
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering function of mechanically interlocked molecules with pyrrole-based macrocyclic platforms
Rafał A. Grzelczak†
 ,
Daniel Wiendlocha†
and
Bartosz Szyszko
*
,
Daniel Wiendlocha†
and
Bartosz Szyszko
*
Faculty of Chemistry, University of Wrocław, 14 F. Joliot-Curie St., 50-383 Wrocław, Poland. E-mail: bartosz.szyszko@uwr.edu.pl; Web: https://www.bszyszko.pl
First published on 25th May 2026
Abstract
This feature article discusses advances in mechanically interlocked molecules comprising porphyrin or porphyrin-like frameworks, as well as systems in which porphyrinoid structural elements were integrated into interlocked architectures to impart specific functions, such as energy and electron transfer, molecular recognition, catalytic activity, and controlled dynamic operation. The review article traces the field's development from early examples of porphyrin-containing catenanes and rotaxanes to more recent systems in which porphyrinoid stations were deliberately engineered to fulfill defined functional roles. In the final section, the group's contributions to the research area are presented. This includes the progressive development of an iminopyrrole-based self-assembly methodology that enabled the construction of increasingly complex structures – from two-dimensional macrocycles, through capsular assemblies, to metal-stabilized mechanically interlocked molecules. Finally, the synthesis of rotaxanes and catenanes comprising calix[4]phyrins is outlined, demonstrating systems that exhibit a specific type of molecular motion termed fluttering.
 Rafał A. Grzelczak | Rafał Grzelczak was born in 1996 in Leszno, Poland. He is currently an Assistant Professor at the Faculty of Chemistry, University of Wrocław. He received his PhD in 2026 under the supervision of Prof. Bartosz Szyszko, conducting research on the synthesis of interlocked molecules. He continues his research with a focus on dynamics and motion control in rotaxanes. In 2026, he received the prestigious START scholarship for outstanding young researchers. |
 Daniel Wiendlocha | Daniel Wiendlocha is a PhD candidate at the Faculty of Chemistry, University of Wrocław, specializing in supramolecular and organic chemistry. He received his BSc degree in 2020 and his MSc degree in 2022 from the University of Wrocław. His research focuses on the synthesis and characterization of mechanically interlocked molecules, particularly rotaxanes and macrocyclic systems. His scientific interests include macrocyclic chemistry, self-assembly processes, and the design of functional molecular architectures. |
 Bartosz Szyszko | Bartosz Szyszko is an Associate Professor at the Faculty of Chemistry, University of Wrocław. He received his PhD in 2014 under the supervision of Prof. Lechosław Latos-Grażyński. In 2015, he completed a postdoctoral fellowship with Prof. Jonathan R. Nitschke at the University of Cambridge, where he investigated the construction of supramolecular capsules through subcomponent self-assembly. His group's research focuses on mechanically interlocked molecules, self-assembly, and macrocyclic chemistry. He has co-authored ca. 45 articles and was honored with several national-level awards, including the National Science Center Award in 2025 and the W. Kołos Award in 2022. |
Introduction
Over the past few decades, mechanically interlocked molecules (MIMs) have progressed from a synthetic curiosity to a broadly applicable class of functional materials.1 Their development has enabled the construction of artificial molecular machines,2–4 switches,5–7 and sensors,8,9 as well as structural motifs capable of exerting controlled influence over macroscopic material properties.10–12Although the fields of pyrrole-based dyes, on the one hand, and mechanically interlocked molecules, on the other, initially developed at different rates and along largely independent trajectories, they began to converge at several key stages.13 This convergence became particularly evident in the context of rotaxane research, where structural motifs were sought that were sufficiently large to function as stoppers, while also fulfilling roles beyond purely structural ones. Porphyrin motifs were intended to impart added value to MIMs by introducing versatile coordination properties and attractive photophysical features.14–16 Owing, in part, to the seminal contributions of Sauvage, Stoddart, Sanders, Hunter, Anderson, and others, the two areas increasingly informed one another, enabling the transfer of concepts and synthetic strategies.17–28
This article focuses on the points of contact between porphyrinoids and the chemistry of rotaxanes and catenanes. Accordingly, mechanically interlocked systems incorporating classic porphyrin macrocycle are discussed only briefly, particularly in light of the extensive reviews and authoritative accounts by Sauvage and others.13–16,29 In contrast, interlocked architectures built from porphyrinoids30,31 – macrocyclic frameworks related to, but distinct from, porphyrins, as well as their derived structural elements – have not yet been systematically reviewed.
This topic has recently become an important line of investigation in our research group at the University of Wrocław. Our interest is motivated by the unusual opportunities that arise from combining pyrrolic macrocycles exhibiting remarkable aromaticity,32,33 conformational flexibility,34 intriguing stereochemistry,35,36 peculiar reactivity,30,37 stabilization of uncommon oxidation states,38–40 chemically robust organometallic motifs,41 and access to multiple electronic states42 – with the defining attributes of mechanically interlocked molecules. Mechanical bonding enables control over molecular motion,43,44 enforces proximity between interacting components,45 and allows tuning of structure, properties, and reactivity that is impossible with conventional covalent design.46
The feature article opens with an overview of the field's historical context and a discussion of the work of research groups that laid the foundations for the area. We then outline what we regard as the most important advances in this field, before presenting selected examples of our recent work on the construction of compounds that may, in a broad sense, be described as mechanically interlocked porphyrinoids (MIPs).
Porphyrin-based MIMs
As noted in the Introduction, the use of porphyrins in the construction of MIMs has already been discussed extensively elsewhere; therefore, this topic will be revisited here only briefly. Nevertheless, any discussion of the connection between pyrrole-based dye chemistry and MIMs would be incomplete without acknowledging the role that porphyrin macrocycles have played – and continue to play – in the supramolecular chemistry of systems stabilized by mechanical bonding.Porphyrins as structural units
Porphyrins began to be used as structural elements of mechanically interlocked molecules in the 1990s, when their size was recognized as suitable for application as rotaxane stoppers.13,17,47,48 Initially, the moieties introduced at the termini of a rotaxane axle served a purely structural role, being intended to prevent dissociation of the macrocycle from the axle, that is, to inhibit the dethreading process. It was later proposed that such entities could exert a far more significant influence, affecting the properties of the interlocked systems through the intrinsic features of porphyrins. From that point onward, the potential of metalloporphyrins was recognized as motifs capable not only of functioning as stoppers but also of introducing intricate features into multicomponent systems.Using their copper(I)-based passive template approach, the Sauvage group constructed [2]rotaxanes featuring a bent axle bearing two metalloporphyrins at its termini (Fig. 1, type A).17 A central 2,9-diphenyl-1,10-phenanthroline recognition site was incorporated into axle 1, enabling the formation of a coordination compound upon reaction with a crown ether 2 functionalized with a complementary phenanthroline motif (Scheme 1). The synthesis required the preformation of a copper(I)-stabilized prerotaxane 3 bearing a benzaldehyde functionality at one end of the thread, followed by its condensation with dipyrromethane and 3,5-di-tert-butylbenzaldehyde in the presence of TFA. Subsequent oxidation of the initially formed porphyrinogen with chloranil, followed by metalation, afforded the conjugated porphyrin stopper in 4. The extension of this methodology afforded higher-order systems, including [3]- 5 and [5]rotaxanes 6 (Fig. 1, types B and C, Scheme 2).18,49 Removal of the Cu(I) template from the [3]rotaxane by complexation with cyanides afforded a molecule featuring a thread terminated with metalloporphyrins.50 The Stoddart group, on the other hand, designed [2]rotaxane 7 composed of the tetracationic blue-box cyclophane assembled around a long polyether chain and terminated with zinc(II) porphyrins connected through their meso-aryl positions (Fig. 1, type D).19 Incorporation of two bis(aryloxo) stations within the thread enabled observation of a shuttling process, characterized by a rate constant of 25 s−1 at −10 °C and a free-energy barrier of 13.6 kcal mol−1 (Scheme 3).
