
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Disentangling effects of pH, potential, and cation concentration in cathodic corrosion of platinum
Mark
Aarts†
 *,
Jamie A.
Trindell†
and
Marc T. M.
Koper
*
*,
Jamie A.
Trindell†
and
Marc T. M.
Koper
*
Leiden Institute of Chemistry, Leiden University, Leiden 2333 CC, The Netherlands. E-mail: m.aarts@lic.leidenuniv.nl; m.koper@lic.leidenuniv.nl
First published on 12th December 2025
Abstract
Cathodic corrosion concerns the electrochemical etching of metals at negative polarization. While this process is detrimental for electrode stability and lifetime, the mechanism responsible for etching has not yet been fully elucidated. In this work, we determine the potential at which signs of cathodic corrosion on platinum electrodes are observed for different aqueous electrolytes, varying bulk pH and cation concentration. From cyclic voltammetry, we find that typical indicators of cathodic corrosion roughening the surface always appear at the same potential with respect to the standard hydrogen electrode, irrespective of the electrolyte. In contrast, we observe from microscopy that the degree of etch pit formation is strongly determined by the cation concentration. This therefore separates potential-induced effects of surface roughening from the rate of cation-mediated dissolution. The electrolyte-independent onset potential reveals aspects of the underlying corrosion mechanism, and our results are discussed in terms of the different roles of the electrolyte components at the interface.
Introduction
Cathodic corrosion is the phenomenon of surface modification or particularly electrode dissolution at potentials more negative than the standard reduction potential of the material, and affects many metal electrodes under different electrolyte conditions.1 Avoiding severe cathodic corrosion is important, as every electrochemical system invariably requires a cathode. While material dissolution of electrodes will limit device lifetime, any modification of the surface can be particularly detrimental for electrocatalysts, that often have surfaces tailored to enhance specific reactivity or selectivity.2 Modification of the electrocatalyst under reducing conditions can therefore affect performance during operation.3,4 Such instability of metals appears to be quite generic in aqueous electrolytes, where cathodic corrosion has been observed on copper, rhodium, gold, and platinum5–8 electrodes under direct current conditions, as well as on additional metals and alloys under alternating polarization.9,10Platinum is of specific interest, as it is widely regarded as a stable electrode particularly under reducing conditions.11 However, cathodic corrosion of platinum in aqueous environments is surprisingly ubiquitous. It has been shown to occur at relatively mild potentials (relative to the reversible hydrogen reaction) in alkaline electrolytes,12 but was also observed in acidic electrolytes upon strong negative polarization.13 A key characteristic of the corrosion process is the etching anisotropy, as evidenced by a strong facet dependence of the resulting etch pits and their morphologies.14 Furthermore, a critical factor for corrosion has been shown to be the presence of irreducible cationic species.5,9,15 In addition to the absence of corrosion pits in electrolytes containing only the acid, the surface structure after corrosion is thought to be influenced by the identity of the cation.6,16,17
Determining the mechanism through which cathodic corrosion occurs has proven to be difficult however, specifically because the required potentials are outside the electrochemical stability window of water. Furthermore, studies on different electrolyte systems imply that the cathodic degradation does not always have to follow the same scheme.1 For example, platinum electrodes display modified surfaces in dry organic solvents,18 but cathodic corrosion of gold in methanol only occurs in the presence of water.19 For platinum in aqueous solutions the current hypothesis is that cathodic platinum dissolution proceeds by the formation of a cation-stabilized anionic species, facilitated by adsorbed hydrogen through a metal hydride intermediate.20,21 In fact, the first experimental evidence of such platinum hydrides under cathodic corrosion conditions has been recently reported.22
To further refine the mechanistic picture of cathodic corrosion, obtaining the conditions at which platinum electrodes are stable is therefore highly valuable. This is clearly also important from a practical point of view. Prior reports have aimed to determine the stability conditions of platinum electrodes at progressively lower potentials in alkaline media using voltammetry,12 tentatively capturing these in a Pourbaix representation. Due to the apparent role of electrolyte cations, it was further suggested to explicitly incorporate their role in updated stability diagrams.20 While these works have investigated only a narrow range of electrolyte compositions,6 such a representation has the benefit of visualizing the dependence of the process on the concentration of the species of interest, with the slope providing information on the underlying reaction.23
To this end we characterize the stability of single-crystal platinum electrodes against cathodic corrosion in aqueous environments in a wide range of electrolytes. Specifically, we investigate the influence of the applied potential, and (bulk) electrolyte pH and cation concentration. Using cyclic voltammetry (CV), we find that the least negative potential where electrochemical signs of corrosion are observed, tentatively referred to as ‘onset potential’, is largely independent of the employed electrolyte and found to be around −1.35 V versus the standard hydrogen electrode (SHE). On the other hand, scanning electron microscopy (SEM) shows that the electrode morphology is highly influenced by the electrolyte cation concentration when keeping the electrode at mildly corroding potentials. Specifically, large scale pitting only occurs at higher interfacial cation concentrations, which is not apparent from the CV signal. These results indicate two distinct steps in the process, separating surface modification through atomic scale roughening evidenced by CV from large scale pitting and dissolution evidenced by SEM. While the former step appears to depend primarily on the applied potential and not on the electrolyte cations, the latter step requires the involvement of non-reducible electrolyte cations. As the near-electrode environment is significantly affected at the corrosion conditions, our observations are discussed in terms of the potential-dependent state of the interfacial region, suggesting the importance of local cation concentration rather than local pH. Apart from outlining the parameter space for electrode stability, the constant onset potential for cathodic corrosion in different electrolytes is indicative of an electrolyte-independent energetic barrier for the cation-mediated dissolution.
