interrupted, and the engine comes to a complete stop. It should also be noted that VeqC(Test) approaches VC arbitrarily closely as α approaches unity.
The dilution of the Test solution up to the point at which the engine stalls, and thereby the determination of the critical dilution factor ψ, can be carried out in a straightforward manner with the osmotic engine. This dilution factor is closely related to the hypothetical osmotic equilibrium volume VeqC(Test) of the Test solution and the cylinder volume VC. Since
| ΔVeqC(Test) = VeqC(Test) − αVC | (38) |
| VeqC(Test) = αVC + ΔVeqC(Test) | (39) |
If the critical dilution factor is defined relative to the initially loaded Test volume,
 | (40) |
| VeqC(Test) = (1 + ψ)·αVC | (41) |
For a more detailed description, the reader is referred to Fig. S9 and to the SI: calibrating the conceptual osmotic engine. Within the present framework of non-reactive binary solutions and the quasi-reversible operating conditions described above, the Gibbs free mixing energy at constant temperature is then evaluated from eqn (34).
In the dilution scenario where the engine stalls, the osmotic pressure of the diluted Test solution is referred to as the critical osmotic pressure, Δπobscri(Test). In this limiting state, the piston cannot advance further within the admissible expansion range, and the system is at quasi-equilibrium with JV → 0. The piston is positioned between the least osmotically diluted liquid column, αVC/AC, and the most osmotically diluted, VC/AC, where AC represents the cross-sectional area.
As shown in the SI: calibrating the conceptual osmotic engine, under the present geometric calibration one obtains
| 0 ≤ Δπobscri(Test) ≤ (1 − α)·P° | (42) |
In the present context, ΔGTestmix(n(A), n(B)) denotes the absolute mixing free energy of the Test composition, whereas ΔGideal,FHmix(n(A), n(B)) denotes the corresponding Flory–Huggins ideal (combinatorial) mixing contribution evaluated for the same composition and within the same reference frame. Under this condition, the excess energy of the Test solution is estimated as
| ΔGE = ΔGTestmix(n(S), n(P)) − ΔGideal,FHmix(n(S), n(P)) | (43) |
Thus, for a real binary polymer–solvent solution treated within the Flory–Huggins framework, the experimentally estimated ΔGTestmix(n(A), n(B)) may be combined with the corresponding ideal Flory–Huggins mixing term to obtain ΔGE, and thereby the interaction parameter χ. In this polymer–solvent specialization, the Flory–Huggins quantities are written as
 | (44) |
 | (45) |
Here, n(S) denotes the number of moles of solvent, and n(P) denotes the number of moles of polymer chains in the Flory–Huggins reference description. Likewise, ϕ(S) and ϕ(P) denote the volume fractions of solvent and polymer, respectively (not to be confused with the osmotic coefficient of the Test solution). Finally, χ = χ(ϕ(P), T) denotes the Flory–Huggins interaction parameter between solvent and polymer.
2.5 van't Hoff ideal solutions
The transport of liquid through a membrane is a non-equilibrium process and is therefore most naturally described within the framework of irreversible thermodynamics. Following the seminal developments by Kedem and Katchalsky12–14 and subsequent contributions,15–19 membrane transport in binary systems is commonly modeled in terms of coupled driving forces. In the near-equilibrium regime, the volumetric flux, JV, is governed by the competition between the osmotic pressure difference, Δπ, and the hydrostatic pressure difference, ΔP. In the present work, JV is considered under PRO conditions; accordingly, the sign of JV follows the PRO flux direction implied by the adopted sign convention for ΔP and Δπ, and may therefore appear reversed relative to the convention often used for RO. For a semipermeable membrane separating two non-reacting components—e.g., a solvent A and a solute B—the volume flux may be written as| JV = Lp·(σΔπ − ΔP) for 0 ≤ ΔP ≤ σΔπ | (46) |
 | (47) |
 | (48) |
In the subsequent section, the van't Hoff relation for the osmotic pressure difference, eqn (12), is adopted as an initial approximation. If this ideal assumption proves inadequate, the framework may be refined through the incorporation of higher-order corrections via MacMillan–Mayer virial coefficients,33 which are expected to constitute the dominant contribution in a more realistic treatment. Accordingly, eqn (59) and (71) are presented as the leading-order results of a first-order virial correction. Furthermore, the osmotic engine may be realized either as a concrete measurement device, for example to determine the Flory–Huggins interaction parameter in eqn (44), or as an idealized engine for theoretical investigations, analogous to the Carnot heat engine.