 | ||
| Fig. 1 Selected geometric types of rotaxanes and catenanes comprising porphyrin macrocycle(s). Various metals incorporated into the porphyrin cavity were depicted as a red sphere. | ||
 | ||
| Scheme 1 The synthesis of [2]rotaxane 4.17 | ||
 | ||
| Scheme 2 (A) [3]Rotaxane 5 and (B) [5]rotaxane 6.18,50 | ||
 | ||
| Scheme 3 [2]Rotaxane-based molecular shuttle 7.19 | ||
The affinity of metalloporphyrins toward N-donor ligands has been exploited in the construction of rotaxanes in which a d-block metal coordination compound of a tetrapyrrole acted as a stopper, coordinatively attached to the termini of a rotaxane axle bearing ligand units such as pyridine (Fig. 1, types E and F). The first example of such a system was reported by Branda and coworkers, who designed a self-assembling [2]rotaxane composed of dibenzo[24]crown-8 and a 1,2-bis(4,4′-dipyridinium)ethane dicationic thread terminated with 4-pyridyl groups. Upon introduction of carbonyl (tetraphenylporphyrinato)ruthenium(II), possessing one vacant axial coordination site, into a solution of the pseudorotaxane, end-capping occurred at the termini of the axle, transforming the pseudorotaxane species into a [2]rotaxane 8 through the formation of hexacoordinate Ru(II) complexes. Addition of a competitive Lewis base, such as pyridine, led to dissociation of the resulting [2]rotaxane (Fig. 1, type E, Scheme 4A).51
 | ||
| Scheme 4 [2]Rotaxanes (A) 8 and (B) 9 incorporating metalloporphyrin stoppers.51,52 | ||
Axial coordination to metalloporphyrins containing Zn(II), Ru(II), and Rh(III) was also exploited to introduce stopper groups onto the axle of a [2]rotaxane self-assembled from a macrocycle incorporating electron-rich 1,5-alkoxynaphthalene motifs and a linear thread featuring a central naphthodiimide moiety.21 Shimizu systematically examined how variations in the porphyrin macrocycle substitution pattern and the dimensions of the thread influence conformations of [2]rotaxanes stoppered with rhodium(III) porphyrins. One such example is rotaxane 9, which in contrary to 8, is terminated with a rhodium(III) porphyrin only on one side (Fig. 1, type F, Scheme 4B).52
Meso substituents oriented at approximately 90° around the rigid, planar porphyrin core enabled this macrocycle to serve as a platform for constructing higher-order rotaxanes. Sauvage and Sour exploited this design by integrating porphyrin meso-aryl substituents into a crown ether framework containing a phenanthroline unit, yielding a Zn(II) porphyrin that enabled the synthesis of a [3]rotaxane 10 via threading of a long axle through macrocycle pairs at the 5,10 and 15,20 positions, followed by stoppering (Fig. 1, type G, Scheme 5A).53 By contrast, the Loeb group introduced linear secondary amine groups at the meso positions of a porphyrin ring. Using a simultaneous threading-followed-by-stoppering strategy, they obtained a [5]rotaxane 11 based on the tetrapyrrolic scaffold (Fig. 1, type H, Scheme 5B).54
 | ||
| Scheme 5 (A) [3]Rotaxane 10 and (B) [5]rotaxane 11.53,54 | ||
Hunter designed a zinc(II) porphyrin bearing a carefully engineered meso substituent incorporating pyridine termini and a pyridine-2,6-dicarboxamide motif. In chloroform, this species existed in dynamic equilibrium with a self-assembled dimer, in which the pyridine unit of one porphyrin acted as an axial ligand to the Zn(II) center of the other. Owing to cooperative binding, the dimer displayed a very high association constant (Kd = 2 × 108 M−1). The presence of hydrogen-bond donors and acceptors within the meso-pyridinedicarboxamide unit further enabled this dimer to form [2]rotaxanes 12 with a range of terephthaloyl derivatives, in which the porphyrins constituted part of the self-assembled macrocyclic component and served as stoppers at the termini of the thread (Fig. 1, type I, Scheme 6).26
 | ||
| Scheme 6 [2]Rotaxane 12.26 | ||
The Anderson group pushed the concept of using porphyrins as components of large macrocyclic architectures to the extreme by designing a [2]rotaxane 13 with a butadiyne axle terminated by two Zn(II) porphyrin stoppers, further functionalized with alkyne groups (Scheme 7).27 The resulting building block was subjected to Pd-catalyzed Glaser coupling in the presence of a hexapyridyl template 14, affording remarkable [4]- 15 and [7]catenane 16 architectures featuring large ring components composed of cyclic porphyrin hexamers and dodecamers in 62% and 6% yield, respectively. Later, the same group demonstrated several other impressive systems in which the porphyrin was incorporated into the axle or ring component of the interlocked molecule.55,56
 | ||
| Scheme 7 Synthesis of [n]catenanes 15 and 16.27 | ||
Photosynthesis-mimicking systems
Molecular systems incorporating metalloporphyrins were recognized early on as valuable models that mimic the action of photosystems, which convert light into chemical energy.13–16 In particular, bis-porphyrin compounds containing a zinc(II) porphyrin unit at one terminus and a gold(III) porphyrin unit at the other were shown to exhibit long-range photoinduced charge separation, with the zinc(II) porphyrin acting as the excited-state electron donor and the gold(III) porphyrin serving as the electron acceptor.57 It was later demonstrated that electron and energy transfer processes can be tuned by incorporating the active species into rotaxane or catenane architectures, which allow for precise spatial separation of the interacting components without close contact through covalent bonds. This structural arrangement suppressed rapid charge recombination, which would otherwise render the processes fast and inefficient.17 The extensive work of the Sauvage group demonstrated that the activity of metalloporphyrin-stoppered rotaxanes can be finely tuned by modifying the MIM architecture, enabling the construction of multicomponent systems with highly desirable photoinduced electron transfer characteristics.58–61 Flamigni, Serroni, and co-workers reported a series of [2]catenanes composed of polyether-strapped zinc(II) porphyrins and cyclobis(paraquat-p-phenylene), which exhibited not only efficient photoinduced electron transfer but also a pronounced dependence of the electron-transfer rate on the strap size.62Multiple systems incorporating C60 fullerene as an electron acceptor were designed, in which photoinduced electron transfer from the excited-state porphyrin led to charge-separated states.63–66 Takata, Ito, and co-workers reported an interesting [2]rotaxane 17 featuring zinc(II) porphyrin donors installed at the termini of the thread and a cyclic component decorated with a C60 fullerene acceptor (Fig. 2). This design enabled the observation of intramolecular electron transfer between the donor metalloporphyrins and the [60]fullerene in benzonitrile.67 Schuster and colleagues reported a [2]rotaxane architecture comprising metalloporphyrin stoppers that acted as electron donors and a macrocycle appended with C60, assembled using the Sauvage bis(phenanthroline)copper(I) motif.68 It was demonstrated that the intramolecular electronic processes observed in this system required the involvement of the [Cu(phen)2]+ complex in the electron transfer events. Later, the role of the same motif was documented for [2]catenanes in which metalloporphyrin and fullerene were spatially significantly separated.69
 | ||
| Fig. 2 Photoinduced electron transfer in C60-appended [2]rotaxane 17.67 | ||
Von Delius and Guldi constructed [2]rotaxane 18 featuring zinc(II) porphyrin stoppers and a C60 fullerene acceptor positioned at the central part of the thread (Scheme 8).70 While electron transfer between the donor and acceptor was observed for the free thread, the introduction of a [10]cycloparaphenylene ring (CPP or its aza-analogue aza[10]CPP) encircling the C60 unit effectively shielded the acceptor. This structural modification suppressed electron transfer and redirected the photophysical pathway toward triplet-state energy transfer between the zinc(II) porphyrin and the fullerene. The Anderson group, on the other hand, described [3]- and [5]-rotaxanes in which multiple energy transfer pathways involving a phenanthroline-based macrocycle, a polyyne thread, and porphyrin stoppers were operative.28
 | ||
| Scheme 8 The [10]CPP-based [2]rotaxane 18 comprising a fullerene moiety.70 | ||
Interlocked receptors and sensors
Porphyrins have several key features – they are large, aromatic, and planar molecules that, upon metalation of their cavity, can bind axially to N- or O-donor ligands.71,72 Combined with the co-conformational flexibility of rotaxanes, these properties enable the formation of adaptable receptors capable of interacting with a wide range of molecular and ionic guests.73 It was this complementarity that enabled Morin and co-workers to design rotaxanes functioning as tweezer-like receptors for fullerenes (Scheme 9).74 The attachment of porphyrin units to two dibenzocrown ether macrocycles led to the formation of a [3]rotaxane 19 in which the distance between the aromatic planes could be modulated. As a result, the porphyrin panels – well known for their efficient interactions with fullerenes – were able to move apart and effectively encapsulate C60, C70, and C84. | ||
| Scheme 9 Binding of C60 by the tweezer-like [3]rotaxane receptor 19.74 | ||
The Sauvage group further explored the dynamic behavior of [3]rotaxanes featuring two zinc(II) porphyrin plates that interacted with bispyridyl podands differing in linker flexibility between the coordinating units.75 The large accessible amplitude of distance variation between the metalloporphyrin panels (10–80 Å), together with the cooperative axial coordination of a single podand molecule to two metal centers, enabled exceptionally high complex stability constants, reaching values of 106–107 M−1.
The Beer group has extensively studied anion binding by porphyrin-based rotaxanes and catenanes. By exploiting anion-templation strategies, they developed a range of interlocked receptors capable of recognizing multiple anionic guests, with the porphyrin macrocycle serving as both the recognition site and a reporter unit.76 The group also designed a receptor based on a porphyrin-strapped macrocycle in which iodotriazole functionalities were incorporated into the cyclic component of the rotaxane to enable anion binding via halogen bonding.77 Axial coordination between a triazole unit on the [2]rotaxane thread and a zinc(II) bound in the porphyrin cavity preorganized the binding pocket and led to a substantial enhancement of anion binding affinities. Notably, quenching of a BODIPY unit installed as a stopper at one terminus of the thread enabled optical detection of the anion recognition event. A similar design principle enabled the construction of a catenane 20 that acted as a host for anionic guests, in which the anions were stabilized within the cavity through a network of hydrogen bonds (Fig. 3).78
 | ||
| Fig. 3 The chloride complex 20 of a porphyrin-incorporating [2]catenane receptor.78 | ||
Switches and molecular machines
Porphyrin-containing MIMs have been extensively explored as platforms for constructing molecular switches and machines.6,19 The Sauvage group demonstrated early on that a [2]rotaxane 21-M assembled from a thread bearing Zn(II) porphyrin stoppers and a macrocycle incorporating an Au(III) porphyrin undergoes a pronounced intercomponent conformational rearrangement (Scheme 10).79 Removal of the copper(I) template induced an approximately 180° rotation of the axle relative to the macrocyclic component, driven by attractive interactions between Zn(II)- and Au(III) porphyrins in 22. The same group designed a [3]rotaxane in which two phenanthroline-based macrocycles fused with metalloporphyrins were threaded onto a linear component bearing a complementary Cu(I)-coordinating motif.80 Upon removal of the template, a shuttling motion was observed, in which the porphyrin plates move closer together or farther apart in response to interactions with guests capable of axial coordination to the Zn(II) porphyrins. | ||
| Scheme 10 Demetallation-induced co-conformational rearrangement of 21-M.79 | ||
The Beer group designed molecular shuttles based on [2]- and [3]rotaxanes in which a Zn(II) porphyrin unit was positioned at the centre of the axle.81 The macrocyclic component incorporated a pyridyl ring. In the absence of competing ligands, shuttling of the macrocycle along the thread was observed, with the Zn(II) porphyrin station influencing the dynamics through axial coordination with the pyridyl unit of the ring component. Upon the addition of an external base, i.e., pyridine, the shuttling was inhibited.