Results
To determine at what applied potentials cathodic corrosion occurs on the platinum electrodes, a protocol similar to that reported earlier was used.12 The experiment is conducted using two cells, a cell containing 100 mM H2SO4 for CV characterization, and a cell containing the corrosion electrolyte (either NaOH or HClO4 + NaClO4). Electrochemical experiments were carried out in a three-electrode configuration, using a reversible hydrogen electrode (RHE) as the reference, and a platinum counter electrode. As a working electrode single crystalline platinum spherical bead electrodes were used (see Methods, and ref. 24). The platinum electrode is subjected to cathodic treatment for 3 minutes using a constant applied current, or chronopotentiometry (CP). Fig. 1a illustrates the CP measurements for corrosion in 10 M NaOH, applying an increasingly cathodic current between −0.3 mA and −10 mA (with the submerged surface area ∼0.25 cm2). In the remainder of the manuscript all potentials applied during the cathodic corrosion experiments are given versus the standard hydrogen electrode (SHE) after 100% correction for the solution potential (IR) drop (see Methods, solution resistance values are shown in S1). After every CP measurement the electrode is thoroughly rinsed and placed in the sulfuric acid cell for CV characterization (50 mV s−1). Fig. 1b shows the resulting positive-going sweep (10th cycle) after each corrosion treatment. Similar to previous work,6,12–14 corrosion is evidenced by a reduction of the peak associated with the (110) steps (∼0.15 V vs. RHE), and most notably by the appearance of a peak characteristic for (100) terraces (∼0.4 V), in addition to a broad peak indicative of (111)-type sites in sulfuric acid (∼0.5 V). The qualitative appearance of these features in the CV, in particular the (100) terrace peak, is used to determine the least negative potential, or tentative ‘onset’, at which corrosion occurs. In 10 M NaOH, from Fig. 1 this is determined to be after applying −5.6 mA yielding a polarization of −1.3 V vs. SHE, in agreement with earlier reports.12 | ||
| Fig. 1 (a) Potential transients for consecutive corrosion steps of the Pt bead electrodes in 10 M NaOH when applying a constant current for 3 minutes. The electrode is rinsed and characterized in sulfuric acid after every corrosion step. The blue dashed line is the RHE potential. (b) CV characterization (50 mV s−1) of the electrode in 0.1 M H2SO4 after every corrosion step, indicated by the legend. The colored bands indicate the main CV features used to evidence corrosion, in particular the formation of (100) terraces. | ||
This protocol is used to obtain the potentials at which the CV indicates electrode corrosion under different conditions, specifically investigating the influence of pH and bulk cation concentration. Fig. 2 shows the potentials on the SHE scale after 100% IR compensation (S1) obtained as a function of bulk Na+ concentration, ranging from 1 mM to 10 M. Potentials were obtained in electrolytes of different bulk pH using different concentrations of NaOH for alkaline conditions, and acidic electrolytes containing 0.1 M HClO4 + x mM NaClO4. The inset shows the same results as a function of bulk pH. In Fig. 2 the error bars denote the standard deviation of the CP potential profile. The grey/red transparent patches indicate the lowest applied potential where no corrosion was observed, or measurement resolution (e.g. for 10 M NaOH this is the potential when applying −3 mA in Fig. 1). The CP and CV transients of all measurements are shown in S2. A key finding is that we observe no strong dependence on either the bulk cation concentration, or the pH. Rather, signs of corrosion are observed as long as the electrode is polarized at potentials below −1.35 ± 0.12 V vs. SHE (mean ± standard deviation of all datapoints).