The osmotic engine consists of a cylindrical configuration, as depicted in Fig. 4(b). The operation of the piston involves oscillating between three pivotal positions: initially resting in the vacant state denoted as P1 and transitioning to the subsequent state containing a pristine, undiluted Test solution labeled as P2. Upon reaching position P2, solvent permeation through a semipermeable membrane is initiated, engaging in PV-work until the system arrives at the concluding position P3, thereby instigating the discharge process. Subsequently, the cylinder returns to position P1 to commence the cycle anew. Throughout the working stroke, the solute amount nC(B) in the initial volume αVC at P2 remains constant. A decrease in the actual osmotic pressure, denoted as [Δπ(Test)], occurs within the range from ΔπvH(Test) to αΔπvH(Test). This decrease is due to an increase in solvent volume, as described by eqn (12), where α represents the initial fraction of the cylinder volume VC filled with undiluted Test solution. The change in Gibbs free energy of mixing is calculated by defining ΔP as directly proportional to [Δπ(Test)], with a constant k, 0 ≤ k < 1, along the dynamic branch. The limiting case k → 1− corresponds to the quasi-static stall limit, whereas k = 0 corresponds to a static membrane-osmometer-like configuration.31,32
The volume is arbitrarily set at αVC at the start of the working stroke, causing an increase in volume and a decrease in actual osmotic pressure. Hence, it follows from eqn (46) and (47) that
 | (49) |
At the time t = 0 the piston initiates its working stroke, V(0)(0) = αVC, and thus eqn (49) is integrated to
 | (50) |
The time needed to complete the working stroke, Γ(0)C, is identified through eqn (50) as
 | (51) |
The time needed to make the engine complete one cycle, Γ(0)E, is identified as the product of the number of engine pistons,  , and the time needed for one piston to complete the working stroke, Γ(0)C, i.e.
, and the time needed for one piston to complete the working stroke, Γ(0)C, i.e.
 | (52) |
The angular velocity, ω(0)E, of the crankshaft is expressed as the reciprocal value of Γ(0)E multiplied by 2π
 | (53) |
 | (54) |
Eqn (54) is valid along the dynamic branch 0 ≤ k < 1. The stall torque is therefore obtained as a limiting value, not by direct substitution at k = 1, namely
 | (55) |
Taking this limit in eqn (54) yields
 | (56) |
 | (57) |
 | (58) |
In the present reference convention, the following expression represents the absolute van't Hoff estimate of the mixing free energy, obtained by adding the residual reference contribution to the extractable osmotic work term. When eqn (25) and (57) are combined, the van't Hoff contribution to the mixing free energy is obtained as
 | (59) |
|
ΔGidealmix = RT(n(A)lnxA + n(B)lnxB)
| (60) |
Hence,
|
ΔGE = ΔGmix − RT(n(A)lnxA + n(B)lnxB)
| (61) |
 | (62) |
Upon substituting eqn (61) and noting that, for constant n(A) with nT = n(A) + n(B)
 | (63) |
 | (64) |
Inserting eqn (64) into eqn (62) yields
 | (65) |
xB is the ideal Raoult-reference term, and deviations from ideality are captured by the remaining term.