The coordination properties of metalloporphyrins were also exploited by Terao and co-workers, who designed a remarkable [3]catenane 23 comprising a large, rigid rectangular macrocycle incorporating Ru(II) porphyrins and two smaller rings, each containing two Zn(II) porphyrin units (Fig. 4).82 Upon introduction of carefully selected ligands such as 3,5-dibromopyridine, DABCO, or bisbipyridyls, multistate switching could be reversibly induced.
 | ||
| Fig. 4 Coordination-induced multistate switching in [3]catenane 23.82 | ||
Catalysts and nanoreactors
In 2003, Nolte and co-workers demonstrated the remarkable potential of MIMs incorporating catalytically active metalloporphyrins.83 A precisely tailored cavity in a hybrid glycoluril–Mn(III) porphyrin system enabled the threading of a polybutadiene polymer strand, which, upon attachment of stoppers, formed a [2]rotaxane 26-Mn with the polymer serving as the axle. Upon addition of a bulky tert-butylpyridine ligand, capable of axial coordination only from outside the cavity, and in the presence of an oxygen source, the double bonds of the threaded polymer were converted into epoxides 27-Mn (Fig. 5). This transformation yielded polybutadiene epoxide, sparking a debate over the mechanism of catalysis.83–87 | ||
| Fig. 5 Epoxidation of polybutadiene polymer in a Mn(III) porphyrin-based rotaxane 26-Mn.83 | ||
The Megiatto group, in contrast, demonstrated remarkable reactivity within nanocavities incorporating cobalt(II) porphyrins, forming rotaxanes via an original active-template synthetic approach.88 In the presence of 3,5-diphenylpyridine, half-threads of the rotaxane bearing diazo and styrene functional groups underwent a radical carbene-transfer reaction, affording the interlocked product of the olefin cyclopropanation in 95% yield. Later, the same group exploited the reactivity of a nanoreactor incorporating a Ru(II) porphyrin system to demonstrate N–H bond carbene insertion, affording [2]rotaxanes.89
A particularly interesting concept involving the use of metalloporphyrin-based capsules in tandem active-metal template synthesis of rotaxanes was described by Hayashi, Weiss, and co-workers.90 Using a ditopic Zn(II) porphyrin–phenanthroline–strapped system, it was demonstrated that azide coordination to the Zn(II) centre, enforced by the nanoreactor design, facilitated the Huisgen cycloaddition between azide- and alkyne-functionalized half-threads. This process afforded [2]rotaxanes and opened new avenues for exploring dual-metal catalysis in active-template syntheses of mechanically interlocked molecules.
MIMs incorporating porphyrinoids as functional platforms
Although many examples of porphyrins used as building blocks for mechanically interlocked molecules have been reported, the past decade has seen the emergence of MIMs based on porphyrinoids, featuring porphyrin-like macrocycles or structural motifs such as bipyrrole, bithiophene, and dipyrromethene. This section focuses on interlocked molecules in which porphyrinoids served as platforms for constructing catenanes and rotaxanes.Calix[4]pyrrole-based rotaxanes
The conjoining of four pyrrole rings through tetrahedral bridges yields calix[4]pyrrole, a nonconjugated macrocycle originally synthesized by Baeyer in the nineteenth century and later shown to act as a highly efficient anion receptor.91,92In 2012, the Ballester group demonstrated that a cage-like species constructed from two calix[4]pyrrole units connected by rigid bisacetylene linkers is capable of encapsulating a linear zwitterionic guest, bis(amidopyridyl-N-oxide), to form a pseudorotaxane 28 (Scheme 11).93 Hydrogen bonding to the pyridinium N-oxide oxygen atom oriented the guest such that the amide functionalities were exposed toward the remaining free space within the cavity. This preorganization facilitated the formation of four-component species upon complexation of tetrabutylammonium cyanate or azide. Installation of bulky stopper groups 29 on the pseudorotaxane 28 using a capping strategy resulted in the formation of a [2]rotaxane 30 in good yield (50%).94 This interlocked receptor was shown to bind a range of anionic guests effectively, including chloride and nitrate.95,96
 | ||
| Scheme 11 Synthesis of bis-calix[4]pyrrole [2]rotaxane 30.94 | ||
Aydogan and co-workers exploited the anion-binding properties of calix[4]pyrroles to construct alternating polymers composed of linear segments with pillar[5]arenes bearing chains of varying lengths.97 The supramolecular polymers were further shown to organize hierarchically into a variety of structures, including spherical aggregates, fibers, microporous films, and three-dimensional materials.
Corrole-embedded interlocked architectures
Corroles are tetrapyrrolic macrocycles that, due to the absence of one methine bridge between the pyrrolic subunits, belong to the so-called contracted porphyrinoids.98,99 Ngo, D’Souza, Goldup, and co-workers employed active-template synthesis to prepare a series of mechanically interlocked porphyrin–corrole systems.100 They demonstrated that the mechanical bond can be exploited to provide steric shielding of porphyrinoid-terminated threads without altering the electronic properties of the porphyrinoids. The copper(I)-catalyzed azide–alkyne cycloaddition reactions between meso-functionalized copper(III) corrole 31 and Zn(II) porphyrin 33 bearing an alkyne group at the meso-position, in the presence of macrocycle 32, afforded a [2]rotaxane 34 (Scheme 12A). This methodology was further extended to construct a series of impressive triads and pentads 35 (Scheme 12B). Upon titration of the latter with DABCO, an altered assembly behavior of the interlocked species into dimeric structures was observed. This effect was attributed to the presence of mechanically interlocked substituents at the central Zn(II) porphyrin framework, which acted as a distinctive picket-fence-like architecture.100 Later, it was demonstrated that a three-component rotaxanation reaction between a metal-free corrole azide, a zinc(II) porphyrin-based alkyne, and a preformed copper(I) complex of a bipyridyl macrocycle, followed by treatment with HBr–PPh3, afforded corrole–porphyrin conjugates with very good yields.101 | ||
| Scheme 12 (A) The active template synthesis of corrole-incorporating rotaxane 34, (B) [5]rotaxane 35.100 | ||
π-Conjugated porphyrinoid catenanes
Cyclic oligothiophenes may be viewed as heteroanalogues of cyclo[n]pyrroles – a class of porphyrinoids composed only of pyrrole rings.102 Given that oligo- and polythiophenes are widely used organic semiconductors with important applications in organic electronics, the properties of interlocked architectures comprising π-conjugated rings are particularly intriguing.103,104 It motivated Bäuerle and co-workers to explore the synthesis of catenanes composed of cyclic oligothiophenes and to examine whether two interlocked π-conjugated rings would exhibit mutual electronic communication.105 The group initially employed a double-templating strategy. Using Cu(I), two oligothiophene-substituted phenanthroline 36 units were arranged perpendicularly in a Sauvage-type geometry 36-Cu (Scheme 13).106 Subsequent complexation of the terminal α-ethynyl groups on the thiophene rings to Pt(II) under high-dilution conditions, followed by iodine-mediated reductive elimination, yielded a new C–C bond enabling catenane 37 formation. Although the target compounds were obtained, the synthetic route was rather elaborate and afforded only small amounts of the desired material. The group therefore further refined the methodology and found that significantly improved yields of conjugated catenanes could be achieved via Pt(II)-catalyzed coupling of the terminal ethynyl groups.107 This development opened access to conjugated catenanes of various sizes. Notably, detailed optical and electrochemical studies indicated that the two macrocycles in the catenane influence one another via through-space donor–acceptor interactions. | ||
| Scheme 13 Synthesis of a conjugated [2]catenane 37.106,107 | ||
In 2025, Zeng, Wu, Ni, and co-workers reported the formation of a [2]catenane 39 composed of two intertwined octaphyrinoid macrocycles incorporating both pyrrole and thiophene units.108 The synthesis began with an α-bromothiophene-substituted dipyrromethene, which, upon coordination to Zn(II), formed a dimeric complex in nearly quantitative yield. Subsequent Suzuki–Miyaura coupling of this intermediate with a dithiophenemethane-derived boronic ester afforded a precursor 38 that was subjected to Yamamoto coupling, yielding the Zn(II)-stabilized catenane 39-Zn in 24% yield (Scheme 14).
 | ||
| Scheme 14 Synthesis of octaphyrinoid-based [2]catenane 39.108 | ||
Finally, oxidative dehydrogenation with DDQ followed by demetalation with HCl provided the fully conjugated, metal-free [2]catenane 39. Single-crystal X-ray diffraction analysis of 39 revealed that the core-modified octaphyrinoid macrocycles were arranged in an almost perpendicular orientation, with all bithiophene subunits adopting cis configuration (Fig. 6). The aromatic character of both rings was supported by harmonic oscillator model of aromaticity (HOMA) calculations, anisotropy of the induced current density (AICD) analysis, and three-dimensional iso-chemical shielding surface (ICSS) maps. Intriguingly, oxidation of 39 to the tetracation induced a pronounced structural distortion, leading to the formation of two antiaromatic octaphyrinoid units with close inter-ring contacts. This arrangement enabled strong through-space electronic coupling between the macrocycles.
 | ||
| Fig. 6 X-ray molecular structure of 39.108 | ||
MIMs embedding porphyrinoids’ structural elements
Several research groups have used not entire porphyrin or porphyrinoid molecules, but rather fragments of these macrocycles, such as pyrrole, bipyrrole, dipyrromethane, and dipyrrin, for the construction of catenanes and rotaxanes. When appropriately functionalized, these units serve as building blocks that act as hydrogen-bond donors/acceptors or as ligands to coordinate metal cations, while simultaneously retaining the ability to form macrocyclic architectures.In 1998, Sessler and Vögtle reported a spectacular example of a self-assembling [2]catenane comprising bipyrrole-based macrocycles.109 The reaction of p-xylylenediamine 40 with a bipyrrole diacyl chloride 41 afforded the interlocked product 42 in 2% yield (Scheme 15). However, the same compound could be obtained in a slightly higher yield (4%) when a sequential synthetic route was employed. The formation of an interlocked architecture was likely facilitated by hydrogen bonds between multiple donor and acceptor sites in the assembling parts of the molecule. The dynamic nature of 42, involving circumrotatory motion of the individual macrocyclic components, was inferred from the broadening of the 1H NMR resonance signals. Upon addition of fluoride, the resonances became significantly sharper, consistent with the anion binding within the catenane cavity. Subsequently, the interlocked receptor was shown to effectively interact with other anions as well.