 | ||
| Fig. 2 Onset potentials obtained by changes in voltammetry as a function of bulk Na+ concentration, varied by using different concentrations of NaOH, or 0.1 M HClO4 + x mM NaClO4. The error bars denote the variation of the potential profile during the corrosion step, with the transparent patch indicating the applied potential in the step before corrosion was observed. Inset: The same data as a function of bulk pH, the blue dashed line is the RHE potential. | ||
We comment briefly on two aspects of the accuracy of these measurements. Firstly, determination of the onset potential is done qualitatively from the CV change (all CV data are shown in S2). Secondly, at potentials much more negative than the RHE, accurate potential control is impeded due to vigorous bubble formation and large solution potential drops.25 As indicated by the error bars this results in variation in the applied potential during the experiment, in particular at lower ionic strength (S1), and could lead to locally increased current densities through dynamic blocking of parts of the surface. Despite these considerations, the measurements and obtained potential values are reproducible over a wide range of bulk cation concentration and pH.
To further investigate the electrode stability near this tentative onset potential, electrodes are polarized in different electrolytes at a fixed potential of −1.5 V vs. SHE for 3 minutes (after 100% IR compensation, see Methods). Indeed, all CVs in Fig. 3a display the characteristics associated with corrosion as mentioned above. Notably, while corrosion in 10 M NaOH shows the largest change, the voltammograms are very similar for the other bulk cation concentrations (10 mM, 1 M) and pH values (1, or 14). With the CV being indicative of changes occurring across the entire surface of the Pt bead, we use scanning electron microscopy (SEM) to investigate facet-specific corrosion features on the surface. In contrast to the similarity in CV, SEM shows that the electrodes undergo a different extent of corrosion. Specifically, Fig. 3b–e show SEM images of the (111) facet on the spherical crystals after corrosion. First, at constant pH 1 increasing the Na+ concentration from 10 mM (Fig. 3b) to 1 M (Fig. 3c) considerably roughens the surface, and characteristic triangular etch pits appear.13,14 Second, at a constant Na+ concentration of 1 M, increasing the pH from pH 1 (Fig. 3c) to pH 14 (Fig. 3d) yields similar triangular pitting in both cases. Only at the highest Na+ concentration of 10 M (Fig. 3e) the surface is corroded much more extensively, corresponding to significant changes in the CV. The (111) facet is most prone to corrosion,14 but similar results are obtained around the other low index planes as shown in (S3). It is worth mentioning that a similar increase of the (100) terraces (CV) without large scale surface modification (SEM) was observed in pure 0.1 M HClO4 with no additional cation present (CV and SEM data shown in S4). Taken together, the results in Fig. 3 indicate that the voltammogram is sensitive to surface changes that are not visible with SEM.7,12,14 These are present in all cases, only leading to pitting under influence of electrolyte cations and unaffected by bulk electrolyte pH.
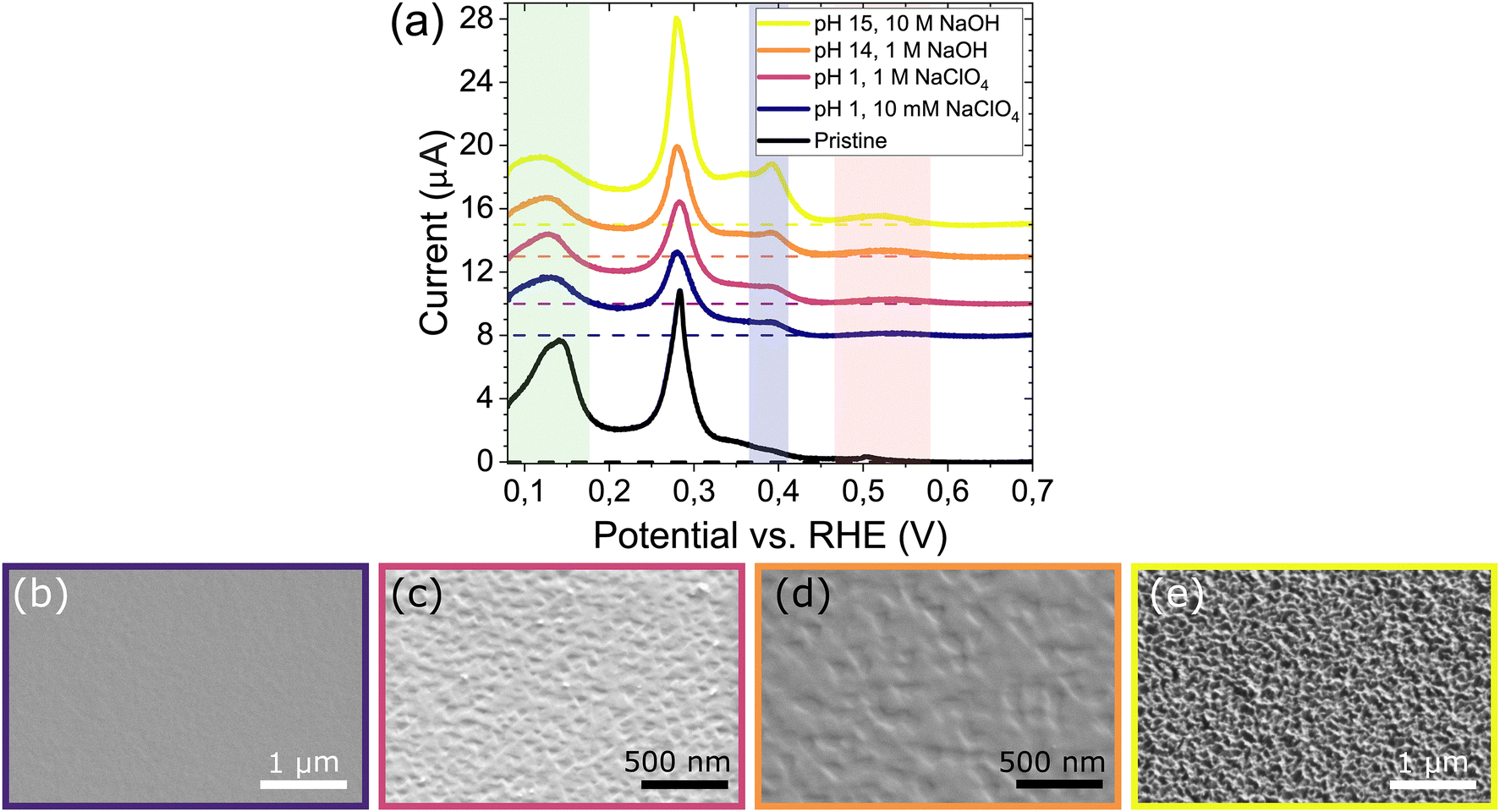 | ||
| Fig. 3 (a) CV characterization in 0.1 M H2SO4 (50 mV s−1) after polarizing electrodes at −1.5 V vs. SHE for 3 minutes in different electrolytes. The colored bands indicate the (100) steps (green), (100) terraces (blue) and (111) terraces (red). (b)–(e) Subsequent SEM images of the (111) planes after corrosion in (b) pH 1, with 10 mM NaClO4. (c) pH 1, with 1 M NaClO4. (d) pH 14, 1 M NaOH. (e) pH 15, 10 M NaOH. | ||
Discussion
