
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heating enables solid-state motion and improves the yield of a [2 + 2] cycloaddition reaction within an organic cocrystal
Gary C.
George
III
 a,
Drew
Owens
b,
Damon M.
Osbourn
c,
Kristin M.
Hutchins
*a and
Ryan H.
Groeneman
*b
a,
Drew
Owens
b,
Damon M.
Osbourn
c,
Kristin M.
Hutchins
*a and
Ryan H.
Groeneman
*b
aDepartment of Chemistry and MU Materials Science & Engineering Institute, University of Missouri, Columbia, MO 65211, USA. E-mail: kristin.hutchins@missouri.edu
bDepartment of Natural Sciences and Mathematics, Webster University, St. Louis, MO 63119, USA. E-mail: ryangroeneman19@webster.edu
cDepartment of Chemistry, Saint Louis University, St. Louis, MO 63103, USA
First published on 26th November 2025
Abstract
The ability to overcome a suppressed yield for a [2 + 2] cycloaddition reaction, due to static disorder, by heating is reported. Increasing the temperature allows the ethylene groups within the reactant to undergo molecular pedal motion, which affords proper alignment of the reactive moieties and enables a quantitative yield for the photocycloaddition. High-temperature single-crystal X-ray diffraction determines the onset temperature for the induced pedal motion and demonstrates alignment at elevated temperature.
Covalent-bond formation within molecular solids continues to be an important yet underutilized area of organic chemistry. In particular, the light-induced [2 + 2] cycloaddition reaction remains an indispensable synthetic approach in the formation of cyclobutane-based molecular targets.1 Seminal work in this area was published by Schmidt where he defined the structural requirements to achieve photocycloaddition reactions in the organic solid state, instead of the more widely used solution state.2 Specifically, Schmidt's topochemical postulate states that a pair of carbon–carbon double bonds (i.e. ethylene groups) should be photoreactive if they lie parallel and within a distance of 4.2 Å, which affords proper overlap of the π-orbitals for the reactive centres. This postulate has been widely used to guide the design of solid-state [2 + 2] and [4 + 4] photocycloaddition reactions.3
Challenges in self-assembly and crystal packing render most single- and multi-component organic solids photostable. To address this problem, molecular templates have been utilized, which, through self-assembly, position the reactants into a suitable orientation and distance for photoreaction.4 In the case of olefin-bearing compounds, although the reactive groups can lie at an appropriate distance, a parallel alignment may still be lacking. A unique feature of ethylene-containing molecules is that they are capable of undergoing temperature-promoted dynamic motion (i.e. ‘pedal motion’) in the solid state.5 This type of molecular motion is often observed in stilbene-based derivatives (Scheme 1), although