 | ||
| Scheme 15 Synthesis of a bipyrrole-based catenane 42.109 | ||
Bozdemir and co-workers exploited the coordination properties of the dipyrrin motif to construct [2]catenanes using Sauvage's passive metal-template methodology.110 Accordingly, a dipyrrin-embedded macrocyclic precursor 43 bearing terminal alkene functionalities appended to ether side chains was synthesized (Scheme 16). Upon treatment with Zn(II) or Cu(II), ligand dimerization around the tetrahedral metal center afforded bis(dipyrrinato)metal complexes 43-M, effectively bringing the terminal double bonds into close proximity. Subsequent ring-closing metathesis catalyzed by the Grubbs 2nd-generation catalyst enabled covalent bond formation between the alkene termini, affording 44-M. Removal of the metal template from 44-M furnished the metal-free [2]catenane, which could subsequently be transformed into bis-BODIPY-like species.
 | ||
| Scheme 16 Synthesis of 44-M.110 | ||
An interesting strategy for constructing interlocked structures from expanded porphyrin-like macrocycles was reported by Nabeshima.111,112 The group described several macrocyclic trimers incorporating dipyrrin motifs separated by meta- and para-substituted phenylene units. Their corresponding tris-BF2 complexes were capable of incorporating protonated secondary amines to form pseudorotaxanes, e.g. 45, via non-classical, bifurcated hydrogen bonds, namely BF2⋯H–N interactions (Fig. 7).
 | ||
| Fig. 7 The pseudorotaxane 45 comprising expanded porphyrin-like ring component.111,112 | ||
The BODIPY motif has been incorporated into rotaxane architectures on several occasions to exploit its outstanding optical properties, including large molar extinction coefficients and high fluorescence quantum yields. Importantly, these features are complemented by excellent chemical stability and synthetic versatility, which allow fine-tuning of the luminescent properties. Akkaya and co-workers designed a [2]rotaxane in which suitably functionalized BODIPY units were attached both to the macrocyclic component – dibenzo-24-crown-8 – and installed as stoppers on the axle bearing a secondary amine at its center.113 Protonation of the amine generated the corresponding ammonium species, triggering spontaneous self-assembly of the [2]rotaxane. The resulting interlocked structure exhibited efficient through-space energy transfer via FRET.
Frasconi, Wasielewski, Stoddart, and co-workers reported a bistable [2]rotaxane composed of a tetrathiafulvalene (TTF)-containing thread terminated with a BODIPY stopper and a cyclobis(paraquat-p-phenylene) macrocycle.20 In the initial redox state, in the absence of an applied potential, the macrocycle resided on the TTF station, allowing free rotation of the BODIPY unit around the Cmeso–Cipso bond and resulting in low fluorescence intensity. Upon oxidation of TTF to the dication TTF2+, the macrocycle shifted toward the BODIPY stopper. This movement restricted the rotation, resulting in a 3.4-fold increase in fluorescence.
Trolez and co-workers described an intriguing route toward interlocked dipyrromethene derivatives. They designed a macrocycle incorporating a central 2,2′-biphenol unit.114,115 When the latter was added to a solution of BODIPY, previously treated with aluminum chloride in anhydrous dichloromethane, substitution of the fluorine atoms by the 2,2′-biphenol moiety took place. This transformation yielded an architecture in which the dipyrromethene unit was coordinated to a boron(III) center within the macrocyclic cavity. Notably, the threaded BODIPY exhibited higher fluorescence quantum yields than the corresponding non-interlocked analogues. This strategy, therefore, provided a promising approach to developing novel, highly emissive dyes. The same group also developed a rotaxane architecture featuring threaded BODIPY units acting as stoppers.116 The resulting system exhibited high molar absorption coefficients and fluorescence quantum yields, as well as remarkable brightness.
Iminopyrrole self-assembly and dynamic supramolecular architectures
From the beginning, our research group has focused on two main areas that complement one another. These themes include the chemistry of mechanically interlocked molecules and the use of self-assembly to construct dynamic supramolecular systems. We are particularly interested in iminopyrrole ligands, an area actively explored by several research groups, including those of Sessler, Love, Fout, and others. What sets our work apart is the approach taken to constructing iminopyrrole architectures. Specifically, we rely on multicomponent reactions operating under thermodynamic control, in a manner conceptually related to the subcomponent self-assembly strategy.117One of the first projects carried out in the group involved the synthesis of an original class of iminopyrrole macrocycles that combined structural features of crown ethers and porphyrins, termed crownphyrins.118,119 A related concept was proposed as early as the 1980s by Sessler; however, the use of tripyrane building block prevented the formation of a conjugated heterocyclic brace within the macrocyclic ring.120,121 Later, Love and co-workers revisited this idea and reported a series of elegant transition-metal “Pac-Man” complexes based on macrocycles incorporating both porphyrinoid and crown ether elements.122,123
Our approach relied on the use of a meso-pentafluorophenyl-substituted diformyldipyrromethane precursor 47, which, upon condensation with diamines bearing oligo(ethylene glycol) motifs 48, formed macrocycles that underwent spontaneous oxidation of the intermediate crownphyrinogens 49 to the corresponding crownphyrins 50 (Scheme 17A).119
 | ||
| Scheme 17 (A) Synthesis and reactivity of crownphyrins.119 (B) Different reactivity of diformylphenanthroline 51 and diformyldipyrromethene 54 in the metal-mediated self-assembly with 53.117,119 | ||
The hybrids turned out to be excellent receptors, as their cavities contain both hydrogen-bond donors and acceptors. As a result, they efficiently bind neutral molecules, such as water, and anions (after reduction), and act as versatile ligands.124 By selecting macrocycles of appropriate size, monomeric mononuclear complexes 50-Pb, dimeric “accordion-type” porphyrinoids,125 as well as figure-eight dimers 55-Zn exhibiting dynamic coordination behaviour in solution, were obtained (Fig. 8). Reactions with Group 1 metal ions were found to engage the ether oxygen atoms only, leading to the formation of intriguing M2(H2O)2-bridged dimers 57-M (M = Na, K).124 Particularly interesting was the reactivity toward Pd(II), which enforced previously unobserved iminopyrrole tautomerism involving proton transfer to a carbon atom. Finally, larger macrocycles displayed Janus-type behaviour: Pd(II) was coordinated within the porphyrinoid core, while alkali metal cation occupied the crown-ether-like pocket.
 | ||
| Fig. 8 The X-ray molecular structure of 56, 50-Pb, 55-Zn, and 57-K.119,124 | ||
Inspired by the work of Nitschke and co-workers,117 who demonstrated that the reaction between phenanthroline-2,9-dicarboxaldehyde 51 and 2,2′-(ethane-1,2-diylbis(oxy))bis(ethan-1-amine) 53 in the presence of a Cu(I) afforded a template-stabilized [2]catenane 52, we set out to investigate a similar transformation employing diformyldipyrromethene 54 (Scheme 17B). Copper(I) proved unsuitable under our reaction conditions, as it was readily oxidized to Cu(II), reflecting the tendency of iminopyrrole ligands to stabilize higher oxidation states of metal ions. At this stage, a fundamental difference between the iminophenanthroline and iminopyrrole systems became apparent. In our case, the sole product formed was the figure-eight complex 55-M.
Although we did not succeed at that time in obtaining a mechanically interlocked architecture, ongoing efforts are aimed at exploiting crownphyrin-based systems toward this goal. Importantly, the observed reactivity prompted us to further explore iminopyrrole ligands in subcomponent self-assembly processes. To evaluate the potential of iminopyrroles for constructing molecular systems of increasing complexity, we next examined their applicability in the assembly of capsular assemblies. Although purely organic cages based on iminopyrroles had already been reported at the time we initiated our studies,126–129 metal–organic assemblies derived from iminopyrroles remained largely unexplored.130,131
In our initial approach, we investigated the reactions between 2,5-diformylpyrrole, tris(2-aminoethyl)amine (tren), and silver(I) salts.132 We found that the outcome strongly depended on the nature of the silver salt, more specifically on the coordinating properties of the counteranion. Two principal types of products were obtained: cascade-type cryptates 58-Ag2F, in which two silver ions were located at opposite poles of a [3+2] cage with a central fluoride (or chloride anion(s)), and so-called “plenates”, e.g., 58-Ag5, i.e., cages whose cavities were filled with a silver(I) cluster composed of three to five metal ions (Fig. 9). Interestingly, theoretical studies have shown that the encapsulated silver clusters exhibit dynamic behaviour, with metal ions continuously rearranging their coordination environments and adopting multiple geometries within the cavity.133 Subsequently, we found that the simple [3+2] cage can encapsulate two metal centers of the same type, such as Zn(II), Cd(II), or Hg(II) (58-Hg2), as well as form heterobinuclear assemblies.134 In these complexes, the cage framework stabilized very short metal–metal contacts, resulting in metallophilic interaction.
 | ||
| Fig. 9 The X-ray molecular structures of metal-stabilized iminopyrrole cages 58-Ag2F, 58-Ag5, 58-Hg2, 59-Zn12, 60-Zn4.132,134,135 | ||
An intriguing observation was made during studies of cages assembled in the presence of Zn(II).135 In this case, the products formed by self-assembly were found to depend strongly on the solvent used. When the reaction was carried out in n-butanol, the expected [3+2] cage incorporating two Zn(II) ions was obtained. In contrast, performing the reaction in chloroform led to the formation of a much larger [12+8] system 59-Zn12 stabilized by the coordination of twelve Zn(II) ions (Fig. 9). The formation of the latter required partial tautomerization of the organic ligand from the diiminopyrrole form to an imino–aminoazafulvene in a tautomerism-coupled self-assembly (Scheme 18A).136 The protons located on the amino groups of the latter were strongly engaged in hydrogen bonding, creating an extensive network of stabilizing interactions. Treatment with a competitive pyridine ligand induced transformation of this assembly into a cage 60-Zn4, in which four octahedral Zn(II) centres occupied the vertices of a tetrahedron.