Our results on the cathodic stability of platinum electrodes can be discussed in terms of the main parameters used in this study: the applied potential, or current, and the bulk electrolyte composition. It should be stressed that our experimental corrosion conditions are far from equilibrium, and that the near-electrode environment varies significantly from the bulk. Specifically for the acidic electrolyte, a (cathodic, 50 mV s−1) linear sweep voltammogram at pH 1 is shown in Fig. 4, indicating three different regions: (I) kinetically controlled proton reduction. Without mass-transport limitation the interface contains both protons and irreducible cations, regulated by their bulk concentrations. (II) Diffusion-limited proton reduction. Protons are severely depleted, such that irreducible cations accumulate at the interface due to the electrode charge. The interfacial pH does not freely increase however, and is limited to approximately 7.26–28 (III) Kinetically controlled water reduction. Local pH increases rapidly and becomes highly alkaline. Positive charge at the interface is almost fully due to accumulation of irreducible electrolyte cations, regulated by the bulk concentration through diffusion and migration. | ||
| Fig. 4 Linear sweep voltammogram at 50 mV s−1 (cathodic scan) for a platinum bead electrode in 0.1 M HClO4 + 10 mM NaClO4. The 3 regions indicate (I) kinetically controlled proton reduction. (II) Diffusion limited proton reduction. (III) Kinetically controlled water reduction. The obtained onset potential for cathodic corrosion is indicated by the red dot and lies in region (III) for a bulk pH of 1. | ||
The red dot in Fig. 4 indicates the cathodic corrosion onset potential obtained earlier, in the third region. While the extent of increased alkalinity is confined to the diffusion layer and not expected to influence our potential measurement, this potential region implies that a possible explanation for the insensitivity of the onset potential is that under these non-equilibrium conditions the local pH and cation concentration become similar in all experiments, irrespective of the corresponding bulk electrolyte. Regardless, we can make some relevant observations from the range of conditions where corrosion is absent in relation to the ongoing discussion whether the cathodic corrosion proceeds through the formation of platinum hydride or platinide species.22
Firstly, with regards to the onset potential value, no corrosion is observed from voltammetry at the (cathodic) end of the second region in Fig. 4, extending to ∼−1 V vs. RHE. For (bulk) pH 1, the interface is still at approximately neutral pH at this potential, while corrosion already occurs at −0.4 V vs. RHE in 10 M NaOH. This means that the cathodic corrosion onset potential actually moves away from the reversible hydrogen electrode with decreasing bulk pH. Further, because cations already accumulate at the interface in this potential region (II) without leading to corrosion, this rather implies that either the potential, or high local alkalinity drives the process. The fact that we obtain a constant onset potential on an absolute scale in all electrolytes around −1.35 V vs. SHE, and that none of the currently proposed mechanisms would benefit from an alkaline environment,1,20,29 makes us believe that the process is driven by the electrode potential. This value for the threshold potential is striking, as it is the same that is found for platinide formation through electron transfer coupled with cation insertion and a concurrent negative oxidation state of platinum in organic electrolytes,30,31 while substantially more positive than the standard potential for Na+ reduction (−2.71 V vs. SHE). Similar to the pH however, we do not observe a change for the onset potential as a function of the bulk Na+ concentration obtained through voltammetry in Fig. 1, as would be expected for such insertion of the cation.