According to eqn (25), for van't Hoff ideal solutions, it follows
| ΔGvHmix(n(A), n(B)) = ΔGrefmix + ΔπvH(Test)·V(Test)·ln(1 + ψ) | (66) |
However, even if ΔGrefmix becomes numerically small in the dilute limit, its composition derivative need not be small. Therefore, ΔGrefmix ≈ 0 does not in itself imply that ∂ΔGrefmix/∂n(B) ≈ 0.
With this choice, eqn (65) can be rewritten as
 | (67) |
In accordance with eqn (S77) it follows that
 | (68) |
 | (69) |
This expression also holds within the working cylinder of a conceptual osmotic engine with a large number of cylinders.34 In the SI, calibrating the conceptual osmotic engine, it is shown that nC(B)∂ψ/∂nC(B) = n(B)∂ψ/∂n(B), yielding
 | (70) |
![[small pi, Greek, circumflex]](https://www.rsc.org/images/entities/char_e1de.gif) (Test) is used in the derivation of eqn (70). Hence, it follows
(Test) is used in the derivation of eqn (70). Hence, it follows
 | (71) |
The Raoult-law activity coefficient, γRB, may be identified for a volatile component B through its fugacity fB(xB,T,P) according to
 | (72) |
 is the vapour pressure of pure B. Consequently, γRB → 1 as xB → 1.
is the vapour pressure of pure B. Consequently, γRB → 1 as xB → 1.
For completeness, one may also introduce a Henry-scale activity coefficient, γHB, through
 | (73) |
 | (74) |
Perspective and limitations: while eqn (71) is algebraically consistent within the present osmotic-engine framework, quantitative use of the resulting estimate of lnγRB requires caution. The dominant sensitivity enters through the treatment of the solution volume in the dilute limit. In particular, the auxiliary assumption of volume additivity, V(Test) = n(A)![[V with combining macron]](https://www.rsc.org/images/entities/i_char_0056_0304.gif) A + n(B)B, is only asymptotically justified as n(B) → 0 if the partial molar solute volume approaches its infinite-dilution value ∞B in a controlled manner. Replacing this limiting partial molar quantity by the pure-component molar volume,
A + n(B)B, is only asymptotically justified as n(B) → 0 if the partial molar solute volume approaches its infinite-dilution value ∞B in a controlled manner. Replacing this limiting partial molar quantity by the pure-component molar volume,  , is a crude closure: for many solutes, ∞B differs substantially from
, is a crude closure: for many solutes, ∞B differs substantially from 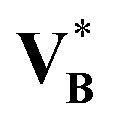 because solvation/structuring effects, compressibility differences, and excess mixing volumes shift the effective volumetric contribution of a single dissolved molecule away from its pure-liquid value. Consequently, the density-based term in eqn (70) and therefore the corrective contribution in eqn (71) can be biased in both magnitude and sign for specific solute–solvent pairs.
because solvation/structuring effects, compressibility differences, and excess mixing volumes shift the effective volumetric contribution of a single dissolved molecule away from its pure-liquid value. Consequently, the density-based term in eqn (70) and therefore the corrective contribution in eqn (71) can be biased in both magnitude and sign for specific solute–solvent pairs.
A second, independent source of fragility is the dilute-solution approximation ψ ≈ Δ(Test) used in deriving eqn (70). This step suppresses higher-order coupling between osmotic work capacity and composition-dependent volume changes, i.e. it effectively truncates the concentration expansion at a stage where real solutions may already exhibit measurable non-ideality in Δπ and in the partial molar volumes. In the same spirit, the van't Hoff ideal input, eqn (66), should be read as a conceptual limiting case rather than a quantitative model for arbitrary dilute mixtures, since real systems may deviate from van't Hoff behaviour due to association, specific interactions, or, for electrolytes, non-colligative contributions that are not captured by the present closure.