 | ||
| Scheme 18 (A) Tautomerism in diiminopyrrole-based ligands.136 (B) Intramolecular dynamics of coordination compounds of diiminopyrroles.139 | ||
Eventually, we sought to determine whether iminopyrroles could serve as building blocks for complex metallo-supramolecular architectures, including mechanically interlocked molecules such as knots and links. To this end, the replacement of 2,6-diformylpyridine by 2,5-diformylpyrrole in the reactions with bipyridyl-containing diamines developed by Stoddart and Trabolsi,137,138 carried out in the presence of Zn(II) or Cd(II), afforded [2]catenane 61-Zn2, trefoil knot 62-Cd3, and Borromean rings 63-Cd6 incorporating diiminopyrrole motif (Fig. 10).139 Interestingly, the X-ray diffraction analysis revealed that, contrary to the high symmetry anticipated based on the 1H NMR spectra recorded at 300 K, the diiminopyrrole motif acted primarily as a bidentate, not a tridentate ligand within the coordination pocket of the assembly. This observation was readily rationalized by considering the intrinsic intramolecular dynamics of the assemblies, including the continuous cleavage and reforming of N–M bonds within diiminopyrrole coordination compound (Scheme 18B). Consequently, such dynamic behavior appeared to be quite a common feature of assemblies incorporating the iminopyrrole coordination motif.
 | ||
| Fig. 10 The X-ray molecular structures of 61-Zn2, 62-Cd3, 63-Cd6.139 | ||
Next, we explored the use of more structurally elaborate pyrrolic ligands in self-assembly processes, going beyond simple iminopyrroles to building blocks reminiscent of those employed in the construction of porphyrins and porphyrinoids.140 In this context, we investigated the reactivity of two aldehydes – diformylbipyrrole 65 and diformyldipyrromethane 67 (Scheme 19). The latter is a direct precursor in the synthesis of porphyrins and numerous other pyrrolic macrocycles; upon oxidation, it forms dipyrrin, often considered as a “half-porphyrin” subunit. In contrast, bipyrrole is found in the frameworks of contracted porphyrinoids such as corroles, as well as in expanded systems including sapphyrins.141 When these aldehydes were subjected to condensation with amine 66 in the presence of Zn(II) or Cd(II) salts, the only products obtained were metal-stabilised [2]catenanes, which we termed catenaphyrins. Interestingly, those two assemblies also demonstrated dynamic behaviour in the solution. Notably, 68-M exhibited interesting optical properties, with an emission centered at 580–620 nm. This exploratory study confirmed that more intricate pyrrole-based building blocks can be successfully employed in self-assembly processes, opening a route to the construction of architectures such as hypothetical knotaphyrins, i.e., molecules that blur the boundaries between molecular knots and expanded porphyrinoids.
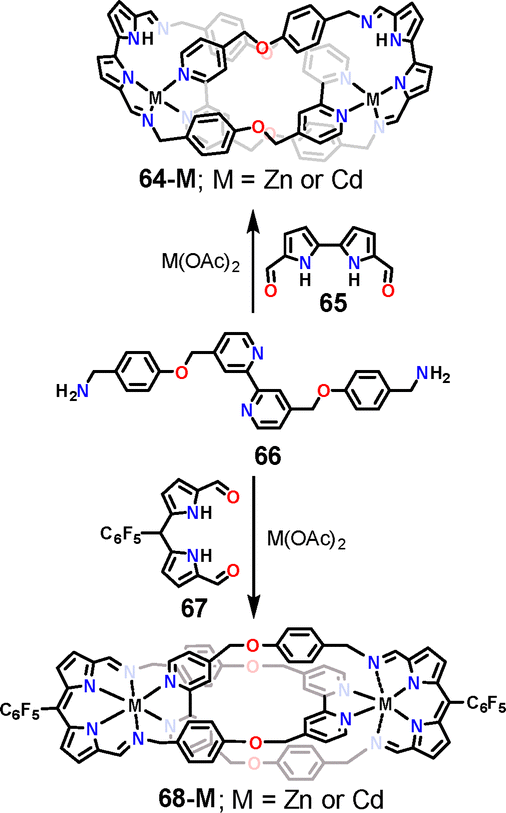 | ||
| Scheme 19 Self-assembly of [2]catenaphyrins 64-M and 68-M.140 | ||
In parallel, we investigated self-assembly reactions leading to molecular knots and links stabilized by silver(I) ions. During these studies, the formation of an intriguing molecular motif, namely, a molecular tweezer 69, was detected. The latter formed from the reaction of 2,2′-bipyridyl-based diamine with 2,6-diformylpyridine in the presence of silver(I) acetate (Fig. 11). The structure consisted of two macrocyclic units incorporating a diiminopyridine motif, with dimerization driven by coordination of Ag(I)⋯Ag(I) pairs.
 | ||
| Fig. 11 The X-ray molecular structures of 69, 70, and 71.142 | ||
Remarkably, a subtle modification of the amine component used in the self-assembly process enabled the selective formation of a Solomon link 70 stabilized by four Ag22+ units. Further investigations revealed that this silver cluster can serve as an effective template, enabling the formation of [2]catenane 71 and trefoil knot 72 when 2,7-diformyl-1,8-naphthyridine was used as the carbonyl component in the self-assembly reaction (Scheme 20). Although compounds 69–72 did not incorporate pyrrole-based ligands, they represent an extension of our earlier work on silver cluster templation, with potential applicability to pyrrolic and other assemblies.
 | ||
| Scheme 20 The Ag(I)⋯Ag(I)-stabilized assemblies 69–72.142 | ||
Calixphyrin-based catenanes and rotaxanes
In search of a general strategy for the synthesis of diverse mechanically interlocked porphyrinoids, we reasoned that access to a versatile and fundamental building block – namely a [2]rotaxane stoppered with a dipyrromethane such as 76 – would enable the preparation of a broad family of such systems.143 This compound was obtained using a macrocyclic bipyridine 73 reported by Goldup and co-workers.144 In a reaction between azide 74 and a meso-4-alkynyl-substituted dipyrromethane 75 under copper(I)-catalyzed active-template conditions,145–147 the desired mechanically interlocked dipyrromethane 76 was formed in high yield (83%) (Scheme 21A). Interestingly, the dipyrromethane stopper was sufficiently large to prevent dethreading under conditions of porphyrinoid condensation. Furthermore, the successful synthesis of a [2]rotaxane featuring a long thread terminated with a dipyrromethane at both ends enabled the subsequent design of the corresponding catenanes. | ||
| Scheme 21 (A) Synthesis of a dipyrromethane-stoppered rotaxane 76 and (B) its condensation products – [3]rotaxane 77 and [4]rotaxane 78.143 | ||
Rotaxane 76 proved to be an excellent building block for the synthesis of porphyrinoids. Upon reaction with pentafluorobenzaldehyde under Lindsey condensation conditions, formation of a [3]rotaxane was observed, featuring an unsubstituted porphyrin unit positioned at the center of the axle. Having confirmed the utility of this building block in porphyrinoid-forming reactions, we next investigated its reaction with acetone. This transformation afforded two products: a [3]rotaxane 77 and a Y-shaped [4]rotaxane 78, incorporating calix[4]phyrin and calix[6]phyrin units, respectively (Scheme 21B). The core calix[n]phyrins represent hybrid macrocycles that bridge nonconjugated calix[n]pyrroles and fully conjugated porphyrins, owing to the presence of meso-sp3 carbon bridges separating the dipyrromethene units within the macrocycle.148,149 As a consequence, calix[4]phyrins adopt a roof-like conformation in the solid state, whereas in solution they exhibit dynamic behavior involving interconversion between V-shaped stereoisomers, leading to time-averaged positions of the methyl groups (Fig. 12A).150 Notably, this conformational switching is retained upon incorporation of the calix[4]phyrin unit into the axle of a rotaxane, thereby inducing motion of the entire mechanically interlocked molecule (Fig. 12B).143 Owing to its resemblance to the motion of bird or butterfly wings during flight, we termed this dynamic behaviour as fluttering. Interestingly, the fluttering motion was suppressed when the calix[4]phyrin unit was incorporated into a [2]catenane, most likely due to an insufficient cavity size that restricts the conformational flexibility. Furthermore, the [3]rotaxane, by virtue of its molecular design, exhibited multimodal motion in response to external stimuli. In addition to fluttering – which can be reversibly switched on and off by (de)protonation of the calixphyrin core – the system also undergoes macrocycle shuttling along the thread and rotational motion of the macrocyclic component. Further strategies to control the fluttering motion are currently being explored in our laboratory.
 | ||
| Fig. 12 (A) The toggling in calix[4]phyrin.149,150 (B) Fluttering in calix[4]phyrin-incorporating [3]rotaxane.143 | ||
The developed strategy for the synthesis of [2]rotaxanes bearing dipyrromethane units at both termini enabled the design of a series of precursors for the construction of [n]catenanes.151 Under optimized conditions, substrates differing in the length and flexibility of the linear component were expected to undergo condensation to form interlocked products. It was ultimately found that the outcome strongly depended on the thread's geometric features. Rotaxanes with sufficiently long and flexible linear components underwent intramolecular self-condensation in the presence of acetone and a catalyst, affording [2]catenanes. In contrast, shorter and more rigid [2]rotaxanes favored intermolecular condensation, leading to the formation of higher-order [3]- and [4]catenanes. These studies demonstrated that the structure of the starting materials plays a decisive role in determining the topology of the final product. Consequently, this methodology is expected to be extendable to the synthesis of higher-order rotaxanes and catenanes based on diverse scaffolds, not limited to the calixphyrin motif.