Secondly, past the onset potential (in region III) we observe that the degree of pitting increases with increasing Na+ concentration for electrodes kept at the same (absolute) potential, in line with the understanding that electrolyte cations increase the rate of cathodic corrosion in aqueous electrolytes.32 Such irreversible material removal through etching contrasts with the reversible topographical changes due to platinide formation through cation insertion observed in organic electrolytes.33 Indeed, this implies that the presence of water could also be a prerequisite for the cathodic dissolution, as was observed on gold electrodes,19 and suggested to be the source of hydrogen for hydride formation.20 Separately, both platinum hydrides22 and platinides34–36 are soluble in aqueous electrolytes. Therefore, it is possible that electrochemical modification of the electrode creates a destabilized surface that is then prone to dissolution with water as a chemical etchant. Such a scenario is analogous to previous reports on the wet etching of silicon.37
To summarize, we observe two distinct phenomena at the onset of cathodic corrosion, separating atomic scale roughening as evidenced by changes in voltammetry from large scale pitting corrosion visible with microscopy.38 The former occurs even in electrolytes with very low or zero bulk cation concentration, not unlike the reversible hydrogen-induced roughening or faceting at (111) step edges after mild hydrogen evolution39 and could potentially be due to recently observed platinum hydride disordering the surface.22 The latter occurs only in the presence of cations at the interface, where disordering of the surface could also provide nucleation sites for the corrosion pits.40 The local alkaline environment seems to be more a consequence of the required potential for cathodic corrosion in aqueous electrolytes than a prerequisite. The observation that the large scale dissolution appears more severe in highly alkaline media would then be related to the higher interfacial cation concentration, which does not seem to influence the onset potential of the cathodic process resulting in atomic roughening, but has a strong effect on the degree and rate of pitting and dissolution. From a practical point of view, as shown in S5, this can lead to severe corrosion occurring even at low bulk cation concentration when driven at sufficiently negative potentials.
Conclusions
To conclude, we investigate the cathodic stability of platinum electrodes in aqueous electrolytes, varying electrode potential, bulk pH, and bulk Na+ concentration. We determine the tentative onset potential of the corrosion process on single crystal platinum bead electrodes using CV characterization, and observe that it is independent of the used bulk electrolyte around −1.35 V vs. SHE. On the other hand, characterization of the surface morphology shows that the extent of corrosion depends heavily on the electrolyte cation concentration. Despite the interface being far from equilibrium at the corrosion potential, our observations further refine the mechanistic picture of cathodic corrosion in aqueous electrolytes. Notably, we observe that the onset corrosion potential is more negative with respect to the reversible hydrogen electrode with decreasing bulk pH, while the potential value with respect to SHE matches with the formation of platinides in organic electrolytes. We further distinguish two separate phenomena. At the onset potential electrodes primarily exhibit atomic scale roughening or surface restructuring, possibly induced by adsorbed hydrogen or hydride formation. In the presence of irreducible Na+ this is accompanied by severe corrosion and pitting, where the exact role of the cation cannot be deconvoluted from the increased interfacial pH under the obtained corrosion conditions. Altogether, both the surface roughening, as well as the dissolution process, were only observed at potentials with an increased pH of the interface. However, electrolyte cations are required for the latter, with the dissolution rate depending on their concentration.Methods
Single crystal bead electrodes