the conditions for enabling or limiting dynamic motion in the solid state are not broadly understood.6 Solid-state pedal motion can be detected by utilizing variable-temperature single-crystal X-ray diffraction (SCXRD) or solid-state nuclear magnetic resonance spectroscopy. Using SCXRD, dynamic motion is detected through molecular disorder, and if the crystallographically determined site occupancies vary as a function of temperature, the disorder is dynamic. However, if there is little to no change in the site occupancies with temperature (e.g., 1–2% over a 50 K range) or if the percentages fluctuate by 1–2% continually, the disorder is defined as static.7
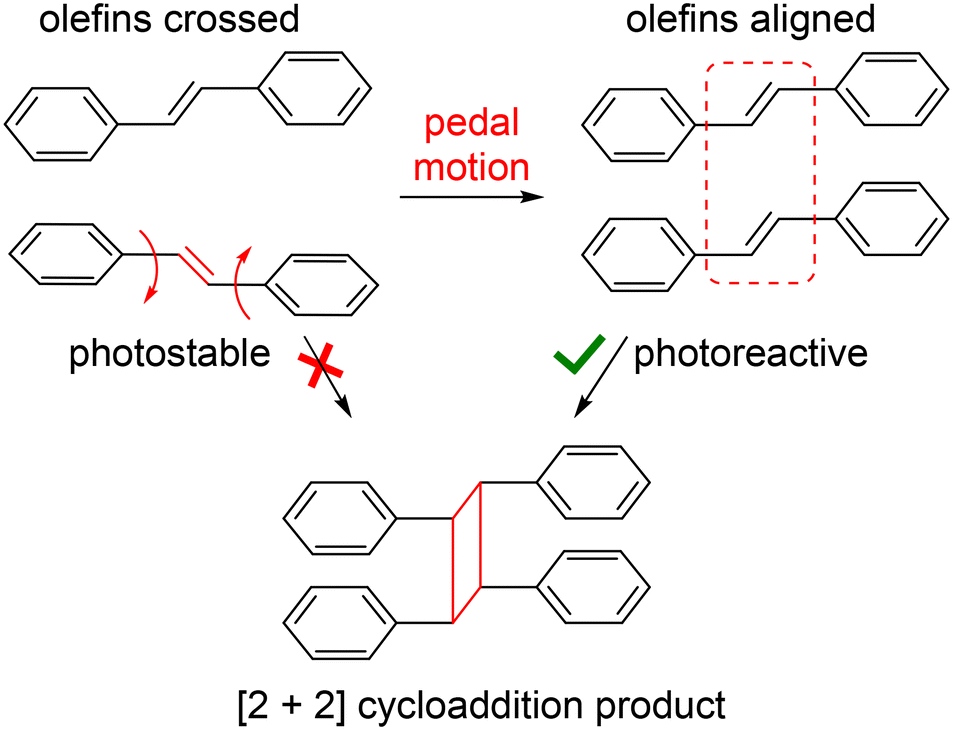 | ||
| Scheme 1 Depiction of dynamic pedal motion within a pair of stilbenes that affords the proper parallel alignment of stacked ethylene groups and allows a [2 + 2] cycloaddition to occur. | ||
The photoreactivity of molecular solids can be influenced by the presence or absence of pedal motion.8 For example, a pair of ethylene groups stacked in a nonparallel arrangement (i.e. criss-crossed) could, in theory, be converted to the photoreactive, parallel orientation through pedal motion. Indeed, high yielding photodimerization reactions have been previously attributed to occurrence of dynamic motion in coordination polymers and cocrystals containing crossed olefins.9 Moreover, for solids that do not meet Schmidt's criteria, topochemical reactions such as the ene–azide, azide–alkyne, and [4 + 2] cycloaddition have been achieved through thermal vibration at room temperature or by heating, which affords a reactive geometry.10
One of us has previously reported a series of photoreactive cocrystals containing trans-methyl-4-[(2-pyridin-4-yl)ethenyl]benzoate (PEB) with various resorcinol derivatives as templates.11 Upon exposure to ultraviolet light, PEB undergoes a stereospecific [2 + 2] cycloaddition reaction to form the photoproduct rctt-1,2-bis(4-pyridyl)-3,4-bis(methylbenzoate)cyclobutane (PBCB, Scheme 2). A photoreaction was observed in ten different cocrystals and was attributed to the ability of PEB to undergo dynamic solid-state pedal motion. However, the cocrystal containing 4,6-dichlororesorcinol (4,6-diCl-Res) as the template, namely (4,6-diCl-Res)·2(PEB), only reached an 85% conversion for the photoreaction (after 50 h irradiation).12 Curiously, cocrystals based on 4,6-dibromoresorcinol and 4,6-diiodoresorcinol afforded quantitative cycloadditions even though these three molecular templates are isosteric. The source of the suppressed yield for (4,6-diCl-Res)·2(PEB) was attributed to static disorder within the ethylene group, wherein the second position was not properly aligned with an adjacent reactant.12 Importantly, the overall yield for the photoreaction was equivalent to the ratio of the parallel aligned ethylene groups within the cocrystal.