Conclusions
The chemistry at the interface between pyrrolic macrocycles and mechanically interlocked molecules, although initiated several decades ago, continues to evolve. While a number of structurally and functionally intriguing architectures have emerged from combining these two classes of molecular systems, the field remains dominated by porphyrin-based examples, with porphyrinoids or their fragments incorporated into MIMs only occasionally. Yet, the potential of such systems appears far from fully realized. Embedding porphyrinoid within the constrained environment of MIMs offers a powerful opportunity to introduce new dimensions of functionality, including controlled modulation of conjugation pathways, the emergence of spatially differentiated magnetic domains, and reversible switching between multiple electronic states. Moreover, mechanically interlocked architectures provide a unique platform for probing how the fundamental properties of pyrrolic macrocycles alter under topological constraints. It is anticipated that deeper exploration of this interface will not only enrich the chemistry of MIMs but also open new conceptual avenues in porphyrinoid chemistry, where topology and electronic structure can be deliberately intertwined.Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results have been included, and no new data were generated or analysed as part of this feature article.Acknowledgements
The National Science Centre of Poland supported this work under the grant agreement no. 2023/51/B/ST4/00040.References
- C. J. Bruns and J. F. Stoddart, The Nature of the Mechanical Bond: From Molecules to Machines, Wiley, 1st edn, 2016 Search PubMed.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed.
- J. M. Abendroth, O. S. Bushuyev, P. S. Weiss and C. J. Barrett, ACS Nano, 2015, 9, 7746–7768 CrossRef CAS PubMed.
- V. Amendola, L. Fabbrizzi, C. Mangano and P. Pallavicini, Acc. Chem. Res., 2001, 34, 488–493 CrossRef CAS PubMed.
- M. Russew and S. Hecht, Adv. Mater., 2010, 22, 3348–3360 CrossRef CAS PubMed.
- R. G. E. Coumans, J. A. A. W. Elemans, A. E. Rowan and R. J. M. Nolte, Chem. – Eur. J., 2013, 19, 7758–7770 CrossRef CAS PubMed.
- B. L. Feringa, N. Koumura, R. A. van Delden and M. K. J. ter Wiel, Appl. Phys. A: Mater. Sci. Process., 2002, 75, 301–308 CrossRef CAS.
- T. L. Mako, J. M. Racicot and M. Levine, Chem. Rev., 2019, 119, 322–477 CrossRef CAS PubMed.
- J. T. Wilmore and P. D. Beer, Adv. Mater., 2024, 36, 2309098 CrossRef CAS PubMed.
- L. F. Hart, J. E. Hertzog, P. M. Rauscher, B. W. Rawe, M. M. Tranquilli and S. J. Rowan, Nat. Rev. Mater., 2021, 6, 508–530 CrossRef CAS.
- S. Mena-Hernando and E. M. Pérez, Chem. Soc. Rev., 2019, 48, 5016–5032 RSC.
- R. A. Grzelczak, A. Władyczyn, A. Białońska, Ł. John and B. Szyszko, Chem. Commun., 2023, 59, 7579–7582 RSC.
- J. A. Faiz, V. Heitz and J.-P. Sauvage, Chem. Soc. Rev., 2009, 38, 422–442 RSC.
- A. Harriman and J.-P. Sauvage, Chem. Soc. Rev., 1996, 25, 41–48 RSC.
- J.-C. Chambron, J.-P. Collin, J.-O. Dalbavie, C. O. Dietrich-Buchecker, V. Heitz, F. Odobel, N. Solladié and J.-P. Sauvage, Coord. Chem. Rev., 1998, 178–180, 1299–1312 CrossRef CAS.
- L. Flamigni, V. Heitz and J.-P. Sauvage, in Non-Covalent Multi-Porphyrin Assemblies, ed. E. Alessio, Springer, Berlin, Heidelberg, 2006, 217–261 Search PubMed.
- J.-C. Chambron, V. Heitz and J.-P. Sauvage, J. Chem. Soc., Chem. Commun., 1992, 1131–1133 RSC.
- J.-C. Chambron, V. Heitz and J.-P. Sauvage, J. Am. Chem. Soc., 1993, 115, 12378–12384 CrossRef CAS.
- P. R. Ashton, M. R. Johnston, J. F. Stoddart, M. S. Tolley and J. W. Wheeler, J. Chem. Soc., Chem. Commun., 1992, 1128–1131 RSC.
- Y. Wu, M. Frasconi, W.-G. Liu, R. M. Young, W. A. Goddard, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2020, 142, 11835–11846 CrossRef CAS PubMed.
- M. J. Gunter, N. Bampos, K. D. Johnstone and J. K. M. Sanders, New J. Chem., 2001, 25, 166–173 RSC.
- K. D. Johnstone, N. Bampos, J. K. M. Sanders and M. J. Gunter, Chem. Commun., 2003, 1396–1397 RSC.
- K. M. Mullen, K. D. Johnstone, D. Nath, N. Bampos, J. K. M. Sanders and M. J. Gunter, Org. Biomol. Chem., 2008, 7, 293–303 RSC.
- K. M. Mullen, K. D. Johnstone, M. Webb, N. Bampos, J. K. M. Sanders and M. J. Gunter, Org. Biomol. Chem., 2008, 6, 278–286 RSC.
- C. A. Hunter and L. D. Sarson, Angew. Chem., Int. Ed. Engl., 1994, 33, 2313–2316 CrossRef.
- C. A. Hunter, C. M. R. Low, M. J. Packer, S. E. Spey, J. G. Vinter, M. O. Vysotsky and C. Zonta, Angew. Chem., Int. Ed., 2001, 40, 2678–2682 CrossRef CAS PubMed.
- M. J. Langton, J. D. Matichak, A. L. Thompson and H. L. Anderson, Chem. Sci., 2011, 2, 1897–1901 RSC.
- D. R. Kohn, L. D. Movsisyan, A. L. Thompson and H. L. Anderson, Org. Lett., 2017, 19, 348–351 CrossRef CAS PubMed.
- J.-P. Sauvage, J.-P. Collin, J. A. Faiz, J. Frey, V. Heitz and C. Tock, J. Porphyr. Phthalocyanines, 2008, 12, 881–905 CrossRef CAS.
- B. Szyszko and L. Latos-Grażyński, Chem. Soc. Rev., 2015, 44, 3588–3616 RSC.
- P. J. Chmielewski and L. Latos-Grażyński, Coord. Chem. Rev., 2005, 249, 2510–2533 CrossRef CAS.
- ed. T. M. Krygowski and M. K. Cyrański, Aromaticity in Heterocyclic Compounds, Springer Berlin Heidelberg, Berlin, Heidelberg, 2009, vol. 19 Search PubMed.
- M. Stępień, N. Sprutta and L. Latos-Grażyński, Angew. Chem., Int. Ed., 2011, 50, 4288–4340 CrossRef PubMed.
- B. Szyszko, M. J. Białek, E. Pacholska-Dudziak and L. Latos-Grażyński, Chem. Rev., 2017, 117, 2839–2909 CrossRef CAS PubMed.
- B. Szyszko, M. Przewoźnik, M. J. Białek, A. Białońska, P. J. Chmielewski and L. Latos-Grażyński, Chem. – Eur. J., 2020, 26, 8555–8566 CrossRef CAS PubMed.
- B. Szyszko, E. Pacholska-Dudziak and L. Latos-Grażyński, J. Org. Chem., 2013, 78, 5090–5095 CrossRef CAS PubMed.
- P. Krzyszowska, A. Burska-Jabłońska, M. Oberski, M. J. Białek, L. Latos-Grażyński and K. Hurej, Angew. Chem., Int. Ed., 2026, 65, e25506 CrossRef CAS PubMed.
- S. Sanfui, M. Usman, S. Sarkar, S. Pramanik, E. Garribba and S. P. Rath, Inorg. Chem., 2022, 61, 8419–8430 CrossRef CAS PubMed.
- M. T. Green, J. H. Dawson and H. B. Gray, Science, 2004, 304, 1653–1656 CrossRef CAS PubMed.
- W. J. Song, M. S. Seo, S. DeBeer George, T. Ohta, R. Song, M.-J. Kang, T. Tosha, T. Kitagawa, E. I. Solomon and W. Nam, J. Am. Chem. Soc., 2007, 129, 1268–1277 CrossRef CAS PubMed.
- M. J. Białek, K. Hurej, H. Furuta and L. Latos-Grażyński, Chem. Soc. Rev., 2023, 52, 2082–2144 RSC.
- T. Higashino, A. Kumagai, S. Sakaki and H. Imahori, Chem. Sci., 2018, 9, 7528–7539 RSC.
- L. Raehm, J.-M. Kern and J.-P. Sauvage, Chem. – Eur. J., 1999, 5, 3310–3317 CrossRef CAS.
- R. A. Bissell, E. Córdova, A. E. Kaifer and J. F. Stoddart, Nature, 1994, 369, 133–137 CrossRef CAS.
- M. J. Power, D. T. J. Morris, I. J. Vitorica-Yrezabal and D. A. Leigh, J. Am. Chem. Soc., 2023, 145, 8593–8599 CrossRef CAS PubMed.
- C. M. Storey, M. R. Gyton, R. E. Andrew and A. B. Chaplin, Angew. Chem., Int. Ed., 2018, 57, 12003–12006 CrossRef CAS PubMed.
- M.-J. Blanco, J.-C. Chambron, V. Heitz and J.-P. Sauvage, Org. Lett., 2000, 2, 3051–3054 CrossRef CAS PubMed.
- J. Wu, F. Fang, W.-Y. Lu, J.-L. Hou, C. Li, Z.-Q. Wu, X.-K. Jiang, Z.-T. Li and Y.-H. Yu, J. Org. Chem., 2007, 72, 2897–2905 CrossRef CAS PubMed.