Single crystal bead electrodes were fabricated by melting the end of a platinum wire (Goodfellow, 1 mm diameter, 99.998%) in a hydrogen (H2, 99.999% Lindegas) oxygen (O2, 99.999% Lindegas) flame, with a coaxial argon (Ar) gas flow (99.999% Lindegas) protecting the flame. The molten bead is slowly crystallized by retracting the flame with a motorized stage. The bead is cooled under Ar flow for 5 minutes after completing the crystallization. Specific advantages of these spherical electrodes for our corrosion studies are (i) the presence of all different crystal orientations, where the bead surface can be projected on the stereographic triangle,14 (ii) the absence of grain boundaries, which have been observed to corrode preferentially,12 (iii) the ability to thoroughly reset the surface through remelting of the crystal.Electrochemistry
Corrosion experiments were conducted in a homemade perfluoroalkoxy cell (PFA) resistant to the alkaline electrolyte, and a glass cell for the characterization in sulfuric acid. The cells were cleaned by overnight soaking in an acidified potassium permanganate solution containing 1 g L−1 KMnO4, >99%, ACS reagent, Emsure), with 0.5 M H2SO4 (98%, ACS reagent, Sigma-Aldrich). The cell was then immersed in dilute piranha solution (35% H2O2, Merck and H2SO4, 98%, ACS reagent, Sigma-Aldrich) to remove residues of manganese oxide and permanganate anions, followed by boiling at least 3 times in ultrapure water (Milli-Q, resistivity ≥ 18.2 MΩ cm). Experiments were conducted using a BioLogic VSP-300 potentiostat, with a platinum counter electrode and a RHE reference electrode (Hydroflex, Gaskatel). Electrolytes were prepared using 30% NaOH (10 M, Suprapur, Merck), HClO4 (60%, Merck, Suprapur®), and H2SO4 (96%, Merck, Suprapur®), diluted with ultrapure water to the desired concentration, and then adding NaClO4·H2O (99.99%, Merck). In both the corrosion and the characterization cells the electrolyte was purged with Ar (99.999%, Lindegas), and Ar was flown over the solution during electrochemical experiments.For every electrolyte condition a remelted bead electrode was used. When sweeping the applied cathodic current for a single electrolyte the bead electrode was flame annealed followed by quenching in ultrapure water after every CV characterization step, prior to the next corrosion step.
IR compensation
For galvanostatic corrosion (Fig. 1, 2 and Fig. S2), 100% IR compensation was applied during the analysis by subtracting the product of the measured resistance (S1) and the applied current. Potentiostatic corrosion experiments in Fig. 3 and Fig. S4 (as well as the LSV in Fig. 4) were done with 85% IR compensation from the software after averaging 4 impedance measurements at 0.4 V vs. RHE at 100 kHz with an amplitude of 20 mV. Since significant current runs at these potentials the residual 15% potential drop was estimated before the experiment, and the setpoint potential was adjusted pre-emptively to reach the same IR-compensated potential values in all electrolytes after this additional 15% correction. Applied setpoint potentials and IR-compensated values are presented in S6.Microscopy
SEM images were obtained on an Apreo2 (Thermo Scientific) with 0.2 nA at 2 kV acceleration voltage.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). The SI includes all electrochemical data used to obtain the onset potential values, supporting experiments under different conditions, as well as additional experimental details. See DOI: https://doi.org/10.1039/d5cp03483c.Acknowledgements
This work received funding support from Hitachi High-Tech Corporation. The authors would like to acknowledge useful discussions with Hitachi High-Tech. We further acknowledge the support of the fine mechanical department at Leiden University and Thijs Hoogenboom for manufacturing the electrochemical cell.References
- T. Wirtanen, T. Prenzel, J.-P. Tessonnier and S. R. Waldvogel, Cathodic Corrosion of Metal Electrodes—How to Prevent It in Electroorganic Synthesis, Chem. Rev., 2021, 121, 10241–10270 CrossRef CAS PubMed.
- B. Roldan Cuenya, Metal Nanoparticle Catalysts Beginning to Shape-up, Acc. Chem. Res., 2013, 46, 1682–1691 CrossRef CAS PubMed.
- P. Grosse, et al., Dynamic Changes in the Structure, Chemical State and Catalytic Selectivity of Cu Nanocubes during CO2 Electroreduction: Size and Support Effects, Angew. Chem., Int. Ed., 2018, 57, 6192–6197 CrossRef CAS PubMed.
- J. Huang, et al., Potential-induced nanoclustering of metallic catalysts during electrochemical CO2 reduction, Nat. Commun., 2018, 9, 3117 CrossRef PubMed.