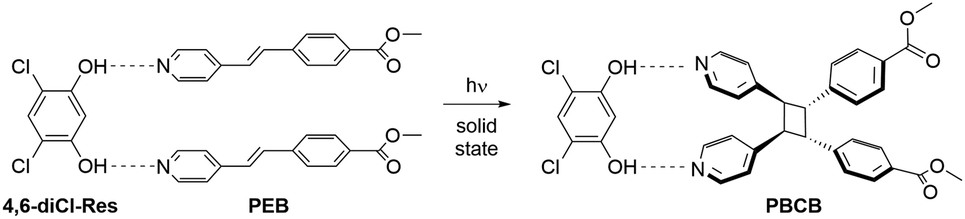 | ||
| Scheme 2 The templated [2 + 2] photocycloaddition of PEB to afford PBCB. | ||
Upon heating, the site occupancy for the minor orientation (where the minor site is less than 50%) within a disordered ethylene-based molecule will generally increase, and more significant pedal motion is often observed at higher temperatures. Thus, we envisioned that heating could cause molecular pedal motion to occur within either a statically disordered or an ordered ethylene-functionalized molecule, thereby enabling dynamic motion, a parallel alignment of ethylene groups, and therefore, an improved yield for a solid-state [2 + 2] photocycloaddition. Here, we describe the ability to improve the yield of a [2 + 2] photocycloaddition to a quantitative level by simply heating the cocrystal in the solid state during light irradiation. By heating (4,6-diCl-Res)·2(PEB), both the statically disordered and ordered ethylene groups become dynamic (Fig. 1), which is supported by high-temperature SCXRD. The resulting dynamic motion enables a nearly quantitative photodimerization. This straightforward strategy is typical in traditional, solution-based, organic synthesis; however, heating has not been commonly applied to solid-state photocycloadditions, likely because the chemical reaction is promoted with light. Here, the simultaneous use of heat and light promotes dynamic motion and olefin alignment for the subsequent chemical transformation.
 | ||
| Fig. 1 X-ray crystal structures of (4,6-diCl-Res)·2(PEB) illustrating the disorder upon heating within PEB. The O–H⋯N hydrogen bonds are shown with yellow dashed lines. The 290 K data is from the original report,12 while the 310 K data is from this work. The red rectangle highlights a change in disorder and motion. | ||
The template 4,6-diCl-Res was purchased, and PEB was synthesized using a literature procedure.13 As previously reported, cocrystallization from ethanol affords (4,6-diCl-Res)·2(PEB), which crystallizes in the monoclinic space group P21/n.12 We obtained identical single crystals for this contribution. Powder X-ray diffraction also confirmed bulk purity of (4,6-diCl-Res)·2(PEB) (Fig. S1).
The cocrystal structure is defined by a three-component hydrogen-bonded assembly held together by O–H⋯N hydrogen bonds and supported by O⋯Cl interactions between template molecules. In the originally reported X-ray data, only one of the two unique ethylene groups was found to be disordered at 290 K, and a free-variable refinement returned an occupancy of 85% for the major site at 290 K (Fig. 1 and Table 1).12 In the prior work, the crystal was slowly cooled, and full crystallographic data sets were collected at 250, 210, and 170 K. After refinement, the major site occupancy for the disordered ethylene group remained around 85–86% concluding that the disorder was static upon cooling (Table 1). Notably, the aromatic rings attached to the ethylene must also be disordered; however, visible electron density and data quality can influence the ability to model ring disorder.
| Temperature (K) | Occupancy for PEB containing N1 (major![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :minor %) :minor %) |
Occupancy for PEB containing N2 (major:minor %) |
|---|---|---|
| Originally reported X-ray data12 (85% yield at 290 K) | ||
| 170 | 86(1):14(1) |
— |
| 210 | 86(1):14(1) |
— |
| 250 | 85(1):15(1) |
— |
| 290 | 85(1):15(1) |
— |
| X-ray data from this work (100% yield at 340 K) | ||
| 290 | 85(1):15(1) |
98(1):2(1) |
| 310 | 84(1):16(1) |
97(1):3(1) |
| 330 | 83(1):17(1) |
96(1):4(1) |
| 350 | 81(1):19(1) |
94(1):6(1) |
| 370 | 81(1):19(1) |
93(1):7(1) |
To determine if either PEB molecule within (4,6-diCl-Res)·2(PEB) would exhibit dynamic motion at higher temperatures, a SCXRD experiment was undertaken at 290, 310, 330, 350, and 370 K (Tables S1 and S2). Upon heating, the cocrystal remains intact, the space group is retained, and the hydrogen and halogen bonds sustain the solid at all temperatures. In this new data, at 290 K, one PEB molecule exhibited the same site occupancies as the previous report; however, we observed a small amount of disorder in the second PEB molecule (minor site ≈ 2%), likely due to advances in detector technology. Upon further heating, the site occupancy values for both ethylene sites continued to change. Ultimately, the values for the minor sites of the ethylene groups reached 19% and 7% at 370 K (Table 1). The increase of these minor orientation occupancies upon heating confirmed both ethylene groups underwent dynamic solid-state pedal motion7 at elevated temperatures.