By analogy with Carnot's ideal heat engine, the osmotic-engine construct remains useful as a unifying conceptual scaffold: one may envisage successive perturbation expansions of ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) γRB in powers of concentration, provided that volumetric inputs are treated in a controlled manner, e.g. via ∞B rather than
γRB in powers of concentration, provided that volumetric inputs are treated in a controlled manner, e.g. via ∞B rather than  . As a natural next step toward a more robust dilute-theory closure, one can re-derive an eqn (71)-type relation using a MacMillan–Mayer virial description of the osmotic pressure,
. As a natural next step toward a more robust dilute-theory closure, one can re-derive an eqn (71)-type relation using a MacMillan–Mayer virial description of the osmotic pressure,
 | (75) |
3 Summary
A membrane-thermodynamic framework, termed the osmotic engine, is developed to determine the excess Gibbs free energy of mixing, ΔGE, in real binary solutions from measurable osmotic and volumetric responses under pressure-retarded osmosis, PRO, conditions. In the van’t Hoff limit, the theory provides a leading-order colligative perspective linking the Henry-reference activity coefficient at infinite dilution to the dimensionless osmotic pressure, while clarifying its sensitivity to volume assumptions and reference-state regularisation. The framework extends to multicomponent mixtures containing electrolytes and non-electrolytes, captures solvent-dependent shifts in reaction equilibria, and supports routine estimation of the Flory–Huggins solvent–polymer interaction parameter.Author contributions
Dennis Wowern Nielsen wrote the first draft, and Dennis Wowern Nielsen and Claus Helix-Nielsen contributed equally to the final version.Conflicts of interest
The authors declare no conflicts of interest.Nomenclature
| AC | Cylindric cross sectional area, [m2] |
| AM | Membrane surface area, [m2] |
| α | Fraction of cylinder volume in osmotic engine, [—] |
| Cn | The nth cylinder, n ∈ {1, 2, 3, …}, [—] |
| En | The nth exhaustion stroke, [—] |
| Fn | The nth filling stroke, [—] |
| Wn | The nth working stroke, [—] |
| ΔG | The change of the Gibbs free energy, [J] |
| ΔGmix | The change of the Gibbs free energy of mixing, [J] |
| ΔGprodmix | The change of the Gibbs free energy of mixing of reaction products, [J] |
| ΔGreacmix | The change of the Gibbs free energy of mixing of reactants, [J] |
| ΔḠmix | The change of the partial molar Gibbs free energy of mixing, [J mol−1] |
| ΔGrxn | The change of the Gibbs free energy of reaction, [J] |
| ΔGExcess | The change of the Gibbs free excess energy, [J] |
| g | The gravitational constant, [9.82 … m s−2] |
| ΓC | The time needed for one single cylinder to perform the working stroke, [s] |
| ΓE | The time needed for one engine revolution, [s] |
| Δh | The travel part between the cylinder volumes αVC and VC, [m] |
| ι | The van’t Hoff factor, [—] |
| JV | The volume flow across the semipermeable membrane, [m3 m−2 s−1] |
| k | The load on the crank shaft, [—] |
| Ka | The chemical affinity constant for an arbitrary addition reaction, [M−1] |
| LP | The solvent permeability coefficient across the semipermeable membrane, [m3 N−1 s−1] |
| μ° | The standard chemical potential of formation, [J mol−1] |
| n | Amount of substance, [mol] |
| N | The number of engine cylinders, [—] |
| ωE | The crank angular velocity, [rad s−1] |
| ΔP | The actual pressure difference of the working cylinder under osmotic dilution, with respect to the pure solvent compartment, [N m−2] |
| ΔPcri | The pressure caused by a column of liquid with the density of cri(Test), [N m−2] |