- N. Solladié, J.-C. Chambron, C. O. Dietrich-Buchecker and J.-P. Sauvage, Angew. Chem., Int. Ed. Engl., 1996, 35, 906–909 CrossRef.
- N. Solladié, J.-C. Chambron and J.-P. Sauvage, J. Am. Chem. Soc., 1999, 121, 3684–3692 CrossRef.
- K. Chichak, M. C. Walsh and N. R. Branda, Chem. Commun., 2000, 847–848 RSC.
- M. Asakawa, T. Ikeda, N. Yui and T. Shimizu, Chem. Lett., 2002, 174–175 CrossRef CAS.
- C. Roche, J.-P. Sauvage, A. Sour and N. L. Strutt, New J. Chem., 2011, 35, 2820–2825 RSC.
- P. Martinez-Bulit, B. H. Wilson and S. J. Loeb, Org. Biomol. Chem., 2020, 18, 4395–4400 RSC.
- L. D. Movsisyan, M. Franz, F. Hampel, A. L. Thompson, R. R. Tykwinski and H. L. Anderson, J. Am. Chem. Soc., 2016, 138, 1366–1376 CrossRef CAS PubMed.
- J. Pruchyathamkorn, W. J. Kendrick, A. T. Frawley, A. Mattioni, F. Caycedo-Soler, S. F. Huelga, M. B. Plenio and H. L. Anderson, Angew. Chem., Int. Ed., 2020, 59, 16455–16458 CrossRef CAS PubMed.
- V. Heitz, S. Chardon-Noblat and J.-P. Sauvage, Tetrahedron Lett., 1991, 32, 197–198 CrossRef CAS.
- J. C. Chambron, A. Harriman, V. Heitz and J. P. Sauvage, J. Am. Chem. Soc., 1993, 115, 6109–6114 CrossRef CAS.
- J. C. Chambron, A. Harriman, V. Heitz and J. P. Sauvage, J. Am. Chem. Soc., 1993, 115, 7419–7425 CrossRef CAS.
- M. Linke, J.-C. Chambron, V. Heitz and J.-P. Sauvage, J. Am. Chem. Soc., 1997, 119, 11329–11330 CrossRef CAS.
- M. Linke, J.-C. Chambron, V. Heitz, J.-P. Sauvage, S. Encinas, F. Barigelletti and L. Flamigni, J. Am. Chem. Soc., 2000, 122, 11834–11844 CrossRef CAS.
- L. Flamigni, A. M. Talarico, S. Serroni, F. Puntoriero, M. J. Gunter, M. R. Johnston and T. P. Jeynes, Chem. – Eur. J., 2003, 9, 2649–2659 CrossRef CAS PubMed.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2001, 34, 40–48 Search PubMed.
- D. Kuciauskas, P. A. Liddell, S. Lin, S. G. Stone, A. L. Moore, T. A. Moore and D. Gust, J. Phys. Chem. B, 2000, 104, 4307–4321 CrossRef CAS.
- D. I. Schuster, K. Li, D. M. Guldi and J. Ramey, Org. Lett., 2004, 6, 1919–1922 CrossRef CAS PubMed.
- A. S. D. Sandanayaka, N. Watanabe, K.-I. Ikeshita, Y. Araki, N. Kihara, Y. Furusho, O. Ito and T. Takata, J. Phys. Chem. B, 2005, 109, 2516–2525 CrossRef CAS PubMed.
- N. Watanabe, N. Kihara, Y. Furusho, T. Takata, Y. Araki and O. Ito, Angew. Chem., Int. Ed., 2003, 42, 681–683 CrossRef CAS PubMed.
- K. Li, D. I. Schuster, D. M. Guldi, M. Á. Herranz and L. Echegoyen, J. Am. Chem. Soc., 2004, 126, 3388–3389 CrossRef CAS PubMed.
- J. D. Megiatto, D. I. Schuster, S. Abwandner, G. De Miguel and D. M. Guldi, J. Am. Chem. Soc., 2010, 132, 3847–3861 CrossRef CAS PubMed.
- F. Schwer, S. Zank, M. Freiberger, F. M. Steudel, N. Geue, L. Ye, P. E. Barran, T. Drewello, D. M. Guldi and M. Von Delius, Angew. Chem., 2025, 137, e202413404 CrossRef.
- M. J. Gunter, T. P. Jeynes and P. Turner, Eur. J. Org. Chem., 2004, 193–208 CrossRef CAS.
- D. P. Cormode, M. G. B. Drew, R. Jagessar and P. D. Beer, Dalton Trans., 2008, 6732–6741 RSC.
- J.-P. Collin, F. Durola, J. Frey, V. Heitz, J.-P. Sauvage, C. Tock and Y. Trolez, Chem. Commun., 2009, 1706–1708 RSC.
- J.-S. Marois, K. Cantin, A. Desmarais and J.-F. Morin, Org. Lett., 2008, 10, 33–36 CrossRef CAS PubMed.
- J. Frey, C. Tock, J.-P. Collin, V. Heitz and J.-P. Sauvage, J. Am. Chem. Soc., 2008, 130, 4592–4593 CrossRef CAS PubMed.
- A. Brown and P. D. Beer, Dalton Trans., 2012, 41, 118–129 RSC.
- Y. Cheong Tse, R. Hein, E. J. Mitchell, Z. Zhang and P. D. Beer, Chem. – Eur. J., 2021, 27, 14550–14559 CrossRef CAS PubMed.
- A. Brown, M. J. Langton, N. L. Kilah, A. L. Thompson and P. D. Beer, Chem. – Eur. J., 2015, 21, 17664–17675 CrossRef CAS PubMed.
- M. Linke, J.-C. Chambron, V. Heitz, J.-P. Sauvage and V. Semetey, Chem. Commun., 1998, 2469–2470 RSC.
- J.-P. Collin, J. Frey, V. Heitz, J.-P. Sauvage, C. Tock and L. Allouche, J. Am. Chem. Soc., 2009, 131, 5609–5620 CrossRef CAS PubMed.
- J. T. Wilmore, Y. Cheong Tse, A. Docker, C. Whitehead, C. K. Williams and P. D. Beer, Chem. – Eur. J., 2023, 29, e202300608 CrossRef CAS PubMed.
- Y. Oka, H. Masai and J. Terao, Angew. Chem., Int. Ed., 2023, 62, e202217002 CrossRef CAS PubMed.
- P. Thordarson, E. J. A. Bijsterveld, A. E. Rowan and R. J. M. Nolte, Nature, 2003, 424, 915–918 CrossRef CAS PubMed.
- R. G. E. Coumans, J. A. A. W. Elemans, R. J. M. Nolte and A. E. Rowan, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19647–19651 CrossRef CAS PubMed.
- A. B. C. Deutman, C. Monnereau, J. A. A. W. Elemans, G. Ercolani, R. J. M. Nolte and A. E. Rowan, Science, 2008, 322, 1668–1671 CrossRef CAS PubMed.
- C. Monnereau, P. H. Ramos, A. B. C. Deutman, J. A. A. W. Elemans, R. J. M. Nolte and A. E. Rowan, J. Am. Chem. Soc., 2010, 132, 1529–1531 CrossRef CAS PubMed.
- R. J. M. Nolte and J. A. A. W. Elemans, Chem. – Eur. J., 2024, 30, e202304230 Search PubMed.
- A. F. P. Alcântara, L. A. Fontana, V. H. Rigolin, Y. F. S. Andrade, M. A. Ribeiro, W. P. Barros, C. Ornelas and J. D. Megiatto, Angew. Chem., Int. Ed., 2018, 57, 8979–8983 CrossRef PubMed.
- L. A. Fontana, M. P. Almeida, A. F. P. Alcântara, V. H. Rigolin, M. A. Ribeiro, W. P. Barros and J. D. Megiatto, Nat. Commun., 2020, 11, 6370 CrossRef CAS PubMed.
- Y. Miyazaki, C. Kahlfuss, A. Ogawa, T. Matsumoto, J. A. Wytko, K. Oohora, T. Hayashi and J. Weiss, Chem. – Eur. J., 2017, 23, 13579–13582 CrossRef CAS PubMed.
- A. Baeyer, Ber. Dtsch. Chem. Ges., 1886, 19, 2184–2185 CrossRef.
- P. A. Gale, J. L. Sessler, V. Král and V. Lynch, J. Am. Chem. Soc., 1996, 118, 5140–5141 CrossRef CAS.
- V. Valderrey, E. C. Escudero-Adán and P. Ballester, J. Am. Chem. Soc., 2012, 134, 10733–10736 CrossRef CAS PubMed.
- J. R. Romero, G. Aragay and P. Ballester, Chem. Sci., 2017, 8, 491–498 RSC.
- R. Molina-Muriel, J. R. Romero, Y. Li, G. Aragay and P. Ballester, Org. Biomol. Chem., 2021, 19, 9986–9995 RSC.
- Y. Li, R. Molina-Muriel, G. Aragay and P. Ballester, Org. Chem. Front., 2024, 11, 5374–5384 RSC.
- D. Memis, N. Bektas and A. Aydogan, ACS Appl. Polym. Mater., 2024, 6, 4339–4348 CrossRef CAS.
- D. T. Gryko and M. Tasior, Fundamentals of Porphyrin Chemistry, John Wiley & Sons, Ltd, 2022, 349–383 Search PubMed.
- C. D. Natale, C. P. Gros and R. Paolesse, Chem. Soc. Rev., 2022, 51, 1277–1335 RSC.
- T. H. Ngo, J. Labuta, G. N. Lim, W. A. Webre, F. D’Souza, P. A. Karr, J. E. M. Lewis, J. P. Hill, K. Ariga and S. M. Goldup, Chem. Sci., 2017, 8, 6679–6685 RSC.
- T. H. Ngo, J. E. M. Lewis, D. T. Payne, F. D’Souza, J. P. Hill, K. Ariga, G. Yoshikawa and S. M. Goldup, Chem. Commun., 2020, 56, 7447–7450 RSC.