A second crystallographic parameter also supports the occurrence of pedal motion. The O⋯Cl interactions between stacked three-component hydrogen-bonded assemblies increase in distance as the solid is heated (Fig. 2). The O⋯Cl distance is 3.173 Å at 290 K and increases to 3.216 Å at 370 K. The addition of thermal energy to the cocrystal allows the pedal motion to occur, which further requires a larger separation between nearest stacked assemblies.
 | ||
| Fig. 2 X-ray crystal structure of (4,6-diCl-Res)·2(PEB) illustrating the stacking of the three-component hydrogen-bonded assembly at 310 K. The O–H⋯N hydrogen bonds and the O⋯Cl interactions are shown with yellow dashed lines. | ||
To determine if the temperature-induced pedal motion within (4,6-diCl-Res)·2(PEB) would increase the yield for the [2 + 2] cycloaddition reaction, a sample was simultaneously heated and exposed to ultraviolet (UV) irradiation within a photochemical cabinet (Fig. S2). A dried crystalline powder of the cocrystal was put between two Pyrex glass plates and placed in the middle of a standard laboratory hotplate (Fig. S3). The temperature of the powder sample was measured by using a Vesogy infrared thermometer. Using the SCXRD data and to ensure pedal motion would occur, the sample was heated to 340 K (∼67 °C) during irradiation. The heated solid was simultaneously exposed to ultraviolet light using a 450 W medium-pressure mercury vapour bulb.
The bulk yield for the [2 + 2] photocycloaddition was determined using 1H nuclear magnetic resonance (1H NMR) spectroscopy. After 20 h of irradiation, the photoreaction reached a quantitative yield. This yield was evident by the complete loss of the ethylene peaks on PEB at 7.43 and 7.65 ppm (Fig. S4) and the concomitant appearance of a pair of cyclobutane signals at 4.69 and 4.75 ppm (Fig. S5), indicative of the formation of the cycloadduct PBCB, stereospecifically (Scheme 2).12 Notably, in the original study, a yield of 85% was achieved after 50 h of irradiation.
As a control experiment, another sample of (4,6-diCl-Res)·2(PEB) was exposed to 20 h of irradiation using an identical experimental setup, except the cocrystal was placed on a shelf in the photoreactor to limit any possible source of heat. The yield based on 1H NMR spectroscopy for the ambient temperature sample was 86% after 20 hours of UV irradiation, which is in agreement with the previously reported data (Fig. S6). Thus, the quantitative yield for the photodimerization above strongly supports that inducing molecular pedal motion within (4,6-diCl-Res)·2(PEB) via heating leads to alignment of ethylene groups and affords an increased reaction yield. To further study the role of heat as a means to increase yield for a [2 + 2] cycloaddition reaction, the sample irradiated at ambient temperature was subsequently placed on the hotplate and heated to 340 K with simultaneous exposure to UV light. After 20 additional hours of UV exposure, the yield for the photodimerization reached 98% as determined by 1H NMR spectroscopy (Fig. S7).
During irradiation, there is an increase in temperature of the hotplate surface (Corning® Pyroceram® glass-ceramic material, Fig. S3), which could alter the sample temperature, even if the plate is not turned on. Thus, the temperature on the hotplate surface was measured using the infrared thermometer after four hours of continuous UV irradiation, and the temperature had risen by 10 K (from 296 to 306 K), which would enable pedal motion in this solid. Thus, an additional sample of (4,6-diCl-Res)·2(PEB) was irradiated for a total of 80 h in the absence of heat, but with the sample placed on the hotplate surface. To our surprise, the increase in temperature on the surface of the hotplate over longer irradiation times (e.g. 80 h) afforded nearly quantitative conversion of PEB to PBCB within the cocrystal (Fig. S8).
For photocycloaddition reactions, the yield can vary based on the amount of sample used and exposure of the entire sample to UV light. For example, if large sample quantities are used and the light does not effectively reach all portions of the sample, the yield and conversion time can be reduced/slower. The experiments described here were conducted using similar scales, and irradiation at 340 K afforded a quantitative conversion in 20 h, while irradiation at 306 K afforded a quantitative conversion in 80 h (Table S3). Thus, when mild heat is applied, the reaction time can be significantly reduced.
Simultaneous thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) was used to investigate the thermal properties of (4,6-diCl-Res)·2(PEB) to ensure that the applied heat was not simply melting the solid, which would enable enhanced molecular movement. Upon heating from room temperature to 250 °C, (4,6-diCl-Res)·2(PEB) exhibits a single endothermic signal indicative of melting at 173 °C (446 K, Fig. S9). The cocrystal was irradiated at a temperature that is 106 K below its melting point; thus, enhanced reactivity is supported by thermal energy enabling dynamic motion, rather than through melting.