| π | The mathematical constant pi, [3.14 …] |
| ψ | The critical dilution factor, which causes the osmotic engine to a complete halt, [—] |
| π | The osmotic pressure of a binary solution, [N m−2] |
| R | The universal gas constant, [8.31 J K−1 mol−1] |
| ρ(Test) | The density of a Test solution, [g cm−3] |
| (Test) | The density of a Test solution after osmotic dilution in the working cylinder, [g cm−3] |
| cri(Test) | The critical density of a Test solution after osmotic dilution in the working cylinder, [g cm−3] |
| T | Kelvin temperature, [K] |
| V(Test) | The volume of a Test solution, [m3] |
| VC | The volume of a single cylinder in the osmotic engine, [m3] |
| VeqC | The osmotic equilibrium volume, [m3] |
| ΔVstop | The increment of volume of a real binary Test solution with its own solvent resulting in an osmotic pressure less than or equal to the critical osmotic pressure, [m3] |
| W | The PV-work done on the surroundings during osmotic dilution, [J] |
| ∂W/∂t | The engine power measured on the crank, [J s−1] |
| HB | Henry's constant for the solute B, [N m−2] |
 | The vapour pressure of pure solute B, [N m−2] |
| γRB | The Raoult-based activity coefficient of solute B, [—] |
| γHB | The Henry-based activity coefficient of solute B, [—] |
| aRB | The Raoult-based activity of solute B, [—] |
| aHB | The Henry-based activity of solute B, [—] |
Data availability
Supplementary information is available. See DOI: https://doi.org/10.1039/d5cp03542b.All arguments and derivations supporting the findings of this study are included within the article. No additional data were generated or analyzed.
References
- M. J. Madden, S. N. Ellis, A. Riabtseva, A. D. Wilson, M. F. Cunningham and P. G. Jessop, Comparison of vapor pressure osmometry, freezing point osmometry and direct membrane osmometry for determining the osmotic pressure of concentrated solutions, Desalination, 2022, 539, 115946 CrossRef CAS.
- M. S. Stevens, Polymer Chemistry – an introduction, Oxford University Press, 1999, pp. 42–58 Search PubMed.
- W. Pfeffer, Osmotisch Untersuchungen, Leipzig, 1891, vol. 344 Search PubMed.
- I. L. Minkov, et al., Equilibrium and Dynamic Osmotic Behaviour of Aqueous Solutions With Varied Concentration at Constant and Variable Volume, Sci. World J., 2013, 876897 CrossRef PubMed.
- R. Berkeley and E. Hartley, A method of measuring directly high osmotic pressures, Proc. R. Soc. London, 1904, 73, 436 Search PubMed.
- M. Elimelech and A. P. William, The future of seawater desalination: energy, technology, and the environment, Science, 2011, 333(6043), 712–717 CrossRef CAS PubMed.
- P. Furmański, S. A.-A. Saleh and P. Łapka, Analysis of a hydro-osmotic power plant using a simple mathematical model, J. Power Technol., 2017, 97(5), 395–405 Search PubMed.
- P. J. Flory, Thermodynamics of high polymer solutions, J. Chem. Phys., 1942, 10, 1 CrossRef.
- J. H. Huggins, Some Properties of Solutions of Long-Chain Compounds, J. Chem. Phys., 1941, 9, 440 CrossRef.
- J. M. Smith and H. C. Van Ness, Introduction to Chemical Engineering Thermodynamics, McGraw-Hill, 4th edn, 1987, p. 351 Search PubMed.
- G. D. C. Kuiken, Thermodynamics of Irreversible Processes: Applications to Diffusion and Rheology, John Wiley & Sons, 1st edn, 1994, pp. 212–213. ISBN 0-471-94844-6 Search PubMed.
- A. Katchalsky and O. Kedem, Thermodynamics of Flow Processes in Biological Systems, J. Biophys., 1962, 2, 53 CrossRef CAS PubMed.
- O. Kedem and A. Katchalsky, Biochim. Biophys. Acta, 1958, 27, 229 CrossRef CAS PubMed.
- O. Kedem and A. Katchalsky, A Physical Interpretation of the Phenomenological Coefficients of Membrane Permeability, J. Gen. Physiol., 1961, 45, 143 CrossRef CAS PubMed.