- S. Liu, et al., Alkali cation-induced cathodic corrosion in Cu electrocatalysts, Nat. Commun., 2024, 15, 5080 CrossRef CAS PubMed.
- T. J. P. Hersbach, et al., Alkali Metal Cation Effects in Structuring Pt, Rh, and Au Surfaces through Cathodic Corrosion, ACS Appl. Mater. Interfaces, 2018, 10, 39363–39379 CrossRef CAS.
- Y. Yang, et al., Elucidating Cathodic Corrosion Mechanisms with Operando Electrochemical Transmission Electron Microscopy, J. Am. Chem. Soc., 2022, 144, 15698–15708 CrossRef CAS PubMed.
- M. M. Elnagar, L. A. Kibler and T. Jacob, Structural Evolution of Au Electrodes during Cathodic Corrosion: Initial Stages of Octahedral-Nanocrystal Growth, J. Electrochem. Soc., 2022, 169, 102509 CrossRef CAS.
- A. I. Yanson, et al., Cathodic Corrosion: A Quick, Clean, and Versatile Method for the Synthesis of Metallic Nanoparticles, Angew. Chem., Int. Ed., 2011, 50, 6346–6350 CrossRef CAS PubMed.
- J. Feng, et al., Cathodic Corrosion of a Bulk Wire to Nonaggregated Functional Nanocrystals and Nanoalloys, ACS Appl. Mater. Interfaces, 2018, 10, 9532–9540 CrossRef CAS PubMed.
- L. Tang, et al., Electrochemical Stability of Nanometer-Scale Pt Particles in Acidic Environments, J. Am. Chem. Soc., 2010, 132, 596–600 CrossRef CAS PubMed.
- T. J. P. Hersbach, A. I. Yanson and M. T. M. Koper, Anisotropic etching of platinum electrodes at the onset of cathodic corrosion, Nat. Commun., 2016, 7, 12653 CrossRef CAS PubMed.
- X. Chen and M. T. M. Koper, In Situ EC-AFM Study of the Initial Stages of Cathodic Corrosion of Pt(111) and Polycrystalline Pt in Acid Solution, J. Phys. Chem. Lett., 2023, 14, 4997–5003 CrossRef CAS PubMed.
- N. Arulmozhi, T. J. P. Hersbach and M. T. M. Koper, Nanoscale morphological evolution of monocrystalline Pt surfaces during cathodic corrosion, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 32267–32277 CrossRef CAS PubMed.
- Y. I. Yanson and A. I. Yanson, Cathodic corrosion. I. Mechanism of corrosion via formation of metal anions in aqueous medium, Low Temp. Phys., 2013, 39, 304–311 CrossRef CAS.
- M. Duca, P. Rodriguez, A. I. Yanson and M. T. M. Koper, Selective Electrocatalysis on Platinum Nanoparticles with Preferential (100) Orientation Prepared by Cathodic Corrosion, Top. Catal., 2014, 57, 255–264 CrossRef CAS.
- M. M. Elnagar, L. A. Kibler and T. Jacob, Effect of Alkali Metal Cations and Trace Metal Impurities on Cathodic Corrosion of Gold Electrode Surfaces, ChemCatChem, 2024, 16, 0–9 Search PubMed.
- J. Simonet, The Platinised Platinum Interface Under Cathodic Polarisation, Platinum Met. Rev., 2006, 50, 180–193 CrossRef CAS.
- M. M. Elnagar, T. Jacob and L. A. Kibler, Cathodic corrosion of Au in aqueous methanolic alkali metal hydroxide electrolytes: Notable role of water, Electrochem. Sci. Adv., 2022, 2, 1–11 Search PubMed.
- T. J. P. Hersbach and M. T. M. Koper, Cathodic corrosion: 21st century insights into a 19th century phenomenon, Curr. Opin. Electrochem., 2021, 26, 100653 CrossRef CAS.
- I. Evazzade, A. Zagalskaya and V. Alexandrov, Revealing Elusive Intermediates of Platinum Cathodic Corrosion through DFT Simulations, J. Phys. Chem. Lett., 2022, 13, 3047–3052 CrossRef CAS PubMed.
- T. J. P. Hersbach, et al., Platinum hydride formation during cathodic corrosion in aqueous solutions, Nat. Mater., 2025, 24, 574–580 CrossRef CAS PubMed.
- A. Zagalskaya, P. Chaudhary and V. Alexandrov, Corrosion of Electrochemical Energy Materials: Stability Analyses Beyond Pourbaix Diagrams, J. Phys. Chem. C, 2023, 127, 14587–14598 CrossRef CAS.