A recent report from Sureshan14 highlights the potential limitations of topochemical postulates for reactions that occur at higher temperatures (e.g., topochemical azide–alkyne cycloaddition). Thus, the authors mentioned that studying non-reactive forms above ambient temperature, but below the melting point, would be of interest.14 The system described here further demonstrates the influence temperature can have on cycloadditions through thermal energy/motion.
In this contribution, the ability to improve the yield for a solid-state [2 + 2] photocycloaddition reaction by heating is reported. Heating (4,6-diCl-Res)·2(PEB) induces pedal motion for both a statically disordered and ordered ethylene, which leads to alignment of the reactive groups in the solid state and a higher yield for the photoreaction in a shorter period of time. For photoreactive solids containing motion-capable functional groups, chemists should routinely consider mild heating with the goal of improving yield, and the process can be accomplished using common laboratory equipment. Currently, additional cocrystalline solids, with lower-than-expected yields, are being explored to test the reliability of this heating approach.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Data availability
The data supporting this article have been included as part of the supplementary information (SI).Supplementary information: experimental details, X-ray data, 1H NMR spectra, and thermal data. See DOI: https://doi.org/10.1039/d5ce00995b.
CCDC 2486480–2486484 contain the supplementary crystallographic data for this paper.15a–e
Acknowledgements
K. M. H. gratefully acknowledges financial support from the National Science Foundation DMR-2411677. R. H. G. gratefully acknowledges financial support from Webster University in the form of various Faculty Research Grants. The authors acknowledge Professor Eric Bosch for the donation of PEB used in the contribution. The authors acknowledge Jane Eilers for collecting PXRD patterns.Notes and references
-
(a) A. D. Richardson, T. R. Vogel, E. F. Traficante, K. J. Glover and C. S. Schindler, Angew. Chem., Int. Ed., 2022, 61, e202201213 CrossRef CAS PubMed
; (b) E. Canales and E. J. Corey, J. Am. Chem. Soc., 2007, 129, 12686 CrossRef CAS PubMed
- G. M. J. Schmidt, Pure Appl. Chem., 1971, 27, 647 CrossRef CAS
-
(a) C. J. Easley, F. Tong, X. Dong, R. O. Al-Kaysi and C. J. Bardeen, Chem. Sci., 2020, 11, 9852 RSC
-
(a) M.-M. Gan, J.-G. Yu, Y.-Y. Wang and Y.-F. Han, Cryst. Growth Des., 2018, 18, 553 CrossRef CAS
- J. Harada and K. Ogawa, J. Am. Chem. Soc., 2001, 123, 10884 CrossRef CAS PubMed
- J. Harada and K. Ogawa, Cryst. Growth Des., 2014, 14, 5182 CrossRef CAS
- J. Harada and K. Ogawa, Chem. Soc. Rev., 2009, 38, 2244 RSC
-
(a) J. Quentin and L. R. MacGillivray, ChemPhysChem, 2020, 21, 154 CrossRef CAS PubMed
-
(a) J. J. Vittal, J. Photochem. Photobiol., C, 2023, 57, 100636 CrossRef CAS
-
(a) R. Khazeber, G. S. Kana and K. M. Sureshan, Angew. Chem., Int. Ed., 2024, 63, e202316513 CrossRef CAS PubMed
- E. Elacqua, P. Kaushik, R. H. Groeneman, J. C. Sumrak, D.-K. Bučar and L. R. MacGillivray, Angew. Chem., Int. Ed., 2012, 51, 1037 CrossRef CAS PubMed
- E. Elacqua, K. A. Kummer, R. H. Groeneman, E. W. Reinheimer and L. R. MacGillivray, J. Photochem. Photobiol., A, 2016, 331, 42 CrossRef CAS
- M. L. Boilot, C. Roux, J. P. Audière, A. Dausse and J. Zarembowitch, Inorg. Chem., 1996, 35, 3975 CrossRef PubMed
- A. Ravi, S. Z. Hassan, S. Bhandary and K. M. Sureshan, Angew. Chem., Int. Ed., 2022, 61, e202200954 CrossRef CAS PubMed
-
(a)
CCDC 2486480: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2pgd1m
| This journal is © The Royal Society of Chemistry 2026 |