- A. Katchalsky and F. Curran, Non-Equilibrium Thermodynamics, Harvard Uni., Press, 1965 Search PubMed.
- H. Vink, Osmotic measurements with solute-permeable membranes, Ark. Kemi, 1960, 15, 149 CAS.
- H. Vink, Diffusion in porous media, Ark. Kemi, 1961, 17, 311 Search PubMed.
- K. S. Spiegler and O. Kedem, Thermodynamics of hyperfiltration (reverse osmosis): criteria for efficient membranes, Desalination, 1966, 1, 311–326 CrossRef CAS.
- N. Lakshminarayanaiah, Transport Phenomena in Membrane, Academic Press Inc., 1972, pp. 292–319 Search PubMed.
- S. Loeb, F. Van Hessen and D. Shahaf, Production of energy from concentrated brines by pressure-retarded osmosis: II. Experimental results and projected energy costs, J. Membr. Sci., 1976, 1, 249–269 CrossRef CAS.
- S. Loeb and S. N. Richard, Osmotic power plants, Science, 1975, 189(4203), 654–655 CrossRef CAS PubMed.
- S. Loeb, Method and apparatus for generating power utilizing pressure-retarded osmosis, US4193267, 1980 Search PubMed.
- S. E. Skilhagen, J. E. Dugstad and R. J. Aaberg, Osmotic power—power production based on the osmotic pressure difference between waters with varying salt gradients, Desalination, 2008, 220(1–3), 476–482 CrossRef CAS.
- K. Gerstandt, et al., Membrane processes in energy supply for an osmotic power plant, Desalination, 2008, 224(1–3), 64–70 CrossRef CAS.
- A. Achilli and A. E. Childress, Pressure retarded osmosis: from the vision of Sidney Loeb to the first prototype installation, Desalination, 2010, 261(3), 205–211 CrossRef CAS.
- M. Tawalbeh, et al., Recent developments in pressure retarded osmosis for desalination and power generation, Renewable Sustainable Energy Rev., 2021, 138, 110492 CrossRef CAS.
- K. L. Lee, R. W. Baker and H. K. Lonsdale, Membranes for power generation by pressure-retarded osmosis, J. Membr. Sci., 1981, 8, 141–171 CrossRef CAS.
- S. Chou, et al., Thin-film composite hollow fiber membranes for pressure retarded osmosis (PRO) process with high power density, J. Membr. Sci., 2012, 389, 25–33 CrossRef CAS.
- G. D. Mehta and S. Loeb, Internal polarization in porous substructure of a semipermeable membrane under pressure-retarded osmosis, J. Membr. Sci., 1978, 4, 261–265 CrossRef CAS.
- G. D. Mehta and S. Loeb, Performance of permasep B-9 and B-10 membranes in various regions and at high osmotic pressures, J. Membr. Sci., 1978, 4, 335–349 CrossRef.
- N. Y. Yip and M. Elimelech, Performance limiting effects in power generation from salinity gradients by pressure retarded osmosis, Environ. Sci. Technol., 2011, 45(23), 10273–10282 CrossRef CAS PubMed.
- A. N. Newby, T. V. Bartholomew and M. S. Mauter, The Economic Infeasibility of Salinity Gradient Energy via Pressure Retarded Osmosis, ACS ES&T Eng., 2021, 1113–1121 Search PubMed.
- T. L. Hill, Theory of Solutions. II. Osmotic Pressure Virial Expansion and Light Scattering in Two Component Solutions, J. Chem. Phys., 1945, 30, 93–97 CrossRef.
- Y. A. Boussouga and A. Lhassini, Study of mass transfer mechanisms for reverse osmosis and nanofiltration membranes intended for desalination, J. Mater. Environ. Sci., 2017, 8, 1128–1138 CAS.
| This journal is © the Owner Societies 2026 |