- D. Seidel, V. Lynch and J. L. Sessler, Angew. Chem., Int. Ed., 2002, 41, 1422–1425 CrossRef CAS PubMed.
- T. Izumi, S. Kobashi, K. Takimiya, Y. Aso and T. Otsubo, J. Am. Chem. Soc., 2003, 125, 5286–5287 CrossRef PubMed.
- P. Bäuerle, J. Becher, J. Lau and P. Mark, Electronic Materials: The Oligomer Approach, John Wiley & Sons, Ltd, 1998, pp. 105–233 Search PubMed.
- M. Ammann, A. Rang, C. A. Schalley and P. Bäuerle, Eur. J. Org. Chem., 2006, 1940–1948 CrossRef CAS.
- P. Bäuerle, M. Ammann, M. Wilde, G. Götz, E. Mena-Osteritz, A. Rang and C. A. Schalley, Angew. Chem., Int. Ed., 2007, 46, 363–368 CrossRef PubMed.
- G. Götz, X. Zhu, A. Mishra, J. Segura, E. Mena-Osteritz and P. Bäuerle, Chem. – Eur. J., 2015, 21, 7193–7210 CrossRef PubMed.
- Y. Sui, Z. Wang, J. Hao, S. Wu, L. Ren, Z. Zeng, J. Wu and Y. Ni, Nat. Synth., 2025, 5, 230–239 CrossRef.
- A. Andrievsky, F. Ahuis, J. L. Sessler, F. Vögtle, D. Gudat and M. Moini, J. Am. Chem. Soc., 1998, 120, 9712–9713 CrossRef CAS.
- B. Nisanci, S. Sahinoglu, E. Tuner, M. Arik, İ. Kani, A. Dastan and Ö. A. Bozdemir, Chem. Commun., 2017, 53, 12418–12421 RSC.
- N. Sakamoto, C. Ikeda and T. Nabeshima, Chem. Commun., 2010, 46, 6732–6734 RSC.
- T. Nakamura, G. Yamaguchi and T. Nabeshima, Angew. Chem., Int. Ed., 2016, 55, 9606–9609 CrossRef CAS PubMed.
- N. Yesilgul, O. Seven, R. Guliyev and E. U. Akkaya, J. Org. Chem., 2018, 83, 13228–13232 CrossRef CAS PubMed.
- M. Hicguet, L. Verrieux, O. Mongin, T. Roisnel, F. Berrée, A. Fihey, B. Le Guennic and Y. Trolez, Angew. Chem., Int. Ed., 2024, 63, e202318297 CrossRef CAS PubMed.
- M. Hicguet, O. Mongin, Y. R. Leroux, T. Roisnel, F. Berrée and Y. Trolez, ChemistryOpen, 2024, 13, e202400196 CrossRef CAS PubMed.
- M. Hicguet, O. Mongin, Y. R. Leroux, F. Berrée and Y. Trolez, Org. Lett., 2025, 27, 12217–12222 CrossRef CAS PubMed.
- M. Hutin, C. A. Schalley, G. Bernardinelli and J. R. Nitschke, Chem. – Eur. J., 2006, 12, 4069–4076 CrossRef CAS PubMed.
- M. Matviyishyn and B. Szyszko, Beilstein J. Org. Chem., 2023, 19, 1630–1650 CrossRef CAS PubMed.
- M. Matviyishyn, A. Białońska and B. Szyszko, Angew. Chem., Int. Ed., 2022, 61, e202211671 CrossRef CAS PubMed.
- J. L. Sessler and A. K. Burrell, Macrocycles, Springer Berlin Heidelberg, Berlin, Heidelberg, 1992, vol. 161, pp. 177–273 Search PubMed.
- J. L. Sessler, M. R. Johnson, V. Lynch and T. Murai, J. Coord. Chem., 1988, 18, 99–104 CrossRef CAS.
- J. W. Leeland, F. J. White and J. B. Love, Chem. Commun., 2011, 47, 4132–4134 RSC.
- J. W. Leeland, C. Finn, B. Escuyer, H. Kawaguchi, G. S. Nichol, A. M. Z. Slawin and J. B. Love, Dalton Trans., 2012, 41, 13815–13831 RSC.
- M. Matviyishyn, K. A. Konieczny, B. Trzaskowski and B. Szyszko, Chem. – Eur. J., 2024, 30, e202402932 CrossRef CAS PubMed.
- W. A. Reiter, A. Gerges, S. Lee, T. Deffo, T. Clifford, A. Danby and K. Bowman-James, Coord. Chem. Rev., 1998, 174, 343–359 CrossRef CAS.
- D. Chen and A. E. Martell, Tetrahedron, 1991, 47, 6895–6902 CrossRef CAS.
- H. J. Han, J. H. Oh, J. L. Sessler and S. K. Kim, Chem. Commun., 2019, 55, 10876–10879 RSC.
- J. H. Oh, J. H. Kim, D. S. Kim, H. J. Han, V. M. Lynch, J. L. Sessler and S. K. Kim, Org. Lett., 2019, 21, 4336–4339 CrossRef CAS PubMed.
- O. D. Fox, T. D. Rolls, P. D. Beer and M. G. B. Drew, Chem. Commun., 2001, 1632–1633 RSC.
- L. Qin, J. Nelson and M. McCann, J. Inorg. Biochem., 1993, 51, 633–639 CrossRef CAS.
- Q. Lu, V. McKee and J. Nelson, J. Chem. Soc., Chem. Commun., 1994, 649–651 RSC.
- A. Sarwa, A. Białońska, M. Garbicz and B. Szyszko, Chem. – Eur. J., 2023, 29, e202203850 CrossRef CAS PubMed.
- B. Trzaskowski, J. P. Martínez, A. Sarwa, B. Szyszko and W. A. Goddard, J. Phys. Chem. A, 2024, 128, 3339–3350 CrossRef CAS PubMed.
- A. Sarwa, P. Krajewski, B. Trzaskowski, J. P. Perdek, M. Siczek and B. Szyszko, Inorg. Chem., 2026, 65, 5280–5285 CrossRef CAS PubMed.
- J. Sukiennik, A. Sarwa, J. P. Perdek, M. Siczek and B. Szyszko, Chem. – Eur. J., 2025, 31, e02714 CrossRef CAS PubMed.
- M. Giri, D. Sahoo, B. P. Samantray, P. P. Sahoo, S. Mishra and T. Guchhait, J. Mol. Struct., 2024, 1308, 138029 CrossRef CAS.
- K. S. Chichak, S. J. Cantrill, A. R. Pease, S.-H. Chiu, G. W. V. Cave, J. L. Atwood and J. F. Stoddart, Science, 2004, 304, 1308–1312 CrossRef CAS PubMed.
- T. Prakasam, M. Lusi, M. Elhabiri, C. Platas-Iglesias, J.-C. Olsen, Z. Asfari, S. Cianférani-Sanglier, F. Debaene, L. J. Charbonnière and A. Trabolsi, Angew. Chem., Int. Ed., 2013, 52, 9956–9960 Search PubMed.
- A. Sarwa, A. Białońska, M. Sobieraj, J. P. Martínez, B. Trzaskowski and B. Szyszko, Angew. Chem., Int. Ed., 2024, 63, e202316489 CrossRef CAS PubMed.
- A. Sarwa, M. Matviyishyn, J. P. Perdek, B. Trzaskowski, D. Kulesza, E. Zych and B. Szyszko, Chem. Commun., 2026, 62, 148–151 RSC.
- V. J. Bauer, D. L. J. Clive, D. Dolphin, J. B. Paine, F. L. Harris, M. M. King, J. Loder, S. W. C. Wang and R. B. Woodward, J. Am. Chem. Soc., 1983, 105, 6429–6436 CrossRef CAS.
- A. Sarwa, A. Khmara, K. A. Konieczny, D. Kulesza, E. Zych, B. Trzaskowski and B. Szyszko, Angew. Chem., Int. Ed., 2025, 64, e202423962 CrossRef CAS PubMed.
- R. A. Grzelczak, T. Basak, B. Trzaskowski, V. Kinzhybalo and B. Szyszko, Angew. Chem., Int. Ed., 2025, 64, e202413579 Search PubMed.
- J. E. M. Lewis, R. J. Bordoli, M. Denis, C. J. Fletcher, M. Galli, E. A. Neal, E. M. Rochette and S. M. Goldup, Chem. Sci., 2016, 7, 3154–3161 Search PubMed.
- V. Aucagne, J. Berná, J. D. Crowley, S. M. Goldup, K. D. Hänni, D. A. Leigh, P. J. Lusby, V. E. Ronaldson, A. M. Z. Slawin, A. Viterisi and D. B. Walker, J. Am. Chem. Soc., 2007, 129, 11950–11963 CrossRef CAS PubMed.
- J. D. Crowley, S. M. Goldup, A.-L. Lee, D. A. Leigh and R. T. McBurney, Chem. Soc. Rev., 2009, 38, 1530–1541 RSC.
- S. M. Goldup, D. A. Leigh, T. Long, P. R. McGonigal, M. D. Symes and J. Wu, J. Am. Chem. Soc., 2009, 131, 15924–15929 CrossRef CAS PubMed.
- C. Bucher, R. S. Zimmerman, V. Lynch, V. Král and J. L. Sessler, J. Am. Chem. Soc., 2001, 123, 2099–2100 CrossRef CAS PubMed.
- V. Král, J. L. Sessler, R. S. Zimmerman, D. Seidel, V. Lynch and B. Andrioletti, Angew. Chem., Int. Ed., 2000, 39, 1055–1058 CrossRef.
- B. Dolenský, J. Kroulík, V. Král, J. L. Sessler, H. Dvořáková, P. Bouř, M. Bernátková, C. Bucher and V. Lynch, J. Am. Chem. Soc., 2004, 126, 13714–13722 CrossRef PubMed.
- R. A. Grzelczak, J. P. Perdek, M. Siczek, P. J. Chmielewski and B. Szyszko, Org. Lett., 2025, 27, 6310–6315 CrossRef PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2026 |