- N. Arulmozhi and G. Jerkiewicz, Design and Development of Instrumentations for the Preparation of Platinum Single Crystals for Electrochemistry and Electrocatalysis Research. Part 1: Semi-Automated Crystal Growth, Electrocatalysis, 2016, 7, 507–518 CrossRef CAS.
- M. M. Elnagar, J. M. Hermann, T. Jacob and L. A. Kibler, An affordable option to Au single crystals through cathodic corrosion of a wire: Fabrication, electrochemical behavior, and applications in electrocatalysis and spectroscopy, Electrochim. Acta, 2021, 372, 137867 CrossRef CAS.
- B. H. Erne, Local pH Change during Diffusion-Limited Proton Reduction Determined by In Situ Infrared Spectroscopy, Electrochem. Solid-State Lett., 1999, 2, 231 CrossRef CAS.
- M. D. J. Gálvez-Vázquez, V. Grozovski, N. Kovács, P. Broekmann and S. Vesztergom, Full Model for the Two-Step Polarization Curves of Hydrogen Evolution, Measured on RDEs in Dilute Acid Solutions, J. Phys. Chem. C, 2020, 124, 3988–4000 CrossRef.
- M. C. O. Monteiro, X. Liu, B. J. L. Hagedoorn, D. D. Snabilié and M. T. M. Koper, Interfacial pH Measurements Using a Rotating Ring-Disc Electrode with a Voltammetric pH Sensor, ChemElectroChem, 2022, 9, e202101223 CrossRef CAS.
- H. P. Sarker, A. Goswami, M. T. Tang and F. Abild-Pedersen, From Micro-environments to Macroscopic Effects: How the Alkaline Hydrogen Evolution Reaction Drives Cu Cathodic Corrosion, ACS Catal., 2025, 15, 8676–8690 CrossRef CAS.
- J. Ghilane, M. Guilloux-Viry, C. Lagrost, J. Simonet and P. Hapiot, Reactivity of Platinum Metal with Organic Radical Anions from Metal to Negative Oxidation States, J. Am. Chem. Soc., 2007, 129, 6654–6661 CrossRef CAS PubMed.
- J. Ghilane, et al., Spectroscopic Evidence of Platinum Negative Oxidation States at Electrochemically Reduced Surfaces, J. Phys. Chem. C, 2007, 111, 5701–5707 CrossRef CAS.
- A. I. Yanson, P. V. Antonov, P. Rodriguez and M. T. M. Koper, Influence of the electrolyte concentration on the size and shape of platinum nanoparticles synthesized by cathodic corrosion, Electrochim. Acta, 2013, 112, 913–918 CrossRef CAS.
- J. Ghilane, M. Guilloux-Viry, C. Lagrost, P. Hapiot and J. Simonet, Cathodic Modifications of Platinum Surfaces in Organic Solvent: Reversibility and Cation Type Effects, J. Phys. Chem. B, 2005, 109, 14925–14931 CrossRef CAS PubMed.
- C. Cougnon, Cathodic reactivity of alkaline metal iodides toward platinum bulk. The formation of new reducing phases, Electrochem. Commun., 2002, 4, 266–271 CrossRef CAS.
- C. Cougnon and J. Simonet, Are tetraalkylammonium cations inserted into palladium cathodes? Formation of new palladium phases involving tetraalkylammonium halides, J. Electroanal. Chem., 2001, 507, 226–233 CrossRef CAS.
- A. Karpov, M. Konuma and M. Jansen, An experimental proof for negative oxidation states of platinum: ESCA-measurements on barium platinides, Chem. Commun., 2006, 838–840 RSC.
- M. A. Gosálvez, I. Zubel and E. Viinikka, Wet etching of silicon, Handbook of Silicon Based MEMS Materials and Technologies, Elsevier, 2010, pp. 447–480 DOI:10.1016/B978-0-12-817786-0.00017-7.
- H. Li, Y. Liang, W. Ju, O. Schneider and U. Stimming, In Situ Monitoring of the Surface Evolution of a Silver Electrode from Polycrystalline to Well-Defined Structures, Langmuir, 2022, 38, 14981–14987 CrossRef CAS PubMed.
- I. T. McCrum, C. J. Bondue and M. T. M. Koper, Hydrogen-Induced Step-Edge Roughening of Platinum Electrode Surfaces, J. Phys. Chem. Lett., 2019, 10, 6842–6849 CrossRef CAS PubMed.
- J. Trindell, M. Aarts and M. Koper, Anisotropic step etching and etch instabilities during cathodic corrosion on platinum, 2025 DOI:10.26434/chemrxiv-2025-vwfdn.
Footnote |
| † M. A. and J. A. T. contributed equally. |
| This journal is © the Owner Societies 2026 |